

Abstract
Hepatic steatosis renders liver more vulnerable to ischemia/reperfusion injury (IRI), which commonly occurs in transplantation, trauma, and liver resection. The underlying mechanism is not fully characterized. We aimed to clarify the role of mechanistic target of rapamycin (mTOR) signaling in hepatic ischemia/reperfusion injury (HIRI) in normal and steatotic liver using Alb-TSC1−/− (AT) and Alb-mTOR−/− (Am) transgenic mice. Steatotic liver induced by high-fat diet was more vulnerable to IRI. Activation of hepatic mTOR in AT mice decreased lipid accumulation attenuated HIRI as measured by terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining, circulating levels of alanine aminotransferase and lactate dehydrogenase, and inflammatory mediators such as monocyte chemoattractant protein 1 (MCP-1), TNF-α, and IL-6 and hepatic cleaved caspase 3 in mice fed either a normal chow diet or a high-fat diet. The effects of mTOR activation on hepatic cleaved caspase 3 were reversed by rapamycin, an inhibitor of mTOR signaling. Inhibition of hepatic mTOR in Am mice increased hepatic lipid deposition and HIRI. The increment in hepatic susceptibility to IRI was significantly attenuated by pretreatment with IKKβ inhibitor. Further, suppression of mTOR facilitated nuclear translocation of NF-κB p65. In conclusion, our study suggests that mTOR activity in hepatocytes decreases hepatic vulnerability to injury through a mechanism dependent on NF-κB proinflammatory cytokine signaling pathway in both normal and steatotic liver.—Li, Z., Zhang, J., Mulholland, M., Zhang, W. mTOR activation protects liver from ischemia/reperfusion-induced injury through NF-κB pathway.
Keywords: hepatic steatosis, IKKβ, nuclear translocation, TSC1
Western countries are experiencing an epidemic of obesity and associated hepatic steatosis. Steatotic livers are particularly vulnerable to damage in the setting of traumatic injury or operative stress, responding with hepatocyte apoptosis and necrosis (1). As a result, liver resection in patients with steatosis is associated with a higher risk of postoperative morbidity. Current therapy for liver injury in the setting of steatosis is limited. Pharmacologic agents such as antioxidants, inflammatory regulators, and microcirculatory mediators have been investigated in attempts to protect steatotic livers from injury in animal models, with mixed success. Defining key endogenous pathways that protect hepatocytes from injury is crucial to provision of safe and effective therapy in the setting of hepatic steatosis.
The mechanistic target of rapamycin (mTOR), a PI3K-related serine/threonine kinase, plays vital roles in cell growth, proliferation, and survival. mTOR is the core component of 2 distinct complexes: mTORC1 (complex 1) and mTORC2 (complex 2). mTORC1 is a central integrator of a wide range of physiologic cues such as amino acid levels and energy status to promote cellular growth and proliferation (2). Through the S6 kinase/S6 axis and 4E-BP1 phosphorylation, mTORC1 signaling controls synthesis of proteins in a wide variety of cells. mTORC1 is the only complex sensitive to acute rapamycin treatment (3). mTORC2 phosphorylates Akt protein kinase B and contains mTOR and rictor (4). mTORC2 was originally identified as an important regulator of the cytoskeleton (5). Recent studies indicate that mTORC2 also regulates cell proliferation and metabolism (6), although its underlying mechanism remains largely unknown.
The role of mTORC1 in energy metabolism, glucose regulation, and lipid homeostasis has been controversial as a result of conflicting results from studies using genetic and pharmacologic interventions. Absence of S6 kinase 1, the mTOR downstream target, protects against diet-induced obesity and improves insulin sensitivity in mice (7). In contrast, administration of rapamycin or its analog, rapalogs, in clinical trials has been reported to cause hyperlipidemia and hypercholesterolemia, as well as activation of gluconeogenesis in liver (8, 9). In rodents, rapamycin administration is associated with the development of nonalcoholic fatty liver disease (10) and impairment of glucose tolerance and insulin sensitivity (11).
The role of mTORC1 in development of steatosis is also uncertain. Hepatic mTORC1 activity has been reported to be elevated in animal models of diet-induced obesity (12) and to be associated with dyslipidemia via increased hepatic very-low-density lipoprotein secretion. Knockdown of hepatic S6 kinase 1 was correlated with reduced steatosis (13). In contrast, genetic manipulation to increase hepatic mTORC1 was not associated with hepatic fat accumulation, even in animals exposed to a high-fat diet (HFD) (14). The effect of mTOR signaling on hepatocellular protection in the setting of steatosis is currently unknown.
In the present study, we demonstrated that hepatic mTOR activity affects hepatocellular injury induced by ischemia/reperfusion in normal and steatotic liver. Activation of hepatic mTOR signaling by deletion of tuberous sclerosis complex 1 (TSC1), the upstream inhibitor of mTOR, decreased lipid accumulation and protected liver from ischemia/reperfusion injury (IRI). Knockdown of hepatic mTOR signaling by deletion of mTOR increased lipid deposition and rendered liver more vulnerable to IRI. Further, mTOR knockdown increased the nuclear translocation of NF-κB p65. Inhibition of IKKβ significantly attenuated hepatic injury induced by ischemia/reperfusion in mTOR-deficient mice. The effects of mTOR signaling on hepatocellular protection may thus be mediated by inhibition of the IKKβ/NF-κB signaling pathway.
MATERIALS AND METHODS
Materials
Phospho-S6 (Ser235/236) and S6 rabbit monoclonal antibodies, phosphor-mTOR (Ser2448) and mTOR rabbit monoclonal antibodies, rabbit anti-TSC1 antibody, and rabbit anti-β-actin antibody were purchased from Cell Signaling Technology (Danvers, MA, USA). Rabbit anti–caspase 3 and NF-κB p65 antibodies were obtained from Abcam (Cambridge, MA, USA). Aprotinin was purchased from Amersham Biosciences (Pittsburgh, PA, USA). Trizol reagent and the reverse transcription system were from Thermo Fisher Scientific (Waltham, MA, USA). Lactate dehydrogenase (LDH) activity assay kit and alanine aminotransferase (ALT) activity assay kit were from Sigma-Aldrich (St. Louis, MO, USA). ELISA kits for TNF-α, IL-6, and monocyte chemoattractant protein 1 (MCP-1) were purchased from R&D Systems (Minneapolis, MN, USA). In situ cell death detection peroxidase kit was from Roche (Basel, Switzerland).
Animals and treatments
Animals
Animals were housed in a temperature-controlled environment with 12-h light/dark cycles, and access to food and water ad libitum. All experimental protocols were approved by the University Committee on Use and Care of Animals of the University of Michigan. Sixteen-week-old male C57BL/6J lean mice and HFD-induced obese mice were used in the present study. Albumin (Alb)-Cre mice, TSC1floxp/floxp mice, and mTORfloxp/floxp mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA).
Generation of Alb-TSC1−/− and Alb-mTOR−/− mice
Alb-TSC1−/− (AT) and Alb-mTOR−/− (Am) mice were generated by cross-breeding Alb-Cre mice with TSC1floxp/floxp mice and mTORfloxp/floxp mice individually. Deletion of hepatic TSC1 or mTOR was confirmed by genotyping and Western blot analysis.
Diet
Where indicated, 4-wk-old mice were assigned to receive standard laboratory chow or a HFD (60% fat, D12492; Research Diets, New Brunswick, NJ, USA) for 12 wk.
Hepatic ischemia/reperfusion-induced injury
Wild-type or genetically engineered male mice (8–16 wk old) were anesthetized with ketamine (100 mg/kg body weight) and xylazine (6 mg/kg body weight) by intraperitoneal injection. The liver was exposed by midline laparotomy, and the hepatic artery and the portal vein were clamped with an atraumatic vascular clip. This procedure has been reported to cause segmental hepatic ischemia and to prevent mesenteric venous congestion by allowing portal decompression throughout the right and caudate lobes of the liver. Blood flow was interrupted for 75 min to induce hepatic ischemia, and reflow was initiated by removal of the vascular clip to allow reperfusion for 6 h. At the end of reperfusion period, plasma and representative samples of the ischemic (middle part of median lobe) and unaffected hepatic lobes were collected and stored at −80°C.
Oil Red O staining
Hepatic tissue slices were washed by 1× PBS 3 times, and then fixed with 4% paraformaldehyde for 10 min. After being washed by water, slices were incubated in 0.3% Oil Red O staining solution for 1 h at room temperature. Samples were then counterstained with hematoxylin for 30 s, followed by washing in running water for more than 30 min. All slides were mounted with 90% glycerol and stored at 4°C before observation. Quantitation of Oil Red O staining was produced by ImageJ software (Image Processing and Analysis in Java; National Institutes of Health, Bethesda, MD, USA; http://imagej.nih.gov/).
In situ cell death detection
Cell death assay was performed according to the manufacturer’s instructions. Hepatic tissue slices were fixed with 4% paraformaldehyde, immersed in 3% H2O2 in methanol to block endogenous peroxidase, and permeabilized with 20 μg/ml proteinase K. TUNEL reaction was performed for 1 h with labeling mix in a 37°C incubator, and fluorescent signal was converted to peroxidase-labeled signal. DAB (3,3′-diaminobenzidine) substrate was added for reaction, and stained cells were analyzed under a light microscope. TUNEL staining quantitation was produced by ImageJ software.
Measurements of hepatic enzymes
Blood samples were transcardially collected after anesthesia in the presence of aprotinin (2 μg/ml) and heparin. Plasma was stored at −70°C before use. ALT and LDH were measured using commercial kits (Sigma-Aldrich) according to the manufacturer's instructions.
Western blot analysis
Liver tissue was isolated and then homogenized in lysis buffer. Proteins were subjected to SDS-PAGE with a 10% running gel, and then transferred to a PVDF membrane. Membranes were incubated for 1 h at room temperature with 5% fat-free milk in Tris-buffered saline containing Tween 20, followed by incubation overnight at 4°C with primary antibodies. Specific reaction was detected using IRDye-conjugated second antibody and visualized using an Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE, USA). ImageJ software was used to quantitate the density of specific bands.
ELISA assay
Hepatic concentrations of TNF-α, IL-6, and MCP-1 were measured using mouse ELISA kits from R&D Systems according to the manufacturer’s instructions.
Statistical analysis
All values were expressed as means ± sem. Statistical differences were evaluated by Student’s t test. A value of P < 0.05 denoted statistical significance. ImageJ software was used for histologic and Western blot analysis.
RESULTS
Steatotic liver injury induced by ischemia/reperfusion
Mice fed an HFD for 12 wk developed hepatic steatosis as measured by significant increases in histologic measures of lipid accumulation (Fig. 1A), hepatic triglyceride content (Fig. 1B), and increased transcription of genes related to hepatic lipogenesis, including nuclear receptors peroxisome proliferator-activated receptor γ2 (pparγ2) and sterol regulatory element-binding protein (srebf1) and key enzymes involved in triglyceride synthesis such as glycerol-3-phosphate acyltransferase 1 (gpam) and acyl CoA:diacylglycerol acyltransferase 1and 2 (dgat1/2) (Fig. 1B). Fatty acid synthase (fasn), one of the key enzymes for de novo synthesis of fatty acids, was decreased by HFD, indicating a negative feedback regulation. Obese mice demonstrated more severe damage after hepatic ischemia/reperfusion injury (HIRI) as measured by significant increases in TUNEL-positive cells (Fig. 1C), levels of serum ALT and LDH (Fig. 1D), and mRNA levels of inflammatory mediators such as MCP-1, TNF-α, and IL-6 (Fig. 1E). HFD also caused greater expression of NF-κB p65 and caspase 3 after HIRI (Fig. 1F).
Figure 1.

IRI in hepatic steatosis. Wild-type C57/BL6J male mice were fed NCD or HFD for 12 wk, then underwent hepatic ischemia (75 min) and reperfusion (6 h) to induce hepatic ischemia/reperfusion injury (HIRI). Results are expressed as means ± sem; n = 8 for each group. *P < 0.05 vs. NCD (Student’s t test). A) Liver histology. Oil Red O staining area and number of lipid droplets were analyzed by ImageJ software. B) Hepatic triglyceride content was measured by colorimetric assay; mRNA levels of lipid metabolism-related genes were determined by real-time quantitative PCR and normalized by glyceraldehyde phosphate dehydrogenase (GAPDH). Fold changes are expressed as means ± sem. C) TUNEL staining area and number of positive hepatocytes at low (×10) and high (×40) magnification. Positive staining area and cell numbers were calculated under ×40 magnification. D) Serum ALT and LDH levels were measured. E) Hepatic mRNA expression of MCP-1, TNF-α, and IL-6 were detected by real-time quantitative PCR and normalized by GAPDH. Fold changes are expressed as means ± sem. F) NF-κB p65 and cleaved caspase 3 were detected by Western blot; GAPDH was used as internal control. Quantitation was performed by ImageJ software, and all values were normalized by GAPDH. Representative results from 3 independent experiments are shown.
Activation of hepatic mTOR signaling decreases hepatic injury
To determine whether mTOR signaling affects the vulnerability of hepatocytes to injury, hepatic injury was induced by ischemia/reperfusion in AT mice fed a normal chow diet (NCD) or HFD for 12 wk. Hepatocyte-specific knockdown of TSC1 caused activation of mTOR signaling as measured by increased phosphorylation of mTOR and S6, the downstream target of mTOR signaling (Fig. 2A). In AT mice fed an NCD, hepatic lipid accumulation was decreased, as was hepatic triglyceride content relative to wild-type littermates (Fig. 2B). These mice demonstrated significant reduction in transcription of lipogenesis-related genes, such as pparγ2, srebf1, acetyl-CoA carboxylase 1 (acaca), fasn, gpam, and dgat1 (Fig. 2B). Activation of hepatic mTOR signaling attenuated HIRI as measured by TUNEL staining (Fig. 2C), circulating levels of ALT and LDH (Fig. 2D), inflammatory mediators, such as MCP-1, TNF-α, and IL-6 (Fig. 2E), and hepatic cleaved caspase 3 (Fig. 2F). The protective effects of mTOR activation are identical between male and female animals (Supplemental Fig. 1). The effects of mTOR activation on hepatic cleaved caspase 3 were reversed by rapamycin (1 mg/kg body weight, administered 15 min before ischemia and immediately before reperfusion), an inhibitor of mTOR signaling. Other pro- and anti-inflammatory cytokines, such as IL-1β and IL-10, were not altered by either mTOR activation or rapamycin injection (Supplemental Fig. 2).
Figure 2.

Effects of mTOR activation on HIRI. AT mice were generated by cross-breeding Alb-Cre mice with TSC1floxp/floxp mice. Animals were fed NCD for 16 wk and challenged with HIRI. Rapamycin (1 mg/kg) was administrated intravenously twice, 15 min before ischemia and then immediately before reperfusion. Results are expressed as means ± sem; n = 10 for each group. *P < 0.05 vs. wild-type mice; #P < 0.05 vs. AT mice (Student’s t test). A) Validation of mTOR activation in liver. Liver protein was extracted and used for Western blot. B) Histologic measurement of hepatic lipid content by Oil Red O staining and quantitated by ImageJ software; hepatic triglyceride content and mRNA levels of hepatic lipid metabolism–related genes are expressed as means ± sem. C) TUNEL staining and number of positive hepatocytes shown at low (×10) and high (×40) magnification. Positive staining area and cell numbers were calculated under ×40 magnification. D) Serum levels of ALT and LDH. E) Hepatic levels of inflammatory factors, including MCP-1, TNF-a, and IL-6, were detected by ELISA assay. F) Cleaved caspase 3 was detected by Western blot. β-Actin was used as internal control. Representative results from 3 independent experiments are shown.
Similar effects were observed in AT mice fed an HFD for 12 wk. Activation of hepatic mTOR by deletion of TSC1 in hepatocytes significantly decreased hepatic steatosis measured histologically, by hepatic triglyceride content, and by expression of genes related to lipid metabolism, such as pparγ2, srebf1, acaca, fasn, gpam, and dgat1 (Fig. 3A–C). Reduction in hepatic lipid was paralleled by attenuated hepatic injury. The number of TUNEL-positive hepatocytes, levels of plasma ALT and LDH, and hepatic cleaved caspase 3 were all suppressed in AT mice relative to wild-type animals (Fig. 3D–F).
Figure 3.
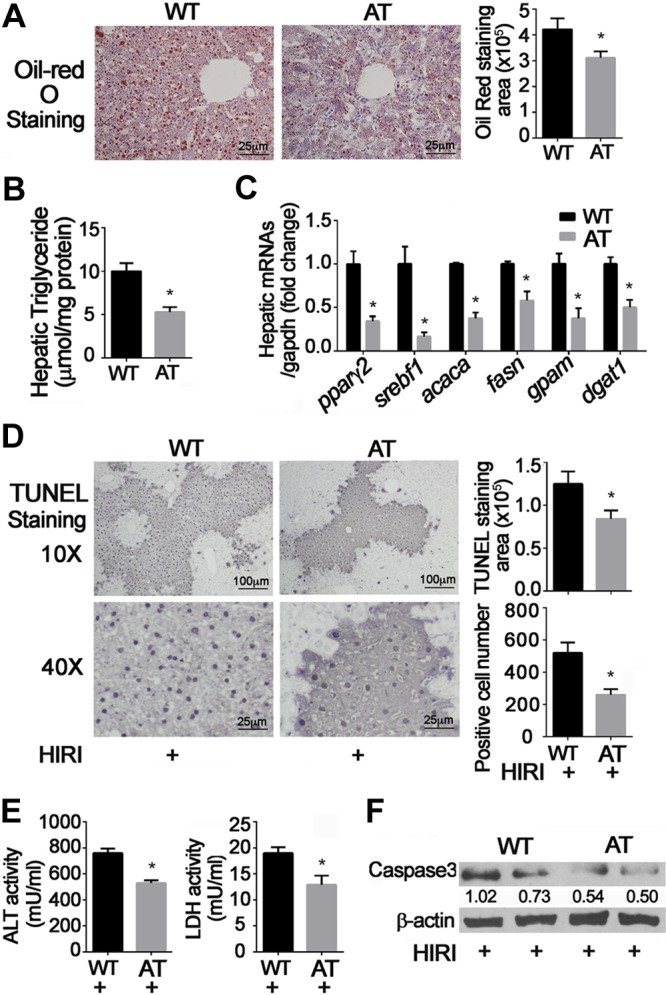
Effects of mTOR activation and HFD on HIRI. AT mice were generated by cross-breeding Alb-Cre mice with TSC1floxp/floxp mice. Four-week-old animals were fed HFD for 12 wk, then challenged with HIRI. Results are expressed as means ± sem; n = 10 for each group. *P < 0.05 vs. wild-type mice; #P < 0.05 vs. AT mice (Student’s t test). A) Hepatic lipid content was measured by Oil Red O staining. B) Hepatic triglyceride content. C) mRNA levels of hepatic lipid metabolism–related genes were detected and normalized by glyceraldehyde phosphate dehydrogenase (GAPDH); D) TUNEL staining and number of positive hepatocytes shown at low (×10) and high (×40) magnification. E) Serum levels of ALT and LDH. F) Cleaved caspase 3 was detected by Western blot. β-Actin was used as internal control. Representative results from 3 independent experiments are shown.
Deletion of hepatic mTOR renders hepatocytes more vulnerable to injury
Cross-breeding Alb-Cre mice with mTORfloxp/floxp mice generated the Am transgenic mice in which hepatocellular mTOR is conditionally deleted. In contrast to the findings in AT mice, Am animals demonstrated greater hepatic lipid accumulation, increased expression of transcription factor pparγ2 and srebf1, and increased transcription of enzymes encoded by acaca, fasn, gpam, and dgat1 (Fig. 4A).
Figure 4.

Effects of mTOR knockdown on HIRI. A–C) Am mice were generated by cross-breeding Alb-Cre mice with mTORfloxp/floxp mice. Animals were fed NCD for 16 wk, and then challenged with HIRI. Results are expressed as means ± sem; n = 10 for each group. *P < 0.05 vs. wild-type mice (Student’s t test). A) Hepatic lipid content measured by Oil Red O staining; hepatic triglyceride content and mRNA levels of hepatic lipid metabolism–related genes. B) TUNEL staining and number of positive hepatocytes; mRNA expression of MCP-1, TNF-α, and IL-6; serum levels of ALT and LDH. C) NF-κB and cleaved caspase 3 detected by Western blot analysis. β-Actin was used as internal control. D, E) Effects of IKKβ inhibition. Sixteen-week-old Am mice were pretreated with IKKβ inhibitor TPCA-1 (6.25 mg/kg body weight) before ischemia/reperfusion. Liver lysates were separated into cytoplasmic and nuclear fractions and used for Western blot analysis; n = 6 for each group. *P < 0.05 vs. wild-type mice under control conditions. #P < 0.05 vs. wild-type mice with HIRI (Student’s t test). D) Representative Western blots. E) TUNEL staining and number of positive hepatocytes; serum levels of ALT and LDH and mRNA expression of IL-6 and MCP-1.
Deletion of mTOR in hepatocytes markedly increased hepatic injury after ischemia/reperfusion. TUNEL staining was increased in Am mice challenged with ischemia/reperfusion relative to wild-type animals (Fig. 4B). HIRI induced greater increments in mRNA of hepatic inflammation-related genes such as MCP-1, TNF-α, and IL-6, and plasma ALT and LDH (Fig. 4B). Hepatic NF-κB p65 and cleaved caspase 3 (Fig. 4C) were also higher in Am mice relative to wild-type animals.
NF-κB-dependent effects were probed by using TPCA-1, an inhibitor of IKKβ. Pretreatment of Am mice with TPCA-1 (6.25 mg/kg body weight) significantly attenuated NF-κB p65 nuclear translocation and cleaved caspase 3 levels compared to wild-type mice (Fig. 4D). Blockage of NF-κB p65 nuclear translocation was associated with significant reduction in hepatocellular damage. Relative to control vehicle, TPCA-1 significantly reduced TUNEL staining and number of TUNEL-positive hepatocytes (Fig. 4E). Increments in plasma ALT and LDH activities, as well as hepatic IL-6 and MCP-1, were also suppressed in Am mice treated with TPCA-1 compared to mice receiving normal saline (Fig. 4E).
Hepatic mTOR signaling regulates nuclear translocation of NF-κB p65
IκBα inhibits NF-κB signaling by masking the nuclear localization signals of NF-κB proteins to keep them sequestered in an inactive state in the cytoplasm (15). IκBα phosphorylation, signal-induced ubiquitination, and proteasomal degradation are required for activation of NF-κB (16). Activation of hepatic mTOR significantly increased levels of IκBα in AT mice relative to wild-type littermates, suggesting a decrement of IκBα degradation. This change is associated with a marked decrement in levels of hepatic nuclear phosphor-NF-κB p65 and NF-κB p65 (Fig. 5A), indicating that activation of mTOR signaling inhibits the nuclear translocation of NF-κB p65. Total NF-κB p65 remained unchanged (Supplemental Fig. 3). Activation of NF-κB after IRI has been demonstrated to increase nuclear translocation of NF-κB p65, leading to subsequent activation of proinflammatory responses in a variety of organs.
Figure 5.

Effects of hepatic mTOR signaling on nuclear translocation of NF-κB p65. A) Hepatic nuclear proteins from AT mice with HIRI were extracted. Western blot analysis was performed to detect alterations of IκBα and NF-κB p65. β-Actin was used as internal control. Representative data from 3 individual experiments are shown. B) Hepatic proteins from AM mice were extracted and separated into cytoplasmic and nuclear fractions. Western blot analysis was performed to validate knockdown of mTOR and nuclear translocation of NF-κB p65. β-Actin was used as internal control of cytoplasmic proteins. Lamin B was used as internal control of nuclear proteins. Western blot was repeated 3 times using distinct samples. C) Hepatocytes isolated from wild-type mice were treated with TNF-α (100 ng/ml) or saline control for 12 h. Western blot was performed to measure changes in IκBα and NF-κB p65. Glyceraldehyde phosphate dehydrogenase (GAPDH) was used as internal control. Experiment was repeated at least 4 times. D) Hepatocytes isolated from mTORfloxp/floxp mice were treated with Ad-GFP or Cre (5 × 106 pfu/well) for 36 h, then exposed to TNF-α (100 ng/ml) for 12 h with saline as control vehicle. Western blot analysis was used to validate knockdown of mTOR and phosphorylation of NF-κB p65 and caspase 3. Lamin B and GAPDH were used as internal control. Experiment was repeated 3 times. E) Hepatocytes from wild-type mice were treated with TNF-α (100 ng/ml) with or without rapamycin (1 nM) for 12 h, and then probed for NF-κB p65 nuclear translocation by immunofluorescent staining. Positive nuclei indicated by arrows. NF-κB p65-positive nuclei were counted and expressed as means ± sem (n = 5). Experiment was repeated twice. P < 0.05 vs. TNF-α alone (Student’s t test).
To determine whether NF-κB signaling contributes to the increased damage of mTOR-deficient hepatocytes, we analyzed the nuclear translocation of NF-κB p65. Cytoplasmic and nuclear fractions of liver proteins were extracted and separated in samples from Am mice. Significant reduction in cytoplasmic and nuclear mTOR was observed in Am mice relative to wild-type animals (Fig. 5B), indicating knockdown of mTOR in hepatocytes. HIRI significantly increased levels of NF-κB p65 in both cytoplasmic and nuclear fractions. Relative to wild-type animals, Am mice demonstrated a significantly higher level of NF-κB p65 after HIRI (Fig. 5B). The change in NF-κB signaling was associated with increased localization of cleaved caspase 3 in nuclear fractions (Fig. 5B). Similar results were also observed in an in vitro model of hepatocellular damage induced by TNF-α (100 ng/ml). Treatment of hepatocytes with TNF-α for 12 h significantly decreased levels of IκBα, and increased phosphor-NF-κB p65 and NF-κB p65 levels (Fig. 5C). Hepatocytes isolated from mTORfloxp/floxp mice were treated with Cre adenovirus (Ad-Cre) or control green fluorescent protein (GFP) adenovirus (Ad) for 36 h, then exposed to TNF-α (100 ng/ml) for 12 h. Knockdown of mTOR led to significant decreases in mTOR signaling measured by phosphorylation of mTOR and S6 in hepatocytes (Fig. 5D). Associated with these changes, levels of phospho-NF-κB p65, total NF-κB p65, and cleaved caspase 3 were significantly increased by TNF-α in cultured hepatocytes treated with Ad-Cre adenovirus relative to Ad-GFP treatment (Fig. 5D). Further, inhibition of mTOR activity by rapamycin markedly increased nuclear translocation of NF-κB p65 induced by TNF-α in primary hepatocytes (Fig. 5E).
DISCUSSION
By using complementary genetic models with gain or loss of function of mTOR signaling in hepatocytes, we demonstrated that mTOR activity substantially alters the susceptibility of hepatocytes to IRI in both normal and steatotic liver. Activation of hepatic mTOR signaling by conditional deletion of TSC1 significantly attenuated hepatic damage induced by ischemia/reperfusion in Alb/TSC1−/− mice fed either standard chow or an HFD. The effects of mTOR activation were reversed by the mTOR inhibitor rapamycin. Conversely, inhibition of mTOR by conditional deletion of mTOR in hepatocytes markedly increased hepatic injury after ischemia/reperfusion in Alb/mTOR−/− mice. Consistent with these findings, previous studies have indicated that melatonin contributes to hepatocellular protection against IRI by activation of mTOR signaling (17). Similarly, NaHS has been shown to ameliorate HIRI by augmenting prosurvival, antiapoptotic, and antiinflammatory signals via mechanisms involving Nrf-2, and by accelerating hepatic regeneration via an Akt-p70S6k-dependent mechanism (18). In contrast, berberine preconditioning has been reported to dramatically attenuate histopathologic damage, restore liver function, and decrease oxidative stress and apoptosis after IRI (19). These alterations were associated with a reduction in mTOR signaling. Rapamycin has been demonstrated to protect liver from ischemia/reperfusion via inhibition of endoplasmic reticulum stress (20).
Our data provide strong evidence for an acute cytoprotective effect of mTOR signaling against hepatic injury. Substantial experimental and clinical evidence supports the efficacy of preventative approaches such as ischemia preconditioning; in addition, such approaches outperform the pharmacologic management of existing hepatic damage using antioxidants, inflammatory regulators, or microcirculatory mediators. The current findings suggest that targeting mTOR might be a new strategy for the prevention of hepatic injury in normal and steatotic liver.
Hepatic steatosis affects almost 30% of adults. Steatotic livers are particularly vulnerable to damage in the settings of traumatic injury and operative stress, although it is currently not known why steatosis renders hepatocytes more susceptible to injury. This study indicates that hepatic mTOR may link steatosis with IRI. Activation of mTOR signaling by conditional deletion of TSC1 in hepatocytes reduced hepatic lipid accumulation and rendered hepatocytes resistant to IRI in both normal and steatotic liver. In contrast, suppression of hepatic mTOR signaling by conditional deletion of mTOR in hepatocytes increased hepatic lipid content and augmented injury of hepatocytes by ischemia/reperfusion.
The relationship between hepatic mTOR activity and lipid metabolism has been controversial. Wang et al. (21) have demonstrated a positive relationship between hepatic mTOR activity and development of nonalcoholic fatty liver disease. Inhibition of mTOR in liver has been reported to impair sterol regulatory element binding protein function and to make mice resistant to development of hepatic steatosis and hypercholesterolemia induced by a high-fat and -cholesterol diet (22). Conversely, inhibition of mTOR signaling by rapamycin has been reported to increase hepatic free fatty acid and triglyceride content by stimulating the expression of PPARγ (10). Differences inherent in genetic and pharmacologic approaches to mTOR signaling may contribute to conflicting experimental results. While pharmacologic agents enable acute and reversible manipulation of mTOR activity, specificity is limited. Genetic manipulation is indispensable for determining the effect of chronic alteration of mTOR activity in hepatic lipid metabolism and cytoprotection of hepatocytes.
Conflicting results on hepatic injury have been reported for rapamycin and analogs (e.g., sirolimus), the mTOR inhibitor commonly used to treat cancer or as immunosuppressants for patients with organ transplants. Studies by Palmes et al. (23) and Fouraschen et al. (24) have shown that rapamycin increases hepatic injury and attenuates liver regeneration by suppressing the proliferation of hepatocytes, endothelial cells, and hepatic stellate cells mostly in the acute phase after hepatectomy and by down-regulating major cytokines and growth factors important for liver regeneration (23, 24). However, other studies have demonstrated the beneficial effect of rapamycin in hepatocellular protection (20, 25). Rapamycin protects hepatocytes from IRI and TNF-α-induced cell death by activation of the mTORC2-Akt signaling pathway. Our studies using transgenic mouse models with both gain and loss of function of mTOR signaling provide solid evidence supporting the critical role of mTOR in the protection of liver against IRI.
Our study identifies NF-κB as a downstream pathway mediating the effects of altered mTOR signaling on the vulnerability of hepatocytes to IRI. NF-κB proteins are composed of several homo- and heterodimer proteins that bind to common DNA elements (26). Phosphorylation of IκB proteins by the IκB kinase (IKK) and their subsequent degradation lead to the release of NF-κB, allowing its translocation to the nucleus to initiate transcription of proinflammatory or antiapoptotic mediators (27). NF-κB mediates both proinflammatory and antiapoptotic responses to inflammatory mediators, ensuring that hepatocytes are protected from cell death while appropriate inflammatory and immune responses are initiated. Early study has demonstrated that NF-κB knockout causes mice death through hepatocyte apoptosis. Our results suggest a proinflammatory effect of NF-κB in the hepatic damage induced by ischemia/reperfusion. One study reported that liver-specific knockout of NF-κB p65 subunit inhibits NF-κB activity but causes no hepatic damage and death (28). We postulate that depending on mTOR activity, the transactivation domain of the p65 subunit of NF-κB in hepatocytes is either activated by suppression of mTOR signaling or attenuated by activation of mTOR signaling. These reciprocal changes lead to the induction or inhibition of apoptotic mechanisms involving caspase 3. In support of this mechanism, inhibition of IKKβ significantly attenuated nuclear translocation of NF-κB p65 and decreased hepatic damage observed with mTOR suppression in hepatocytes. Consistent with the current observations, other investigators have reported that conditional deletion of IKKβ in hepatocytes impairs NF-κB activation with ischemia/reperfusion, which is associated with attenuation of cellular necrosis and lower levels of serum aminotransferases and decreased inflammatory infiltrate (29). The molecular mechanisms by which mTOR affects NF-κB activation remain to be explored. Our study suggests that mTOR signaling in hepatocytes may modulate NF-κB activity by altering levels of IκBα. Further study should explore whether mTOR signaling regulates IκBα degradation in hepatocytes.
In summary, this study indicates that mTOR activity in hepatocytes decreases hepatic vulnerability to injury through a mechanism dependent on NF-κB signaling pathway in both normal and steatotic liver. Targeting mTOR may thus provide a novel strategy for the prevention and therapy of hepatic injury.
ACKNOWLEDGMENTS
This work was supported, in part, by grants from the National Natural Science Foundation of China (81330010 and 81390354; to J.Z. and W.Z.), the American Diabetes Association (1-13-BS-225; to Z.L.), and the U.S. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (1R01DK110273-01A1; to M.M. and W.Z.). The authors declare no conflicts of interest.
Glossary
- acaca
acetyl-CoA carboxylase 1
- Ad
adenovirus
- Alb
albumin
- ALT
alanine aminotransferase
- Am
Alb-mTOR−/− transgenic mice
- AT
Alb-TSC1−/− transgenic mice
- dgat1/2
acyl CoA:diacylglycerol acyltransferase 1/2
- fasn
fatty acid synthase
- GFP
green fluorescent protein
- gpam
glycerol-3-phosphate acyltransferase 1
- HFD
high-fat diet
- HIRI
hepatic ischemia/reperfusion injury
- IRI
ischemia/reperfusion injury
- LDH
lactate dehydrogenase
- MCP-1
monocyte chemoattractant protein 1
- mTOR
mechanistic target of rapamycin
- NCD
normal chow diet
- pparγ2
peroxisome proliferator-activated receptor γ2
- srebf1
sterol regulatory element-binding protein
- TSC1
tuberous sclerosis complex 1
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick-end labeling
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
Z. Li and J. Zhang researched data; Z. Li, M. Mulholland, and W. Zhang contributed to discussion and reviewed and edited the article; and M. Mulholland and W. Zhang designed and supervised the experiments.
REFERENCES
- 1.Selzner M., Clavien P. A. (2001) Fatty liver in liver transplantation and surgery. Semin. Liver Dis. 21, 105–113 [DOI] [PubMed] [Google Scholar]
- 2.Gulati P., Thomas G. (2007) Nutrient sensing in the mTOR/S6K1 signalling pathway. Biochem. Soc. Trans. 35, 236–238 [DOI] [PubMed] [Google Scholar]
- 3.Wullschleger S., Loewith R., Hall M. N. (2006) TOR signaling in growth and metabolism. Cell 124, 471–484 [DOI] [PubMed] [Google Scholar]
- 4.Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 5.Jacinto E., Loewith R., Schmidt A., Lin S., Rüegg M. A., Hall A., Hall M. N. (2004) Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 6, 1122–1128 [DOI] [PubMed] [Google Scholar]
- 6.Laplante M., Sabatini D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Um S. H., Frigerio F., Watanabe M., Picard F., Joaquin M., Sticker M., Fumagalli S., Allegrini P. R., Kozma S. C., Auwerx J., Thomas G. (2004) Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431, 200–205 [DOI] [PubMed] [Google Scholar]
- 8.Houde V. P., Brûlé S., Festuccia W. T., Blanchard P. G., Bellmann K., Deshaies Y., Marette A. (2010) Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes 59, 1338–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Busaidy N. L., Farooki A., Dowlati A., Perentesis J. P., Dancey J. E., Doyle L. A., Brell J. M., Siu L. L. (2012) Management of metabolic effects associated with anticancer agents targeting the PI3K-Akt-mTOR pathway. J. Clin. Oncol. 30, 2919–2928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaveroux C., Eichner L. J., Dufour C. R., Shatnawi A., Khoutorsky A., Bourque G., Sonenberg N., Giguère V. (2013) Molecular and genetic crosstalks between mTOR and ERRα are key determinants of rapamycin-induced nonalcoholic fatty liver. Cell Metab. 17, 586–598 [DOI] [PubMed] [Google Scholar]
- 11.Xu G., Wang Z., Li Y., Li Z., Tang H., Zhao J., Xiang X., Ding L., Ma L., Yuan F., Fei J., Wang W., Wang N., Guan Y., Tang C., Mulholland M., Zhang W. (2012) Ghrelin contributes to derangements of glucose metabolism induced by rapamycin in mice. Diabetologia 55, 1813–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ai D., Baez J. M., Jiang H., Conlon D. M., Hernandez-Ono A., Frank-Kamenetsky M., Milstein S., Fitzgerald K., Murphy A. J., Woo C. W., Strong A., Ginsberg H. N., Tabas I., Rader D. J., Tall A. R. (2012) Activation of ER stress and mTORC1 suppresses hepatic sortilin-1 levels in obese mice. J. Clin. Invest. 122, 1677–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bae E. J., Xu J., Oh D. Y., Bandyopadhyay G., Lagakos W. S., Keshwani M., Olefsky J. M. (2012) Liver-specific p70 S6 kinase depletion protects against hepatic steatosis and systemic insulin resistance. J. Biol. Chem. 287, 18769–18780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kenerson H. L., Subramanian S., McIntyre R., Kazami M., Yeung R. S. (2015) Livers with constitutive mTORC1 activity resist steatosis independent of feedback suppression of Akt. PLoS One 10, e0117000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacobs M. D., Harrison S. C. (1998) Structure of an IkappaBalpha/NF-kappaB complex. Cell 95, 749–758 [DOI] [PubMed] [Google Scholar]
- 16.Palombella V. J., Rando O. J., Goldberg A. L., Maniatis T. (1994) The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell 78, 773–785 [DOI] [PubMed] [Google Scholar]
- 17.Kang J. W., Cho H. I., Lee S. M. (2014) Melatonin inhibits mTOR-dependent autophagy during liver ischemia/reperfusion. Cell. Physiol. Biochem. 33, 23–36 [DOI] [PubMed] [Google Scholar]
- 18.Shimada S., Fukai M., Wakayama K., Ishikawa T., Kobayashi N., Kimura T., Yamashita K., Kamiyama T., Shimamura T., Taketomi A., Todo S. (2015) Hydrogen sulfide augments survival signals in warm ischemia and reperfusion of the mouse liver. Surg. Today 45, 892–903 [DOI] [PubMed] [Google Scholar]
- 19.Sheng M., Zhou Y., Yu W., Weng Y., Xu R., Du H. (2015) Protective effect of Berberine pretreatment in hepatic ischemia/reperfusion injury of rat. Transplant. Proc. 47, 275–282 [DOI] [PubMed] [Google Scholar]
- 20.Zhu J., Hua X., Li D., Zhang J., Xia Q. (2015) Rapamycin attenuates mouse liver ischemia and reperfusion injury by inhibiting endoplasmic reticulum stress. Transplant. Proc. 47, 1646–1652 [DOI] [PubMed] [Google Scholar]
- 21.Wang Y., Shi M., Fu H., Xu H., Wei J., Wang T., Wang X. (2010) Mammalian target of the rapamycin pathway is involved in non-alcoholic fatty liver disease. Mol. Med. Rep. 3, 909–915 [DOI] [PubMed] [Google Scholar]
- 22.Wang Y., Viscarra J., Kim S. J., Sul H. S. (2015) Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 16, 678–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palmes D., Zibert A., Budny T., Bahde R., Minin E., Kebschull L., Holzen J., Schmidt H., Spiegel H. U. (2008) Impact of rapamycin on liver regeneration. Virchows Arch. 452, 545–557 [DOI] [PubMed] [Google Scholar]
- 24.Fouraschen S. M., de Ruiter P. E., Kwekkeboom J., de Bruin R. W., Kazemier G., Metselaar H. J., Tilanus H. W., van der Laan L. J., de Jonge J. (2013) mTOR signaling in liver regeneration: rapamycin combined with growth factor treatment. World J. Transplant. 3, 36–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu J., Lu T., Yue S., Shen X., Gao F., Busuttil R. W., Kupiec-Weglinski J. W., Xia Q., Zhai Y. (2015) Rapamycin protection of livers from ischemia and reperfusion injury is dependent on both autophagy induction and mammalian target of rapamycin complex 2-Akt activation. Transplantation 99, 48–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wan F., Lenardo M. J. (2009) Specification of DNA binding activity of NF-kappaB proteins. Cold Spring Harb. Perspect. Biol. 1, a000067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wertz I. E., Dixit V. M. (2010) Signaling to NF-kappaB: regulation by ubiquitination. Cold Spring Harb. Perspect. Biol. 2, a003350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ringelhan M., Schmid R. M., Geisler F. (2012) The NF-κB subunit RelA/p65 is dispensable for successful liver regeneration after partial hepatectomy in mice. PLoS One 7, e46469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luedde T., Assmus U., Wüstefeld T., Meyer zu Vilsendorf A., Roskams T., Schmidt-Supprian M., Rajewsky K., Brenner D. A., Manns M. P., Pasparakis M., Trautwein C. (2005) Deletion of IKK2 in hepatocytes does not sensitize these cells to TNF-induced apoptosis but protects from ischemia/reperfusion injury. J. Clin. Invest. 115, 849–859 [DOI] [PMC free article] [PubMed] [Google Scholar]