

Abstract
Ca2+ and mitochondria are inextricably linked to cardiac function and dysfunction. Ca2+ is central to cardiac excitation–contraction coupling and stimulates mitochondrial energy production to fuel contraction. Under pathological conditions of dysregulated Ca2+ cycling, mitochondrial Ca2+ overload activates cellular death pathways. Thus, in the cardiomyocyte, the mitochondrial Ca2+ microdomain is where contraction, energy and death collide. A key component of mitochondrial Ca2+ signalling is the mitochondrial Ca2+ uniporter complex (uniplex), an inner membrane Ca2+ transporter and major pathway of mitochondrial Ca2+ entry. Once known only as the unidentified target for ruthenium red and related compounds, in recent years, the uniplex has evolved into a complex multiprotein assembly. The identification of the molecular constituents of the uniplex has made possible the generation of targeted genetic models to interrogate uniplex function in vivo. This review will summarize our current understanding of the molecular structure of the uniplex, its impact on mitochondrial energetics and cardiac physiology, its contribution to cardiomyocyte death, and its expanding roles in cardiac biology.
Keywords: calcium mitochondria, cell death, energy, heart, mitochondria
Abbreviations
- EMRE
essential MCU regulator
- MICU1
mitochondrial calcium uptake 1
- MICU2
mitochondrial calcium uptake 2
- MCU
mitochondrial calcium uniporter
- MCUb
mitochondrial calcium uniporter b
- MCUR1
mitochondrial calcium uniporter regulator 1
- MPTP
mitochondrial permeability transition pore complex
- ROS
reactive oxygen species
- RyR1
ryanodine receptor
- TRPC3
transient receptor potential canonical 3
- SLC25a23
solute carrier family 25 member 23
Introduction
Ca2+ is a ubiquitous intracellular second messenger with pleiotropic functions ranging from controlling gene expression to regulating cardiac contraction, activating metabolism and initiating cell death (Orrenius & Nicotera, 1994; van Haasteren et al. 1999; Bers, 2008; Balaban, 2009). It has long been recognized that at the mitochondria, Ca2+ signalling can drive both cellular metabolism and death. Under physiological conditions, mitochondrial Ca2+ serves as a signal to enhance energy production by activating three dehydrogenases of the tricarboxylic acid (TCA) cycle (pyruvate dehydrogenase, isocitrate dehydrogenase and α‐ketoglutarate dehydrogenase) (Denton, 2009), as well as the ATP synthase (Jouaville et al. 1999) (Fig. 1). Under pathological conditions of cytosolic Ca2+ overload, however, Ca2+ signalling at the mitochondria, instead of upregulating metabolism, actually engages mitochondrial death pathways. Mitochondrial Ca2+ overload triggers the opening of the mitochondrial permeability transition pore (MPTP), leading to permeabilization of the mitochondrial inner membrane, mitochondrial dysfunction and cell death (Haworth & Hunter, 1979; Hunter & Haworth, 1979; Kwong & Molkentin, 2015) (Fig. 2). In addition to its role relating to energy and the permeability transition, mitochondrial Ca2+ plays an important part in regulating the cellular redox state. Ca2+ activation of TCA cycle dehydrogenases controls NADH production, which in turn impacts cellular anti‐oxidative capacity regeneration and mitochondrial reactive oxygen species (ROS) production (Kohlhaas et al. 2010). Thus, Ca2+ signalling at the mitochondria converges on a number of cellular life and death pathways.
Figure 1. Mitochondrial Ca2+ signalling in cardiac metabolic contraction coupling.
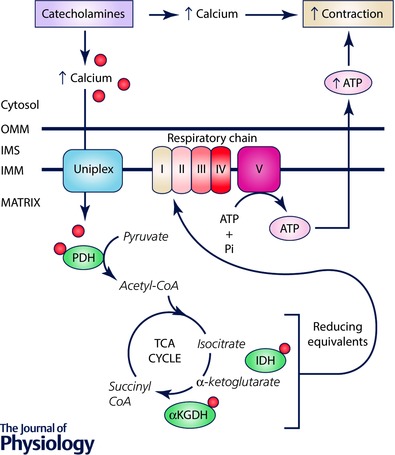
In the cardiac fight or flight response, catecholamine signalling leads to increased cytosolic Ca2+ that drives enhanced contraction. This enhanced contraction is coupled to elevated mitochondrial energetic output as the uniplex transports Ca2+ into the mitochondrial matrix. Ca2+ activates the TCA cycle enzymes pyruvate dehydrogenase (PDH), isocitrate dehydrogenase (IDH), and α‐ketoglutarate dehydrogenase (αKGDH), as well as the mitochondrial ATP synthase (complex V). Cumulatively, this results in increased mitochondrial ATP production that fuels contraction. IMM, inner mitochondrial membrane; IMS, intermembrane space; OMM, outer mitochondrial membrane.
Figure 2. Ca2+ overload‐induced cell death in the heart.
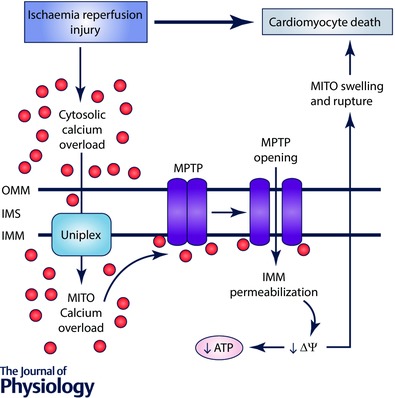
Pathological conditions such as cardiac ischaemia–reperfusion injury result in cytosolic Ca2+ overload. The uniplex transports Ca2+ into the mitochondrial matrix, resulting in activation and opening of the MPTP. MPTP opening causes inner mitochondrial membrane (IMM) permeabilization, loss of mitochondrial membrane potential (ΔΨ) and impaired ATP synthesis. IMM permeabilization also causes mitochondrial (MITO) swelling and rupture, which ultimately results in cardiomyocyte death. IMS, intermembrane space; OMM, outer mitochondrial membrane.
Ca2+ and mitochondria have special relevance to the heart, as Ca2+ is at the core of cardiac excitation–contraction coupling (Bers, 2002), and mitochondria contribute largely to both cardiac physiology and pathophysiology. The heart is heavily reliant on mitochondria as a source of ATP. Indeed, mitochondria constitute ∼30% of the cardiomyocyte's volume and supply > 90% of the ATP required for cardiac contraction (Harris & Das, 1991; Piquereau et al. 2013). Given that Ca2+ underlies both contraction and enhancement of mitochondrial ATP production, mitochondrial Ca2+ microdomains – sites of high local cytosolic Ca2+ established by the close apposition of mitochondria to regions of Ca2+ release, such as the sarcoplasmic reticulum, which facilitate mitochondrial Ca2+ transport – are a critical integration point where ATP supply can be coordinated to meet the energetic demands of contraction (Balaban, 2009). In the face of pathological levels of Ca2+, however, mitochondria serve as a platform whereby death pathways are engaged, and the mitochondrial Ca2+ overload–MPTP axis of cell death has been shown to contribute largely to the cardiomyocyte death observed following cardiac ischaemia–reperfusion injury (Griffiths & Halestrap, 1995; Murphy & Steenbergen, 2008) (Fig. 2). Moreover, during heart failure, alterations in cytosolic Na+ and Ca2+ handling impair mitochondrial Ca2+ influx, thereby potentiating bioenergetic mismatch and aberrant ROS production, which may collectively secondarily engage the MPTP and contribute to adverse cardiac remodelling and the progression to heart failure (Kohlhaas et al. 2010; more comprehensively reviewed in this issue by Maack et al.). Thus, the mitochondrial Ca2+ microdomain presents an intriguing target to control cardiac function through modulating energetics and death.
It has long been appreciated that mitochondrial Ca2+ influx can be inhibited by the drugs ruthenium red and its derivative Ru360 (Zazueta et al. 1999). Indeed, ruthenium red and Ru360 have been demonstrated to inhibit Ca2+ overload‐induced cell death in numerous models including neuronal excitotoxicity (Dessi et al. 1995) and cardiac ischaemia–reperfusion injury (Garcia‐Rivas et al. 2006; Zhang et al. 2006), underscoring the power of modulating mitochondrial Ca2+ handling. The molecular identification of the target of ruthenium red and Ru360, the mitochondrial calcium uniporter (MCU), has generated intense interest in the study of the mitochondrial Ca2+ dynamics (Baughman et al. 2011; De Stefani et al. 2011). MCU is the core component of the multi‐protein MCU holocomplex (uniplex), an inner membrane Ca2+ transporter and a major mode of mitochondrial Ca2+ entry (Kamer & Mootha, 2015). This discovery has paved the way for the genetic manipulation of mitochondrial Ca2+ influx and identification of molecular regulators, and importantly, has set the stage for the study of mitochondrial Ca2+ signalling in vivo.
This review will focus on MCU and the uniplex in the regulation of cardiac function and dysfunction. We will (1) summarize the current understanding of the molecular architecture of the uniplex, (2) review lessons learned from genetically modified mice on MCU's contribution to cardiac energetics and cardiac physiology, (3) define the contribution of MCU to the regulation of cardiac pathology, and (4) explore new pathways that MCU signalling may impact.
The mitochondrial calcium uniporter holocomplex
The mitochondrial Ca2+ uniporter holocomplex (uniplex) is a major pathway of mitochondrial Ca2+ influx that facilitates the membrane potential‐dependent transport of Ca2+ ions across the mitochondrial inner membrane into the matrix (Pradhan et al. 2010; Kamer et al. 2014). The uniplex is a 480 kDa multimeric channel consisting of pore‐forming MCU subunits (Baughman et al. 2011; De Stefani et al. 2011) as well as the regulatory elements MICU1 (Perocchi et al. 2010; Mallilankaraman et al. 2012b), MICU2 (Plovanich et al. 2013), EMRE (Sancak et al. 2013), MCUb (Raffaello et al. 2013) and SLC25a23 (Hoffman et al. 2014). MCU, a dual pass transmembrane protein of the inner mitochondrial membrane, is central to the uniplex as it oligomerizes to form the Ca2+ transducing pore of the complex (Baughman et al. 2011; De Stefani et al. 2011; Raffaello et al. 2013). The Ca2+ selectivity filter of MCU lies within a highly conserved intermembrane space facing a loop containing a DIME motif that links the two transmembrane helices (Baughman et al. 2011). Key acidic residues within this motif are critical for Ca2+ transport as mutations at these sites render the uniplex inactive (Baughman et al. 2011). Computational studies have suggested that the uniplex channel is composed of MCU tetramers (Raffaello et al. 2013). Interestingly, however, a recent structural analysis by nuclear magnetic resonance in combination with electron microscopy of the Caenorhabditis elegans uniplex revealed a pentameric organization (Oxenoid et al. 2016). This study in C. elegans may have important implications for our understanding of uniplex architecture, and highlights the need for additional studies to understand the composition of this transporter in mammals and in vivo.
As mentioned above, in addition to MCU oligomers, models of the uniplex also include the regulatory proteins MICU1, MICU2, EMRE, SLC25a23 and MCUb. MICU1 and MICU2 are thought to serve as Ca2+‐sensing proteins that modulate channel opening at low and high cytosolic Ca2+ concentrations (Patron et al. 2014). EMRE has been found to regulate uniplex assembly by linking MCU to MICU1/2 (Sancak et al. 2013), and recent studies have also shown that EMRE can also regulate uniplex activity by serving as a matrix Ca2+ sensor (Vais et al. 2016). SLC25a23, an inner membrane Mg‐ATP/Pi transporter is thought to modulate Ca2+ influx through interaction with MCU and MICU1 (Hoffman et al. 2014). MCUb was identified as an MCU paralogue that shares high sequence and structure similarity with MCU but lacks the key acidic residues that confer Ca2+ transport ability (Raffaello et al. 2013). Thus, MCUb may function by integrating into the MCU oligomeric pore to act as an endogenous repressor of uniplex‐dependent Ca2+ transport (Raffaello et al. 2013). Finally, MCUR1 has also been proposed to function as a uniplex regulator (Mallilankaraman et al. 2012a), but this finding has been controversial (Paupe et al. 2015) and MCUR1 may serve as a link between the uniplex and the respiratory chain (Tomar et al. 2016).
In an added layer of complexity, the molecular composition of the uniplex – the expression levels of regulatory subunits, and stoichiometry of Ca2+ transducing to inhibitory subunits within the uniplex pore – may vary between tissues (Raffaello et al. 2013). Thus, to date, the precise molecular composition of the cardiac uniplex is unknown. Clearly, work aimed at understanding the complete molecular architecture of the uniplex is needed, as MCU's oligomerization states may have important implications for the stoichiometry and interactions with regulatory subunits, thereby impacting the design of pharmacological agents to modulate uniplex function in the heart.
The MCU and the regulation of cardiac energetics
Cardiac function is driven by Ca2+ cycling and excitation–contraction coupling that require energy (Bers, 2002). The fact that mitochondria are the major suppliers of ATP for contraction, combined with the fact that Ca2+ signalling regulates mitochondrial energy output, points to the importance of the mitochondrial Ca2+ microdomain in linking cardiac energy supply to contractile demand (Balaban, 2009). In vivo models of MCU deletion and inactivation have greatly illuminated the contribution of MCU, the uniplex and mitochondrial Ca2+ dynamics to the regulation of cardiac energetics.
Mouse models of uniplex inhibition, either by constitutive gene disruption using a genetrap strategy (MCU‐constitutive knockout (KO); Pan et al. 2013), or via a targeted loxP‐Cre strategy allowing for the cardiac specific induction of MCU deletion (MCU cardiac KO; Kwong et al. 2015; Luongo et al. 2015), or through transgenic overexpression of a dominant negative MCU in the heart (dnMCU; Wu et al. 2015), have resulted in inhibition of mitochondrial Ca2+ influx. Despite this across‐the‐board inhibition of uniplex‐dependent Ca2+ influx observed amongst the models, interestingly, the effects of in vivo MCU deletion on matrix Ca2+ content varied. MCU‐constitutive KO mitochondria displayed depressed matrix Ca2+ content (Pan et al. 2013) while matrix Ca2+ levels in the MCU cardiac KO mitochondria were unchanged (Kwong et al. 2015; Luongo et al. 2015), suggesting differential effects of long term versus acute MCU deletion on mitochondrial Ca2+ homeostasis.
With regards to mitochondrial bioenergetics, MCU deletion from cardiac mitochondria did not affect mitochondrial energetics at baseline, suggesting that MCU is dispensable for mitochondrial function in unstimulated conditions (Pan et al. 2013; Kwong et al. 2015). The impact of MCU deletion on mitochondrial energetics was revealed in the presence of Ca2+. While control mitochondria increased oxygen consumption with Ca2+ stimulation, this response is abrogated with MCU deletion (Pan et al. 2013; Kwong et al. 2015). Significantly, this impairment in Ca2+‐stimulated respiration translated into an inability of MCU‐deleted cardiac mitochondria to upregulate ATP synthesis in response to Ca2+ signalling (Kwong et al. 2015).
When does the heart require Ca2+‐stimulated upregulation of mitochondrial energy production? During the fight or flight response, catecholamine stimulation initiates a cascade of events that ultimately lead to elevated cytosolic Ca2+ that drives enhanced contraction (Katz & Lorell, 2000). Thus, increased mitochondrial energy production is required to support this elevated contractile response. Studies with dnMCU transgenic expression revealed that MCU plays an important role in cardiac pacemaker cell function. With dnMCU‐mediated MCU inhibition, stimulation of pacemaker cells with the β‐adrenergic agonist isoproterenol resulted in inhibited mitochondrial Ca2+ influx and impaired heart rate acceleration, suggesting an impairment in the coordination of the fight or flight response (Wu et al. 2015). In addition to pacemaker cells, MCU was shown to have an expanded role in cardiac fight or flight regulation as cardiomyocyte‐specific deletion of MCU resulted in impaired isoproterenol‐induced increase in mitochondrial matrix Ca2+ content (Kwong et al. 2015), which resulted in blunted isoproterenol‐stimulated respiration (Kwong et al. 2015; Luongo et al. 2015), decreased isoproterenol‐induced NADH production (Luongo et al. 2015) and depressed cardiac contractility in response to acute isoproterenol challenge (Kwong et al. 2015; Luongo et al. 2015). It should be noted, however, that the MCU‐constitutive KO animals did not demonstrate similar functional deficits in response to adrenergic stress (Holmstrom et al. 2015), once again highlighting the differential effects of constitutive global MCU deletion versus induced acute MCU deletion in the adult heart.
Importantly, with prolonged catecholamine stimulation, the differences between control and MCU cardiac KO animals were abolished (Kwong et al. 2015) as over an expanded time frame, MCU cardiac KO animals were able to match the matrix Ca2+ accumulation, mitochondrial respiration, and cardiac function of controls (Kwong et al. 2015). Interestingly, reminiscent of the observations in mice, isolated guinea pig cardiomyocytes treated with Ru360 and treated with isoproterenol displayed a slight trend for reduced cytosolic Ca2+ with acute adrenergic stimulation, but this difference was abrogated over time (Kohlhaas et al. 2010). Strikingly, however, guinea pig cardiomyocytes displayed a sustained depressed NADPH production with prolonged isoproterenol, suggesting a sustained bioenergetic mismatch (Kohlhaas et al. 2010), a difference that could potentially be attributed to the differences in model systems (discussed further below). Nevertheless, translating the deficit in the acute isoproterenol response observed in the mouse heart to the whole animal, MCU cardiac KO animals displayed impaired running capacity when challenged by an enforced immediate sprint protocol, yet performed like controls when afforded a long and slow warm‐up (Kwong et al. 2015).
Together, the studies using targeted genetic approaches support the concept that MCU and the uniplex transduce a fast Ca2+ signal that allows mitochondria to match energy output to increased contractile demand and highlight the possibility of modulating mitochondrial Ca2+ influx to enhance cardiac function. An important point of consideration, however, as we grow our understanding of MCU's role in cardiac physiology, is that at present, much of our knowledge is based on mouse modelling. As the resting heart rate in mice (450–750 beats min–1) is much higher than that of larger animals like guinea pigs (200–300 beats min–1) and humans (60–100 beats min–1), murine cardiac mitochondria are challenged with much more Ca2+ than that of larger animals. As such, the functions of mitochondrial Ca2+ signalling, and perhaps even uniplex activity and regulation, may be very different between excitable versus non‐excitable tissues, and also across species. Indeed, it has been demonstrated that uniplex activity varies between tissues and that cardiac mitochondria display low current density as compared to mitochondria from other sources (Fieni et al. 2012). This decreased current density may be one mechanism whereby cardiac mitochondria can withstand the cyclical Ca2+ elevations of excitation–contraction coupling to prevent mitochondrial Ca2+ overload (Fieni et al. 2012). As an extension of this idea, one possibility might be that cardiac uniplex activity and regulation differ amongst species, thereby accounting for species‐specific differences in cardiac function. At present, data from the mouse models point to the possibility that uniplex function is restricted to acute mitochondrial Ca2+ influx, which opens the door for alternative MCU‐independent mechanisms of mitochondrial Ca2+ import. What are the molecular identities of these putative additional mitochondrial Ca2+ transporters? Indeed, mitochondrial Ca2+ current has been detected in cells with MCU knockdown (Bondarenko et al. 2013) and both the transient receptor potential canonical 3 (TRPC3) and mitochondria localized ryanodine receptor 1 (RyR1) have been suggested to play a role in mitochondrial Ca2+ influx (Ryu et al. 2010; Feng et al. 2013). But certainly, additional studies are needed to determine whether TRPC3 and RyR1 contribute to cardiac mitochondrial Ca2+ handling, whether additional yet unidentified mechanisms contribute to mitochondrial Ca2+ influx and cardiac energetics, and finally, how these mechanisms might interact with the uniplex.
MCU's role in calcium overload‐induced death in the heart
As mentioned above, mitochondrial Ca2+ signalling not only impacts energetics but can also engage cellular death pathways. Under physiological conditions, the mitochondrial inner membrane is impermeable to most small molecules and ions, and the electron transport chain establishes an electrochemical gradient that is harnessed by the ATP synthase to generate ATP. Under pathological conditions, however, it is recognized that mitochondrial Ca2+ overload triggers the opening of the mitochondrial permeability transition pore, which allows for free passage of solutes < 1.5 kDa in size (Haworth & Hunter, 1979; Hunter & Haworth, 1979; Kwong & Molkentin, 2015). This results in inner membrane permeabilization, membrane potential collapse, ATP synthesis impairment, mitochondrial swelling, rupture and cell death (Kwong & Molkentin, 2015). This mitochondrial Ca2+–MPTP signalling axis has long been postulated to contribute significantly to the cardiomyocyte death observed following ischaemia–reperfusion injury. MPTP inhibition either pharmacologically or via genetic ablation of MPTP constituents has shown great promise in preventing cardiomyocyte death (reviewed in Kwong & Molkentin, 2015). Since the uniplex is a major mode of mitochondrial Ca2+ influx, uniplex inhibition represents an attractive alternative means to prevent Ca2+ overload activation of the MPTP and subsequent death in the heart. Indeed, pharmacological inhibition of the uniplex with the MCU‐specific inhibitors ruthenium red and its derivative Ru360 have been highly effective in limiting cardiac ischaemic injury (Garcia‐Rivas et al. 2006; Zhang et al. 2006).
While studies using ruthenium red and Ru360 to inhibit mitochondrial Ca2+ have been overwhelming in their support for uniplex inhibition as a means to prevent Ca2+ overload‐induced cell death both in vitro and in vivo (Groskreutz et al. 1992; Dessi et al. 1995; Garcia‐Rivas et al. 2006; Zhang et al. 2006; Qiu et al. 2013), studies using gene targeted mouse models of uniplex inactivation have been less clear. MCU‐constitutive KO mice display resistance to Ca2+ overload‐induced MPTP activation, but surprisingly, no protection against in vivo cardiac ischaemia–reperfusion injury (Pan et al. 2013). In contrast, the MCU cardiac KO mice with adult induction of MCU ablation not only displayed inhibited Ca2+‐stimulated MPTP opening, but also showed greatly reduced cardiomyocyte death following in vivo cardiac ischaemia–reperfusion injury (Kwong et al. 2015; Luongo et al. 2015).
What accounts for the differences in death between these two animal models? The answer may lie in the consequences of constitutive versus acute MCU deletion – which to date, are not fully understood. The MCU‐constitutive KO mice display changes in metabolism that cause a shift away from oxidative pathways, as well as insensitivity to the MPTP inhibitor cyclosporine A (Pan et al. 2013). Similar alterations were not observed in an MCU cardiac KO model (Kwong et al. 2015; Luongo et al. 2015). These findings hint at the possibility that long‐term MCU deletion may cause global gene expression changes that we have not fully catalogued, via mechanisms that to date are unknown. Since acute MCU inhibition is highly protective against Ca2+ overload‐induced death, controlled and reversible inhibition of mitochondrial Ca2+ influx may be a strategy to prevent cardiomyocyte demise following ischaemia–reperfusion injury, and studies directly comparing the two models will be important in the design of uniplex‐targeted therapeutics.
A role for MCU beyond mitochondria?
As discussed above, the uniplex has well recognized roles in regulating cardiac biology through its immediate actions on mitochondrial energetics and mitochondrial death pathways. In addition to proximal effects on mitochondria, however, does uniplex signalling extend beyond the confines of mitochondrial biology to regulate broader aspects of cellular function? Hints at a wider role come from the MCU‐constitutive KO mice, as these were 30% smaller, and displayed depressed pyruvate dehydrogenase activity and chronic acidosis (Pan et al. 2013), suggesting the possibility that chronic inhibition of MCU signalling results in widespread metabolic changes that intersect with global growth pathways. Recent work on MCU in skeletal muscle also supports a connection between the uniporter and growth pathways as adeno‐associated virus (AAV)‐mediated MCU overexpression enhanced mitochondrial Ca2+ influx and caused myofibre hypertrophy, while AAV delivery of MCU shRNA inhibited matrix Ca2+ influx and resulted in myofibre atrophy (Mammucari et al. 2015). Further, AAV–MCU activated both the Akt–GSK3 α/β–4E‐BP1 growth signalling axis and the PCG1α mitochondriogenesis pathway, while AAV–shMCU caused the opposite effect (Mammucari et al. 2015). Collectively, these findings suggest that the uniplex may link mitochondrial energetics to global growth programmes. These findings, however, need to be validated in the MCU global knockout and loxP‐targeted mouse models, and it remains to be determined if the uniplex can control similar pathways in the heart.
Perspectives
The study of mitochondrial Ca2+ dynamics has undergone a molecular revolution. Once known only as a phenomenon that could be inhibited by ruthenium red and its derivatives, the mitochondrial Ca2+ import machinery has now grown into the multiprotein assembly we now know as the uniplex. Yet, the molecular landscape of the uniplex may still be evolving. With a growing list of uniplex regulatory subunits and regulators, as well as a potential new framework for the uniplex pore structure observed in invertebrates, understanding the precise molecular architecture of the uniplex in the mammalian heart, and defining regulators that impact cardiac mitochondrial function will be of great importance as we move to developing new tools to modulate uniplex function. The mouse models of MCU inactivation have revealed roles for the uniplex in regulating two very different pathways: cardiac metabolic contraction coupling, and Ca2+ overload‐induced death. Moving forward, it will be important to understand if the uniplex's role in physiological Ca2+ signalling can be separated from its pathological roles and if there are regulatory elements that are specific for energy production versus death. Studies on the constitutive and inducible cardiac MCU deletion models have also illuminated differences between long‐term and short‐term inhibition of uniplex‐dependent mitochondrial Ca2+ influx. Certainly studies thus far support acute uniplex inhibition as a therapeutic avenue of great interest to restrict cardiomyocyte loss following ischaemia–reperfusion injury. Therefore, understanding the ramifications of long term uniplex inhibition as well as how mitochondrial Ca2+ signalling influences global cardiac gene expression will be critical as we move towards the goal of developing new strategies to modulate uniplex function to enhance cardiac function by augmenting mitochondrial energetic output while limiting cardiomyocyte death.
Additional information
Competing interests
None declared.
Funding
This work is supported by grant funding from the American Heart Association (16SDG26420043).
Biography
Jennifer Kwong is an Assistant Professor in the Division of Cardiovascular Biology and Department of Pediatrics at Emory University School of Medicine. She received her doctoral degree in neuroscience from Cornell University where she focused on primary mitochondrial disorders and neurodegenerative diseases. She conducted her postdoctoral training in cardiovascular biology with Dr Jeffery Molkentin at Cincinnati Children's Hospital Medical Center, where she focused on mitochondrial control of cell death in the heart. The research in her laboratory at Emory focuses on mitochondrial signalling and regulation of the physiology and pathology of excitable tissues.

References
- Balaban RS (2009). The role of Ca2+ signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim Biophys Acta 1787, 1334–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher‐Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V & Mootha VK (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM (2002). Cardiac excitation–contraction coupling. Nature 415, 198–205. [DOI] [PubMed] [Google Scholar]
- Bers DM (2008). Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 70, 23–49. [DOI] [PubMed] [Google Scholar]
- Bondarenko AI, Jean‐Quartier C, Malli R & Graier WF (2013). Characterization of distinct single‐channel properties of Ca2+ inward currents in mitochondria. Pflugers Arch 465, 997–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton RM (2009). Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta 1787, 1309–1316. [DOI] [PubMed] [Google Scholar]
- Dessi F, Ben‐Ari Y & Charriaut‐Marlangue C (1995). Ruthenium red protects against glutamate‐induced neuronal death in cerebellar culture. Neurosci Lett 201, 53–56. [DOI] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabo I & Rizzuto R (2011). A forty‐kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Li H, Tai Y, Huang J, Su Y, Abramowitz J, Zhu MX, Birnbaumer L & Wang Y (2013). Canonical transient receptor potential 3 channels regulate mitochondrial calcium uptake. Proc Natl Acad Sci USA 110, 11011–11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieni F, Lee SB, Jan YN & Kirichok Y (2012). Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat Commun 3, 1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Rivas GdeJ, Carvajal K, Correa F & Zazueta C (2006). Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post‐ischaemic functional recovery in rats in vivo. Br J Pharmacol 149, 829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths EJ & Halestrap AP (1995). Mitochondrial non‐specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J 307, 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groskreutz JL, Bronk SF & Gores GJ (1992). Ruthenium red delays the onset of cell death during oxidative stress of rat hepatocytes. Gastroenterology 102, 1030–1038. [DOI] [PubMed] [Google Scholar]
- Harris DA & Das AM (1991). Control of mitochondrial ATP synthesis in the heart. Biochem J 280, 561–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haworth RA & Hunter DR (1979). The Ca2+‐induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys 195, 460–467. [DOI] [PubMed] [Google Scholar]
- Hoffman NE, Chandramoorthy HC, Shanmughapriya S, Zhang XQ, Vallem S, Doonan PJ, Malliankaraman K, Guo S, Rajan S, Elrod JW, Koch WJ, Cheung JY & Madesh M (2014). SLC25A23 augments mitochondrial Ca2+ uptake, interacts with MCU, and induces oxidative stress‐mediated cell death. Mol Biol Cell 25, 936–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmstrom KM, Pan X, Liu JC, Menazza S, Liu J, Nguyen TT, Pan H, Parks RJ, Anderson S, Noguchi A, Springer D, Murphy E & Finkel T (2015). Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J Mol Cell Cardiol 85, 178–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter DR & Haworth RA (1979). The Ca2+‐induced membrane transition in mitochondria. I. The protective mechanisms. Arch Biochem Biophys 195, 453–459. [DOI] [PubMed] [Google Scholar]
- Jouaville LS, Pinton P, Bastianutto C, Rutter GA & Rizzuto R (1999). Regulation of mitochondrial ATP synthesis by calcium: evidence for a long‐term metabolic priming. Proc Natl Acad Sci USA 96, 13807–13812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamer KJ & Mootha VK (2015). The molecular era of the mitochondrial calcium uniporter. Nat Rev Mol Cell Biol 16, 545–553. [DOI] [PubMed] [Google Scholar]
- Kamer KJ, Sancak Y & Mootha VK (2014). The uniporter: from newly identified parts to function. Biochem Biophys Res Commun 449, 370–372. [DOI] [PubMed] [Google Scholar]
- Katz AM & Lorell BH (2000). Regulation of cardiac contraction and relaxation. Circulation 102, IV69–IV74. [DOI] [PubMed] [Google Scholar]
- Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Bohm M, O'Rourke B & Maack C (2010). Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 121, 1606–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM & Molkentin JD (2015). The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep 12, 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JQ & Molkentin JD (2015). Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab 21, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, Koch WJ, Cheung JY, Madesh M & Elrod JW (2015). The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep 12, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallilankaraman K, Cardenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenar T, Csordas G, Madireddi P, Yang J, Muller M, Miller R, Kolesar JE, Molgo J, Kaufman B, Hajnoczky G, Foskett JK & Madesh M (2012a). MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol 14, 1336–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, Rothberg BS, Mak DO, Foskett JK & Madesh M (2012b). MICU1 is an essential gatekeeper for MCU‐mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell 151, 630–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammucari C, Gherardi G, Zamparo I, Raffaello A, Boncompagni S, Chemello F, Cagnin S, Braga A, Zanin S, Pallafacchina G, Zentilin L, Sandri M, De Stefani D, Protasi F, Lanfranchi G & Rizzuto R (2015). The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep 10, 1269–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E & Steenbergen C (2008). Mechanisms underlying acute protection from cardiac ischemia‐reperfusion injury. Physiol Rev 88, 581–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrenius S & Nicotera P (1994). The calcium ion and cell death. J Neural Transm Suppl 43, 1–11. [PubMed] [Google Scholar]
- Oxenoid K, Dong Y, Cao C, Cui T, Sancak Y, Markhard AL, Grabarek Z, Kong L, Liu Z, Ouyang B, Cong Y, Mootha VK & Chou JJ (2016). Architecture of the mitochondrial calcium uniporter. Nature 533, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E & Finkel T (2013). The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol 15, 1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabo I, De Stefani D & Rizzuto R (2014). MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell 53, 726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paupe V, Prudent J, Dassa EP, Rendon OZ & Shoubridge EA (2015). CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab 21, 109–116. [DOI] [PubMed] [Google Scholar]
- Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE & Mootha VK (2010). MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 467, 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piquereau J, Caffin F, Novotova M, Lemaire C, Veksler V, Garnier A, Ventura‐Clapier R & Joubert F (2013). Mitochondrial dynamics in the adult cardiomyocytes: which roles for a highly specialized cell? Front Physiol 4, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, Taneja N, Oshea J, Koteliansky V & Mootha VK (2013). MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One 8, e55785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan RK, Qi F, Beard DA & Dash RK (2010). Characterization of membrane potential dependency of mitochondrial Ca2+ uptake by an improved biophysical model of mitochondrial Ca2+ uniporter. PLoS One 5, e13278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Tan YW, Hagenston AM, Martel MA, Kneisel N, Skehel PA, Wyllie DJ, Bading H & Hardingham GE (2013). Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun 4, 2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabo I & Rizzuto R (2013). The mitochondrial calcium uniporter is a multimer that can include a dominant‐negative pore‐forming subunit. EMBO J 32, 2362–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu SY, Beutner G, Dirksen RT, Kinnally KW & Sheu SS (2010). Mitochondrial ryanodine receptors and other mitochondrial Ca2+ permeable channels. FEBS Lett 584, 1948–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Markhard AL, Kitami T, Kovacs‐Bogdan E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O & Mootha VK (2013). EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342, 1379–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, Timbalia SA, Goldman SJ, Breves SL, Corbally DP, Nemani N, Fairweather JP, Cutri AR, Zhang X, Song J, Jana F, Huang J, Barrero C, Rabinowitz JE, Luongo TS, Schumacher SM, Rockman ME, Dietrich A, Merali S, Caplan J, Stathopulos P, Ahima RS, Cheung JY, Houser SR, Koch WJ, Patel V, Gohil VM, Elrod JW, Rajan S & Madesh M (2016). MCUR1 is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep 15, 1673–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vais H, Mallilankaraman K, Mak DO, Hoff H, Payne R, Tanis JE & Foskett JK (2016). EMRE is a matrix Ca2+ sensor that governs gatekeeping of the mitochondrial Ca2+ uniporter. Cell Rep 14, 403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Haasteren G, Li S, Muda M, Susini S & Schlegel W (1999). Calcium signalling and gene expression. J Recept Signal Transduct Res 19, 481–492. [DOI] [PubMed] [Google Scholar]
- Wu Y, Rasmussen TP, Koval OM, Joiner ML, Hall DD, Chen B, Luczak ED, Wang Q, Rokita AG, Wehrens XH, Song LS & Anderson ME (2015). The mitochondrial uniporter controls fight or flight heart rate increases. Nat Commun 6, 6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zazueta C, Sosa‐Torres ME, Correa F & Garza‐Ortiz A (1999). Inhibitory properties of ruthenium amine complexes on mitochondrial calcium uptake. J Bioenerg Biomembr 31, 551–557. [DOI] [PubMed] [Google Scholar]
- Zhang SZ, Gao Q, Cao CM, Bruce IC & Xia Q (2006). Involvement of the mitochondrial calcium uniporter in cardioprotection by ischemic preconditioning. Life Sci 78, 738–745. [DOI] [PubMed] [Google Scholar]