

Abstract
The innate immune response is a central element of the initial defense against bacterial and viral pathogens. Macrophages are key innate immune cells that upon encountering pathogen associated molecular patterns respond by producing cytokines, including Interferon-β (IFNβ). In this study, we identify a novel role for RIPK1 and RIPK3, a pair of homologous serine/threonine kinases previously implicated in the regulation of necroptosis and pathologic tissue injury, in directing IFNβ production in macrophages. Using genetic and pharmacologic tools we show that catalytic activity of RIPK1 directs IFNβ synthesis induced by lipopolysaccharide (LPS) in mice. Additionally, we report that RIPK1 kinase-dependent IFNβ production may be elicited in an analogous fashion using LPS in bone-marrow derived macrophages (BMDMs) upon inhibition of caspases. Notably, this regulation requires kinase activities of both RIPK1 and RIPK3, but not the necroptosis effector protein, MLKL. Mechanistically, we provide evidence that a necrosome-like RIPK1 and RIPK3 aggregates facilitate canonical TRIF-dependent IFNβ production downstream of the LPS receptor TLR4. Intriguingly, we also show that RIPK1 and RIPK3 kinase-dependent synthesis of IFNβ is markedly induced by avirulent strains of gram-negative bacteria, Yersinia and Klebsiella, and less-so by their wild-type counterparts. Overall, these observations identify unexpected roles for RIPK1 and RIPK3 kinases in the production of IFNβ during the host inflammatory responses to bacterial infection and suggest that the axis in which these kinases operate may represent a target for bacterial virulence factors.
Introduction
Receptor-Interacting Protein Kinases 1 and 3 (RIPK1 and RIPK3) are homologous serine/threonine kinases that are critical regulators of necroptosis, a form of necrotic cell death associated with pathologic tissue injury (1–6). RIPK1 and RIPK3 are components of the signaling machinery downstream of innate immune pathogen recognition receptors (PRRs), Toll-like receptors 3 and 4 (TLR3 and TLR4) (7–11). Catalytic activity of these kinases, induced by activation of TLR3 or TLR4 in the presence of pan-caspase inhibitor, zVAD.fmk (zVAD), promote necroptosis in primary macrophages (9, 12–14). This regulation is dependent on the TLR3/4 Rip-Homotypic Interacting Motif (RHIM)-domain containing adapter protein, Toll-interleukin-receptor-domain-containing-adapter-inducing-Interferon-β (TRIF) (9, 12–14). Upon activation, RIPK1 and RIPK3 form insoluble cellular aggregates, termed ‘necrosomes,’ which function to allow phosphorylation and activate of the target pseudokinase, MLKL, by RIPK3 (15–18). Phosphorylation of MLKL promotes its oligomerization and translocation to the cell surface where it functions to increase membrane permeability and facilitate necrotic cell death through a mechanism that is not yet fully understood (19–23).
New evidence suggests existence of direct links of RIPK1 and RIPK3 to inflammation that occur independently of cell death. For example, in vitro, RIPK1 kinase activation has been linked to TNFα production in contexts of caspase inhibition and DNA damage (24–27). We have recently demonstrated the existence of a TRIF-mediated cell death-independent signaling pathway downstream of RIPK1 and RIPK3 that involves activation of Erk1/2 and NFkB and directs synthesis of acute inflammatory cytokines, including TNFa, following TLR4 activation (14). In another example, RIPK1 kinase-dependent IL1α expression and tissue inflammation, occurring in the absence of necroptosis, was reported in the Ptpn(spin) model of human neutrophilic dermatitis (28). In macrophages, RIPK3 is required for activation of NFκB and IL1β release both in kinase-independent and dependent manner conditional on caspase-8 inhibition (29–31).
Here, we report a novel role for the kinase activity of RIPK1 in directing lipopolysaccharide (LPS) induced IFNβ synthesis in mice and in vitro in primary mouse macrophages when caspases are inhibited. We report that this regulation requires TRIF and occurs by way of a canonical Type I interferon (IFN-I) response pathway, requiring TBK1, IKKε, and IRF3/7. These canonical IFN-I inducing factors co-localize to RIPK1/RIPK3 kinase containing detergent-insoluble cellular fractions following TLR4 activation, and this localization was disrupted by the selective RIPK1 kinase inhibitor, Nec-1s. Together, these data demonstrate that a necrosome-like RIPK1 and RIPK3 signaling platform mediates TRIF-dependent IFNβ production. Moreover, enhanced RIPK1 and RIPK3 kinase-dependent IFNβ synthesis was observed after challenge with attenuated mutant strains of Yersinia pseudotuberculosis and Klebsiella pneumoniae in the presence of caspase inhibition, suggesting that this pathway may represent a new target of bacterial virulence factors.
Materials and Methods
Animals
Female Balb/c mice at 6–8 weeks of age were used for LPS experiments (Charles River Labs). Ripk3−/− (on C57BL/6 background) and matched controls were previously described (Newton et al., 2004) and provided to us by Dr. Vishva Dixit (Genentech). Sting−/− mice were a gift from Dr. Alexander Poltorak. TRIF−/− (Ticam1−/−) (C57BL/6J-Ticam1Lps2/J) mice and corresponding control mice (C57BL/6J, B6.129P2) were purchased from Jackson labs. D138N RIPK1, K45A RIPK1, K51A RIPK3, and Mlkl−/− mice were previously described (30, 32–34).
All use of animals was approved by the Tufts University, UMASS, and Fox Chase Cancer Center Institutional Animal Care and Use Committees. Mice were maintained in Tufts animal facility in cages with light/dark cycle and experiments performed according to the protocol with all efforts to minimize the number and suffering of the animals.
Bacteria
Yersinia strains (Yptb and ΔyscF) were on the IP2666 background and have been previously described (35). Yersinia bacteria were cultured overnight in 2XYT at 26C. On the day of infection, cultures were diluted 1:40 in 5mM CaCl2 containing 2XYT for 2 hours and then moved to 37C for two hours prior to infection. Klebsiella strains (Kp and ΔcpsB) were on the Kp ATCC 43816 background. ΔcpsB was generated and gifted by Michelle Paczosa and will be reported in a future publication. Klebsiella bacteria were grown overnight at 37C in L broth.
Infections
Macrophages were infected in antibiotic free media (10% L929 conditioned media + 10% FBS in RPMI) bacteria at an MOI of 40–60 and bacterial were spun down at 300g for 3 minutes. Two hours post-infection, 100μg/mL Gentamicin was administered and macrophages were harvested 6 hours post-infection. Prior to infection, bacterial counts were estimated using an optical density read-out and confirmed by colony-forming unit assay.
In vivo LPS challenge and Nec-1s treatment
Mice were injected intravenously or intraperitoneally with 30 mg/kg optimized Nec-1s (7-Cl-O-Nec-1) (36), 15 min prior to intraperitoneal injection of 50 μg/kg LPS (Sigma). CD11b+ bone marrow cells were collected 1 hour post-injection from femurs and stained at a dilution of 1:300 using PE conjugated anti-mouse/human CD11b+ (BioLegend, 101207) for 1 hour in FACS buffer (2% FBS and 1μM EDTA in PBS). 20–40% of total cells were CD11b+ and these were sorted by FACS. Sorted cells were used for qRT-PCR or RNA-Seq.
Reagents
Lipopolysaccharide (LPS) (Escherichia coli 0111:B4) was purchased from Sigma. Optimized Necrostatin-1s (Nec-1s) (5-[(7-chloro-1H-indol-3-yl) methyl]-3-methyl-2,4-imidazolidinedione) was synthesized as previously described (Teng et al., 2005). For in vivo administration, Nec-1s was dissolved in PBS containing 25% Polyethylene Glycol 400 (Spectrum labs). RIPK3 inhibitor, GSK’872, was also previously described (9, 37). TBK1/IKKε inhibitors, MRT67307 and BX795 were previously described (38, 39). IDN6556 was previously described (40). zVAD.fmk was purchased from ApexBio.
Cells
Bone marrow derived macrophages (BMDMs) were prepared by flushing bone marrows from femurs and tibias. BMDMs were allowed to differentiate for 7 days in the presence of conditioned media from L929 cells, containing Macrophage Colony Stimulating Factor (M-CSF) - 30% L929 media, 20% FBS and 1% PSA (penicillin/streptomycin/antimycotic solution) in RPMI1640. Medium was replenished on day 3. 48 hours before treating the cells, the medium was exchanged to 10% L929 media, 10% FBS and 1% PSA in RPMI1640. On day 7, the adherent cells were collected using PBS and centrifuged at 430 g for 5 min and replated for the experiments. For mRNA and Western blot experiments, 2 × 106 BMDMs were seeded into 35 mm2 dishes. For cell viability experiments, 50,000 cells per well were seeded in 96 well plates. RAW cells (RAW264.7) were grown in DMEM containing 10% FBS and 1% PSA. For experiments, cells were treated with all reagents (stimulants and inhibitors) simultaneously. For example, inhibitors (ie: Nec-1s) were added to cells with LPS and zVAD or IDN at time zero.
Western blotting
Cells were lysed and harvested in RIPA buffer (Cell Signaling) supplemented with Phenymethanesulfonylfluoride (PMSF) (5 μg/ml, Sigma), leupeptine (1μg/ml), pepstatin (1μg/ml) and aproprotinin (1μg/ml). The protein concentrations were determined using Bio-Rad Protein Assay reagent, and equal amounts of protein were subjected to Western blotting. Samples were separated by SDS/PAGE and transferred to PVDF membranes. After being blocking in Protein-free T20 (TBS) blocking buffer (Fisher Scientific) at room temperature for 1 h, the membranes were rinsed with TBST (0.1%) and incubated at 4 °C overnight with primary antibodies (1:1000 dilution). The membranes were washed and incubated with secondary antibody (1:5000 dilution) at room temperature for 1 h. Signals were developed using Luminata Classico or Forte HRP substrates (Millipore).
Antibodies
The following antibodies acquired from Cell Signaling Technologies were used: TBK1 (#3504), p-TBK1 (#5483), IKKε (#3416), p-IKKε (#8766), IRF3 (#4302), and p-IRF3 (4947), RIPK1 (#3493), αTubulin (#3873), anti-mouse IgG HRP-linked antibody, and anti-rabbit IgG HRP-linked antibody. RIPK3 antibody was purchased from Prosci (#2283). Primary antibodies were used at a dilution of 1:1000 and secondary antibodies were used at a dilution of 1:5000.
Cell Viability Assays
The cells were seeded as described above in 100 μL of media. Typically, cells were treated with 10 ng/ml LPS and 50 μM zVAD for 24 hours. Cell viability was determined using CellTiter-Glo viability assay kit (Promega). Each independent experiment was performed in duplicate and repeated three times. Viability of the cells, relative to an untreated control, was determined and plotted.
Measurement of IFNβ by ELISA
Mouse interferon beta (IFNβ) was measured using colorimetric ELISA. 96 well plate was incubated at 4C overnight after coating wells with 50 μL of monoclonal rat anti-mouse IFNβ (Santa Cruz, SC-57201) diluted 1:500 (final concentration: 0.2μg/mL) in 0.1M carbonate buffer. The following day, wells were incubated with 200 μL of blocking buffer (10% FBS in PBS) for 2 hours at 37C. After blocking, wells were incubated with 50μL of standard or sample (diluted 1:1, 1:5, and/or 1:25 in blocking buffer) and incubated overnight at 4C. Interferon-beta protein purchased from PBL (product number: 12400) and standard curves were generated using concentrations 0–1000IU/mL (0–2ng/mL). The following day, wells were incubated with 50μL of polyclonal rabbit anti-mouse IFN-β detecting antibody from PBL (product number: 32400) diluted 1:2000 (final concentration: 0.5μg/mL) in blocking buffer and incubated overnight at 4C. The following day, 50μL of goat anti-rabbit-HRP secondary antibody (Cell Signaling) diluted 1:2000 was added to the wells. Plates were incubated for 2–3 hours at room temperature. Subsequently, 50 μL of the TMB substrate was added. The reaction was stopped by the addition of 50 μL 2N H2SO4. Absorbance was measured at 450 nM. A washing step with buffer (PBS 0.05% TWEEN) was performed after each step and before the addition the substrate.
RNA extraction, cDNA synthesis and qRT-PCR
For RNA extraction, cells were seeded as described above. Cells were stimulated with 10 ng/ml LPS, 50 μM zVAD and 30 μM Nec-1s or other inhibitors as indicated. Total RNA was isolated using RNA MiniPrep kit (ZYMO Research) according to the manufacturer’s protocol. 50 ng to 1 μg of RNA was converted to cDNA using iScript cDNA Synthesis kit (BioRad). 1 μL of cDNA was used with 500 pM primers in 20 μL qPCR reactions using VeriQuest SYBR Green master mix (Affymetrix). qRT-PCR reactions were performed in LightCycler 480 II using the following program: 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of amplification (95°C for 15 sec, 60°C for 1 min). GAPDH was analyzed as a housekeeping gene.
The primer sequences used to amplify murine genes are as following:
Mouse GAPDH: forward 5′-TGTGTCCGTCGTGGATCTGA-3′, reverse: 5′- GGTCCTCAGTGTAGCCCAAG3′.
Mouse IFNb1: forward 5′-CAGCTCCAAGAAAGGACGAAC-3′, reverse 5′-GGCAGTGTAACTCTTCTGCAT-3′.
Mouse MX2: forward 5′-GAGGCTCTTCAGAATGAGCAA -3′, reverse 5′-CTC TGCGGTCAGTCTCTCT-3′.
Mouse IFIT1: forward 5′- CTGAGATGTCACTTCACATGG AA -3′, reverse 5′-GTGCATCCCCAATGGGTTCT -3′.
Necrosome formation assay
Isolation of NP40 soluble and insoluble fractions was performed as previously described (15, 41). Cells were seeded into 35 cm2 dishes at 2 × 106 cells/well and stimulated with 10 ng/ml LPS, 50 μM zVAD and 30 μM Nec-1s for up to 3 hr. Cells were lysed in 1%TritonX100 or 1%NP-40 lysis buffer (150mM NaCl, 20mM Tris-Cl (pH 7.5), 1% TritonX100 or 1%NP-40, 1mM EDTA, 3mM Na-fluoride, 1mM B-glycerophosphate, 1mM Sodium Orthovanadate, 5uM Idoacetamide, 2uM N-ethylmaleimide and Phosphatase and 5μg/ml PMSF and 1μg/ml leupeptine, 1μg/ml pepstatin and 1μg/ml aprotinin. Lysates were flash frozen on dry-ice, thawed on ice and vortexed for 10 sec followed by centrifugation at 1000g for 15 minutes in the refrigerated table-top centrifuge to remove nuclei. Supernatants were collected and centrifuged at 34,400g for 15 minutes to precipitate detergent-insoluble cellular fraction (NP40 or Triton insoluble). Supernatants were collected (NP40 or Triton soluble fractions) and pellets were boiled in 1x SDS-PAGE buffer.
Sucrose-gradient velocity sedimentation
RAW264.7 macrophages were treated for 3–4 hrs and lysates collected as in the necrosome formation assay. Samples were flash frozen, thawed, and nuclei were precipitated and discarded as in necrosome formation assay. Protein concentration normalized, and placed upon a 10–50% linear sucrose gradient. Samples were subject to velocity sedimentation at ~250,000g for 2.5–3 hours and samples were collected in 13–14 × 1 mL aliquots. Samples were subjected to Chloroform/Methanol protein precipitation to eliminate sucrose and/or concentrate protein samples prior resuspension in Laemmli buffer for Western Analysis.
Co-immunoprecipitation studies
RAW264.7 macrophages were treated for 3–4 hrs and lysates collected as in the necrosome formation assay. Samples were vortexed and incubated on ice prior to precipitating nuclei. Lysate protein concentration was normalized to 2mg/mL across samples and lysates were incubated overnight with Rabbit anti-RIPK1 or Rabbit anti-IKKε antibody. Antibody bound protein was captured using Protein A-conjugated magnetic beads per sample (ThermoFisher Scientific, Pierce Protein A Magnetic Beads #88845) and eluted by boiling in 1× Laemmli buffer for 5 min.
RNA Sequencing
For RNA-Seq analysis, mice were divided intro 3 groups - control (n=2), LPS (n=2) and LPS/Nec-1s (n=2). Bone marrow cells were isolated by FACS as described for qPCR analysis. Total RNAs were isolated using Qiagen RNeasy kit according to the manufacturer’s protocol. Input RNA samples were first analysis by Agilent BioAnalyzer 2100 to asset the integrity and quantity. The RNA samples were then amplified using NuGen Ovation RNA System V2. The resulting cDNA samples were fragmented on Covaris M220 Focused Solicitor, followed by purification and concentration with a Qiagen MiniElulte Spin Column. Following this step, S1 Nuclease (Promega) was used according to the manufacture’s protocol. Then, the amplified and fragmented cDNA samples from RNA amplification was used as input for library preparation, using Illumina TruSeq DNA Sample Preparation Kit per the manufacturer’s instruction. The resulted libraries were quantified and pooled at equal molar concentration for sequencing. The sequencing was done on a lane of High Output single read 100 bases on an Illumina HiSeq 2500 using SBS V3 chemistry. The base calling and demultiplexing was performed with CASAVA v1.8. The resulting data were then aligned to mouse mm10 reference genome with Tophat 2 and different gene expression analysis with Cuffdiff. Gene expression profiles were analyzed using Ingenuity pathway analysis (IPA) software to identify pathways regulated by LPS and Nec-1s. http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE73836
Statistics
For in vivo studies, statistics were analyzed by two-tailed Student t-test and significance was determined using an alpha value of 0.05. Error bars reflect standard error (SE) from the mean. For in vitro studies, experiments were repeated with at least 2, but in most cases 3 biological replicates. In each figure, one representative dataset is shown due to the variability in the general responsiveness to LPS in independent cell preparations. Error bars reflect standard deviation (SD) from the mean with measurements made in duplicate.
Author Contributions
D.S. performed cell viability, Western analyses, and ELISAs. M.N., D.S., and S.S. performed RNA work and qRT-PCRs. D.S., M.N. and M.Z. performed in vivo experiments. D.S. and N.S. fractionated lysates using sucrose gradient and velocity sedimentation. A.T. performed the RNA-Seq analysis. D.S. prepared the fractionation samples. J.B. and P.G. generated and provided the K45A RIPK1 and K51A RIPK3 mice. A.P. and M.P. developed RIPK1 D138N mice. M.Z., M.P., and M.K. provided the D138N RIPK1 cells. M.W. provided Ripk3−/− cells. S.N. and S.B. provided Mlkl−/− cells. K.F provided Irf3−/−/Irf7−/− cells. J.M. and M.P. provided bacterial strains. M.Z., J.B., P.G, M.P., K.F., N.S., S.B., and J.M. helped revise the manuscript. D.S. and A.D. wrote and revised the manuscript.
Results
Kinase activity of RIPK1 is required for IFNβ synthesis induced by LPS in mice
In a previous study, we reported that kinase activity of RIPK1 was required for acute inflammatory cytokine production induced by LPS in mice, including TNFα, IL6, CCL3/4 and CXCL1/2, suggesting that the catalytic activity of RIPK1 might serve as an important role in the innate immune response induced by TLR4 (Najjar et al. 2016). Synthesis of Type I interferons, including Interferon-beta (IFNβ) (42–44), is elicited by TLR4 activation and plays an important role in innate and adaptive immunity against infections (45). To inquire whether the catalytic activity of RIPK1 may also be required for LPS-induced IFNβ synthesis, we first tested the effect of the selective RIPK1 inhibitor, Nec-1s, on this process. Nec-1s injection prior to the challenge with LPS abolished IFNβ mRNA synthesis in CD11b+ monocytic cells in vivo (Figure 1a). In addition, we observed that LPS-driven induction of a panel of interferon-stimulated genes (ISGs) was similarly abolished by Nec-1s in CD11b+ cells (Figure 1b). LPS-induced IFNβ synthesis was also attenuated in monocytic CD11b+ cells from D138N RIPK1 kinase-inactive mice (D138N RIPK1 or Ripk1D138N/D138N) (Figure 1c). Together, these pharmacologic and genetic approaches suggest that RIPK1 kinase is indeed required for LPS-induced upregulation of IFNβ expression in vivo.
Figure 1. LPS induced IFN-I production is dependent on kinase-activity of RIPK1.
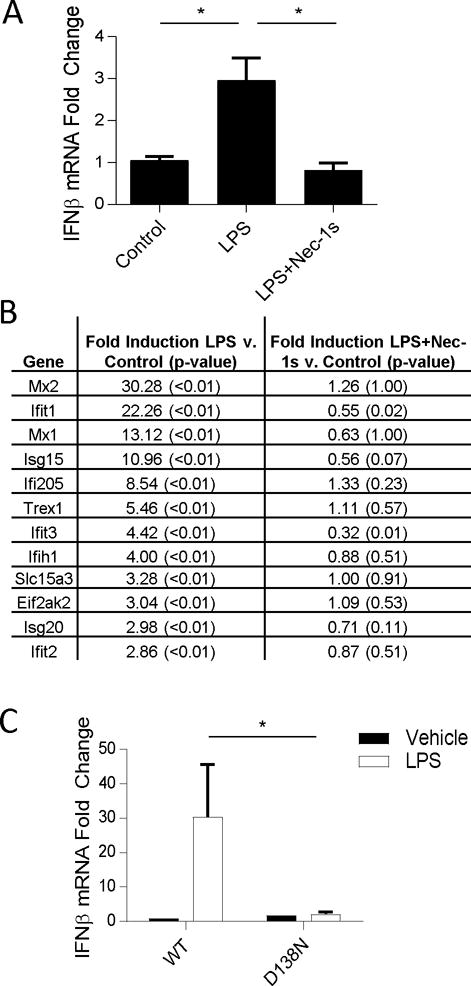
(A) qRT-PCR analysis of Ifnb mRNA expression in wild type (WT) mice injected with Nec-1s (iv) 15 min prior to LPS (ip). n= 6 animals per group and *p<0.05. Values reflect mean ± SE. (B) RNA seq analysis of a panel of IFN-I response genes in CD11b+ cells isolated from mice injected as in (A). Values reflect mean. P values marked in parentheses. GSE73836. (C) qRT-PCR analysis of Ifnb mRNA expression WT and D138N mice injected with LPS (ip). n= 3–7 animals per group and *p<0.05. Values reflect mean across biological variants and error bars reflect SE.
LPS with zVAD.fmk induces RIPK1 kinase-dependent IFNβ synthesis in BMDMs
Deletion of caspase-8 or use of pan-caspase inhibitor, zVAD, is sufficient to facilitate RIPK1 kinase activation by LPS in vitro (9, 12, 14, 46). Within 24 hours, LPS and zVAD treatment, but not LPS alone induced RIPK1 kinase-dependent cell death in bone marrow derived macrophages (BMDMs) (Figure S1a and S1b). Requirement for caspase inhibition for RIPK1 activation in vitro is also supported by our previously published phopsho-RIPK1 ELISA data (14). To address the possible role of RIPK1 in IFN-I production, we first examined whether zVAD promoted LPS-induced IFNβ synthesis in BMDMs. Indeed, LPS paired with zVAD greatly increased synthesis of IFNβ compared to LPS alone over an 8 hour time course (Figure 2a). Similarly, mRNA expression of interferon stimulated genes (ISGs), MX2 and IFIT1, was augmented by LPS with zVAD compared to LPS alone (Figure 2b, 2c).
Figure 2. LPS with zVAD induces IFNβ synthesis in a RIPK1 kinase-dependent manner.
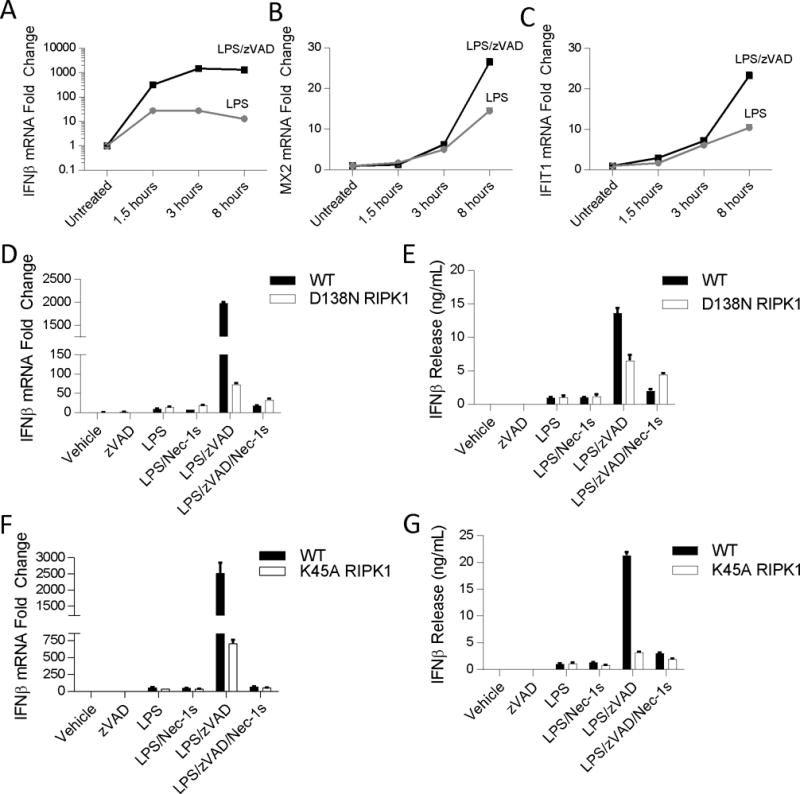
(A–C) Time course of mRNA expression changes in Ifnb (A), Mx2 (B), and Ifit1 (C) evaluated by qRT-PCR in wild type BMDMs. Black squares – LPS/zVAD; Grey circles – LPS. (D-E) qRT-PCR and ELISA analysis of Ifnb mRNA expression and IFNβ protein release in WT and D138N BMDMs treated for 5–7 hrs. (F-G) qRT-PCR and ELISA analysis of Ifnb mRNA expression and IFNβ protein release in WT and K45A BMDMs treated for 5–7 hrs. Data are representative. Error bars reflect SD from the mean. BMDMs were treated with LPS=10 ng/mL, zVAD=50 μM, and/or Nec-1s=30μM where indicated.
Next, to evaluate whether this regulation was dependent on the catalytic activity of RIPK1 kinase, we examined IFNβ synthesis in BMDMs generated from two different mouse strains expressing kinase-dead mutants of RIPK1 (D138N RIPK1 or K45A RIPK1 (Ripk1K45A/K45A) mice (32, 33). Consistent with our hypothesis, we observed that in the absence of the kinase activity of RIPK1, increases in IFNβ mRNA synthesis and protein release in response to LPS with zVAD was abolished or attenuated, respectively (Figure 2d–g). Nec-1s also blocked IFNβ production by LPS with zVAD (Figure 2d–g). Conversely, catalytic activity of RIPK1 was not essential for the responses to LPS alone, confirming that activation of RIPK1 in the presence of caspase inhibition greatly promotes IFNβ synthesis in BMDMs.
IFNβ synthesis induced by LPS with zVAD requires kinase activity of RIPK3 but not MLKL
RIPK3 is a central component of the necroptosis signaling cascade and is required for necroptosis induced by LPS with zVAD (47). Catalytic activity of RIPK3 is required for the formation of detergent-insoluble RIPK1-RIPK3 ‘necrosome’ complexes and consequent phosphorylation of MLKL, which ultimately leads to the execution of necroptosis (4, 14, 17, 19, 20, 23, 48). Consistently, we observed that K51A RIPK3 BMDMs (Ripk3K51/K51A), expressing low levels of a kinase-dead mutant of RIPK3 (37), and RIPK3 knockout (Ripk3−/−) BMDMs were protected from cell death induced by LPS with zVAD (Figure S2a, Fig S2b).
We next sought to determine whether RIPK1 kinase-dependent IFNβ production required these additional necroptotic factors. Stimulation of RIPK3 K51A BMDMs by LPS with zVAD failed to induce IFNβ production above the response by LPS alone, indicating that RIPK3 was required for RIPK1 kinase-dependent IFNβ production (Figure 3a). To further evaluate the role of catalytic activity of RIPK3, we used the specific RIPK3 kinase-inhibitor GSK’872 (9, 37). We observed that GSK’872 blocked IFNβ synthesis induced by LPS with zVAD, confirming that the catalytic activity of RIPK3 is also required for RIPK1 kinase-dependent IFNβ production in BMDMs (Figure 3b). In contrast, deletion of Mlkl only minimally impaired IFNβ mRNA synthesis induced by LPS with zVAD, even though these BMDMs were, expectedly, completely resistant to necroptosis (Figure 3c, 3d) (14). These observations demonstrated that RIPK1 and RIPK3 kinase-dependent IFNβ synthesis is independent of necroptosis pathway and that bifurcation of the respective signaling cascades occurs upstream of MLKL. Furthermore, given the paucity of known targets of RIPK1 and RIPK3, these findings highlight the possibility of new cell death-independent enzymatic targets for these kinases that remain to be defined.
Figure 3. Kinase-activity of RIPK3, but not MLKL, is required for RIPK1 kinase-dependent IFNβ synthesis.
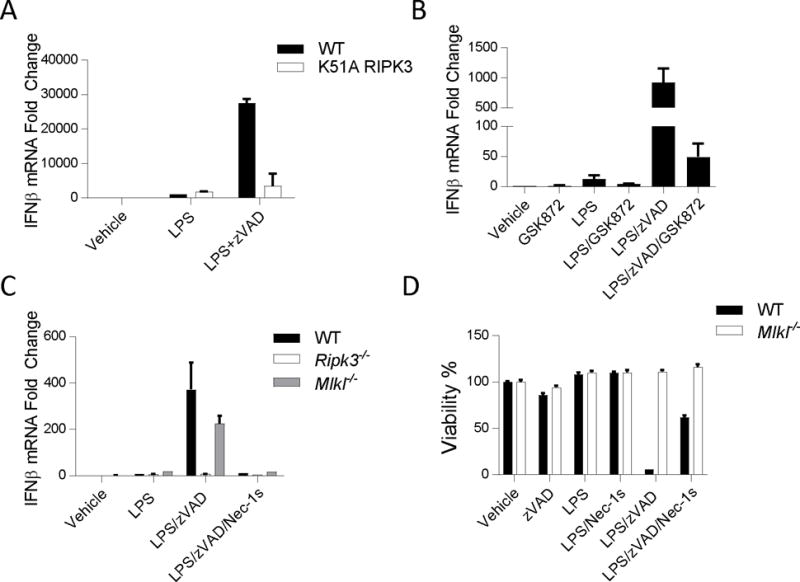
(A) qRT-PCR analysis of Ifnb mRNA expression in WT and K51A BMDMs treated for 5–7 hrs. (B) qRT-PCR of Ifnb mRNA expression in wild type BMDMs treated with RIPK3 kinase inhibitor (GSK872= 5μM) for 5–7 hrs. (C) qRT-PCR of Ifnb mRNA expression in wild type (WT), RIPK3 knockout (Ripk3−/−) and MLKL knockout (Mlkl−/−) BMDMs treated for 7 hrs. (D) Cell viability of WT and Mlkl−/− BMDMs treated for 24 hours and evaluated by CellTiterGlo ATP assay. Experiments were repeated 3 times. Data are representative. Error bars reflect SD from the mean. BMDMs were treated with LPS=10 ng/mL, zVAD=50 μM, GSK’872=5μM, and/or Nec-1s=30μM where indicated.
IFNβ production by LPS with zVAD requires TRIF, STING, TBK1/IKKε, and IRF3/7
IFNβ production, induced by LPS alone, is dependent on a number of key intermediaries starting with the adapter protein TRIF, which mediates signaling through canonical downstream IFN-I pathway intermediaries, including homologous kinases TANK-binding kinase (TBK1), and Inhibitor of Kappa b Kinase ε (IKKε); and the transcription factors, Interferon Regulatory Factors (IRFs) (42, 49–52). As TRIF (Ticam 1) is required for RIPK1 and RIPK3 dependent necroptosis (13), we evaluated IFNβ mRNA synthesis in Ticam1−/− BMDMs to examine whether TRIF is similarly required for the RIPK1 and RIPK3 kinase-dependent IFNβ synthesis. Not surprisingly, in the absence of TRIF, neither LPS nor LPS with zVAD induced IFNβ mRNA synthesis (Figure 4a). Therefore, next we sought to determine whether increased IFNβ synthesis upon RIPK1 and RIPK3 activation, reflects induction of the canonical IFN-I axis (TBK1, IKKε, and IRF3) or whether a new TRIF-dependent signaling mechanism is engaged by RIPK1 and RIPK3. Over an extended timecourse, we found that LPS with zVAD enhanced phosphorylation of TBK1, IKKε, and IRF3 compared to LPS alone, suggesting IFN-I pathway hyper-activation (Figure 4b, S3a–c). Additionally, TBK1, IKKε, and IRF3 phosphorylation in the cells stimulated with either LPS alone or LPS with zVAD was reduced in Ticam1−/− BMDMs, confirming that TRIF was required for IFN-I pathway activation by LPS and zVAD (Figure 4c). By comparison, early TRIF-dependent activation of the IFN-I pathway (1 hour post-treatment) was not affected by zVAD or blocked by Nec-1s, suggesting that the catalytic function of RIPK1 is not required for early signaling events in this pathway (Figure 4d). A recent study demonstrated that TRIF directly associated with STING, an intracellular nucleotide sensor, and was also required for STING dependent IFN-I synthesis (53). Consistent with a role for STING as a co-driver for TRIF-dependent IFNβ synthesis, we found that STING was also required for TRIF/RIPK1-dependent IFNβ synthesis by LPS with zVAD (Figure 4e).
Figure 4. RIPK1 kinase-dependent IFNβ synthesis requires TRIF, STING, TBK1/IKKε, and IRF3/7.
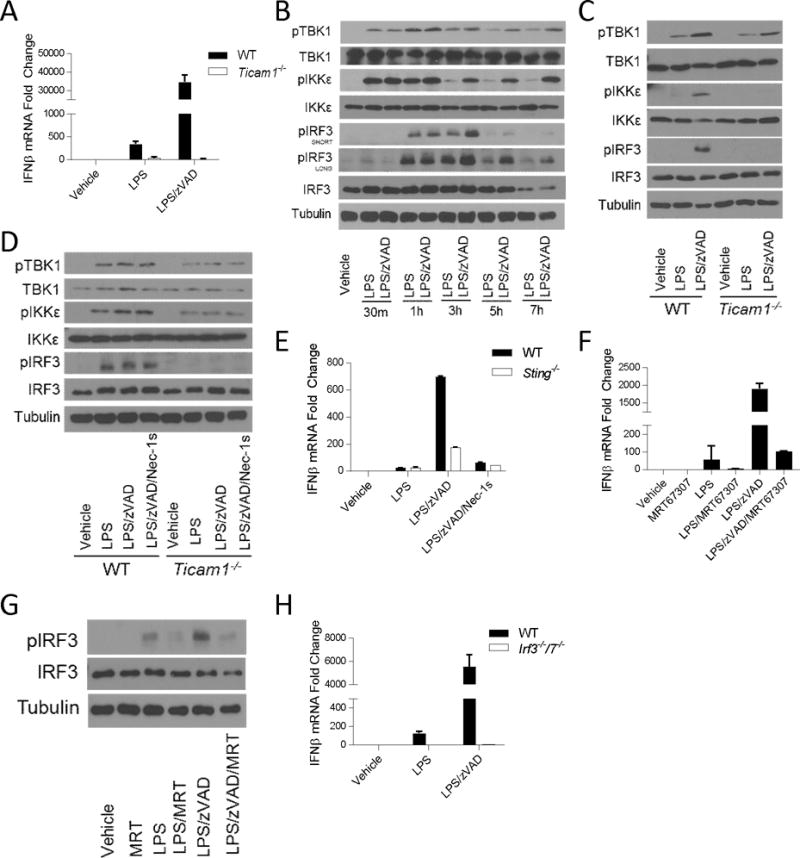
(A) qRT-PCR of Ifnb mRNA expression in WT and Ticam1−/− BMDMs treated for 5–7 hrs. (B) Time course of TBK1, IKKε, and IRF3 phosphorylation evaluated by Western analysis in wild type BMDMs. (C) Western analysis of TBK1, IKKε, and IRF3 phosphorylation in wild type (WT) and Ticam1−/− BMDMs treated for 3–4 hours. (D) Western analysis of TBK1, IKKε, and IRF3 phosphorylation in wild type (WT) and Ticam1−/− BMDMs treated for 1 hour. (E) qRT-PCR analysis of Ifnb mRNA expression in WT and Sting−/− BMDMs treated for 5–7 hrs. (F) qRT-PCR of Ifnb mRNA expression in wild type BMDMs treated with TBK1/IKKε inhibitor (MRT67307, 2μM) for 5–7 hrs. (G) Western analysis of IRF3 phosphorylation in wild type BMDMs treated with MRT67307 for 3–4 hrs. (H) qRT-PCR of Ifnb mRNA expression in WT and Irf3−/−7−/− BMDMs treated for 5–7 hours. Data are representative. Error bars reflect SD from the mean. BMDMs were treated with LPS=10 ng/mL, zVAD=50 μM, Nec-1=30μM and/or MRT67307=2μM where indicated.
To test the functional roles of TBK1 and IKKε in RIPK1 and RIPK3 kinase-dependent IFNβ synthesis, two dual TBK1/IKKε inhibitors (MRT67307 and BX795) were used (38, 39). These inhibitors blocked both LPS and LPS with zVAD induced IFNβ mRNA synthesis (Figure 4f and S3d), and we confirmed that MRT67307 also blocked phosphorylation of IRF3 induced by LPS and LPS with zVAD (Figure 4g).
To examine the contribution of IRFs to RIPK1 and RIPK3 kinase-dependent IFNβ production, we evaluated responses in Irf3−/−/Irf7−/− BMDMs. Again, IFNβ mRNA synthesis induced by LPS or LPS with zVAD were completely abolished in the absence of IRF3 and IRF7, demonstrating the IRFs are also required for RIPK1 and RIPK3 kinase-dependent IFNβ synthesis (Figure 4h). Altogether, these data suggest that TRIF plays a dual role in promoting both signaling by RIPK1 and RIPK3 kinases and TBK1/IKKε as well as allows cross-talk between the two pathways to enhance activation of the TBK1, IKKε, and IRF3 signaling cascade.
Lastly, we examined whether activation of IFN-I pathway intermediaries was also regulated by catalytic activities of RIPK1 and RIPK3. Importantly, phosphorylation of TBK1, IKKε, and IRF3 was attenuated in K45A RIPK1 BMDMs and D138N RIPK1 BMDMs and inhibited by Nec-1s in wild-type BMDMs, confirming the requirement for RIPK1 kinase in this regulation (Figure 5a and 5b). Similarly, these events were abolished in K51A RIPK3 BMDMs, in Ripk3−/− BMDMs and by GSK’872 (Figure 5c, 5d and S4). Notably, RIPKI kinase was specifically required for TRIF-dependent signaling events that were augmented by LPS with zVAD as early activation of TBK1, IKKε, and IRF3 at 1 hour post-treatment did not require RIPK1 kinase (Figure 4d). Together, these observations suggest that RIPK1 and RIPK3 kinase-dependent IFNβ synthesis is mediated by the canonical TBK1/IKKε/IRF signaling cascade and that this regulation is also dependent on the adapter TRIF.
Figure 5. IFN-I pathway activation by LPS with zVAD is RIPK1 and RIPK3 kinase-dependent.
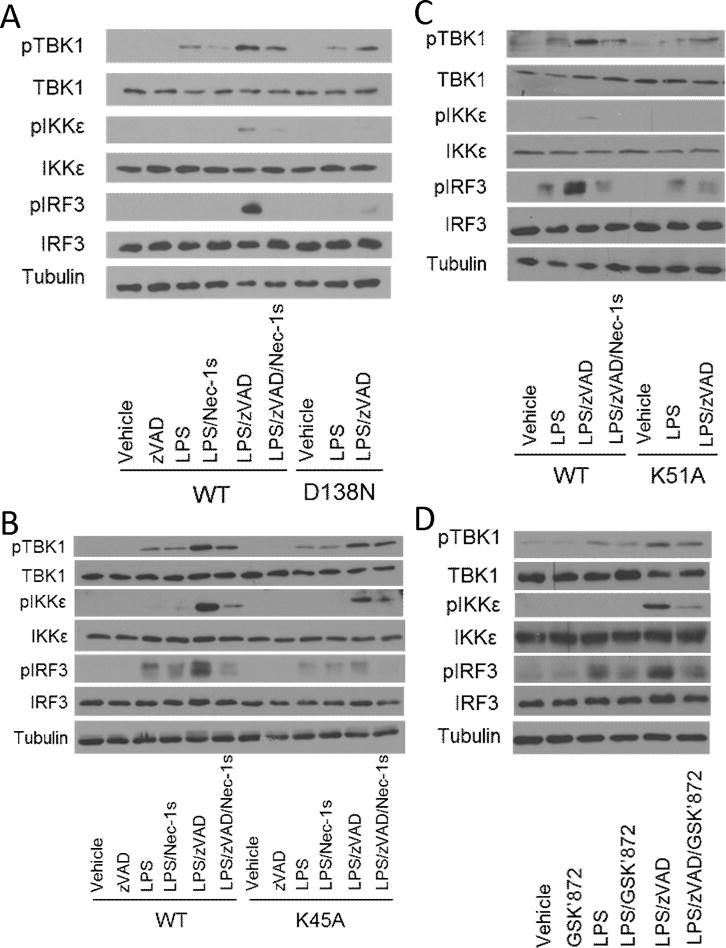
(A) Western analysis of TBK1, IKKε, and IRF3 phosphorylation in wild type (WT) and D138N RIPK1 (D138N) BMDMs treated for 3–4 hrs. (B) Western analysis of TBK1, IKKε, and IRF3 phosphorylation in wild type (WT) and K45A RIPK1 (K45A) BMDMs treated for 3–4 hrs. (C) Western analysis of TBK1, IKKε, and IRF3 phosphorylation in wild type (WT) and K51A RIPK3 (K51A) BMDMs treated for 3–4 hrs. (D) Western analysis of TBK1, IKKε, and IRF3 phosphorylation in wild type BMDMs treated with RIPK3 kinase inhibitor (GSK’872) for 3–4 hrs. BMDMs were treated with LPS=10 ng/mL, zVAD=50 μM, Nec-1=30μM and/or GSK’872=5μM where indicated.
LPS with zVAD promotes localization and activation of TBK1, IKKε, and IRF3 in RIPK1 and RIPK3-containing detergent-insoluble cellular fractions
Observing that IFN-I pathway intermediaries were required for RIPK1 and RIPK3 kinase-dependent IFNβ synthesis, we considered the possibility that these intermediaries associate with RIPK1 and RIPK3 ‘necrosomes’. Activation of RIPK1 and RIPK3 kinase-dependent cell death is associated with post-translational modification and enrichment of the kinases in detergent insoluble cellular fractions, representing necrosome complexes (14, 15, 41, 54). LPS and zVAD treatment of BMDMs resulted in the co-enrichment of modified forms of RIPK1 and RIPK3 in detergent insoluble cellular fractions that was blocked by Nec-1s (Figure 6a), consistent with our previous report of the formation of detergent-insoluble necrosome-like aggregates in LPS and zVAD-treated BMDMs (14). We next analyzed co-enrichment of IFN-I pathway intermediaries with RIPK1 and RIPK3 under these conditions. Notably, we observed that TBK1, IKKε, and IRF3 were enriched in detergent insoluble fractions (Figure 6b). Moreover, we found that phosphorylated or activated forms of these intermediaries were enriched in these cellular compartments (Figure 6b). This regulation was dependent on the catalytic function of RIPK1 and RIPK3 as enrichment of these factors was abolished in the presence of Nec-1s or in K51A RIPK3 BMDMs (Figure 6b,c). We further examined co-enrichment with RIPK1 and RIPK3 using additional high molecular weight protein complex fractionation by sucrose-gradient-based velocity sedimentation. These experiments were performed in an immortalized macrophage cell line (RAW264.7 cells), providing a more abundant source of material than BMDMs, using an optimized pan-caspase inhibitor, IDN6556 (40). RIPK1 and RIPK3 were enriched in fractions 12 and 13, indicating the formation of a high molecular weight complex, when cells were treated by LPS and a pan-caspase-inhibitor that was abolished in the presence of Nec-1s (Figure 6d). Consistent with the previous data, TBK1, IKKε, and IRF3 were again co-enriched in these same fractions (Figure 6d). Intriguingly, additional co-immunoprecipitation studies revealed that TBK1 and IKKε directly complexed with RIPK1, as did RIPK3, when cells were treated with LPS with IDN and this was abolished in the presence of Nec-1s (Figure 6e). We also found that TRIF association with IKKε was not modified in a RIPK1 kinase-dependent manner, suggesting that RIPK1 kinase-dependent signaling may be mediated by post-translational modification as opposed changes in molecular association (Figure 6f). Lastly, to confirm that RIPK1 and RIPK3 kinase-dependent IFN-I pathway signaling did not also require MLKL, we examined enrichment of IFN-I pathway intermediaries in detergent-insoluble fractions in Mlkl−/− BMDMs. Consistent with our previous observation that MLKL was not required for RIPK1 and RIPK3 kinase-dependent IFNβ synthesis, we observed that MLKL was also dispensable for co-enrichment and activation of TBK1, IKKε, and IRF3 in necrosome containing cellular fractions (Figure 6g). Together, these data support the model that IFN-I pathway intermediaries may be recruited and directly activated by RIPK1 and RIPK3 necrosome complexes.
Figure 6. Kinase activity of RIPK1 and RIPK3 are required for localization and activation of IFN-I pathway intermediaries in detergent-insoluble cellular fractions.
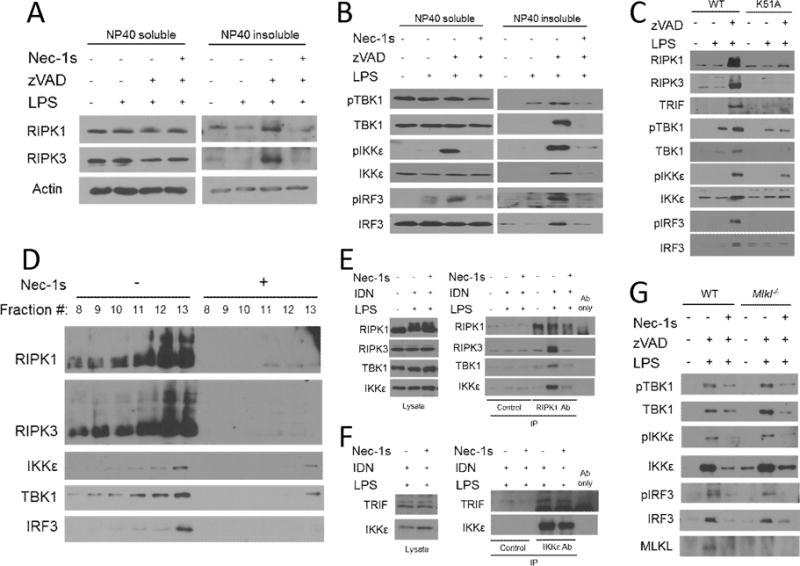
(A) Western analysis of localization and modification of RIPK1 and RIPK3 in NP40-soluble and NP40-insoluble fractions from wild type BMDMs treated for 3–4 hrs. (B) Western analysis of localization and phosphorylation of TRIF, TBK1, IKKε, IRF3 in NP40-soluble and NP40-insoluble fractions from wild type BMDMs treated for 3–4 hrs. (C) Western analysis of RIPK1, RIPK3, TRIF, TBK1, IKKε, and IRF3 in NP40-insoluble fractions from wild type (WT) and K51A RIPK3 (K51A) BMDMs treated for 3–4 hrs.(D) Western analysis of RIPK1, RIPK3, TBK1, IKKε, and IRF3 in RAW264.7 macrophages treated with LPS and IDN6556 (10μM) +/− Nec-1s for 3–4 hrs. Lysates were fractionated on a linear sucrose-gradient by velocity sedimentation and protein was collected following chloroform-methanol precipitation. (E) Co-immunoprecipitation analysis of RIPK3, TBK1, and IKKε following immunoprecipitation of RIPK1 from RAW264.7 macrophages treated for 3–4 hrs. (F) Co-immunoprecipitation analysis of TRIF following immunoprecipitation of IKKε from RAW264.7 macrophages treated for 3–4 hrs. Control=Beads only. (G) Western analysis of RIPK1, RIPK3, TRIF, TBK1, IKKε, and IRF3 in NP40-insoluble fractions from wild type (WT) and Mlkl−/− BMDMs treated for 3–4 hrs. BMDMs were treated with LPS=10 ng/mL, zVAD=50 μM, IDN6556=10μM, and/or Nec-1s=30μM where indicated.
RIPK1 and RIPK3 kinase-dependent IFNβ synthesis in macrophages is induced by avirulent strains of gram-negative bacteria
Several studies have demonstrated that TRIF-dependent IFNβ production is an important feature of the host-response that aids in the resolution of gram negative bacterial infections (55–57). However, gram negative bacteria pathogens such as Yersinia and Klebsiella have evolved an armament of virulence factors to manipulate host-responses and facilitate their pathogenesis. In case of Yersinia, Type-III Secretion System (T3SS) Yop proteins are injected directly into host cells (58–60). Several of these Yops impede host-cell defenses by blocking host pro-inflammatory signaling. Similarly, Klebsiella is equipped with an outer polysaccharide capsule that facilitates immune evasion and enhances bacterial virulence (61, 62). We evaluated whether these pathogens use their virulence factors to dampen sensing of their LPS and subsequent IFNβ induction by RIPK1 and RIPK3.
We examined IFNβ mRNA synthesis in the presence of zVAD following infection with WT or avirulent forms of Yersinia pseudotuberculosis (Yptb) and Klebsiella pneumonia (Kp). Notably, the avirulent strain of Yptb, lacking the ability to inject Yops into host cells (ΔyscF), and the avirulent strain of Kp (ΔcpsB), unable to produce an outer polysaccharide capsule, induced a robust increase in IFNβ mRNA synthesis compared to WT or pathogenic counterparts in the presence of zVAD (Figure 7a, 7b). To verify that IFNβ induction was dependent on RIPK1 and RIPK3 kinases, we also determined that the response was absent in K45A RIPK1 and K51A RIPK3 BMDMs (Figure 7a, 7b). Furthermore, consistent with the previous data, we again observed robust RIPK1 kinase activity-dependent co-accumulation of RIPK1, RIPK3 and IFN-I pathway components in the insoluble fractions of cells infected with ΔyscF or ΔcpsB in the presence of zVAD (Figure 7c, 7d). In sum, these data provide direct demonstration that gram negative bacteria are capable of inducing RIPK1 and RIPK3 kinase-dependent IFNβ synthesis in vitro in the presence of zVAD, and that this induction may be under negative regulation by bacterial virulence factors. Accordingly, these data, paired with our in vivo observations of LPS-induced RIPK1 kinase-dependent IFNβ synthesis, suggest a potential role for RIPK1 kinase-dependent IFNβ synthesis as part of a physiologic host-response in bacterial infection.
Figure 7. RIPK1 and RIPK3 kinase-dependent IFNβ synthesis is augmented by attenuated strains of Yersinia pseudotuberculosis and Klebsiella pneumonia.
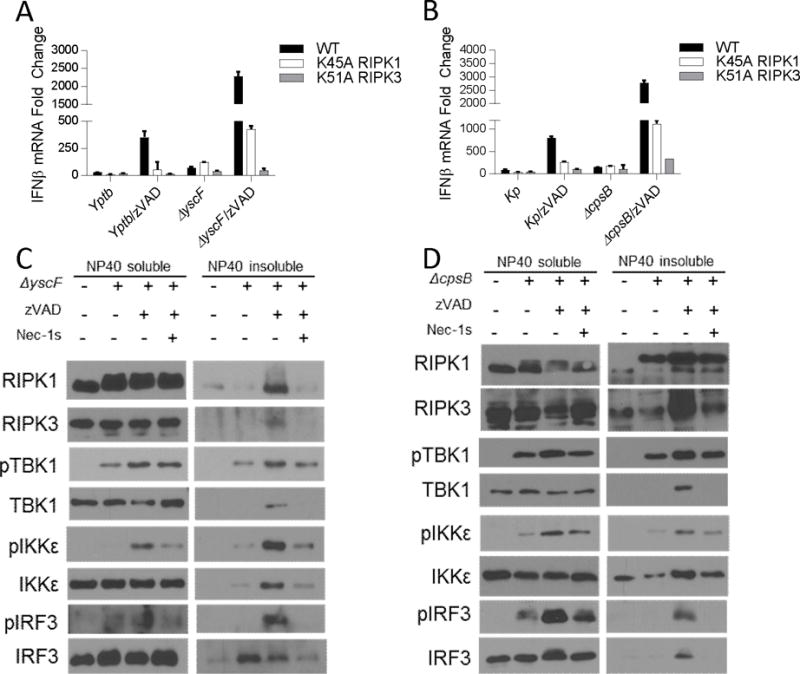
(A) qRT-PCR of Ifnb mRNA expression in wild type (WT), K45A RIPK1 (K45A), and K51A RIPK3 (K51A) BMDMs infected with wild type Yersinia pseudotuberculosis (Yptb) or a mutant strain unable to inject pathogenicity factors into macrophages (ΔyscF), at an MOI of 40–60. (B) qRT-PCR of Ifnb mRNA expression in WT, K45A, and K51A BMDMs infected with wild type Klebsiella pneumonia (Kp), or a mutant strain lacking its out polysaccharide capsule virulence factor (ΔcpsB) at an MOI of 40–60. (C) Western analysis of localization and phosphorylation of TRIF, TBK1, IKKε, IRF3 in NP40-soluble and NP40-insoluble fractions from wild type BMDMs treated as described in (A). (D) Western analysis of localization and phosphorylation of TRIF, TBK1, IKKε, IRF3 in NP40-soluble and NP40-insoluble fractions from wild type BMDMs treated as described in (B). Data are representative. Error bars reflect SD from the mean. BMDMs were treated with zVAD=50 μM and/or Nec-1s=30μM where indicated. Gentamicin (100μg/mL) was added 2 hours post-infection in each experiment.
Discussion
RIPK1 and RIPK3 activation and necrosis are generally believed to be associated with acute and pathologic injury; however, emerging evidence suggests RIPK1 and RIPK3 may play significant roles in physiologic host-defense responses to viral and bacterial infections (63, 64). To date, studies have focused on the roles of RIPK1 and RIPK3 in the context of pathogen-induced cell death. For example, RIPK1 and caspase-8-dependent apoptosis has been described as a central feature of the innate immune response to gram-negative bacterial pathogen, Yersinia (65, 66). Similarly, RIPK1 and RIPK3 kinase-dependent necroptosis have been implicated in macrophage cell death by Salmonella and local-tissue damage in necrotizing pneumonia caused by Methicillin-Resistant Staphylococcus Aureus (MRSA) (67, 68). However, inflammatory signaling is also a critical component of the initial innate immune response that has not been as closely examined in RIPK1 and RIPK3 biology.
Previous work suggested that kinase activities of RIPK1 and RIPK3 may not contribute to cytokine synthesis downstream of TLR3 and TLR4 (7, 8, 69, 70). However, these studies were performed in the absence of caspase inhibition, which is a requisite to elicit kinase functions of RIPK1 and RIPK3 in vitro (9, 12–14). In contrast, data in this study as well as other recent reports (14, 24, 27, 71) describe RIPK1 and RIPK3 kinase-dependent cytokine responses that manifest independent of MLKL-dependent cell death signaling. In a prior study we reported that RIPK1 and RIPK3 kinase-dependent cytokine profile evaluated in vitro, in the presence of zVAD, correlated strongly with the inflammatory profile observed in vivo, in the absence of exogenous caspase manipulation (14). We observed the dichotomy between RIPK1 kinase regulation in vitro and in vivo was linked to limited caspase-8 activation in bone marrow monocytic cells collected from mice injected with LPS (14). Similarly, in this study we find that exogenous inhibition of caspases is not requisite for RIPK1 and RIPK3 kinase-dependent IFNβ synthesis in vivo following LPS challenge. These findings suggest that use of zVAD in vitro may aid in uncovering new pro-inflammatory roles for RIPK1 and RIPK3 in vivo.
In vitro observation of detergent-insoluble, amyloid-like, RIPK1 and RIPK3 complexes, also known as necrosomes, have been linked to RIPK1 and RIPK3 kinase-dependent signaling (14, 15, 41). Indeed, it is well established that caspase inhibition is essential the induction of necroptosis and that MLKL is recruited to necrosome signaling platforms (9, 12–14). Similarly, in the presence of caspase inhibition, we also observe enrichment and activation of IFN-I pathway intermediaries in necrosome containing cellular fraction and, remarkably, this did not require MLKL. Although the necrosome was originally defined as a cell-death signaling complex, our data here and in our previous study suggest that the necrosome may additionally serve as a signaling platform for the induction of IFNβ and other cytokines (14). It is important to note that the precise details of necrosome signaling are poorly understood and it remains to be determined whether a singular ‘necrosome’ platform is capable of driving cytokine and pro-death signaling independent of one another. Alternatively, the ‘necrosome’ might reflect physically and/or compositionally heterogenous RIPK1 and RIPK3 containing platforms that are defined and influenced by their immediate molecular context to promote cytokine synthesis or pro-death signaling.
Our data found that STING contributes to TRIF/RIPK1-dependent IFNβ synthesis (Fig. S4e). Recent work by Wang X. et al. suggested that TRIF is required for STING to activate IFNβ synthesis. Thus, it is possible that STING and TRIF may similarly cooperate in promoting RIPK1 activation and IFNβ synthesis. STING has also been proposed to respond to damaged DNA to induce IFNβ synthesis. This might serve as an indirect mechanism of STING participation in TRIF-RIPK1 kinase-dependent events. However, we believe this latter regulation is unlikely to be a major mechanism contributing RIPK1 kinase-dependent IFNβ synthesis because we observe considerable induction in IFNβ by LPS with zVAD in Mlkl−/− BMDMs, a system without measurable loss in cell viability (Figure 3c, 3d). Nevertheless, further experiments are needed to further clarify the mechanism of STING involvement.
Protective host defenses mediated specifically by TRIF have been described in response to several gram-negative bacterial pathogens (55–57). For instance, pulmonary infection by Klebsiella has been found to become significantly more deadly in TRIF-deficient mice (55). Conversely, pre-administration of TLR3 agonist, poly(I:C), which engages TRIF-dependent signaling exclusively, has been shown to promote clearance of gram negative bacterial pathogens (57). Given these observations, our data raise the question as to whether previously described TRIF-dependent protective host responses are mediated by RIPK1/RIPK3 in vivo. Furthermore, our studies in macrophages suggest that TRIF/RIPK1/RIPK3-dependent responses may be either broadly or specifically targeted by bacterial virulence factors. While this has been addressed in various viral infections (72–76), the regulation of RIPK1 and RIPK3 signaling by gram negative bacterial pathogen virulence factors is still poorly understood.
Accordingly, to our knowledge, our work is the first to demonstrate the capacity of RIPK1 and RIPK3 kinases to induce cytokine synthesis in response to gram negative bacteria. We report activation of RIPK1 and RIPK3 kinase-dependent IFNβ synthesis by Klebsiella and Yersinia mutants in vitro in the presence of zVAD. This observation is analogous to the regulation of RIPK1 previously noted in viral-induced RIG-I signaling in which caspase-8-dependent cleavage of RIPK1 attenuated IRF3 activation and IFN-I production (77). However, significantly, our data additionally reveals the propensity of avirulent bacterial strains to promote more efficient RIPK1 and RIPK3 kinase-dependent IFNβ induction compared to their wild type pathogenic counterparts in vitro.
Data by Nogusa et al. showed that RIPK3 was dispensable for the IFN-I transcriptional response induced by RIG-like receptor (RLR) activation or Influenza virus infection in mouse embryonic fibroblasts (78), in contrast to the central role of this protein in cell-death induction during influenza infection in the same cell types (34). These data suggest that there may be important differences in the activation and consequence of the RIPK1 and RIPK3 kinase-dependent cytokine responses in cases of gram-negative bacterial infections, which often occur extracellularly, versus intracellular viral infections, where cell death may be necessary to eliminate infected host cells and prevent viral replication. However, broader sets of pathogens and the roles of cell type specific factors remain to be examined.
In this study we describe a novel role for RIPK1 kinase in directing IFNβ synthesis in response to LPS in vivo. We find that RIPK1 kinase-dependent IFNβ synthesis may be elicited in analogous fashion in BMDMs using LPS with zVAD. Notably, we observed that RIPK1 kinase-dependent IFNβ synthesis in BMDMs also required RIPK3 kinase but not MLKL, a requisite executioner of necroptosis pathway. Conversely, RIPK1 kinase-dependent IFNβ synthesis required the TLR4 adapter protein, TRIF, and downstream canonical IFN-I pathway intermediaries, including TBK1, IKKε and IRF3. It is important to note that RIPK1 and RIPK3 kinase-dependent IFN-I pathway activation in vitro was a delayed feature of TRIF-dependent signaling, suggesting that RIPK1 and RIPK3 kinases are not required for early endosomal translocation of TLR4 (79) (Figure 8). Interestingly, we also noted that the intracellular nucleotide sensor, STING, likely participates in this regulation. Examination of detergent-insoluble cellular fractions or ‘necrosome’-like fractions in RIPK1 kinase-activating conditions suggested that the ‘necrosome’ may represent a scaffold for the TRIF-IRF3 signaling axis, and/or that one or multiple IFN-I pathway intermediaries might be direct targets of RIPK1 and/or RIPK3. These data were further supported by co-immunoprecipitation studies. Finally, our observation that avirulent bacteria are capable of inducing RIPK1 and RIPK3 kinase-dependent IFNβ synthesis to a greater extent than their pathogenic counterparts in vitro, prompts further examination of the role that RIPK1 and RIPK3 kinase-dependent IFNβ synthesis may play in the host response against bacterial infection.
Figure 8.
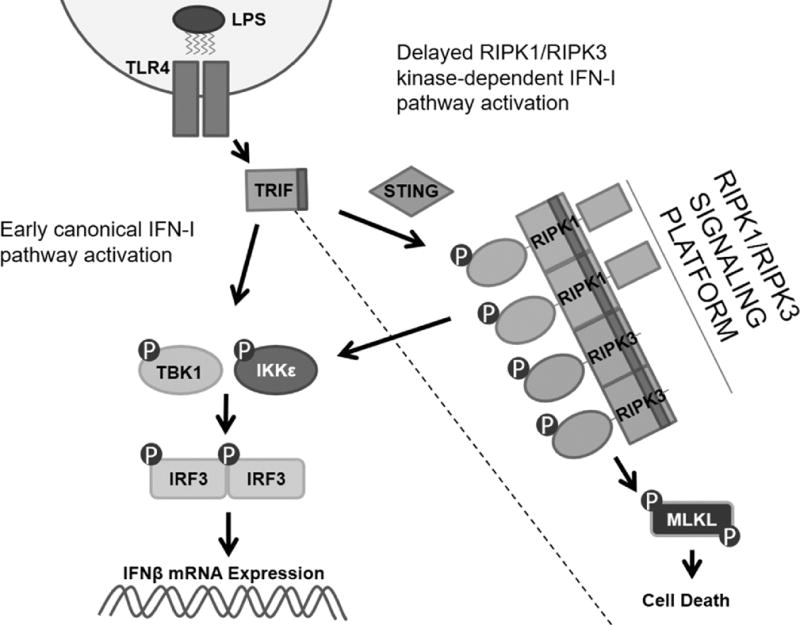
RIPK1 and RIPK3 kinases engage the IFN-I pathway downstream of the adapter protein TRIF.
Supplementary Material
Acknowledgments
We thank Drs. Vishva Dixit, Stan Krajewski, Sergei Nedospasov, and Alexander Poltorak for providing the reagents. P.J.G. and J.B are employees of GlaxoSmithKline. A.D. is a consultant for Denali Therapeutics.
This work was supported in part by grants from NIH to A.D. (RO1GM080356 and RO1GM084205), to S.B. and A.D. (RO1CA190542), to J.M. (RO1 AI113166), to M.K. (RO1AI07118), and to M.W. (RO1NS047447).
References
- 1.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–21. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Christofferson DE, Li Y, Yuan J. Control of Life-or-Death Decisions by RIP1 Kinase. Annu Rev Physiol. 2014;76:129–50. doi: 10.1146/annurev-physiol-021113-170259. [DOI] [PubMed] [Google Scholar]
- 3.Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–23. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 4.Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–47. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 5.Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370:455–65. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 7.Cusson-Hermance N, Khurana S, Lee TH, Fitzgerald KA, Kelliher MA. Rip1 mediates the Trif-dependent toll-like receptor 3- and 4-induced NF-{kappa}B activation but does not contribute to interferon regulatory factor 3 activation. J Biol Chem. 2005;280:36560–6. doi: 10.1074/jbc.M506831200. [DOI] [PubMed] [Google Scholar]
- 8.Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, Tschopp J. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat Immunol. 2004;5:503–7. doi: 10.1038/ni1061. [DOI] [PubMed] [Google Scholar]
- 9.Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, Mocarski ES. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288:31268–79. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaiser WJ, Offermann MK. Apoptosis Induced by the Toll-Like Receptor Adaptor TRIF Is Dependent on Its Receptor Interacting Protein Homotypic Interaction Motif. J Immunol. 2005;174:4942–4952. doi: 10.4049/jimmunol.174.8.4942. [DOI] [PubMed] [Google Scholar]
- 11.Vivarelli MS. RIP Links TLR4 to Akt and Is Essential for Cell Survival in Response to LPS Stimulation. J Exp Med. 2004;200:399–404. doi: 10.1084/jem.20040446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schworer SA, Smirnova II, Kurbatova I, Bagina U, Churova M, Fowler T, Roy AL, Degterev A, Poltorak A. Toll-like Receptor-Mediated Downregulation of the Deubiquitinase CYLD Protects Macrophages from Necroptosis in Wild-Derived Mice. J Biol Chem. 2014 doi: 10.1074/jbc.M114.547547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci. 2011;108:20054–20059. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Najjar M, Saleh D, Zelic M, Nogusa S, Shah S, Tai A, Finger JN, Polykratis A, Gough PJ, Bertin J, Whalen MJ, Pasparakis M, Balachandran S, Kelliher M, Poltorak A, Degterev A. RIPK1 and RIPK3 Kinases Promote Cell-Death-Independent Inflammation by Toll-like Receptor 4. Immunity. 2016;45:46–59. doi: 10.1016/j.immuni.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, Chan FKM, Wu H. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339–50. doi: 10.1016/j.cell.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho Y, Challa S, Moquin D, Genga R, Dutta T, Guildford M, Chan FK. NIH Public Access. 2010;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cook WD, Moujalled DM, Ralph TJ, Lock P, Young SN, Murphy JM, Vaux DL. RIPK1- and RIPK3-induced cell death mode is determined by target availability. Cell Death Differ. 2014:1–13. doi: 10.1038/cdd.2014.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orozco S, Yatim N, Werner MR, Tran H, Gunja SY, Tait SW, Albert ML, Green DR, Oberst A. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 2014;21:1511–1521. doi: 10.1038/cdd.2014.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang J-G, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, Young SN, Varghese LN, Tannahill GM, Hatchell EC, Majewski IJ, Okamoto T, Dobson RCJ, Hilton DJ, Babon JJ, Nicola NA, Strasser A, Silke J, Alexander WS. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443–53. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 20.Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–27. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 22.Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, Ma J, Chen W, Zhang Y, Zhou X, Yang Z, Wu SQ, Chen L, Han J. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013;23:994–1006. doi: 10.1038/cr.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, Pierotti C, Garnier JM, Dobson RCJ, Webb AI, Tripaydonis A, Babon JJ, Mulcair MD, Scanlon MJ, Alexander WS, Wilks AF, Czabotar PE, Lessene G, Murphy JM, Silke J. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci U S A. 2014;111 doi: 10.1073/pnas.1408987111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christofferson DE, Li Y, Hitomi J, Zhou W, Upperman C, Zhu H, Gerber SA, Gygi S, Yuan J. A novel role for RIP1 kinase in mediating TNFα production. Cell Death Dis. 2012;3:e320. doi: 10.1038/cddis.2012.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biton S, Ashkenazi A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-α feedforward signaling. Cell. 2011;145:92–103. doi: 10.1016/j.cell.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 26.Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, Yuan J. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–23. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McNamara CR, Ahuja R, Osafo-Addo AD, Barrows D, Kettenbach A, Skidan I, Teng X, Cuny GD, Gerber S, Degterev A. Akt Regulates TNFα synthesis downstream of RIP1 kinase activation during necroptosis. PLoS One. 2013;8:e56576. doi: 10.1371/journal.pone.0056576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lukens JR, Vogel P, Johnson GR, Kelliher MA, Iwakura Y, Lamkanfi M, Kanneganti T-D. RIP1-driven autoinflammation targets IL-1α independently of inflammasomes and RIP3. Nature. 2013;498:224–7. doi: 10.1038/nature12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moriwaki K, Balaji S, McQuade T, Malhotra N, Kang J, Chan FKM. The Necroptosis Adaptor RIPK3 Promotes Injury-Induced Cytokine Expression and Tissue Repair. Immunity. 2014;41:567–578. doi: 10.1016/j.immuni.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moriwaki K, Bertin J, Gough PJ, Chan FKM. A RIPK3-caspase 8 complex mediates atypical pro-IL-1β processing. J Immunol. 2015;194:1938–44. doi: 10.4049/jimmunol.1402167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, Hall C, Kaur Spall S, Anderton H, Masters SL, Rashidi M, Wicks IP, Alexander WS, Mitsuuchi Y, Benetatos CA, Condon SM, Wong WW-L, Silke J, Vaux DL, Vince JE. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun. 2015;6:6282. doi: 10.1038/ncomms7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berger SB, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, Capriotti C, Cook M, Finger J, Hughes-Earle A, Harris PA, Kaiser WJ, Mocarski ES, Bertin J, Gough PJ. Cutting Edge: RIP1 Kinase Activity Is Dispensable for Normal Development but Is a Key Regulator of Inflammation in SHARPIN-Deficient Mice. J Immunol. 2014 doi: 10.4049/jimmunol.1400499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van T-M, Lee TH, Chan FKM, Pasparakis M, Kelliher MA. Cutting Edge: RIPK1 Kinase Inactive Mice Are Viable and Protected from TNF-Induced Necroptosis In Vivo. J Immunol. 2014 doi: 10.4049/jimmunol.1400590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nogusa S, Thapa RJ, Dillon CP, Oberst A, Green DR, Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Iii THO, Ingram JP, Rodriguez DA, Kosoff R, Sharma S, Sturm O, Verbist K, Gough PJ, Bertin J, Hartmann BM, Sealfon SC, Kaiser WJ, Mocarski ES, Thomas PG, Oberst A, Green DR, Balachandran S. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus Article RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis. Cell Host Microbe. 2016:1–12. doi: 10.1016/j.chom.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis AJ, Mecsas J. Mutations in the Yersinia pseudotuberculosis type III secretion system needle protein, YscF, that specifically abrogate effector translocation into host cells. J Bacteriol. 2007;189:83–97. doi: 10.1128/JB.01396-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Degterev A, Maki JL, Yuan J. Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase. Cell Death Differ. 2013;20:366. doi: 10.1038/cdd.2012.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, Ouellette M, King BW, Wisnoski D, Lakdawala AS, DeMartino MP, Casillas LN, Haile PA, Sehon CA, Marquis RW, Upton J, Daley-Bauer LP, Roback L, Ramia N, Dovey CM, Carette JE, Chan FM, Bertin J, Gough PJ, Mocarski ES, Kaiser WJ. RIP3 Induces Apoptosis Independent of Pronecrotic Kinase Activity. Mol Cell. 2014;56:481–495. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clark K, Peggie M, Plater L, Sorcek RJ, Young ERR, Madwed JB, Hough J, McIver EG, Cohen P. Novel cross-talk within the IKK family controls innate immunity. Biochem J. 2011;434:93–104. doi: 10.1042/BJ20101701. [DOI] [PubMed] [Google Scholar]
- 39.Clark K, Plater L, Peggie M, Cohen P. Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IκB Kinase ε:A distinct upstream kinase mediates ser-172 phosphorylation and activation. J Biol Chem. 2009;284:14136–14146. doi: 10.1074/jbc.M109.000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brumatti G, Ma C, Lalaoui N, Nguyen N, Navarro M, Tanzer MC, Richmond J, Ghisi M, Salmon JM, Silke N, Pomilio G, Glaser SP, De Valle E, Gugasyan R, Gurthridge MA, Condon SM, Johnstone RW, Lock R, Salvesen G, Wei A, Vaux DL, Ekert PG, Silke J. The caspase-8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. 2016;8:1–12. doi: 10.1126/scitranslmed.aad3099. [DOI] [PubMed] [Google Scholar]
- 41.Moquin DM, McQuade T, Chan FKM. CYLD deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS One. 2013;8:e76841. doi: 10.1371/journal.pone.0076841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Doyle S, Vaidya S, O’Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, Cheng G. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–63. doi: 10.1016/s1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 43.Sakaguchi S, Negishi H, Asagiri M, Nakajima C, Mizutani T, Takaoka A, Honda K, Taniguchi T. Essential role of IRF-3 in lipopolysaccharide-induced interferon-β gene expression and endotoxin shock. Biochem Biophys Res Commun. 2003;306:860–866. doi: 10.1016/s0006-291x(03)01049-0. [DOI] [PubMed] [Google Scholar]
- 44.Power MR, Li B, Yamamoto M, Akira S, Lin TJ. A role of Toll-IL-1 receptor domain-containing adaptor-inducing IFN-beta in the host response to Pseudomonas aeruginosa lung infection in mice. J Immunol. 2007;178:3170–3176. doi: 10.4049/jimmunol.178.5.3170. [DOI] [PubMed] [Google Scholar]
- 45.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2013;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–11. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 47.Kim SJ, Li J. Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis. 2013;4:e716–12. doi: 10.1038/cddis.2013.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, Liu ZG. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A. 2012;109:5322–7. doi: 10.1073/pnas.1200012109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–3. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 50.Solis M, Romieu-Mourez R, Goubau D, Grandvaux N, Mesplede T, Julkunen I, Nardin A, Salcedo M, Hiscott J. Involvement of TBK1 and IKKepsilon in lipopolysaccharide-induced activation of the interferon response in primary human macrophages. Eur J Immunol. 2007;37:528–39. doi: 10.1002/eji.200636090. [DOI] [PubMed] [Google Scholar]
- 51.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao S-M, Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–6. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 52.Gatot JS, Gioia R, Chau TL, Patrascu F, Warnier M, Close P, Chapelle JP, Muraille E, Brown K, Siebenlist U, Piette J, Dejardin E, Chariot A. Lipopolysaccharide-mediated interferon regulatory factor activation involves TBK1-IKKepsilon-dependent Lys(63)-linked polyubiquitination and phosphorylation of TANK/I-TRAF. J Biol Chem. 2007;282:31131–46. doi: 10.1074/jbc.M701690200. [DOI] [PubMed] [Google Scholar]
- 53.Wang X, Majumdar T, Kessler P, Ozhegov E, Zhang Y, Chattopadhyay S, Barik S, Sen GC. STING Requires the Adaptor TRIF to Trigger Innate Immune Responses to Microbial Infection. Cell Host Microbe. 2016;20:329–341. doi: 10.1016/j.chom.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt JP, Ye J, Zhang X, Chang A, Vakifahmetoglu-Norberg H, Geng J, Py B, Zhou W, Amin P, Lima JB, Qi C, Yu Q, Trapp B, Yuan J. Activation of Necroptosis in Multiple Sclerosis. Cell Rep. 2015;10:1836–1849. doi: 10.1016/j.celrep.2015.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cai S, Batra S, Shen L, Wakamatsu N, Jeyaseelan S. Both TRIF- and MyD88-dependent signaling contribute to host defense against pulmonary Klebsiella infection. J Immunol. 2009;183:6629–38. doi: 10.4049/jimmunol.0901033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sotolongo J, España C, Echeverry A, Siefker D, Altman N, Zaias J, Santaolalla R, Ruiz J, Schesser K, Adkins B, Fukata M. Host innate recognition of an intestinal bacterial pathogen induces TRIF-dependent protective immunity. J Exp Med. 2011;208:2705–16. doi: 10.1084/jem.20110547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ruiz J, Kanagavelu S, Flores C, Romero L, Riveron R, Shih DQ, Fukata M. Systemic Activation of TLR3-Dependent TRIF Signaling Confers Host Defense against Gram-Negative Bacteria in the Intestine. Front Cell Infect Microbiol. 2016;5:1–11. doi: 10.3389/fcimb.2015.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davis AJ, Díaz DADJ, Mecsas J. A dominant-negative needle mutant blocks type III secretion of early but not late substrates in Yersinia. Mol Microbiol. 2010;76:236–59. doi: 10.1111/j.1365-2958.2010.07096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Viboud GI, Bliska JB. Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu Rev Microbiol. 2005;59:69–89. doi: 10.1146/annurev.micro.59.030804.121320. [DOI] [PubMed] [Google Scholar]
- 60.Zhang Y, Romanov G, Bliska JB. Type III secretion system-dependent translocation of ectopically expressed Yop effectors into macrophages by intracellular Yersinia pseudotuberculosis. Infect Immun. 2011;79:4322–31. doi: 10.1128/IAI.05396-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lawlor MS, Handley SA, Miller VL. Comparison of the host responses to wild-type and cpsB mutant Klebsiella pneumoniae infections. Infect Immun. 2006;74:5402–7. doi: 10.1128/IAI.00244-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lawlor MS, Hsu J, Rick PD, Miller VL. Identification of Klebsiella pneumoniae virulence determinants using an intranasal infection model. Mol Microbiol. 2005;58:1054–73. doi: 10.1111/j.1365-2958.2005.04918.x. [DOI] [PubMed] [Google Scholar]
- 63.Saleh D, Degterev A. Emerging Roles for RIPK1 and RIPK3 in Pathogen-Induced Cell Death and Host Immunity. Current topics in microbiology and immunology. 2015 doi: 10.1007/82_2015_449. [DOI] [PubMed] [Google Scholar]
- 64.Sridharan H, Upton JW. Programmed necrosis in microbial pathogenesis. Trends Microbiol. 2014;22:199–207. doi: 10.1016/j.tim.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 65.Weng D, Marty-Roix R, Ganesan S, Proulx MK, Vladimer GI, Kaiser WJ, Mocarski ES, Pouliot K, Chan FK-M, Kelliher MA, Harris PA, Bertin J, Gough PJ, Shayakhmetov DM, Goguen JD, Fitzgerald KA, Silverman N, Lien E. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc Natl Acad Sci U S A. 2014;111:7391–6. doi: 10.1073/pnas.1403477111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Philip NH, Dillon CP, Snyder AG, Fitzgerald P, Wynosky-Dolfi MA, Zwack EE, Hu B, Fitzgerald L, Mauldin EA, Copenhaver AM, Shin S, Wei L, Parker M, Zhang J, Oberst A, Green DR, Brodsky IE. Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-κB and MAPK signaling. Proc Natl Acad Sci U S A. 2014;111:7385–90. doi: 10.1073/pnas.1403252111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kitur K, Parker D, Nieto P, Ahn DS, Cohen TS, Chung S, Wachtel S, Bueno S, Prince A. Toxin-Induced Necroptosis Is a Major Mechanism of Staphylococcus aureus Lung Damage. PLoS Pathog. 2015;11:e1004820. doi: 10.1371/journal.ppat.1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol. 2012;13:954–62. doi: 10.1038/ni.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The Death Domain Kinase RIP Mediates the TNF-Induced NF- κ B Signal. 1998;8:297–303. doi: 10.1016/s1074-7613(00)80535-x. [DOI] [PubMed] [Google Scholar]
- 70.Newton K, Sun X, Dixit VM. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol. 2004;24:1464–9. doi: 10.1128/MCB.24.4.1464-1469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wong WWL, Vince JE, Lalaoui N, Lawlor KE, Chau D, Bankovacki A, Anderton H, Metcalf D, O’Reilly L, Jost PJ, Murphy JM, Alexander WS, Strasser A, Vaux DL, Silke J. cIAPs and XIAP regulate myelopoiesis through cytokine production in an RIPK1- And RIPK3-dependent manner. Blood. 2014;123:2562–2572. doi: 10.1182/blood-2013-06-510743. [DOI] [PubMed] [Google Scholar]
- 72.Huang Z, Wu SQ, Liang Y, Zhou X, Chen W, Li L, Wu J, Zhuang Q, Chen C, Li J, Zhong CQ, Xia W, Zhou R, Zheng C, Han J. RIP1/RIP3 Binding to HSV-1 ICP6 Initiates Necroptosis to Restrict Virus Propagation in Mice. Cell Host Microbe. 2015;17:229–42. doi: 10.1016/j.chom.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 73.Wang X, Li Y, Liu S, Yu X, Li L, Shi C, He W, Li J, Xu L, Hu Z, Yu L, Yang Z, Chen Q, Ge L, Zhang Z, Zhou B, Jiang X, Chen S, He S. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci U S A. 2014;111:15438–43. doi: 10.1073/pnas.1412767111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, Kaiser WJ, Mocarski ES. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe. 2015;17:243–51. doi: 10.1016/j.chom.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11:290–7. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Omoto S, Guo H, Talekar GR, Roback L, Kaiser WJ, Mocarski E. Suppression of RIP3-dependent Necroptosis by Human Cytomegalovirus. J Biol Chem. 2015 doi: 10.1074/jbc.M115.646042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rajput A, Kovalenko A, Bogdanov K, Yang SH, Kang TB, Kim JC, Du J, Wallach D. RIG-I RNA helicase activation of IRF3 transcription factor is negatively regulated by caspase-8-mediated cleavage of the RIP1 protein. Immunity. 2011;34:340–51. doi: 10.1016/j.immuni.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 78.Nogusa S, Slifker MJ, Ingram JP, Thapa RJ, Balachandran S. RIPK3 Is Largely Dispensable for RIG-I-Like Receptor- and Type I Interferon-Driven Transcriptional Responses to Influenza A Virus in Murine Fibroblasts. PLoS One. 2016;11:e0158774. doi: 10.1371/journal.pone.0158774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. 2008;9:361–8. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.