

Abstract
Key points
Heart size increases with age (called hypertrophy), and its ability to contract declines. However, these reflect average changes that may not be present, or present to the same extent, in all older individuals.
That aging happens at different rates is well accepted clinically. People who are aging rapidly are frail and frailty is measured with a ‘frailty index’.
We quantified frailty with a validated mouse frailty index tool and evaluated the impacts of age and frailty on cardiac hypertrophy and contractile dysfunction.
Hypertrophy increased with age, while contractions, calcium currents and calcium transients declined; these changes were graded by frailty scores.
Overall health status, quantified as frailty, may promote maladaptive changes associated with cardiac aging and facilitate the development of diseases such as heart failure.
To understand age‐related changes in heart structure and function, it is essential to know both chronological age and the health status of the animal.
Abstract
On average, cardiac hypertrophy and contractile dysfunction increase with age. Still, individuals age at different rates and their health status varies from fit to frail. We investigated the influence of frailty on age‐dependent ventricular remodelling. Frailty was quantified as deficit accumulation in adult (≈7 months) and aged (≈27 months) C57BL/6J mice by adapting a validated frailty index (FI) tool. Hypertrophy and contractile function were evaluated in Langendorff‐perfused hearts; cellular correlates/mechanisms were investigated in ventricular myocytes. FI scores increased with age. Mean cardiac hypertrophy increased with age, but values in the adult and aged groups overlapped. When plotted as a function of frailty, hypertrophy was graded by FI score (r = 0.67–0.55, P < 0.0003). Myocyte area also correlated positively with FI (r = 0.34, P = 0.03). Left ventricular developed pressure (LVDP) plus rates of pressure development (+dP/dt) and decay (−dP/dt) declined with age and this was graded by frailty (r = −0.51, P = 0.0007; r = −0.48, P = 0.002; r = −0.56, P = 0.0002 for LVDP, +dP/dt and −dP/dt). Smaller, slower contractions graded by FI score were also seen in ventricular myocytes. Contractile dysfunction in cardiomyocytes isolated from frail mice was attributable to parallel changes in underlying Ca2+ transients. These changes were not due to reduced sarcoplasmic reticulum stores, but were graded by smaller Ca2+ currents (r = −0.40, P = 0.008), lower gain (r = −0.37, P = 0.02) and reduced expression of Cav1.2 protein (r = −0.68, P = 0.003). These results show that cardiac hypertrophy and contractile dysfunction in naturally aging mice are graded by overall health and suggest that frailty, in addition to chronological age, can help explain heterogeneity in cardiac aging.
Keywords: ageing, excitation‐contraction coupling, frailty, ventricular myocyte
Key points
Heart size increases with age (called hypertrophy), and its ability to contract declines. However, these reflect average changes that may not be present, or present to the same extent, in all older individuals.
That aging happens at different rates is well accepted clinically. People who are aging rapidly are frail and frailty is measured with a ‘frailty index’.
We quantified frailty with a validated mouse frailty index tool and evaluated the impacts of age and frailty on cardiac hypertrophy and contractile dysfunction.
Hypertrophy increased with age, while contractions, calcium currents and calcium transients declined; these changes were graded by frailty scores.
Overall health status, quantified as frailty, may promote maladaptive changes associated with cardiac aging and facilitate the development of diseases such as heart failure.
To understand age‐related changes in heart structure and function, it is essential to know both chronological age and the health status of the animal.
Abbreviations
- BW
body weight
- FI
frailty index
- HW
heart weight
- LVDP
left ventricular developed pressure
- +dP/dt
rate of pressure development
- −dP/dt
rate of pressure decay
- SR
sarcoplasmic reticulum
- TL
tibia length
Introduction
Studies in both humans and animals have shown that age modifies ventricular structure and function, even in the absence of overt cardiovascular disease (Fares & Howlett 2010; Feridooni et al. 2015a; Keller & Howlett, 2016). Indeed, there is strong evidence that contractile function, especially during exercise, declines with age and older individuals exhibit less complete relaxation than younger adults (Fleg & Strait, 2012; Strait & Lakatta, 2012). The occurrence of left ventricular hypertrophy also increases with age in people (Chen & Frangogiannis, 2010; Fleg & Strait, 2012; Strait & Lakatta, 2012). These changes are characteristic of the aging heart, as they are seen in both humans and in animal models of aging (Feridooni et al. 2015a; Keller & Howlett, 2016). Pre‐clinical studies have shown that age‐dependent ventricular hypertrophy and contractile dysfunction are present not only at the organ level, but also in isolated ventricular myocytes (Feridooni et al. 2015a). These changes associated with cardiac aging are important, as they are typically maladaptive and predispose the heart towards the development of diseases such as heart failure (Keller & Howlett, 2016).
Despite growing evidence for age‐dependent modifications in the heart, these reflect average responses that may not be present, or present to the same degree, in all individuals of the same age (Howlett & Rockwood, 2013). Indeed, there is strong evidence for heterogeneity in a wide range of cardiovascular responses in healthy older individuals. For example, while left ventricular contractility, cardiac index and ejection fraction decline with age on average, some older individuals perform at the same levels as young adults (Lakatta & Levy, 2003). The idea that people age at different rates is well accepted clinically (Clegg et al. 2013). The term frailty, introduced into modern use in 1979, refers to the unmeasured heterogeneity in mortality risk of people of the same age (Vaupel et al. 1979). This concept has been expanded to capture unmeasured heterogeneity in the risk for and expression of a large number of age‐related adverse features in people and, more recently, in animals of the same chronological age (Rockwood et al. 2017). Individuals at greater risk, or with more aging features, are said to be frail (Clegg et al. 2013). Age‐related frailty increases the risk of developing various diseases, including cardiovascular disease, in older adults (Afilalo et al. 2014; Singh et al. 2014). Furthermore, when frail older adults do develop diseases such as heart failure, they experience worse outcomes and higher mortality rates than non‐frail patients (Boxer et al. 2014; Uchmanowicz et al. 2014, 2015; Goldwater & Pinney, 2015; Jermyn & Patel, 2015). Still, why frailty increases susceptibility to heart disease is not understood, in part because very little is known about the impact of frailty itself on the heart.
Studies in humans have shown that frailty can be quantified using different methods (de Vries et al. 2011; Bouillon et al. 2013). One commonly used approach is to construct a ‘frailty index’ (FI) by counting accumulation of health deficits, such as clinically apparent signs, symptoms and laboratory abnormalities (Mitnitski et al. 2001; Searle et al. 2008; Howlett et al. 2014; Rockwood, 2016). The number of deficits in an individual is divided by the total number measured to yield an FI score between 0 (no deficits) and 1 (all deficits). Based on the deficit accumulation approach validated in humans, we developed novel tools to quantify frailty with an FI in naturally aging mice (Parks et al. 2012; Whitehead et al. 2014; Feridooni et al. 2015b; Kane et al. 2016; Moghtadaei et al. 2016; Rockwood et al. 2017). We showed that the relationship between FI scores and normalized age are similar in mice and humans and that the rates of deficit accumulation are similar in aging mice and people (Whitehead et al. 2014). We also found that there is marked heterogeneity in individual mouse FI scores at any given age (Feridooni et al. 2015b), as seen in clinical studies (Mitnitski et al. 2001; Kim & Jazwinski, 2015). Thus, the mouse FI is a powerful new instrument that can be used to investigate the links between aging, frailty and disease in an animal model.
The overall objective of this study was to investigate the impact of frailty on age‐dependent remodelling in the mouse ventricle. We quantified frailty with our newly developed FI tool in adult and aged C57BL/6J mice and investigated age‐ and frailty‐dependent changes in ventricular structure and function in Langendorff‐perfused hearts. Cellular correlates and underlying mechanisms were explored in isolated ventricular myocytes. We demonstrate that age‐related changes in ventricular structure and function are graded by FI scores and suggest that frailty can help explain heterogeneity in cardiac aging.
Methods
Animals
All animal experiments followed guidelines of the Canadian Council on Animal Care Guide to the Care and Use of Experimental Animals (CCAC, Ottawa, ON: Vol. 1, 2nd edn. 1993; Vol. 2, 1984). All animal protocols were approved by the Dalhousie University Committee on Laboratory Animals. This work complies with the principles outlined by the Journal of Physiology and follows the ethics checklist provided by the journal. Male C57BL/6J mice were purchased at 3–4 weeks of age from Charles River Laboratories (St. Constant, QC, Canada) and were housed in groups (3–5 littermates per cage) in the Dalhousie University Carlton Animal Care Facility. Mice were maintained on a 12 h light/dark cycle and they had free access to water and food (ProLab RMH 3500, Purina LabDiet, Aberfoyle, Ontario, Canada). Two groups of mice were evaluated, an adult group (≈7 months; n = 24) and an aged group (≈27 months; n = 54).
Frailty assessment
Frailty was assessed once in each individual mouse on the day of the experiment with techniques that have been described previously (Whitehead et al. 2014; Feridooni et al. 2015b). Briefly, mice were weighed and their body surface temperature was measured with an infrared probe (Infrascan; La Crosse Technology, La Crosse, WI, USA). A clinical FI score for each mouse was calculated using a published checklist (Whitehead et al. 2014; Feridooni et al. 2015b). This involved the evaluation of 31 variables representative of various health domains including the integument, musculoskeletal system, vestibulocochlear and auditory systems, ocular and nasal systems, digestive system, urogenital system, respiratory system, signs of discomfort, as well as the body weight (g) and body surface temperature (°C) as shown in Table 1. Note that cardiac and vascular items were excluded; this ability to adapt the FI to set specific organ changes in the context of overall deficit accumulation is seen as a strength of this approach (Rockwood, 2016). FI scores were computed from these data as follows: for each parameter, a score of 0 was given for no deficit, 0.5 for a mild deficit and 1 for a severe deficit (Table 1). Body weight (g) and body surface temperature (°C) were scored differently, based on deviation from average reference values obtained from the appropriate, age‐matched cohort, as described previously (Whitehead et al. 2014). Variables that were within 1 SD of the reference values were coded as 0, >1 SD were scored 0.25, >2 SD = 0.5, >3 SD = 0.75 and >4 SD received a score of 1, the maximum score for the item (Table 1). Individual scores for each of the 31 parameters were added and divided by the number of deficits measured to produce an FI score between 0 and 1 for each animal.
Table 1.
Scoring of individual deficits in adult and aged mice
| Body system* | Frailty index item | Deficit score | ||||
|---|---|---|---|---|---|---|
| 0 | 0.5 | 1.0 | ||||
| Integument | Alopecia | |||||
| Loss of fur colour | ||||||
| Dermatitis | ||||||
| Loss of whiskers | ||||||
| Coat condition | ||||||
| Physical/musculoskeletal | Tumours | |||||
| Distended abdomen | ||||||
| Kyphosis | ||||||
| Tail stiffening | ||||||
| Gait disorders | ||||||
| Tremor | ||||||
| Forelimb grip strength | ||||||
| Body condition score | ||||||
| Vestibulocochlear/auditory | Vestibular disturbance | |||||
| Hearing loss | ||||||
| Ocular/nasal | Cataracts | |||||
| Eye discharge/swelling | ||||||
| Microphthalmia | ||||||
| Corneal opacity | ||||||
| Vision loss | ||||||
| Menace reflex | ||||||
| Nasal discharge | ||||||
| Digestive/urogenital | Malocclusions | |||||
| Rectal prolapse | ||||||
| Vaginal/uterine/penile prolapse | ||||||
| Diarrhoea | ||||||
| Respiratory | Breathing rate/depth | |||||
| Discomfort | Mouse grimace scale | |||||
| Piloerection | ||||||
| 0 | 0.25 | 0.50 | 0.75 | 1.0 | ||
| Other† | Temperature | |||||
| Weight | ||||||
| Frailty index score‡ | ||||||
*Frailty scoring system, adapted from Whitehead et al. (2014). All deficits except temperature and weight were scored as 0 if absent, 0.5 if present but mild and 1.0 if present and severe.
†Temperature and weight were scored based on deviation from mean reference values from age‐matched mice. Values that differed from the reference by >1 SD were scored as 0.25, >2 SD were 0.5, >3 SD were 0.75 and >4 SD received a score of 1.
‡Deficit scores were added and divided by 31 (total number of items evaluated) to produce a frailty index score for each mouse.
Previous studies by our group and others have validated the mouse clinical FI approach and shown that there is a high degree of inter‐rater reliability when different raters are used to complete the FI on the same mouse (Feridooni et al. 2015b; Kane et al. 2015, 2017). In the present study, two raters were used to complete FI assessments. To verify that FI scores obtained by these two raters were similar, we compared their ratings of the same mice in a subset of the animals used in this study (n = 31). We found excellent inter‐rater agreement, with an intra‐class correlation coefficient of 0.82. We also confirmed that FI scores did not vary significantly in an individual mouse over a short period of time. FI assessments were completed twice in a subset of animals (n = 8), 3 days apart. There was no significant difference between FI scores in the two sets of measurements (mean ± SEM values were 0.185 ± 0.015 and 0.181 ± 0.012 on day 0 and day 3, respectively).
Langendorff‐perfused heart studies
Mice were deeply anaesthetized with an intraperitoneal injection of sodium pentobarbital (200 mg kg−1); heparin (3000 U kg−1) was also injected to inhibit blood coagulation. The depth of anaesthesia was evaluated with a toe pinch test. If no pedal withdrawal reflex was observed following the toe pinch test, a thoracotomy was performed, and hearts were quickly excised and placed in a Petri dish containing ice‐cold Tyrode's buffer (described below). The aorta was cannulated with a 25 5/8‐gauge needle on a Radnoti Langendorff apparatus (Radnoti LLC, Monrovia, CA, USA). The heart was perfused at a constant pressure of 80 ± 0.5 mmHg with a buffer solution (37°C) containing (in mm): 126 NaCl, 0.9 NaH2PO4, 4 KCl, 20 NaHCO3, 0.5 MgSO4, 5.5 glucose and 1.8 CaCl2. The perfusate was aerated with 95% O2/5% CO2 to achieve a pH of 7.4. The perfusate was filtered with in‐line 1 μm glass fibre filters (Radnoti Glass Technology).
A fluid‐filled balloon was constructed from GLAD Cling Wrap (The Clorox Company of Canada Ltd, Brampton, ON, Canada), inserted into the left ventricle through an incision made in the left atrium and inflated to a minimum pressure of 5–10 mmHg. The balloon was attached to a pressure transducer (ADInstruments, Colorado Springs, CO, USA). A PowerLab 8/35 data acquisition system (ADInstruments) converted the changes in balloon pressure into a digital signal. LabChart 7 software (ADInstruments) was used to analyse data. Recordings of left ventricular pressure were used to quantify developed pressure (LVDP), and the maximum rates of pressure development and decay (+dP/dt and –dP/dt, respectively). Spontaneous beating rate data were analysed by measuring the time from peak‐to‐peak and calculating the rate for each heart. Hearts were allowed to spontaneously beat and stabilize for 20 min before recordings were made and then responses recorded during a 10 min period were averaged. Right tibia length and heart weight were measured at the end of each experiment.
Ventricular myocyte studies
Ventricular myocytes were isolated with our established techniques (Li et al. 1995; Ferrier et al. 2000; Parks et al. 2014). In brief, mice were anaesthetized as described above. The aorta was cannulated and hearts were perfused for 10 min (2 ml min−1) with Ca2+‐free solution (mm): 105 NaCl, 5 KCl, 25 Hepes, 0.33 NaH2PO4, 1 MgCl2, 20 glucose, 3 sodium pyruvate, 1 lactic acid (100% O2; pH 7.4; 37°C) followed by perfusion (8–10 min) with the same solution plus 50 μm Ca2+, collagenase (8 mg/30 ml, Worthington Type I, 250 U mg−1; Lakewood, NJ, USA), dispase II (3.5 mg; Roche, Indianapolis, IN, USA) and trypsin (0.5 mg). The ventricles were removed and minced in high K+ buffer (mm): 30 KCl, 50 l‐glutamic acid, 30 KH2PO4, 20 taurine, 10 Hepes, 10 glucose, 3 MgSO4 and 0.5 EGTA (pH 7.4). The supernatant was filtered (225 μm polyethylene filter; Spectra/Mesh) and stored at room temperature. Rod‐shaped, quiescent cells with distinct striations were used in experiments.
Contractions, Ca2+ transients and Ca2+ currents were recorded simultaneously. Cells were loaded with fura‐2 acetoxymethyl (AM) (2.5 μm; 20 min in the dark; Invitrogen, Burlington, ON, Canada). An aliquot of the cell suspension was placed on the stage of an inverted microscope (Nikon Eclipse, TE200, Nikon Canada, Mississauga, ON, Canada) and superfused (3 ml min−1) with buffer (mm): 145 NaCl, 10 glucose, 10 Hepes, 4 KCl, 1 CaCl2, 1 MgCl2 (pH 7.4; 37°C) supplemented with 4‐aminopyridine (4 mm) to block transient outward K+ currents and lidocaine (0.3 mm) to block Na+ currents. Cells were voltage clamped with high resistance microelectrodes (18–25 MΩ; 2.7 m KCl) and discontinuous single electrode voltage clamp (5–6 kHz) techniques (Axoclamp 2B amplifier; Molecular Devices, Sunnyvale, CA, USA). ClampEx v8.2 software (Molecular Devices) was used to generate voltage clamp protocols. Cells were stimulated with five, 50 ms conditioning pulses from −80 to 0 mV (pulse frequency = 2 Hz) and then repolarized to −40 mV for 450 ms. Ca2+ currents, contractions and Ca2+ transients were recorded simultaneously during test steps (250 ms) to potentials between −40 and +80 mV.
Contractions (unloaded cell shortening) were recorded with a charge‐coupled device camera (120 Hz; model TM‐640, Pulnix America, Sunnyvale, CA, USA) coupled to a video edge detector (Crescent Electronics, Sandy, UT, USA). Ca2+ concentrations were recorded with a PTI (Photon Technology International, Birmingham, NJ, USA) fluorescence system. Fura‐2 was alternately excited with 340 and 380 nm light; fluorescence emission was measured at 510 nm (5 ms sampling interval) with a DeltaRam fluorescence system and Felix v1.4 software (Photon Technologies International). Fluorescence measurements were corrected for background and converted to Ca2+ concentrations with established techniques (Parks et al. 2014). To allow recording of contractions and fluorescence simultaneously, a dichroic cube directed red light to the camera/video edge detector while remaining light was sent to the photomultiplier tube.
Sarcoplasmic reticulum (SR) Ca2+ content was measured in voltage clamped cells exposed to 10 mm caffeine, applied 450 ms after a series of five conditioning pulses (conditioning pulse duration = 50 ms) delivered at a frequency of 2 Hz. Caffeine was rapidly applied for 1 s in a solution containing (mm): 10 caffeine, 140 LiCl, 4 KCl, 10 glucose, 5 Hepes, 4 MgCl2, 4 4‐aminopyridine and 0.3 lidocaine. Caffeine solution was nominally Ca2+ and Na+ free to inhibit Ca2+ extrusion by the Na+–Ca2+ exchanger. The peak of the caffeine‐induced Ca2+ transient was used to quantify SR Ca2+ content.
Current and contractions were analysed with Clampfit 8.2 (Molecular Devices). Ca2+ currents were measured as the difference between the peak and the end of the test step and were normalized to cell capacitance. Cell capacitance was measured by integrating capacitive transients with Clampfit software and cell length, width and area were measured with a video edge detector. Contractions were measured as the difference between the peak contraction and resting cell length. Contractions were normalized to diastolic cell length. All fluorescence data were analysed with Felix (PTI) and Clampfit software. Peak Ca2+ transient amplitudes were quantified as the difference between diastolic and peak systolic Ca2+. The ratio of the rate of rise of the Ca2+ transient (nm s–1) per unit of normalized Ca2+ current (pA pF–1) was calculated to measure EC‐coupling gain. The caffeine‐induced Ca2+ transient was measured as the difference between systolic and diastolic Ca2+ levels. The fractional release of SR Ca2+ was calculated by dividing peak Ca2+ transient by the caffeine‐induced Ca2+ transient.
Western blot studies
Left ventricular tissue was homogenized with a Dounce homogenizer in lysis buffer [RIPA buffer supplemented with EDTA, protease inhibitor cocktail (Thermofisher #78430) and phosphatase inhibitor cocktail (Thermofisher #78420)]. The homogenate was then centrifuged at 8000 r.p.m. for 15 min at 4°C, and the supernatant was collected and used in Western blot experiments. Protein levels were measured with the Bio‐Rad RC DC protein kit (#500–0122). Thirty micrograms of protein was separated by SDS‐PAGE under reducing conditions and then transferred to 0.45 μm polyvinylidene fluoride (PVDF) membranes. Membranes were stained with Ponceau S (0.2% in 5% acetic acid), blocked in 5% skimmed milk for 1 h at room temperature, and then incubated overnight at 4°C with the Cav1.2 primary antibody (Alomone labs #ACC‐003, 1:2000 dilution; Jerusalem, Israel) in 5% skimmed milk. Membranes were then incubated in secondary antibody solution (5% skimmed milk, goat secondary antibody 1:10,000, Abcam, Cambridge, MA, USA) for 1 h at room temperature. Bands were detected by exposure to Clarity Western ECL substrate (Bio‐Rad, Hercules, CA, USA) and subsequent chemiluminescence detection with a Biorad Chemidoc XRS system. Densitometry was performed using ImageJ (v 1.41).
Ponceau S was used as the loading control, and each band was normalized to the Ponceau staining at 250 kDa for that lane. Results were then presented as arbitrary densitometric values relative to an internal control sample as 1, to allow comparisons across membranes. Ponceau S was used as the loading control as there is evidence to suggest that standard housekeeping proteins, such as GAPDH and β‐actin, are affected by age (Lowe et al. 2000; Li & Shen, 2013; Mennes et al. 2014). Ponceau S has been shown to reliably correlate with protein loading in a number of studies (Li & Shen, 2013), and in fact exhibits enhanced correlation at higher protein levels than β‐actin (Romero‐Calvo et al 2010). Normalization of Western blots to Ponceau S has been published in many aging research studies (e.g. Mennes et al. 2014; Mitchell et al. 2016).
Statistics
Sigmaplot software (v11.0, Systat Software Inc., Chicago, IL, USA) and SPSS software (v21.0) were used for all statistical analyses and Sigmaplot was used to create the figures. Data from adult and aged mice are presented as scatterplots as well as box and whisker plots. Differences between means were evaluated with either a Student's t test, for normally distributed data, or a Mann–Whitney rank sum test, when data were not normally distributed. We evaluated relationships between various parameters and frailty with linear regression analysis. When both age and frailty were significantly related to the parameter under study we used multivariable regression and calculated semi‐partial correlations to assess their separate contributions. Values of P < 0.05 was considered statistically significant.
Chemicals
All chemicals were purchased from Sigma‐Aldrich (Oakville, ON, Canada) or BDH Inc. (Toronto, ON, Canada). Fura‐2 AM was prepared in anhydrous DMSO and stored at −20°C until use. All other chemicals were prepared in deionized water.
Results
FI scores in adult and aging mice
Mice were assessed clinically to obtain individual FI scores prior to each experiment. Table 2 shows mean (± SEM) ages and FI scores for all the mice used in this study, separated into two age groups. Aged mice were older than adult mice (203 ± 21 vs. 811 ± 11 days of age; P < 0.001) and their FI scores were significantly higher when compared to the younger group (0.170 ± 0.012 for adult vs. 0.373 ± 0.012 for aged mice; n = 24 adult and 54 aged mice; P < 0.001). Figure 1 shows FI scores for all animals used in the study, separated by age. The scatterplots illustrate all individual FI scores, the box plots show the median plus 25th and 75th percentiles and the whiskers show the 10th and 90th percentiles. These data demonstrate that mice of similar ages have a wide range of FI scores and this heterogeneity is particularly pronounced in the older group. Figure 1 also shows that, although frailty scores on average increased with age, values for the younger and older groups overlapped despite substantial differences in their ages (Fig. 1). This indicates that mice exhibit differences in health status, which can be quantified according to FI score.
Table 2.
Physical characteristics of mice used in this study
| Parameter† | Adult | Aged | P value |
|---|---|---|---|
| Age (days) | 203 ± 21 | 811 ± 11* | <0.001 |
| Frailty index | 0.170 ± 0.012 | 0.373 ± 0.012* | <0.001 |
| Body weight (BW, g) | 39.12 ± 1.45 | 34.82 ± 0.83* | 0.012 |
| Heart weight (HW, mg) | 233.8 ± 7.4 | 286.0 ± 9.3* | 0.003 |
| Tibia length (TL, mm) | 18.95 ± 0.16 | 18.65 ± 0.11 | 0.173 |
| HW:BW (mg g–1) | 5.96 ± 0.2 | 8.33 ± 0.31* | <0.001 |
| HW:TL (mg mm–1) | 12.5 ± 0.4 | 15.3 ± 0.5* | 0.003 |
†Numbers represent the mean ± SEM. Values of n = 24 adult mice and 54 aged mice for ages and FI scores. For measures of cardiac hypertrophy, n = 11 adult mice (n = 10 for the HW and TL groups) and 30 aged mice. An asterisk denotes significantly different from the adult group. Differences between age groups were assessed using a Student's t test or a Mann–Whitney Rank Sum test for data that were non‐parametric.
Figure 1. Frailty increases with age.
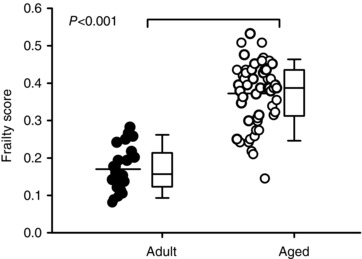
The distribution of frailty index (FI) scores in adult (203 ± 21 days; n = 24; filled symbols) and aged (811 ± 11 days; n = 54; open symbols) C57BL/6J mice are shown as a scatterplot (left) and as box plots. Mean FI scores, indicated by a horizontal line in the scatterplot, were higher in aged mice compared to adult mice (P < 0.001) although there was overlap in FI scores in the two groups. The box and whisker plot illustrates, from top to bottom: the 90th percentile, the 75th percentile, the median, the 25th percentile and the 10th percentile. Differences between age groups were assessed with a Mann–Whitney Rank Sum test.
We next determined whether the contributions of one or a few deficits dominated the FI score in either age group. Tables 3 and 4 illustrate the number of adult and aged mice, respectively, that exhibited each deficit in the index (left side of each table). The percentage of mice that exhibited each deficit are shown on right side of each table. Table 3 clearly demonstrates that, even in the adult group, mice express different specific deficits across a range of diverse body systems. The number of deficits, along with the percentage of mice expressing these deficits, clearly increases with age across a wide ranges of systems (Table 4). It is also clear from these tables that no one item dominates the FI in either age group.
Table 3.
Individual frailty index deficits for all adult mice
| Number of mice with deficit* | Per cent of mice with deficit | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Score | 0 | 0.25 | 0.5 | 0.75 | 1 | 0 | 0.25 | 0.5 | 0.75 | 1 |
| Temperature | 15 | 6 | 3 | 0 | 0 | 62.5 | 25 | 12.5 | 0 | 0 |
| Body weight | 13 | 4 | 4 | 0 | 3 | 54.2 | 16.7 | 16.7 | 0.0 | 12.5 |
| Score | 0 | 0.5 | 1 | 0 | 0.5 | 1 | ||||
| Alopecia | 17 | 6 | 1 | 70.8 | 25.0 | 4.2 | ||||
| Loss of fur colour | 9 | 12 | 3 | 37.5 | 50.0 | 12.5 | ||||
| Dermatitis | 19 | 4 | 1 | 79.2 | 16.7 | 4.2 | ||||
| Loss of whiskers | 20 | 1 | 3 | 83.3 | 4.2 | 12.5 | ||||
| Coat condition | 12 | 10 | 2 | 50.0 | 41.7 | 8.3 | ||||
| Tumours | 24 | 0 | 0 | 100 | 0.0 | 0.0 | ||||
| Distended abdomen | 15 | 3 | 6 | 62.5 | 12.5 | 25.0 | ||||
| Kyphosis | 21 | 3 | 0 | 87.5 | 12.5 | 0.0 | ||||
| Tail stiffening | 22 | 2 | 0 | 91.7 | 8.3 | 0.0 | ||||
| Gait disorders | 18 | 6 | 0 | 75.0 | 25.0 | 0.0 | ||||
| Tremor | 23 | 1 | 0 | 95.8 | 4.2 | 0.0 | ||||
| Forelimb grip strength | 7 | 13 | 4 | 29.2 | 54.2 | 16.7 | ||||
| Body condition score | 14 | 4 | 6 | 58.3 | 16.7 | 25.0 | ||||
| Vestibular disturbance | 23 | 1 | 0 | 95.8 | 4.2 | 0.0 | ||||
| Hearing loss | 12 | 11 | 1 | 50.0 | 45.8 | 4.2 | ||||
| Cataracts | 24 | 0 | 0 | 100 | 0.0 | 0.0 | ||||
| Corneal capacity | 24 | 0 | 0 | 100 | 0.0 | 0.0 | ||||
| Eye discharge/swelling | 22 | 2 | 0 | 91.7 | 8.3 | 0.0 | ||||
| Microphthalmia | 22 | 2 | 0 | 91.7 | 8.3 | 0.0 | ||||
| Vision loss | 3 | 21 | 0 | 12.5 | 87.5 | 0.0 | ||||
| Menace reflex | 0 | 13 | 11 | 0.0 | 54.2 | 45.8 | ||||
| Nasal discharge | 23 | 1 | 0 | 95.8 | 4.2 | 0.0 | ||||
| Malocclusions | 22 | 2 | 0 | 91.7 | 8.3 | 0.0 | ||||
| Rectal prolapse | 24 | 0 | 0 | 100 | 0.0 | 0.0 | ||||
| Penile/uterine prolapse | 23 | 1 | 0 | 95.8 | 4.2 | 0.0 | ||||
| Diarrhoea | 24 | 0 | 0 | 100 | 0.0 | 0.0 | ||||
| Breathing rate/depth | 24 | 0 | 0 | 100 | 0.0 | 0.0 | ||||
| Mouse grimace scale | 21 | 3 | 0 | 87.5 | 12.5 | 0.0 | ||||
| Piloerection | 4 | 11 | 9 | 16.7 | 45.8 | 37.5 | ||||
*Individual deficits used to create FI scores for 24 adult mice.
Table 4.
Individual frailty index deficits for all aged mice
| Number of mice with deficit* | Per cent of mice with deficit | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Score | 0 | 0.25 | 0.5 | 0.75 | 1 | 0 | 0.25 | 0.5 | 0.75 | 1 |
| Temperature | 32 | 19 | 0 | 0 | 0 | 62.7 | 37.3 | 0.0 | 0.0 | 0.0 |
| Body weight | 11 | 31 | 8 | 1 | 0 | 21.6 | 60.8 | 15.7 | 2.0 | 0.0 |
| Score | 0 | 0.5 | 1 | 0 | 0.5 | 1 | ||||
| Alopecia | 16 | 26 | 9 | 31.4 | 51.0 | 17.6 | ||||
| Loss of fur colour | 9 | 32 | 10 | 17.6 | 62.7 | 19.6 | ||||
| Dermatitis | 29 | 9 | 13 | 56.9 | 17.6 | 25.5 | ||||
| Loss of whiskers | 41 | 4 | 6 | 80.4 | 7.8 | 11.8 | ||||
| Coat condition | 8 | 27 | 16 | 15.7 | 52.9 | 31.4 | ||||
| Tumours | 25 | 24 | 2 | 49.0 | 47.1 | 3.9 | ||||
| Distended abdomen | 9 | 14 | 28 | 17.6 | 27.5 | 54.9 | ||||
| Kyphosis | 3 | 12 | 36 | 5.9 | 23.5 | 70.6 | ||||
| Tail stiffening | 27 | 20 | 4 | 52.9 | 39.2 | 7.8 | ||||
| Gait disorders | 3 | 27 | 21 | 5.9 | 52.9 | 41.2 | ||||
| Tremor | 16 | 21 | 14 | 31.4 | 41.2 | 27.5 | ||||
| Forelimb grip strength | 3 | 35 | 13 | 5.9 | 68.6 | 25.5 | ||||
| Body condition score | 1 | 17 | 33 | 2.0 | 33.3 | 64.7 | ||||
| Vestibular disturbance | 26 | 23 | 2 | 51.0 | 45.1 | 3.9 | ||||
| Hearing loss | 3 | 7 | 41 | 5.9 | 13.7 | 80.4 | ||||
| Cataracts | 50 | 1 | 0 | 98.0 | 2.0 | 0.0 | ||||
| Corneal capacity | 47 | 3 | 1 | 92.2 | 5.9 | 2.0 | ||||
| Eye discharge/swelling | 38 | 10 | 3 | 74.5 | 19.6 | 5.9 | ||||
| Microphthalmia | 45 | 6 | 0 | 88.2 | 11.8 | 0.0 | ||||
| Vision loss | 1 | 16 | 34 | 2.0 | 31.4 | 66.7 | ||||
| Menace reflex | 2 | 3 | 46 | 3.9 | 5.9 | 90.2 | ||||
| Nasal discharge | 47 | 4 | 0 | 92.2 | 7.8 | 0.0 | ||||
| Malocclusions | 47 | 4 | 0 | 92.2 | 7.8 | 0.0 | ||||
| Rectal prolapse | 50 | 1 | 0 | 98.0 | 2.0 | 0.0 | ||||
| Penile/uterine prolapse | 50 | 1 | 0 | 98.0 | 2.0 | 0.0 | ||||
| Diarrhoea | 51 | 0 | 0 | 100 | 0.0 | 0.0 | ||||
| Breathing rate/depth | 17 | 28 | 6 | 33.3 | 54.9 | 11.8 | ||||
| Mouse grimace scale | 36 | 15 | 0 | 70.6 | 29.4 | 0.0 | ||||
| Piloerection | 0 | 0 | 51 | 0.0 | 0.0 | 100 | ||||
*Individual deficits used to create FI scores for 51 aged mice.
Impact of age and frailty on cardiac hypertrophy
To evaluate the relationship between age, frailty and cardiac hypertrophy, body weight (BW), heart weight (HW) and tibia length (TL) were recorded in the perfused heart experiments. Mean values (± SEM) for these parameters, along with average HW:BW and HW:TL ratios, are shown in Table 2. Although HW and BW both increased with age, TL did not. Results showed that HW:BW and HW:TL ratios increased with age (Table 2), consistent with age‐dependent cardiac hypertrophy. Scatterplots plus box and whisker plots for these data are shown in Fig. 2. On average, HW, HW:BW ratios and HW:TL ratios increased with age, but there was marked heterogeneity in the older group and in many cases individual values from the adult and aged groups overlapped (Fig 2 A–C). To determine the relationship between hypertrophy and frailty, we plotted HW, HW:BW and HW:TL as a function of FI score for all mice from both age groups (Fig. 2 D–F). We found that HW (P = 0.0003), HW:BW (P < 0.0001) and HW:TL (P = 0.0003) showed strong positive correlations with FI score (Fig. 2 D–F). So too did age, where HW (P = 0.003), HW:BW (P < 0.001) and HW:TL (P = 0.003) all increased (Fig. 2 A–C). In the semi‐partial correlations, however, only the FI score, and not age, was significant in each case. These analyses demonstrate that cardiac hypertrophy increased linearly as a function of frailty and indicate that cardiac hypertrophy was graded by FI score.
Figure 2. Cardiac hypertrophy increases with age and is directly proportional to FI scores.
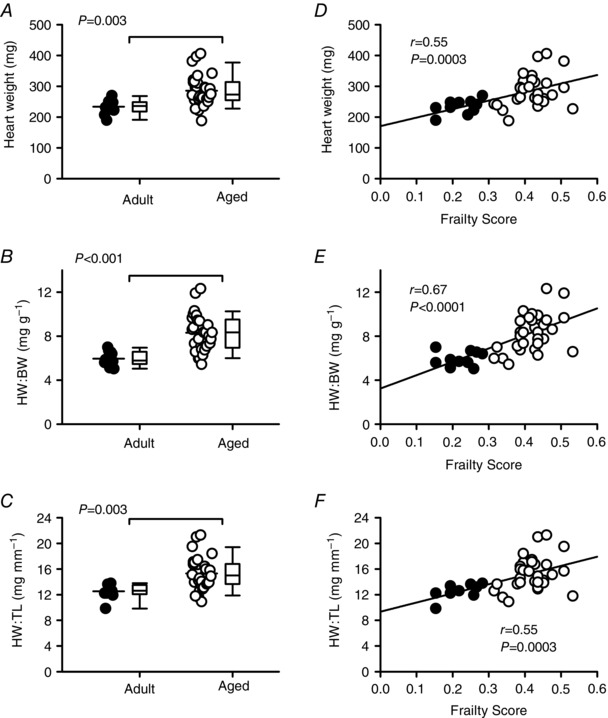
A–C, mean HW, HW:BW and HW:TL ratios were higher in aged mice compared to adult mice (P < 0.003 for all). D–F, scatterplots show that FI scores are positively correlated with HW, HW:BW and HW:TL ratios (n = 10 adult and 30 aged mice). Differences between age groups were assessed with a t test or Mann–Whitney Rank Sum test; correlations were evaluated with linear regression analysis. HW = heart weight; HW:BW = heart weight to body weight ratio; HW:TL = heart weight to tibia length ratio. Filled symbols indicate adult mice and open symbols indicate aged mice.
To determine whether age and frailty correlated with hypertrophy at the cellular level, we quantified the length, width and cross‐sectional area of isolated ventricular myocytes. Figure 3 A shows that age had no significant effect on cell length. Cell width and cross‐sectional area did increase slightly with age (Fig. 3 B and C), although this was not statistically significant. We then analysed these data by FI score (Fig. 3 D–F). Interestingly, while cell length (P = 0.19) was not related to FI score, cell width (P = 0.03) and cell area (P = 0.03) were positively correlated with frailty. By contrast, there was no effect of age and no correlation with frailty score for cell capacitance (mean ± SEM values for age were 261.5 ± 19.4 vs. 273.6 ± 12.0 pF for adult vs. aged; the correlation with FI score was r = 0.16; P = 0.31). Thus, although there is no link between myocyte hypertrophy and age, frailty analysis provides some evidence for a link between cellular hypertrophy and overall health as indicated by FI score.
Figure 3. Myocyte hypertrophy, characterized by larger cell widths and areas, increases as FI scores increase.
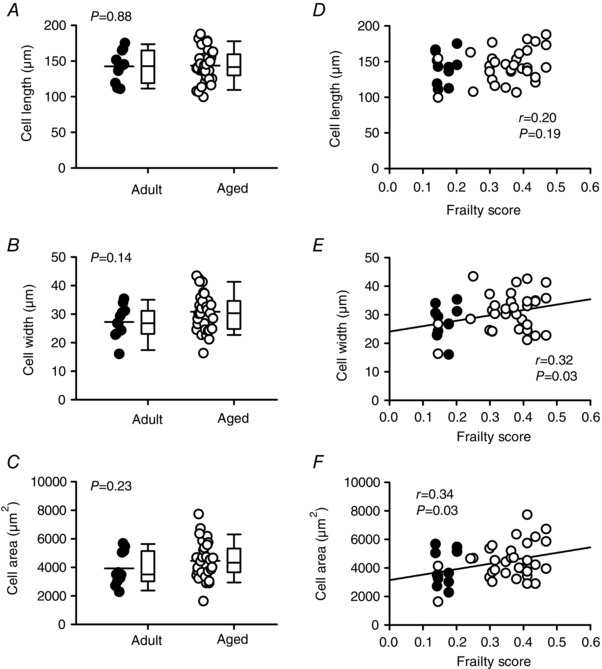
A–C, scatterplots and box and whisker plots indicate that cell length, width and area were not affected by age. D, cell length was not correlated with FI scores. E–F, scatterplots show that cell width and area were positively correlated with frailty (n = 11 adult and 32 aged myocytes). Differences between age groups were assessed with a t test; the relationship between FI and indices of hypertrophy were evaluated with linear regression. Filled symbols indicate adult mice and open symbols indicate aged mice.
Influence of age and frailty on contractile function in intact hearts
We next investigated the relationship between age, frailty and myocardial function in Langendorff‐perfused hearts. Figure 4 A and B shows pressure recordings from mice with three different FI scores; these recordings were selected to be representative of the mean data. Initially, we plotted LVDP, +dP/dt and −dP/dt as a function of age, as shown in Fig. 4 C–E. We found that LVDP (P < 0.001), +dP/dt (P < 0.001) and –dP/dt (P < 0.001) declined with age, although there was considerable heterogeneity in all three measures, especially in the older group. These data demonstrate that, on average, contractions were smaller and slower in aged hearts compared to younger hearts, but this was highly variable (Fig. 4 C–E). To determine whether these age‐dependent changes in contractile function were graded by frailty, we next plotted these parameters as a function of FI score. Figure 4 F shows that LVDP was highly correlated with FI score (P = 0.0007), such that LVDP declined as frailty increased. Similarly, both +dP/dt (P = 0.002) and –dP/dt (P = 0.0002) declined as frailty increased (Fig. 4 G and H). In the semi‐partial correlations, age was significant for LVDP and –dP/dT, while both FI score and age contributed to +dP/dT. These observations demonstrate that the rates of cardiac contraction and relaxation declined with age and FI score. By contrast, spontaneous beating rates were not affected by age (300 ± 14 vs. 331 ± 13 beats per minute in the young and old groups, respectively) and were not correlated with frailty (r = 0.17, P = 0.28).
Figure 4. Left ventricular contractile function declines with age and is graded by frailty.
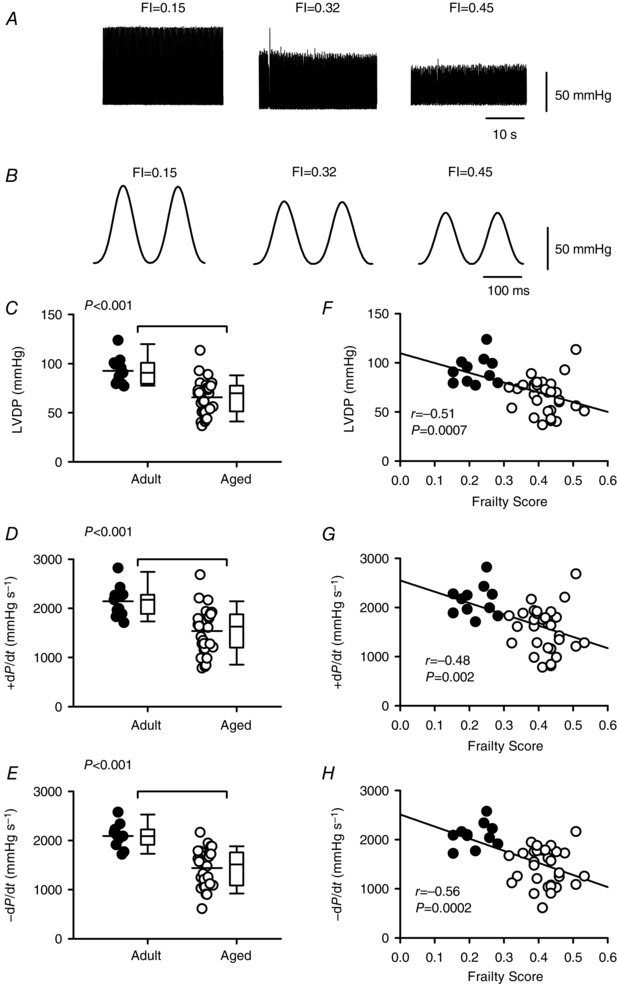
A, representative examples of pressure recorded from perfused hearts isolated from mice with different FI scores. B, pressure recordings illustrated in A shown at an expanded time scale to illustrate differences in +dP/dt and –dP/dt. C–E, scatterplots plus box and whisker plots demonstrate that LVDP, +dP/dt and –dP/dt decreased with age. F–H, regression lines show that LVDP, +dP/dt and –dP/dt were graded by FI score and declined as frailty increased (n = 11 adult and 30 aged mice). Differences between age groups were assessed with either a t test or a Mann–Whitney Rank Sum test; correlations were performed with linear regression analysis. Filled symbols indicate adult mice and open symbols indicate aged mice.
Impact of age and frailty on contractions, Ca2+ transients and SR Ca2+ content
The next set of experiments investigated whether contractile dysfunction observed in the aging intact hearts was also present in isolated ventricular myocytes. Figure 5 A shows representative examples of contractions recorded in voltage‐clamped myocytes isolated from mice with varying FI scores. The mean data clearly show that peak contraction declined with age (Fig. 5 B; P < 0.0001), consistent with our observations in the intact heart. Further analyses demonstrated that the average velocity of contraction (P = 0.007) and relaxation (P = 0.001) slowed with age (Fig. 5 C and D), in agreement with the perfused heart experiments (Fig. 4). When frailty analyses were performed, we found that peak contraction as well as the speed of contraction and relaxation were inversely related to FI score (Fig. 5 E–G). In the semi‐partial correlations, age was significant for peak contraction as seen for LVDP, while both FI score and age contributed to changes in the velocity of contraction and relaxation. Thus, older mice with high levels of frailty exhibited smaller, slower contractions indicative of contractile dysfunction at the cellular level.
Figure 5. The age‐dependent decline and slowing of contraction is graded by frailty in voltage‐clamped ventricular myocytes.
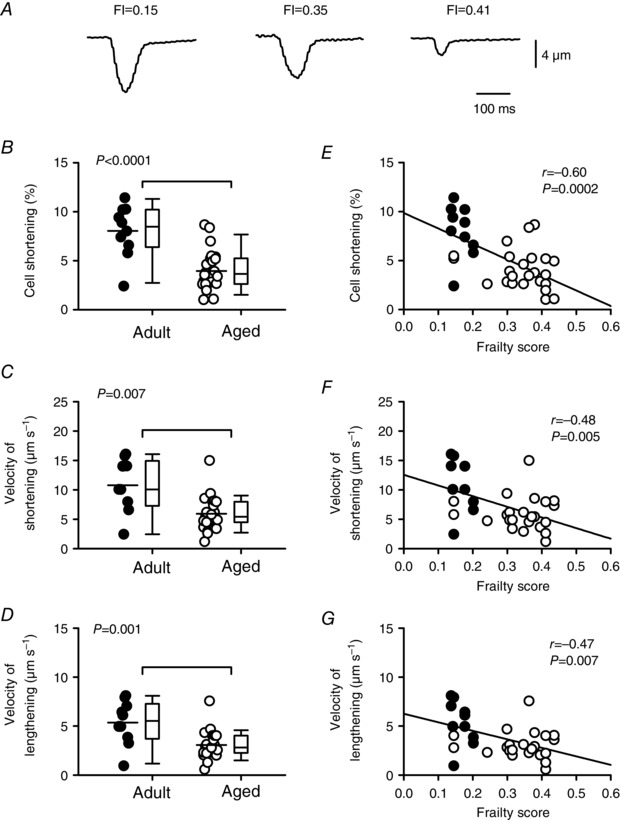
A, representative recordings of myocyte contractions recorded from mice with different FI scores. B, peak contractions normalized to cell size were smaller in aged animals compared to adults. C and D, velocities of shortening and lengthening were slower in the aged group compared to the adult group. E–G, scatterplots show that peak contractions, as well as the velocities of shortening and lengthening, were inversely proportional to FI score. Differences between age groups were assessed with a t test and correlations were assessed by linear regression (n = 10 adult and 24 aged myocytes). Filled symbols indicate adult mice and open symbols indicate aged mice.
Cardiac contractions are initiated by Ca2+ current, which triggers the release of Ca2+ from the SR and results in a Ca2+ transient (Bers, 2014). To determine whether contractile dysfunction in aged and frail hearts was associated with changes in the underlying Ca2+ transients, we measured the amplitudes and time courses of Ca2+ transients recorded simultaneously with the contractions in voltage‐clamped myocytes. Figure 6 A shows representative Ca2+ transients in adult and aged myocytes. Mean data indicate that peak Ca2+ transients (P = 0.02) and the rate of rise of the Ca2+ transient (P = 0.001) both declined with age, although the decay rate (tau) was not affected by age (Fig. 6 B–D). Frailty analysis revealed that FI score was a powerful predictor of the decline in peak transients (Fig. 6 E) as well as the decline in their rate of rise (Fig. 6 F). By contrast, there was no correlation between frailty and decay rate (Fig. 6 G). When semi‐partial correlations were investigated to evaluate the contributions of age and FI score, FI score alone contributed to the decline in peak Ca2+ transients while age alone contributed to changes in the rate of rise. These data indicate that smaller and slower contractions in aged and frail hearts are attributable, in part, to changes in the magnitude and speed of Ca2+ transients.
Figure 6. The peaks and rates of rise of the Ca2+ transients are attenuated by age and graded by FI score in voltage‐clamped ventricular myocytes.
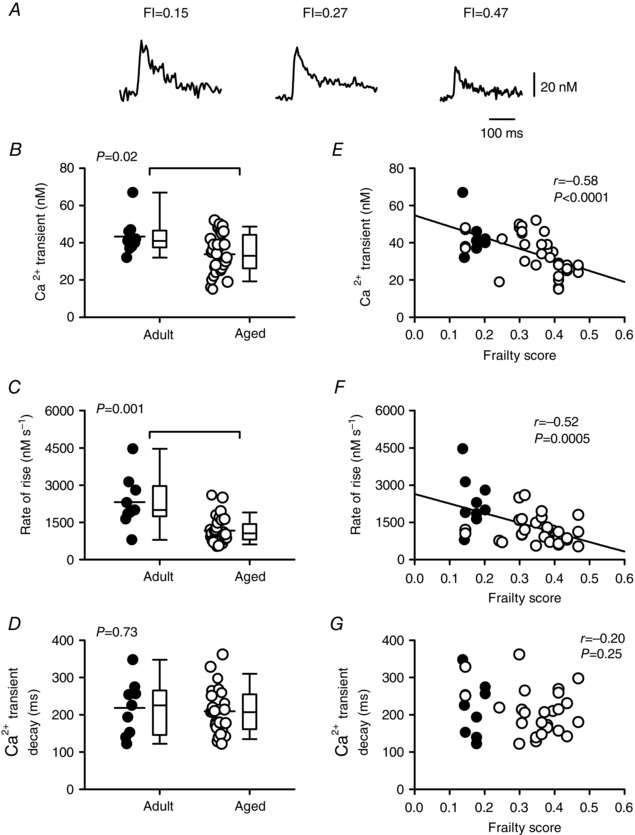
A, representative recordings of Ca2+ transients from myocytes isolated from mice with different FI scores. B, scatterplots plus box and whisker plots show that peak Ca2+ transients declined with age. C and D, the Ca2+ transient rates of rise declined with age, but decay rates were not affected. E–G, Ca2+ transient amplitudes and rates of rise were graded by FI, but decay rates were not. Differences between age groups were assessed using a Mann–Whitney Rank Sum test and correlations were evaluated with linear regression (n = 9 adult and 32 aged myocytes). Filled symbols indicate adult mice and open symbols indicate aged mice.
To determine whether smaller Ca2+ transients in frail hearts are the result of changes in the amount of SR Ca2+ available for release, we measured caffeine‐induced Ca2+ transients in voltage‐clamped myocytes from mice with varying ages and FI scores (Fig. 7). Representative caffeine‐induced Ca2+ transients from mice with different FI scores are shown in Fig. 7 A. The data show that there was no difference in the magnitude of caffeine‐induced Ca2+ transients as a function of age or FI score (Fig. 7 B and D). We measured the rate of rise of the caffeine‐induced Ca2+ transients and found that there was no effect of age and no correlation with frailty score for this parameter (mean ± SEM values were 103.3 ± 16.9 vs. 106.5 ± 19.9 nm s−1; r = 0.01; P = 0.97). Similarly, fractional release (the amount of SR Ca2+ released as a fraction of total available SR Ca2+) was not affected by either age or frailty (Fig. 7 C and E). Together, these observations demonstrate that the smaller Ca2+ transients characteristic of aged and frail hearts are due to a smaller trigger Ca2+ current, along with less SR Ca2+ release per unit Ca2+ current, rather than limited SR Ca2+ stores.
Figure 7. Frailty and age have no effect on either fractional SR Ca2+ release or SR Ca2+ content.
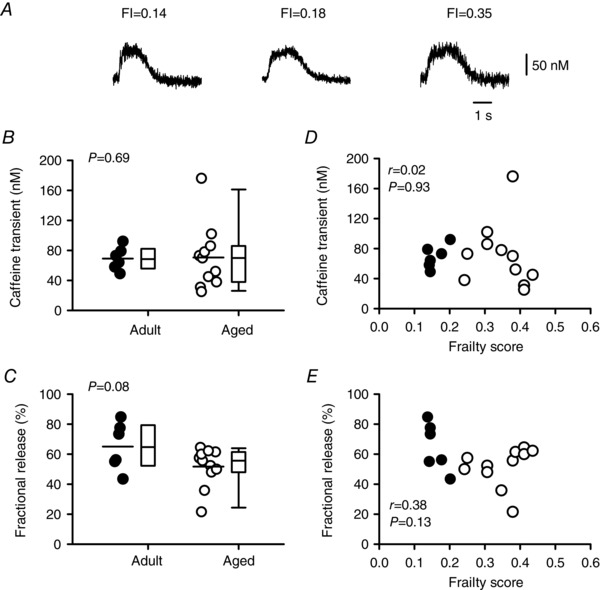
A, representative examples of caffeine‐induced Ca2+ transients recorded from myocytes isolated from mice with different FI scores. B and D, SR Ca2+ content was similar in myocytes from the adult and aged groups and showed no obvious relationship with FI score. C and E, fractional SR Ca2+ release was not affected by either age or frailty. Differences between age groups were assessed using a t test and Mann–Whitney Rank Sum test and linear regression analysis was used to evaluate the impact of FI score (n = 6 adult and 11 aged myocytes). Filled symbols indicate adult mice and open symbols indicate aged mice.
The effect of age and frailty on L‐type Ca2+ current density and Cav1.2 expression
As SR Ca2+ release is triggered by Ca2+ influx via L‐type Ca2+ channels, we investigated whether a decrease in Ca2+ current contributed to the smaller Ca2+ transients seen in frail cardiomyocytes. We recorded Ca2+ currents at the same time as we recorded Ca2+ transients and contractions. Figure 8 A shows representative examples of Ca2+ current recordings from mice with varying FI scores. Mean data showed that peak Ca2+ currents were smaller in cells from aged mice when compared to cells from younger animals (Fig. 8 B; P = 0.008). The integral of the Ca2+ current (Ca2+ flux) also declined with age (Fig. 8 C; P = 0.03). We quantified the impact of age on the gain of SR Ca2+ release, expressed as the amount of Ca2+ released from the SR per unit of Ca2+ current. Results showed that gain decreased with age (Fig. 8 D; P = 0.04). Next, we investigated whether these average age‐dependent changes in Ca2+ current were graded by FI score. Results showed that the declines in peak Ca2+ current and Ca2+ flux were graded by FI score (Fig. 8 E and F). Frailty analysis also showed that the decrease in gain of SR Ca2+ release was graded by the overall health of the animal, measured as FI score (Fig. 8 G). Assessment of the semi‐partial correlations revealed that both FI score and age contributed to these changes in Ca2+ current, Ca2+ flux and gain.
Figure 8. Frailty grades the age‐dependent reduction in peak Ca2+ current, Ca2+ flux and the gain of SR Ca2+ release in voltage‐clamped myocytes.
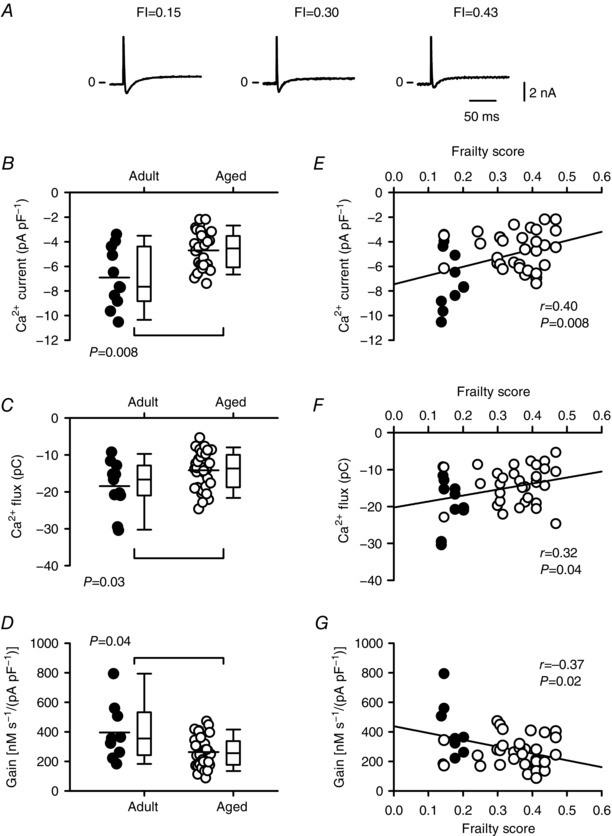
A, representative Ca2+ currents recorded from cells from mice with different FI scores. B and C, peak Ca2+ current and the integral of the Ca2+ current (Ca2+ flux) declined with age. D, the gain of SR Ca2+ release decreased with age. E and F, the age‐related declines in peak Ca2+ current and Ca2+ flux were graded by FI score. G, the gain of SR Ca2+ release was negatively correlated with frailty score. Differences between age groups were assessed using a Mann–Whitney Rank Sum test and correlations were evaluated with linear regression analysis (n = 11 adult and 32 aged myocytes). Filled symbols indicate adult mice and open symbols indicate aged mice.
The voltage‐clamp experiments presented in Figs 6 and 8 investigated contractions, Ca2+ transients, Ca2+ currents and gain in cardiomyocytes paced with pre‐pulses delivered at a frequency of 2 Hz. As previous studies have shown that age‐dependent changes in cardiomyocyte contraction are frequency‐dependent (Janczweski & Lakatta, 2010; Feridooni et al. 2015a), we investigated whether contractions and underlying mechanisms were graded by frailty when cells were paced at 4 Hz. These data are presented in Fig. 9. In cells paced at 4 Hz, peak contractions (P = 0.005) and Ca2+ currents (P = 0.03) declined with age whereas Ca2+ transients and gain did not (data not shown). By contrast, peak contractions (Fig. 9 A), Ca2+ transients (Fig. 9 B), Ca2+ currents (Fig. 9 C) and the gain of SR Ca2+ release (Fig. 9 D) were all graded by frailty when the pacing frequency was increased from 2 to 4 Hz. In the semi‐partial correlations, FI score alone explained the change in peak Ca2+ transients whereas both age and FI score contributed to the changes in contractions, Ca2+ currents and gain. These data demonstrate that similar relationships between key Ca2+ handling mechanisms, age and frailty scores are seen, regardless of whether cells were paced at 2 or 4 Hz.
Figure 9. Frailty scores grade the age‐related decline in peak contractions, Ca2+ transients, Ca2+ currents and gain when the pacing rate is increased to 4 Hz in voltage‐clamped myocytes.
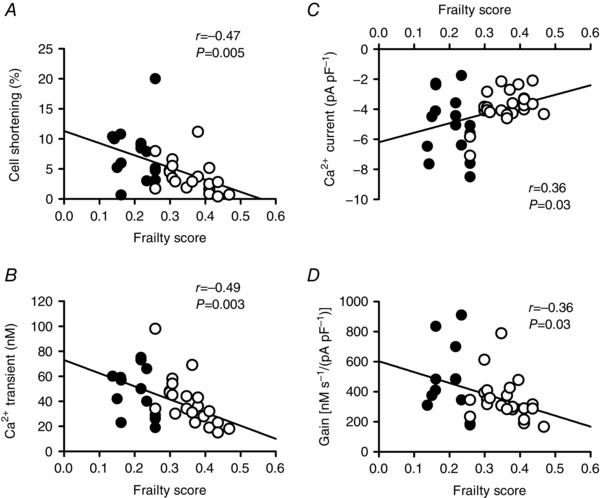
A–D, peak contractions, Ca2+ transients, Ca2+ currents and the gain of SR Ca2+ release were graded by FI score when the pacing rate was increased from 2 to 4 Hz. Contractions (P = 0.005) and Ca2+ currents (P = 0.03) also declined with age at 4 Hz, whereas Ca2+ transients and gain did not (data not shown). Correlations were evaluated with linear regression analysis (n = 14–15 adult and 19–23 aged myocytes). Filled symbols indicate adult mice and open symbols indicate aged mice.
We next investigated whether a reduction in the expression of L‐type Ca2+ channels contributed to the smaller Ca2+ currents characteristic of cardiomyocytes from frail mice. To investigate this, we quantified the expression of Cav1.2 protein (the major pore‐forming α subunit of the L‐channel) with Western blot analysis. Figure 10 A shows representative Western blots for Cav1.2 expression from mice with a wide range of FI scores. Figure 10 B shows that mean values for Cav1.2 protein expression were significantly lower in cells from aged mice when compared to cells from adult animals (P = 0.03). Interestingly, expression of Cav1.2 was clearly negatively correlated with, and graded by, frailty (Fig. 10 C). In the semi‐partial correlations, both age and FI contributed. These observations demonstrate that age‐dependent changes in the expression of Cav1.2 protein at the cellular level are graded by the overall health of the animal, as quantified with an FI score.
Figure 10. Frailty grades the age‐dependent decline in expression of Cav1.2 protein in the mouse heart.
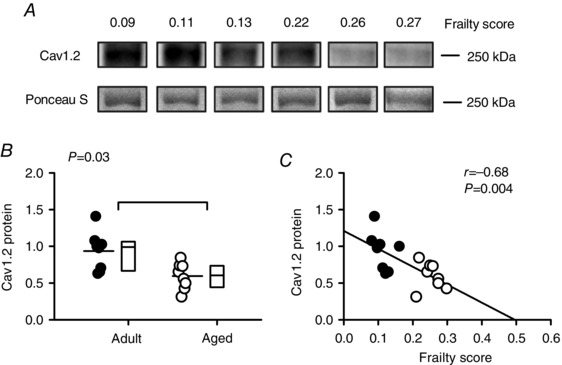
A, representative Western blots for Cav1.2 protein expression in mice with varying FIs. Ponceau S staining was used as a loading control in all experiments (lower panels). B, mean expression of Cav1.2 decreased with age in the mouse heart. C, cardiac Cav1.2 expression was closely graded by frailty score in the mouse. Differences between age groups were assessed using a Mann–Whitney Rank Sum test and correlations were evaluated with linear regression analysis (n = 8 adult and 8 aged hearts). Filled symbols indicate adult mice and open symbols indicate aged mice.
Discussion
This study investigated the impact of age and frailty on ventricular structure and function using a novel FI tool to measure frailty in adult and aged mice. Results showed that while on average FI scores increased with age, there was considerable heterogeneity. Cardiac hypertrophy also increased with age, although the degree of hypertrophy varied, especially in the older mice. When measures of hypertrophy were plotted as a function of FI score, it was clear that hypertrophy was graded by frailty at both the macroscopic and the microscopic levels. Functional changes associated with frailty also occurred at the level of the heart and the individual ventricular myocyte. LVDP, +dP/dt and –dP/dt declined with age and were negatively correlated with FI score. Myocytes from aged mice exhibited smaller, slower contractions that were graded by frailty and this contractile dysfunction was largely attributable to changes in underlying Ca2+ transients. The age‐ and frailty‐dependent reduction in SR Ca2+ release was due to a reduction in peak Ca2+ current, reduced Ca2+ flux and a decrease in the gain of SR Ca2+ release. There was also a clear decline in the expression of Cav1.2 protein in the heart with age and this was graded by FI score. Our results demonstrate that there is considerable heterogeneity in the impact of age on cardiac structure and function in animals of the same chronological age. Although in many cases both frailty and chronological age contribute significantly to the variance, in some cases only one or the other is significant. These observations suggest that differences in overall health status, quantified as age‐related deficit accumulation, contribute importantly to the impact of age on the heart.
Our group has developed several different tools to quantify frailty with an FI based on deficit accumulation (Parks et al. 2012; Whitehead et al. 2014; Feridooni et al. 2015b: Rockwood et al. 2017). In our initial study, we developed an FI that included parameters linked to cardiovascular health, such as heart rate, blood pressure, blood volume, as well as overall activity levels, body composition and metabolism (Parks et al. 2012). More recently we created a simplified FI composed of clinically evident deficits in health across a wide variety of systems that are not direct measures of cardiac function per se. Deficits were evaluated in systems including: the integument, musculoskeletal system, vestibulocochlear/auditory systems, ocular/nasal systems, digestive system, urogenital system, respiratory system and signs of discomfort (Whitehead et al. 2014; Feridooni et al. 2015b; Kane et al. 2016; Rockwood et al. 2017). In the present study, we used our newly developed mouse clinical FI tool to quantify frailty as a measure of overall health (Howlett & Rockwood, 2014) and we compared FI scores in mice of two very different ages. We investigated an adult group, with an average age of ≈7 months and an aged group, with a mean age of ≈27 months. We found that, on average, FI scores increased with age as reported previously (Parks et al. 2012; Whitehead et al. 2014; Feridooni et al. 2015b; Rockwood et al. 2017). We also showed that there was considerable heterogeneity in FI scores from mice of the same chronological age. Some adult mice had high scores and some aged mice had low scores, so the distributions for these two groups overlapped despite their marked difference in age. Together, these observations indicate that there is a high degree of variability in the overall health status of naturally aging C57BL/6J mice and that we can identify these differences using our FI approach.
The present study clearly showed that mice expressed a range of different deficits across a wide variety of bodily systems. We also found that no single deficit (e.g. body weight), or group of deficits, dominated the FI in either age group. This is potentially important, as an earlier longitudinal study of cardiac hypertrophy in aging rats reported that a drop in body weight beyond a given threshold predicted mortality (Yin et al. 1982). The results of the present cross‐sectional study in aging mice show that the FI score is not heavily influenced by changes in body weight. We have also recently shown, in a longitudinal study of frailty in aging mice, that individual deficits accumulate with age at varying rates in both mice and in humans (Rockwood et al. 2017). This study also demonstrates that high FI scores predict mortality in both mice and in humans (Rockwood et al. 2017). Together with the results of the present study, these observations strongly suggest that frailty reflects the accumulation of diverse deficits in multiple body systems.
Here, we found evidence for age‐dependent cardiac hypertrophy, consistent with previous reports in different pre‐clinical models of aging (Feridooni et al. 2015a; Keller & Howlett, 2016) and in older people (Chen & Frangogiannis, 2010; Fleg & Strait, 2012; Strait & Lakatta, 2012). This was evident when HW was normalized to BW, and even when HW was normalized to TL, which did not change with age. Still, there was substantial variability in the degree of hypertrophy at both the organ and the cellular levels, especially in older mice. In an earlier cross‐sectional study we used a different FI tool to show that ventricular myocyte hypertrophy varied with FI score in 30‐month‐old mice (Parks et al. 2012). Our earlier study, however, examined a very small group of mice at one age only and used an FI composed of a number of parameters linked to cardiovascular health (Parks et al. 2012). Here, we used our new FI (Whitehead et al. 2014; Feridooni et al. 2015b; Rockwood et al. 2017) composed of clinically evident deficits in health across a wide range of body systems. We report the novel observation that cardiac hypertrophy is clearly graded by frailty at both the organ and the cellular levels. These data strongly suggest that the overall health of the animal, as measured with a FI composed of items that are not closely linked to cardiovascular health, is a major determinant of cardiac hypertrophy in aging.
Here, we found that age did not significantly affect ventricular myocyte dimensions. This contrasts with results of a number of studies in aging rats and mice (e.g. Fraticelli et al. 1989; Feridooni et al. 2015a). On the other hand, other investigators have reported that there is no change in cell length or cell width with age in these models (Guo & Ren, 2006; Ren et al. 2007; Mellor et al. 2014). In the present study, we report the novel observation that cell width and cross‐sectional area are positively correlated with, and graded by, FI score. It is possible that studies where ventricular myocyte dimensions increase with age evaluated larger numbers of old and frail animals compared to studies that report no difference. In contrast to our findings with cell size, we did not see a link between FI score and cell capacitance. Similar results have been reported in ventricular myocytes from older animals, although some studies report that cell size and capacitance increase in parallel with age (Feridooni et al. 2015a). Further exploration of the links between cardiac hypertrophy, age and frailty at the organ and cellular levels are now warranted.
We found that cardiac contractions are smaller and slower in hearts from aged mice when compared to younger adults, consistent with reports in pre‐clinical models (Feridooni et al. 2015a; Keller & Howlett, 2016) and in humans (Fleg & Strait, 2012; Strait & Lakatta, 2012). We previously found an inverse relationship between FI score and peak ventricular myocyte contraction in a small cross‐sectional study with an FI that included items that linked to cardiovascular health (Parks et al. 2012). We have also recently reported that age‐dependent sino‐atrial node dysfunction is graded by frailty in the mouse model (Moghtadaei et al. 2016). We have now extended these observations to examine the relationship between overall health and contraction, in both myocytes and intact hearts, in a large cohort of mice of varying ages and FI scores with an FI based on deficits not closely linked to the cardiovascular system. When our data were stratified by frailty, we found that age‐related changes in the speed and amplitude of contraction were graded by FI score at both the macroscopic and the microscopic levels. These observations indicate that overall health, as evaluated with a FI based on the accumulation of deficits in a range of body systems, is a strong predictor of contractile dysfunction in the aging heart.
It is well established that cardiac contraction is activated by a transient rise in intracellular Ca2+ (Bers, 2014). To determine underlying cellular mechanisms involved in contractile dysfunction in frailty, we investigated links between age, frailty and Ca2+ handling in the heart. We found that both the peak and the rate of rise of the Ca2+ transient were negatively correlated with FI score. These observations strongly suggest that the slower rates of shortening and smaller contractions seen in the frail heart are attributable to changes in the underlying Ca2+ transients. Despite the significant negative correlation between peak Ca2+ transient and frailty score, we found that there was no clear relationship between frailty and Ca2+ transient decay rates. These observations suggest that the prolonged relaxation of contraction seen in hearts from frail animals is not due to prolonged SR Ca2+ release. It is possible that age‐associated changes in myofilaments, such as a shift from the fast α myosin heavy chain (MHC) isoform to the slower β isoform, or reduced phosphorylation of troponin I (Feridooni et al. 2015a) could contribute to slowing of relaxation in older frail hearts. Modifications in the sarcomeric protein titin in the aging heart may also increase myocyte stiffness and slow relaxation (Hamdani et al. 2013) and this would be interesting to explore experimentally.
The smaller Ca2+ transients observed in our study were not due to a reduction in SR Ca2+ stores, as neither age nor FI score affected SR content. However, peak Ca2+ currents recorded simultaneously with the Ca2+ transients were clearly graded by frailty, as both declined in parallel as FI scores increased. Ca2+ flux and the gain of SR Ca2+ release were negatively correlated with FI score. These age‐associated changes in peak contractions, Ca2+ transients, Ca2+ currents and the gain of SR Ca2+ release were graded by frailty, regardless of whether the cells were paced at 2 or 4 Hz. This is important, as previous studies have reported that age‐dependent contractile dysfunction is exacerbated by faster pacing frequencies (Janczweski & Lakatta, 2010; Feridooni et al. 2015a). These findings indicate that both Ca2+ influx and SR Ca2+ release are attenuated in ventricular myocytes from frail mice, which can explain the smaller contractions seen in animals with high FI scores.
Interestingly, although most previous studies report that Ca2+ current declines with age, some studies have found that it does not change and some report an age‐dependent increase (Janczweski & Lakatta, 2010; Feridooni et al. 2015a). Reasons for the differences in results between studies are not clear, but the use of different aging models (e.g. mice, rats, sheep), diverse age ranges and dissimilar experimental conditions (e.g. temperature, pacing rates) have been implicated (Janczweski & Lakatta, 2010; Feridooni et al. 2015a). The present study clearly showed that Ca2+ currents, measured as either peak current or Ca2+ flux, were smaller in ventricular myocytes from aged (≈27 months) mice when compared to younger adults (≈7 months), as we have reported previously in the mouse model (Grandy & Howlett, 2006). We also report the novel observation that Cav1.2 protein expression is lower in hearts from aged mice compared to adult animals. These findings strongly suggest that the mechanism responsible for the age‐dependent decline in Ca2+ current is a decrease in the expression of L‐type Ca2+ channels. Our study is also the first to demonstrate that the expression of Cav1.2 protein is graded by frailty score. These observations are important, as they demonstrate that age and frailty‐dependent changes at the protein level scale up to affect function at both the cellular and the organ levels.
Here we found that the spontaneous beating rate in Langendorff‐perfused hearts was not affected by either age or frailty. Our finding that the beating rate was not affected by age agrees with results of most prior studies in the Langendorff‐perfused mouse heart (Stein et al. 2008; Guzadhur et al. 2012; Porter et al. 2014), although one study reported an age‐dependent decline in heart rate (Headrick et al. 2003). Our group recently conducted a study designed to specifically investigate the impacts of age and frailty on heart rate and sinoatrial node function in mice (Moghtadaei et al. 2016). We used a range of techniques, including intra‐cardiac electrophysiology and optical mapping, to show that frailty predicted a decline in heart rate and sinoatrial node dysfunction the aging mouse atrium (Moghtadaei et al. 2016). The reasons we did not observe lower rates in the present study are unclear, although the present study was not designed to directly evaluate heart rate and sinoatrial node function in a detailed manner. It is possible that differences in experimental conditions (e.g. differences in experimental preparations, ionic composition of buffer solutions, different anaesthetics) between the two studies might be important. Additional work in this area would be of interest.
There are some limitations to this study. The peak Ca2+ transients reported in our study were relatively small, although are similar to those reported in our previous studies under similar experimental conditions, including physiological temperature (37°C), 2–4 Hz pacing rates and 1 mm external Ca2+ (e.g. Howlett, 2010; Parks et al. 2014). Previous studies have shown that external Ca2+ concentrations, pacing frequencies and temperature have a major impact on intracellular Ca2+ (Shattock & Bers, 1987; Shutt & Howlett, 2008; Gattoni et al. 2016). It would be interesting to explore the impact of frailty and age on Ca2+ handling in ventricular myocytes with a wider range of experimental conditions. The range of FI scores for the Cav1.2 expression experiments only included mice with FI scores up to 0.30. Although we found that both age and FI were linked to lower Cav1.2 expression, it would be interesting to explore the impact of age and frailty on mice with a wider range of FI scores. We investigated fewer adult mice than aged mice, so the groups were asymmetrical. Although theoretically we might have seen more adult mice with higher FI scores if we increased the sample size, we did not see this in our recent large study of frailty in 251 mice followed to extinction (Rockwood et al. 2017). The FI approach allows the investigation of a homogeneous population of animals under conditions that minimize environmental and potential epigenetic differences, which could facilitate the identification of genetic differences within the commonly used C57BL6/J strain of mice. This line of inquiry was not pursued in the present study, but could be in future studies designed to investigate genetic links between frailty and cardiovascular function.
Our observation that cardiac hypertrophy and contractile dysfunction in aging are graded by FI score has important implications. Previous studies in humans have shown that frailty increases the risk of cardiovascular diseases including heart failure in older adults (Afilalo et al. 2014; Singh et al. 2014; Uchmanowicz et al. 2014; 2015; Boxer et al. 2014; Goldwater & Pinney, 2015; Jermyn & Patel, 2015). The reasons for this are unclear. Indeed, little is known about the impact of frailty on the heart, in part because until recently there has been no model of frailty in naturally aging animals. We have clearly shown that myocardial hypertrophy and contractile dysfunction are closely associated with frailty in an animal model. These data indicate that maladaptive changes associated with cardiac aging are prominent in animals with high frailty scores and suggest that frailty may increase susceptibility to diseases such as heart failure in older adults.
We have demonstrated that adverse remodelling in the aging heart is graded by FI score, which suggests a close link between cardiac aging and overall health. We found evidence for a clear connection between frailty and adverse cardiac remodelling at the level of the intact heart, in individual ventricular myocytes and at the subcellular level, as indicated by the negative correlation between FI score and peak Ca2+ currents. These observations strongly support the emerging view that aging and ultimately frailty arises as a consequence of the accumulation of widespread molecular and cellular deficits that eventually scale up to produce detectable macroscopic deficits at the organ and systems levels (Howlett & Rockwood, 2013; Lakatta, 2015; Rockwood et al. 2015). The mouse FI provides a powerful new tool that can be used to investigate how subcellular deficits may accumulate and scale up to produce overt disease in frail older adults, as well as to design interventions to attenuate such damage.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
These studies were conducted at Dalhousie University in the laboratory of SEH. SEH, HAF and RAR contributed to the conception or design of the work; SEH, HAF, RAR, AEK, OA, AB, NP and RGT contributed to the acquisition, analysis or interpretation of data for the work; SEH, HAF, RAR, AEK, OA, AB, NP and RGT helped to draft the work or revise it critically for important intellectual content. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was funded by the Canadian Institutes for Health Research (grant number MOP 126018 to SEH and MOP 93718 and 142486 to RAR).
Acknowledgements
The authors express their appreciation for excellent technical assistance provided by Dr Jie‐quan Zhu and Peter Nicholl. The authors are also grateful to Drs O. Theou and J. Godin for statistical advice.
References
- Afilalo J, Alexander KP, Mack MJ, Maurer MS, Green P, Allen LA, Popma JJ, Ferrucci L & Forman DE (2014). Frailty assessment in the cardiovascular care of older adults. J Am Coll Cardiol 63, 747–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM (2014). Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu Rev Physiol 76, 107–127. [DOI] [PubMed] [Google Scholar]
- Bouillon K, Kivimaki M, Hamer M, Sabia S, Fransson EI, Singh‐Manoux A, Gale CR & Batty GD (2013). Measures of frailty in population‐based studies: an overview. BMC Geriatr 13, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer RS, Shah KB & Kenny AM (2014). Frailty and prognosis in advanced heart failure. Curr Opin Support Palliat Care 8, 25–29. [DOI] [PubMed] [Google Scholar]
- Chen W & Frangogiannis NG (2010). The role of inflammatory and fibrogenic pathways in heart failure associated with aging. Heart Fail Rev 15, 415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg A, Young J, Iliffe S, Rikkert MO & Rockwood K (2013). Frailty in elderly people. Lancet 381, 752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries NM, Staal JB, van Ravensberg CD, Hobbelen JS, Olde Rikkert MG & Nijhuis‐van der Sanden MW (2011). Outcome instruments to measure frailty: a systematic review. Ageing Res Rev 10, 104–114. [DOI] [PubMed] [Google Scholar]
- Fares E & Howlett SE (2010). Effect of age on cardiac excitation–contraction coupling. Clin Exp Pharmacol Physiol 37, 1–7. [DOI] [PubMed] [Google Scholar]
- Feridooni HA, Dibb KM & Howlett SE (2015a). How cardiomyocyte excitation, calcium release and contraction become altered with age. J Mol Cell Cardiol 83, 62–72. [DOI] [PubMed] [Google Scholar]
- Feridooni HA, Sun MH, Rockwood K & Howlett SE (2015b). Reliability of a frailty index based on the clinical assessment of health deficits in male C57BL/6J mice. J Gerontol A Biol Sci Med Sci 70, 686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrier GR, Redondo IM, Mason CA, Mapplebeck C & Howlett SE (2000). Regulation of contraction and relaxation by membrane potential in cardiac ventricular myocytes. Am J Physiol Heart Circ Physiol 278, H1618–1626. [DOI] [PubMed] [Google Scholar]
- Fleg JL & Strait J (2012). Age‐associated changes in cardiovascular structure and function: a fertile milieu for future disease. Heart Fail Rev 17, 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraticelli A, Josephson R, Danziger R, Lakatta E & Spurgeon H (1989). Morphological and contractile characteristics of rat cardiac myocytes from maturation to senescence. Am J Physiol Heart Circ Physiol 257, H259–265. [DOI] [PubMed] [Google Scholar]
- Gattoni S, ÅT Røe, Frisk M, Louch WE, Niederer SA & Smith NP (2016). The calcium‐frequency response in the rat ventricular myocyte: an experimental and modelling study. J Physiol 594, 4193–4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandy SA & Howlett SE (2006). Cardiac excitation–contraction coupling is altered in myocytes from aged male mice but not in cells from aged female mice. Am J Physiol Heart Circ Physiol 291, H2362–2370. [DOI] [PubMed] [Google Scholar]
- Goldwater DS1 & Pinney SP (2015). Frailty in advanced heart failure: a consequence of aging or a separate entity? Clin Med Insights Cardiol 9, 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo KK & Ren J (2006). Cardiac overexpression of alcohol dehydrogenase (ADH) alleviates aging‐associated cardiomyocyte contractile dysfunction: role of intracellular Ca2+ cycling proteins. Aging Cell 5, 259–265. [DOI] [PubMed] [Google Scholar]
- Guzadhur L, Jiang W, Pearcey SM, Jeevaratnam K, Duehmke RM, Grace AA, Lei M & Huang CL (2012). The age‐dependence of atrial arrhythmogenicity in Scn5a+/‐ murine hearts reflects alterations in action potential propagation and recovery. Clin Exp Pharmacol Physiol 39, 518–527. [DOI] [PubMed] [Google Scholar]
- Hamdani N, Bishu KG, von Frieling‐Salewsky M, Redfield MM & Linke WA (2013). Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc Res 97, 464–471. [DOI] [PubMed] [Google Scholar]
- Headrick JP, Willems L, Ashton KJ, Holmgren K, Peart J & Matherne GP (2003). Ischaemic tolerance in aged mouse myocardium: the role of adenosine and effects of A1 adenosine receptor overexpression. J Physiol 549, 823–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett SE (2010). Age‐associated changes in excitation‐contraction coupling are more prominent in ventricular myocytes from male rats than in myocytes from female rats. Am J Physiol Heart Circ Physiol 298, H659–670. [DOI] [PubMed] [Google Scholar]
- Howlett SE & Rockwood K (2013). New horizons in frailty: ageing and the deficit‐scaling problem. Age Ageing 42, 416–423. [DOI] [PubMed] [Google Scholar]
- Howlett SE, Rockwood MR, Mitnitski A & Rockwood K (2014). Standard laboratory tests to identify older adults at increased risk of death. BMC Med 12, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett SE & Rockwood K (2014). Ageing: develop models of frailty. Nature 512, 253. [DOI] [PubMed] [Google Scholar]
- Janczewski AM & Lakatta EG (2010). Modulation of sarcoplasmic reticulum Ca2+ cycling in systolic and diastolic heart failure associated with aging. Heart Fail Rev 15, 431–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jermyn R & Patel S (2015). The biologic syndrome of frailty in heart failure. Clin Med Insights Cardiol 8, 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane AE, Hilmer SN, Boyer D, Gavin K, Nines D, Howlett SE, de Cabo R & Mitchell SJ (2016). Impact of longevity interventions on a validated mouse clinical frailty index. J Gerontol A Biol Sci Med Sci 71, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane AE, Hilmer SN, Huizer‐Pajkos A, Mach J, Nines D, Boyer D, Gavin K, Mitchell SJ & de Cabo R (2015). Factors that impact on interrater reliability of the mouse clinical frailty index. J Gerontol A Biol Sci Med Sci 70, 694–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane AE, Ayaz O, Ghimire A, Feridooni HA & Howlett SE (2017). Implementation of the mouse frailty index. Can J Physiol Pharmacol, DOI: 10.1139/cjpp-2017-0025. [DOI] [PubMed] [Google Scholar]
- Keller K & Howlett SE (2016). Sex differences in the biology and pathology of the ageing heart. Can J Cardiol 32, 1065–1073. [DOI] [PubMed] [Google Scholar]
- Kim S & Jazwinski SM (2015). Quantitative measures of healthy aging and biological age. Healthy Aging Res 4, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakatta EG & Levy D (2003). Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part II: the aging heart in health: links to heart disease. Circulation 107, 346–354. [DOI] [PubMed] [Google Scholar]
- Lakatta EG (2015). So! What's aging? Is cardiovascular aging a disease? J Mol Cell Cardiol 83, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GR, Ferrier GR & Howlett SE (1995). Calcium currents in ventricular myocytes of prehypertrophic cardiomyopathic hamsters. Am J Physiol Heart Circ Physiol 268, H999–1005. [DOI] [PubMed] [Google Scholar]
- Li R & Shen Y (2013). An old method facing a new challenge: re‐visiting housekeeping proteins as internal reference control for neuroscience research. Life Sci 92, 747–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe DA, Degens H, Chen KD & Alway SE (2000). Glyceraldehyde‐3‐phosphate dehydrogenase varies with age in glycolytic muscles of rats. J Gerontol A Biol Sci Med Sci 55, B160–164. [DOI] [PubMed] [Google Scholar]
- Mellor KM, Curl CL, Chandramouli C, Pedrazzini T, Wendt IR & Delbridge LM (2014). Ageing‐related cardiomyocyte functional decline is sex and angiotensin II dependent. Age (Dordr) 36, 9630–9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennes E, Dungan CM, Frendo‐Cumbo S, Williamson DL & Wright DC (2014). Aging‐associated reductions in lipolytic and mitochondrial proteins in mouse adipose tissue are not rescued by metformin treatment. J Gerontol A Biol Sci Med Sci 69, 1060–1068. [DOI] [PubMed] [Google Scholar]
- Mitchell SJ, Madrigal‐Matute J, Scheibye‐Knudsen M, Fang E, Aon M, González‐Reyes JA, Cortassa S, Kaushik S, Gonzalez‐Freire M, Patel B, Wahl D, Ali A, Calvo‐Rubio M, Burón MI, Guiterrez V, Ward TM, Palacios HH, Cai H, Frederick DW, Hine C, Broeskamp F, Habering L, Dawson J, Beasley TM, Wan J, Ikeno Y, Hubbard G, Becker KG, Zhang Y, Bohr VA, Longo DL, Navas P, Ferrucci L, Sinclair DA, Cohen P, Egan JM, Mitchell JR, Baur JA, Allison DB, Anson RM, Villalba JM, Madeo F, Cuervo AM, Pearson KJ, Ingram DK, Bernier M & de Cabo R (2016). Effects of sex, strain, and energy intake on hallmarks of aging in mice. Cell Metab 23, 1093–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitnitski AB, Mogilner AJ & Rockwood K (2001). Accumulation of deficits as a proxy measure of aging. ScientificWorldJournal 1, 323–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghtadaei M, Jansen HJ, Mackasey M, Rafferty SA, Bogachev O, Sapp JL, Howlett SE, and Robert A & Rose RA (2016). The impacts of age and frailty on rate and sinoatrial node function. J Physiol 594, 7105–7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks RJ, Fares E, Macdonald JK, Ernst MC, Sinal CJ, Rockwood K & Howlett SE (2012). A procedure for creating a frailty index based on deficit accumulation in aging mice. J Gerontol A Biol Sci Med Sci 67, 217–227. [DOI] [PubMed] [Google Scholar]
- Parks RJ, Ray G, Bienvenu LA, Rose RA & Howlett SE (2014). Sex differences in SR Ca release in murine ventricular myocytes are regulated by the cAMP/PKA pathway. J Mol Cell Cardiol 75, 162–173. [DOI] [PubMed] [Google Scholar]
- Porter GA, Urciuoli WR, Brookes PS & Nadtochiy SM (2014). SIRT3 deficiency exacerbates ischemia‐reperfusion injury: implication for aged hearts. Am J Physiol Heart Circ Physiol 306, H1602–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Li Q, Wu S, Li SY & Babcock SA (2007). Cardiac overexpression of antioxidant catalase attenuates aging‐induced cardiomyocyte relaxation dysfunction. Mech Ageing Dev 128, 276–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockwood K, Mitnitski A & Howlett SE (2015). Frailty: scaling from cellular deficit accumulation? Interdiscip Top Gerontol Geriatr 41, 1–14. [DOI] [PubMed] [Google Scholar]
- Rockwood K, Blodgett JM, Theou O, Sun MH, Feridooni HA, Mitnitski A, Rose RA, Godin J, Gregson E & Howlett SE (2017). A frailty index based on deficit accumulation quantifies mortality risk in humans and in mice. Sci Rep 7, 43068–43078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockwood K (2016). Conceptual models of frailty: accumulation of deficits. Can J Cardiol 32, 1046–1050. [DOI] [PubMed] [Google Scholar]
- Romero‐Calvo I, Ocón B, Martínez‐Moya P, Suárez MD, Zarzuelo A, Martínez‐Augustin O & de Medina FS (2010). Reversible Ponceau staining as a loading control alternative to actin in Western blots. Anal Biochem 401, 318–320. [DOI] [PubMed] [Google Scholar]
- Searle SD, Mitnitski A, Gahbauer EA, Gill TM & Rockwood K (2008). A standard procedure for creating a frailty index. BMC Geriatr 8, 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shattock MJ & Bers DM (1987). Inotropic response to hypothermia and the temperature‐dependence of ryanodine action in isolated rabbit and rat ventricular muscle: implications for excitation‐contraction coupling. Circ Res 61, 761–771. [DOI] [PubMed] [Google Scholar]
- Shutt RH & Howlett SE (2008). Hypothermia increases the gain of excitation‐contraction coupling in guinea pig ventricular myocytes. Am J Physiol Cell Physiol 295, C692–700. [DOI] [PubMed] [Google Scholar]
- Singh M, Stewart R & White H (2014). Importance of frailty in patients with cardiovascular disease. Eur Heart J 35, 1726–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein M, Noorman M, van Veen TA, Herold E, Engelen MA, Boulaksil M, Antoons G, Jansen JA, van Oosterhout MF, Hauer RN, de Bakker JM & van Rijen HV (2008). Dominant arrhythmia vulnerability of the right ventricle in senescent mice. Heart Rhythm 5, 438–448. [DOI] [PubMed] [Google Scholar]
- Strait JB & Lakatta EG (2012). Aging‐associated cardiovascular changes and their relationship to heart failure. Heart Fail Clin 8, 143–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchmanowicz I, Loboz‐Rudnicka M, Szelag P, Jankowska‐Polanska B & Loboz‐Grudzien K (2014). Frailty in heart failure. Curr Heart Fail Rep 11, 266–273. [DOI] [PubMed] [Google Scholar]
- Vaupel JW, Manton KG & Stallard E (1979). The impact of heterogeneity in individual frailty on the dynamics of mortality. Demography 16, 439–454. [PubMed] [Google Scholar]
- Whitehead JC, Hildebrand BA, Sun M, Rockwood MR, Rose RA, Rockwood K & Howlett SE (2014). A clinical frailty index in aging mice: comparisons with frailty index data in humans. J Gerontol A Biol Sci Med Sci 69, 621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin FC, Spurgeon HA, Rakusan K, Weisfeldt ML & Lakatta EG (1982). Use of tibial length to quantify cardiac hypertrophy: application in the aging rat. Am J Physiol Hear Circ Physiol 243, H941–947. [DOI] [PubMed] [Google Scholar]