

Abstract
In experimental physiological research, a common study design for examining the functional role of a gene or a genetic variant is to introduce that genetic variant into a model organism (such as yeast or mouse) and then to search for phenotypic consequences. The development of DNA biobanks linked to dense phenotypic information enables such an experiment to be applied to human subjects in the form of a phenome‐wide association study (PheWAS). The PheWAS paradigm takes advantage of a curated medical phenome, often derived from electronic health records, to search for associations between ‘input functions’ and phenotypes in an unbiased fashion. The most commonly studied input function to date has been single nucleotide polymorphisms (SNPs), but other inputs, such as sets of SNPs or a disease or drug exposure, are now being explored to probe the genetic and phenotypic architecture of human traits. Potential outcomes of these approaches include defining subsets of complex diseases (that can then be targeted by specific therapies) and drug repurposing.
Keywords: biobanks, electronic health records, genomics, GWAS, phenome, PheWAS, polygenic risk score
Abbreviations
- EHR
electronic health record
- eMERGE
Electronic Medical Records and Genomics Network
- GWAS
genome‐wide association study
- PheWAS
phenome‐wide association study
- PPV
positive predictive value
- SNP
single nucleotide polymorphism
Human studies using forward or reverse genetics
A typical ‘forward genetics’ experiment seeks to identify the genetic basis of a trait. In human genetics, examples of traits studied include Mendelian diseases, common diseases, laboratory values and physiological characteristics such as hair colour. The methods used range from linkage analysis in families to studies of candidate genes (often chosen from an understanding of underlying physiology) to unbiased approaches such as genome‐wide association study (GWAS). While the candidate gene approach feels intuitively appealing, replication of initially positive association results often fails. The GWAS approach is less subject to the biases of the candidate‐gene approach, but also requires replication, often in very large datasets. The GWAS approach examines associations with common genetic variants and as a result generally generates modest odds ratios for single nucleotide polymorphisms (SNPs) associated with phenotypes of interest. There are exceptions to this general rule, often for diseases with onset after reproductive age or conditions requiring a major environmental input, such as variable drug responses.
In contrast to human genetics, physiological research commonly uses a ‘reverse genetics’ experimental paradigm to understand the function of genes or phenotypic consequences of genetic variation: a conventional reverse genetics study design is to introduce a genetic variant into a model organism (such as yeast or mouse) and then to search for phenotypic consequences. The phenome‐wide association study (PheWAS) is a method that enables a ‘reverse genetics’ experiment to be applied to human subjects (Abstract Figure). PheWAS takes advantage of increasingly large sets of human genetic variation coupled to dense phenotypic information often from electronic health records (EHRs) to analyse genotype–phenotype associations (Bush et al. 2016; Denny et al. 2016; Roden & Denny, 2016).
Large resources linking human phenotypes to genotypes
As discussed further below, the key resource that enables PheWAS is dense phenotypic data coupled to DNA repositories. Examples of such resources that have been created around the world and that contain > 200,000 samples include the UK Biobank (Allen et al. 2014), the Chinese Kadoorie biobank (Chen et al. 2011), Vanderbilt BioVU (Roden et al. 2008), the Electronic Medical Records and Genomics Network (eMERGE) (Gottesman et al. 2013; Crawford et al. 2014; Rasmussen‐Torvik et al. 2014), deCODE in Iceland (Gulcher & Stefansson, 1998), Kaiser's Genes, Environment, and Health Project (Banda et al. 2015), and the US Veterans Administration Million Veterans Program (MVP) (Gaziano et al. 2016). All of these include EHR data as one source of phenotypes; some, like BioVU and eMERGE (of which BioVU is a component), rely exclusively on dense EHR data, while others (e.g. UK Biobank) currently include sparse EHR data but dense prospectively obtained phenotypic data. The upcoming US Precision Medicine Initiative (PMI) Cohort Study (Collins & Varmus, 2015) will recruit at least 1,000,000 participants and use both EHR data and data prospectively collected by questionnaires, examinations, and smartphones as sources of phenotypic information. There are important methodological challenges that need to be addressed as phenome scanning approaches become more widely tested and their place in precision medicine becomes better defined; these include improving definitions of cases and controls from inherently ‘messy’ EHR data and developing appropriate statistical methods (Wei & Denny, 2015; Bush et al. 2016).
Curating the medical phenome
The creation of electronic records, and in particular the ability to structure information in EHRs to make it more receptive to research efforts, is enabling for phenome scanning applications. A GWAS is enabled by dense genomic information and at each locus the specific genotype (reference or variant) is known. By contrast, even in the well curated EHR‐based phenome, the ability to definitively exclude specific conditions or to define them based simply on billing codes may be limited. EHR data can also require expert clinical input for appropriate interpretation. A patient with well controlled hypertension may have consistently normal blood pressure readings and yet should still carry the diagnosis. On the other hand, a patient without hypertension who presents with a broken bone may have severely elevated blood pressure at the time of that encounter (Teixeira et al. 2016). Current efforts in EHR‐based phenotyping use a combination of billing codes, laboratory data, medications and natural language processing of free text (e.g. in clinical notes) to develop algorithms for establishing case or control status for a particular trait (Wei et al. 2016). Records meeting algorithmic definitions are manually reviewed and a positive predictive value (PPV) established. For most traits, a positive predictive value greater than 95% is achievable and experience across eMERGE has shown that algorithms developed at one site can be exported to other sites with very good PPVs (Denny et al. 2011).
Initial studies: associations with single SNPs
In the mid‐2000s, a number of groups suggested that increasing the availability of dense phenotypic information, derived from encounters that patients had with a healthcare system, coupled to increasingly inexpensive genotyping could enable the concept of ‘phenome scanning’ to identify phenotypes associated with specific genetic variants (Jones et al. 2005; Ghebranious et al. 2007). Initial studies testing this idea focused on single SNPs, generally identified by GWAS; however, the genetic architecture of most common diseases includes contributions by common and rare variants across multiple genes, some of which influence other disease susceptibilities. One outcome of the PheWAS paradigm is to probe such pleiotropic effects and thus to identify in the universe of patients with common diseases subsets with genomic (and phenomic) architecture that predict clinically important variability in disease susceptibility, disease progression and development of complications, or response to specific therapies.
The actual term ‘PheWAS’ was coined by Denny and colleagues in 2010 in the first demonstration that the concept could, in fact, be executed (Denny et al. 2010). They developed software based on disease codes to define 776 sets of cases and controls from EHR data. They genotyped 6005 European American subjects for SNPs previously associated by GWAS with seven common diseases and showed that their PheWAS replicated those associations in 4/7 cases. The algorithm also identified previously unreported associations between the target SNPs and diseases at a nominal P‐value < 0.01. The same group, working with collaborators in the eMERGE network, then went on to perform a GWAS and PheWAS using cardiac conduction assessed by QRS duration on the electrocardiogram as the target phenotype (Ritchie et al. 2013). The GWAS identified SNPs near a group of sodium channel genes (a biologically plausible candidate locus, also found by others studying this phenotype; Sotoodehnia et al. 2010) and the PheWAS of these SNPs indicated they also increase risk for a common arrhythmia, atrial fibrillation, ascertained over more than two decades across EHRs. These findings highlight the potential for PheWAS to uncover clinically interesting and potentially important signals for disease susceptibility.
Investigators in the eMERGE network then went on to apply this approach to validate a network‐wide GWAS that implicated variants near FOXE1 as predisposing to hypothyroidism (Denny et al. 2011). The initial GWAS result was obtained in 1317 cases and 5053 European ancestry controls across five EHRs. The algorithms to identify hypothyroidism were developed at one site and deployed across the others with positive predictive values > 90% for both case and control algorithms. The FOXE1 signal was replicated in an independent dataset and variants near this gene have previously been associated with thyroid cancer. The eMERGE team went on to perform a PheWAS in 13,617 individuals to search for other or additional phenotypes associated with the FOXE1 variants. The strongest association replicated the hypothyroidism phenotype, with an odds ratio of 0.76 (P = 2.17 × 10−13), nearly identical to the odds ratio (0.74, P = 4 × 10−9) generated by the original GWAS. The PheWAS also identified signals for some subtypes of thyroid disease (thyroiditis, nodular and multinodular goitre, and thyrotoxicosis) but not with others, like Graves’ disease or thyroid cancer. Other interesting associations that did achieve nominal statistical significance included atrial flutter (there is a known association between thyroid disorders and atrial arrhythmias) and pernicious anaemia, a disease sometimes associated with hypothyroidism. This study highlighted the potential that PheWAS can identify subsets that carry different prognosis within a universe of patients with the same clinical diagnosis.
Validating PheWAS in the GWAS catalogue
This approach was then tested on a large scale (Denny et al. 2013) across the eMERGE network to examine the extent to which PheWAS could replicate the results of previous GWAS in the catalogue maintained by the National Human Genome Research Institute (Hindorff et al. 2015). Some traits in the GWAS catalogue (such as baldness or the ability to smell asparagus metabolites in urine) are not captured in the EHR and so could not be studied. PheWAS in 13,835 subjects replicated 66% of previously reported associations from well‐powered GWAS. In addition, there were 63 new associations with P‐values < 4.6 × 10−6 with specific SNPs studied, further highlighting the potential that the PheWAS approach can help define pleiotropic genetic effects. For example, variants in IRF4, previously associated with hair and eye colour, were strongly associated with non‐melanoma skin cancer (P = 3.8 × 10−17) and with a benign skin disease, actinic keratosis (P = 4.1 × 10−26). A more recent demonstration of the potential for PheWAS to identify pleiotropic effects of individual SNPs was an evaluation of traits associated with polymorphisms thought to be of Neanderthal origin in an eMERGE population of approximately 28,000 European ancestry subjects (Simonti et al. 2016). Traits associated with Neanderthal alleles in both discovery and replication sets included hypercoagulable state, protein calorie malnutrition, urinary system symptoms and tobacco use.
Input functions beyond single SNPs
These initial applications of the PheWAS methodology focused on associations with single SNPs, generally drawn from GWASs, and thus have the drawback that they are often associated with small effect sizes. Therefore, recent applications of PheWAS have started to move beyond searching for associations with single SNPs as input functions to probe the curated medical phenome (Fig. 1).
Figure 1. The phenome‐wide association study.
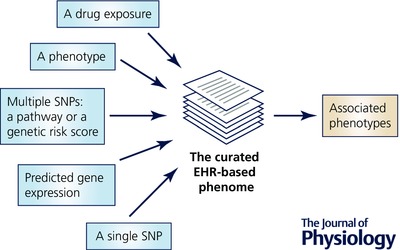
Once a curated medical phenome is in place, investigators can use a wide variety of input functions, ranging from single nucleotide polymorphisms (SNPs) to more complex genetic constructs to diseases or drug exposures, to probe associated phenotypes.
Most GWASs search for associations with a single phenotype, although recent work also highlights to potential power of a multivariate GWAS that searches for associations across multiple related phenotypes (Galesloot et al. 2014). In one sense, therefore, the PheWAS approach, by searching across thousands of phenotypes, represents an extension of this logic although the PheWAS starting point has been genetic variation whereas the starting point for a GWAS is single (or multiple) phenotypes.
One example of a different input function is a set of SNPs, such as a genetic risk score, a weighted set of SNPs derived from GWAS data and used to predict a phenotype in a population (Kathiresan et al. 2008). In one study, genetic risk scores using thousands of SNPs derived from GWAS of psychiatric traits were able to distinguish extremes across phenotypes for ∼50 traits in the ‘behavioral phenome’ in 3152 individuals (Krapohl et al. 2016), although the effect sizes were still small (< 2% of total variance). This study also highlights the potential for extending phenotypic analysis from case–control studies to examine continuous traits such as psychiatric phenotypes or laboratory data. Probing the phenotypic associations with SNPs in genes encoding known drug targets may reveal new drug indications or potentially predict on‐target adverse drug effects (Rastegar‐Mojarad et al. 2015). Along the same lines, a study in the Kadoorie biobank demonstrated that loss of function alleles in PLA2, encoding a potential drug target, were not associated with disease risk, providing a reassuring safety signal for further candidate blocker development (Millwood et al. 2016).
Another potentially interesting approach is to use predicted gene expression as the ‘input function’ for PheWAS. Gamazon et al. (2015) have described PrediXcan, a method that couples tissue‐specific expression data with dense genotyping to develop predictors of tissue‐specific gene expression. Using this approach, it should be feasible to identify human traits associated with predicted increased or decreased expression of any gene. An initial evaluation of this hypothesis using the Wellcome Trust Case Control Consortium dataset and BioVU replicated autoimmune signals associated with diseases such as rheumatoid arthritis, ulcerative colitis, Crohn's disease and type 1 diabetes.
The focus of PheWAS experiments to date has been on searching for phenotypic associations with common SNP, or sets of SNPs, often assayed using dense and increasingly inexpensive genotyping platforms. The cost of genome sequencing is also dropping, and the pending availability of large sets of whole genome sequences will pose new analytical challenges and opportunities to the field. One example is developing best methods to integrate rare and common variation into genetic risk scores. Another is to develop datasets that are sufficiently large that associations with rare SNPs can be reliably identified.
The input function need not be a genetic variant. For example, Warner & Alterovitz (2012) showed that very high white cell counts were most often associated with Clostridium difficile infection or septic shock, a finding that could eventually guide clinical care. The input can be a disease and PheWAS can be used to identify mechanistic subsets (Denny et al. 2016): PheWAS of rheumatoid arthritis cases identified a significant association between antinuclear antibodies and Sjogren's syndrome (Liao et al. 2013).
Conclusion
PheWAS has been enabled by accrual of massive healthcare information in EHRs, development of methods to extract believable sets of cases and controls, increasingly inexpensive genotyping, and methods to analyse the relationship between genetic variation and phenotypes. The initial results outlined here are highly promising and in some ways are reminiscent of the ‘early days’ of GWAS, in which interesting signals were generated in thousands of subjects and with time have now been validated in tens or hundreds of thousands of subjects. Analysis of such very large datasets will help distinguish between true and false PheWAS signals and increase the validity of the PheWAS results. One can envision that PheWAS will become a powerful tool used by both clinicians and basic scientists. For the clinician the development of very large collections coupling DNA, other potential biomarkers, EHRs and sociocultural information will enable phenome scanning across diverse ancestries and health conditions and thus help propel the development of personalized treatments. For the basic physiologist, PheWAS will complement experimental research and help validate insight gained from studying genes and gene variants in model organisms.
Additional information
Competing interests
None.
Funding
This work was supported by grants from the US National Institutes of Health (P50GM115305, U01HG008672, and UL1TR000445).
Biography
Dan Roden received his medical degree and training in Internal Medicine from McGill University. He then went to Vanderbilt where he trained in Clinical Pharmacology and Cardiology, and has been a faculty member there since. His initial career focus – that he has maintained – was studies of the clinical, genetic, cellular, and molecular basis of arrhythmia susceptibility and variability responses to arrhythmia therapies. In 2006, he was appointed Senior Vice President for Personalized Medicine and charged with coordinating and advancing an agenda for personalized medicine at Vanderbilt. He directs BioVU, the Vanderbilt DNA databank that as of fall 2016 links DNA samples from >225,000 patients to deidentified electronic medical records. He currently acts as co‐PI for the Vanderbilt sites for the Pharmacogenomics Research Network and the Electronic Medical Records and Genomics (eMERGE) Network, and serves on the FDA's Science Board and the NIH's National Advisory Council for Human Genome Research.

References
- Allen NE, Sudlow C, Peakman T, Collins R & UK Biobank (2014). UK biobank data: come and get it. Sci Transl Med 6, 224ed224. [DOI] [PubMed] [Google Scholar]
- Banda Y, Kvale MN, Hoffmann TJ, Hesselson SE, Ranatunga D, Tang H, Sabatti C, Croen LA, Dispensa BP, Henderson M, Iribarren C, Jorgenson E, Kushi LH, Ludwig D, Olberg D, Quesenberry CP Jr, Rowell S, Sadler M, Sakoda LC, Sciortino S, Shen L, Smethurst D, Somkin CP, Van Den Eeden SK, Walter L, Whitmer RA, Kwok PY, Schaefer C & Risch N (2015). Characterizing race/ethnicity and genetic ancestry for 100,000 subjects in the genetic epidemiology research on adult health and aging (GERA) cohort. Genetics 200, 1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush WS, Oetjens MT & Crawford DC (2016). Unravelling the human genome‐phenome relationship using phenome‐wide association studies. Nat Rev Genet 17, 129–145. [DOI] [PubMed] [Google Scholar]
- Chen Z, Chen J, Collins R, Guo Y, Peto R, Wu F & Li L (2011). China kadoorie biobank of 0.5 million people: survey methods, baseline characteristics and long‐term follow‐up. Int J Epidemiol 40, 1652–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS & Varmus H (2015). A new initiative on precision medicine. N Engl J Med 372, 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford DC, Crosslin DR, Tromp G, Kullo IJ, Kuivaniemi H, Hayes MG, Denny JC, Bush WS, Haines JL, Roden DM, McCarty CA, Jarvik GP & Ritchie MD (2014). eMERGEing progress in genomics—the first seven years. Front Genet 5, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny JC, Bastarache L, Ritchie MD, Carroll RJ, Zink R, Mosley JD, Field JR, Pulley JM, Ramirez AH, Bowton E, Basford MA, Carrell DS, Peissig PL, Kho AN, Pacheco JA, Rasmussen LV, Crosslin DR, Crane PK, Pathak J, Bielinski SJ, Pendergrass SA, Xu H, Hindorff LA, Li R, Manolio TA, Chute CG, Chisholm RL, Larson EB, Jarvik GP, Brilliant MH, McCarty CA, Kullo IJ, Haines JL, Crawford DC, Masys DR & Roden DM (2013). Systematic comparison of phenome‐wide association study of electronic medical record data and genome‐wide association study data. Nat Biotechnol 31, 1102–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny JC, Bastarache L & Roden DM (2016). Phenome‐wide association studies as a tool to advance precision medicine. Annu Rev Genomics Hum Genet 17, 353–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny JC, Crawford DC, Ritchie MD, Bielinski SJ, Basford MA, Bradford Y, Chai HS, Bastarache L, Zuvich R, Peissig P, Carrell D, Ramirez AH, Pathak J, Wilke RA, Rasmussen L, Wang X, Pacheco JA, Kho AN, Hayes MG, Weston N, Matsumoto M, Kopp PA, Newton KM, Jarvik GP, Li R, Manolio TA, Kullo IJ, Chute CG, Chisholm RL, Larson EB, McCarty CA, Masys DR, Roden DM & de Andrade M (2011). Variants near FOXE1 are associated with hypothyroidism and other thyroid conditions: using electronic medical records for genome‐ and phenome‐wide studies. Am J Hum Genet 89, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny JC, Ritchie MD, Basford MA, Pulley JM, Bastarache L, Brown‐Gentry K, Wang D, Masys DR, Roden DM & Crawford DC (2010). PheWAS: demonstrating the feasibility of a phenome‐wide scan to discover gene‐disease associations. Bioinformatics 26, 1205–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galesloot TE, van Steen K, Kiemeney LA, Janss LL & Vermeulen SH (2014). A comparison of multivariate genome‐wide association methods. PloS One 9, e95923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino‐Michaels K, Carroll RJ, Eyler AE, Denny JC, GTEx Consortium , Nicolae DL, Cox NJ & Im HK (2015). A gene‐based association method for mapping traits using reference transcriptome data. Nat Genet 47, 1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaziano JM, Concato J, Brophy M, Fiore L, Pyarajan S, Breeling J, Whitbourne S, Deen J, Shannon C, Humphries D, Guarino P, Aslan M, Anderson D, LaFleur R, Hammond T, Schaa K, Moser J, Huang G, Muralidhar S, Przygodzki R & O'Leary TJ (2016). Million veteran program: a mega‐biobank to study genetic influences on health and disease. J Clin Epidemiol 70, 214–223. [DOI] [PubMed] [Google Scholar]
- Ghebranious N, McCarty CA & Wilke RA (2007). Clinical phenome scanning. Personal Med 4, 175–182. [DOI] [PubMed] [Google Scholar]
- Gottesman O, Kuivaniemi H, Tromp G, Faucett WA, Li R, Manolio TA, Sanderson SC, Kannry J, Zinberg R, Basford MA, Brilliant M, Carey DJ, Chisholm RL, Chute CG, Connolly JJ, Crosslin D, Denny JC, Gallego CJ, Haines JL, Hakonarson H, Harley J, Jarvik GP, Kohane I, Kullo IJ, Larson EB, McCarty C, Ritchie MD, Roden DM, Smith ME, Bottinger EP & Williams MS (2013). The electronic medical records and genomics (eMERGE) network: past, present, and future. Genet Med 15, 761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulcher J & Stefansson K (1998). Population genomics: laying the groundwork for genetic disease modeling and targeting. Clin Chem Lab Med 36, 523–527. [DOI] [PubMed] [Google Scholar]
- Hindorff LA, MacArthur J, Morales J, Junkins HA, Hall PN, Klemm AK & Manolio TA (2015). A Catalog of Published Genome‐Wide Association Studies. www.genome.gov/gwastudies (accessed 30 December, 2015).
- Jones R, Pembrey M, Golding J & Herrick D (2005). The search for genenotype/phenotype associations and the phenome scan. Paediatr Perinat Epidemiol 19, 264–275. [DOI] [PubMed] [Google Scholar]
- Kathiresan S, Melander O, Anevski D, Guiducci C, Burtt NP, Roos C, Hirschhorn JN, Berglund G, Hedblad B, Groop L, Altshuler DM, Newton‐Cheh C & Orho‐Melander M (2008). Polymorphisms associated with cholesterol and risk of cardiovascular events. New Engl J Med 358, 1240–1249. [DOI] [PubMed] [Google Scholar]
- Krapohl E, Euesden J, Zabaneh D, Pingault JB, Rimfeld K, von Stumm S, Dale PS, Breen G, O'Reilly PF & Plomin R (2016). Phenome‐wide analysis of genome‐wide polygenic scores. Mol Psychiatry 21, 1188–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao KP, Kurreeman F, Li G, Duclos G, Murphy S, Guzman R, Cai T, Gupta N, Gainer V, Schur P, Cui J, Denny JC, Szolovits P, Churchill S, Kohane I, Karlson EW & Plenge RM (2013). Associations of autoantibodies, autoimmune risk alleles, and clinical diagnoses from the electronic medical records in rheumatoid arthritis cases and non‐rheumatoid arthritis controls. Arthritis Rheum 65, 571–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millwood IY, Bennett DA, Walters RG, Clarke R, Waterworth D, Johnson T, Chen Y, Yang L, Guo Y, Bian Z, Hacker A, Yeo A, Parish S, Hill MR, Chissoe S, Peto R, Cardon L, Collins R, Li L & Chen Z (2016). A phenome‐wide association study of a lipoprotein‐associated phospholipase A2 loss‐of‐function variant in 90 000 Chinese adults. Int J Epidemiol 45, 1588–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen‐Torvik LJ, Stallings SC, Gordon AS, Almoguera B, Basford MA, Bielinski SJ, Brautbar A, Brilliant MH, Carrell DS, Connolly JJ, Crosslin DR, Doheny KF, Gallego CJ, Gottesman O, Kim DS, Leppig KA, Li R, Lin S, Manzi S, Mejia AR, Pacheco JA, Pan V, Pathak J, Perry CL, Peterson JF, Prows CA, Ralston J, Rasmussen LV, Ritchie MD, Sadhasivam S, Scott SA, Smith M, Vega A, Vinks AA, Volpi S, Wolf WA, Bottinger E, Chisholm RL, Chute CG, Haines JL, Harley JB, Keating B, Holm IA, Kullo IJ, Jarvik GP, Larson EB, Manolio T, McCarty CA, Nickerson DA, Scherer SE, Williams MS, Roden DM & Denny JC (2014). Design and anticipated outcomes of the eMERGE‐PGx project: a multicenter pilot for preemptive pharmacogenomics in electronic health record systems. Clin Pharmacol Ther 96, 482–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastegar‐Mojarad M, Ye Z, Kolesar JM, Hebbring SJ & Lin SM (2015). Opportunities for drug repositioning from phenome‐wide association studies. Nat Biotechnol 33, 342–345. [DOI] [PubMed] [Google Scholar]
- Ritchie MD, Denny JC, Zuvich RL, Crawford DC, Schildcrout JS, Bastarache L, Ramirez AH, Mosley JD, Pulley JM, Basford MA, Bradford Y, Rasmussen LV, Pathak J, Chute CG, Kullo IJ, McCarty CA, Chisholm RL, Kho AN, Carlson CS, Larson EB, Jarvik GP, Sotoodehnia N, Cohorts for H, Aging Research in Genomic Epidemiology QRSG , Manolio TA, Li R, Masys DR, Haines JL & Roden DM (2013). Genome‐ and phenome‐wide analyses of cardiac conduction identifies markers of arrhythmia risk. Circulation 127, 1377–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden DM & Denny JC (2016). Integrating electronic health record genotype and phenotype datasets to transform patient care. Clin Pharmacol Ther 99, 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden DM, Pulley JM, Basford MA, Bernard GR, Clayton EW, Balser JR & Masys DR (2008). Development of a large‐scale de‐identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther 84, 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonti CN, Vernot B, Bastarache L, Bottinger E, Carrell DS, Chisholm RL, Crosslin DR, Hebbring SJ, Jarvik GP, Kullo IJ, Li R, Pathak J, Ritchie MD, Roden DM, Verma SS, Tromp G, Prato JD, Bush WS, Akey JM, Denny JC & Capra JA (2016). The phenotypic legacy of admixture between modern humans and Neandertals. Science 351, 737–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotoodehnia N, Isaacs A, de Bakker PI, Dorr M, Newton‐Cheh C, Nolte IM, van der HP, Muller M, Eijgelsheim M, Alonso A, Hicks AA, Padmanabhan S, Hayward C, Smith AV, Polasek O, Giovannone S, Fu J, Magnani JW, Marciante KD, Pfeufer A, Gharib SA, Teumer A, Li M, Bis JC, Rivadeneira F, Aspelund T, Kottgen A, Johnson T, Rice K, Sie MP, Wang YA, Klopp N, Fuchsberger C, Wild SH, Mateo LI, Estrada K, Volker U, Wright AF, Asselbergs FW, Qu J, Chakravarti A, Sinner MF, Kors JA, Petersmann A, Harris TB, Soliman EZ, Munroe PB, Psaty BM, Oostra BA, Cupples LA, Perz S, de Boer RA, Uitterlinden AG, Volzke H, Spector TD, Liu FY, Boerwinkle E, Dominiczak AF, Rotter JI, van HG, Levy D, Wichmann HE, Van Gilst WH, Witteman JC, Kroemer HK, Kao WH, Heckbert SR, Meitinger T, Hofman A, Campbell H, Folsom AR, van Veldhuisen DJ, Schwienbacher C, O'Donnell CJ, Volpato CB, Caulfield MJ, Connell JM, Launer L, Lu X, Franke L, Fehrmann RS, te Meerman G, Groen HJ, Weersma RK, van den Berg LH, Wijmenga C, Ophoff RA, Navis G, Rudan I, Snieder H, Wilson JF, Pramstaller PP, Siscovick DS, Wang TJ, Gudnason V, van Duijn CM, Felix SB, Fishman GI, Jamshidi Y, Stricker BH, Samani NJ, Kaab S & Arking DE (2010). Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet 42, 1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira PL, Wei WQ, Cronin RM, Mo H, VanHouten JP, Carroll RJ, LaRose E, Bastarache LA, Rosenbloom ST, Edwards TL, Roden DM, Lasko TA, Dart RA, Nikolai AM, Peissig PL & Denny JC (2016). Evaluating electronic health record data sources and algorithmic approaches to identify hypertensive individuals. J Am Med Inform Assoc 24, 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner JL & Alterovitz G (2012). Phenome based analysis as a means for discovering context dependent clinical reference ranges. AMIA Annu Symp Proc 2012, 1441–1449. [PMC free article] [PubMed] [Google Scholar]
- Wei WQ & Denny JC (2015). Extracting research‐quality phenotypes from electronic health records to support precision medicine. Genome Med 7, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei WQ, Teixeira PL, Mo H, Cronin RM, Warner JL & Denny JC (2016). Combining billing codes, clinical notes, and medications from electronic health records provides superior phenotyping performance. J Am Med Inform Assoc 23, e20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]