

Abstract
Mature peripheral double negative (DN) T cells expressing αβ TCR but lacking CD4/CD8 co-receptors play protective as well as pathogenic roles. To better understand their development and functioning in vivo, we concomitantly inactivated CD4 and CD8 genes in mice with intact MHC class I and class II molecules with the hypothesis that this would enable the development of DNT cells. We also envisaged that these DNT cells could be activated by bacterial superantigens in vivo as activation of T cells by superantigens does not require CD4 and CD8 coreceptors. Since HLA class II molecules present superantigens more efficiently than murine MHC class II molecules, CD4 CD8 double knockout (DKO) mice transgenically expressing HLA-DR3 or HLA-DQ8 molecules were generated. While thymic cellularity was comparable between wild type (WT) and DKO mice, CD3+ αβ TCR+ thymocytes were significantly reduced in DKO mice implying defects in thymic positive selection. Splenic CD3+ αβ TCR+ cells and FoxP3+ T regulatory cells were present in DKO mice but significantly reduced. However, the in vivo inflammatory responses and immunopathology elicited by acute challenge with the superantigen staphylococcal enterotoxin B (SEB) were comparable between WT and DKO mice. Choric exposure to SEB precipitated a lupus-like inflammatory disease with characteristic lympho-monocytic infiltration in lungs, livers and kidneys, along with production of anti-nuclear antibodies in DKO mice as in WT mice. Overall, our results suggest that DNT cells can develop efficiently in vivo and chronic exposure to bacterial superantigens may precipitate a lupus-like autoimmune disease through activation of DNT cells.
Keywords: Double negative T lymphocytes, HLA class II transgenic mice, Superantigen, inflammation, Lupus
Introduction
Peripheral T cells bearing the αβ TCR are broadly classified into CD4+ and CD8+ T cells based on their co-receptor expression (1, 2). CD4+ T cells recognize processed exogenous peptides presented by MHC class II molecules and are designated as T helper (Th) cells (3). The CD8+ T cells on the other hand, respond to endogenous antigenic peptides presented in the context of MHC class I molecules and mediate cell-mediated cytotoxicity (4). However, mature T cells expressing αβ TCR and lacking CD4/CD8 co-receptors, referred to as double negative (DN) T cells, have been demonstrated in the periphery of healthy mice and humans (5-7) [reviewed in ref (8)]. Studies have shown that DNT cells play a protective role in immunity to intracellular pathogens (9, 10) and have a regulatory role in type 1 diabetes and transplantation (11-15). However, several reports have demonstrated a pathogenic role for DNT cells in diseases such as spondyloarthropathy, autoimmune lymphoproliferative syndrome (ALPS), aplastic anemia and lupus (16-20). Thus, there are a lot of ambiguities regarding the functions of DNT cells in vivo. This could be attributed to lack of good animal models for studying the functions of DNT cells.
The current knowledge on the immunobiology of DNT cells is largely derived from studies done on mice that have spontaneous mutations in the Fas (Faslpr) or FasL (Faslgld) genes (21-23). In these mice, due to the defective Fas-FasL mediated apoptosis, a majority of CD4-CD8 DN thymocytes that are reactive to the self-antigens, fail to undergo thymic negative selection and escape into periphery (24-26). In peripheral tissues, these autoreactive immature DNT cells undergo significant expansion causing lymphadenopathy and generalized systemic autoimmune diseases such as lupus (27). Due to the profound defects in T cell negative selection, many experts in this field do not consider lpr/gld models as ideal models for studying the functions of mature DNT cells (28, 29). Therefore, we realized the need for a good animal model to understand the development of DNT cells and to study their functions in vivo. We hypothesized that genetically inactivating CD4 and CD8 genes concurrently in mice with intact MHC class I and class II molecules may facilitate the generation of DNT cells expressing αβ TCR.
In this report, we discuss the generation of CD4 CD8 double knockout mice (DKO) on the HLA-DR3/HLA-DQ8 background and development/functioning of DNT cells in them. The reasons for choosing HLA-DR3 or HLA-DQ8 background are two folds. One, this would allow us to test the functions of DNT cells in vivo using staphylococcal superantigens (SSAgs). Unlike conventional antigens, SSAgs robustly activate αβ TCR+ T cells without involving the engagement of CD4 and CD8 coreceptors (30). As HLA class II molecules present SSAgs more efficiently than mouse MHC class II molecules (31), we could challenge CD4 CD8 DKO expressing HLA-DR3 or HLA-DQ8 with SSAgs and study a variety of DNT cell functions in vivo. Two, HLA-class II molecules (including HLA-DR3 and HLA-DQ8) are strongly associated with certain autoimmune diseases such as systemic lupus erythematosus, arthritis, type 1 diabetes and multiple sclerosis (32, 33). Therefore, in the future we could investigate the role of DNT cells in various autoimmune inflammatory diseases using these HLA-DR3/HLA-DQ8 DKO mice (34).
Mateirals and Methods
Mice
Transgenic mice expressing HLA-DR3 and HLA-DQ8 molecules in the absence of endogenous mouse MHC class II molecules have been previously described (35, 36). HLA-DR3 and HLA-DQ8 transgenic mice with targeted disruption of CD4 or CD8 genes were generated by mating them with CD4-/- and CD8-/- mice (a generous gift from Dr. Tak Mak), respectively (37). Subsequently, HLA-DR3 and HLA-DQ8 transgenic mice lacking both CD4 and CD8 molecules were generated by intercrossing respective CD4-/- and CD8-/- mice. Hereafter, mice lacking both CD4 and CD8 coreceptors are designated as DKO mice. Non-obese diabetic Severe combined immunodeficient (NOD-SCID) mice obtained from The Jackson Laboratory (Bar Harbor, ME, USA) were maintained in our mouse colony. All mice were bred within the barrier facility of Mayo Clinic Immunogenetics Mouse Colony (Rochester, MN, USA) and moved to a conventional facility after weaning. All the experiments were approved by the Mayo Clinic Institutional Animal Care and Use Committee.
Reagents, antibodies and Flow cytometry
Endotoxin-reduced, highly purified staphylococcal enterotoxin B (SEB, Toxin Laboratories, Sarasota, FL) was dissolved in PBS at 1 mg/ml and stored frozen at -80°C in aliquots. The purity of SEB was verified by SDS-PAGE followed by Coomassie blue staining and the absence of other staphylococcal SAg was verified using staphylococcal enterotoxin identification visual immunoassay (SET VIA™, 3M, MN, USA). The following antibodies were used for flow cytometry (BD biosciences) CD4 - GK1.5, CD8 - 53-6.7, CD19 -1D3, B220 - RA3-6B2, Mac-1 - M 1/70, CD44 - M7, CD25 - 3C7, CD62L - MEL-14, and isotype control. The following anti-mouse TCR Vβ antibodies were used. Vβ2 (clone - B20.6), Vβ3 (Clone KJ25), Vβ4 (Clone KT4), Vβ5 (Clone MR9-4), Vβ6 (Clone RR4-7), Vβ7 (Clone TR310), Vβ8 (Clone F23.1), Vβ9 (Clone MR10-2), Vβ10 (Clone B21.5), Vβ11 (Clone RR3-15), Vβ12 (Clone MR11-1), Vβ13 (Clone MR12-3), Vβ14 (Clone 14-2) and Vβ17 (Clone KJ23), the pan-TCRβ chain antibody (Clone H57-597), γδ TCR - GL-3, NK1.1 - PK136 and CD49b - DX5. FoxP3+ T cells were enumerated using the intracellular staining kit from eBioscience (San Diego, CA, USA). Splenic mononuclear cells were prepared as per standard procedure (38). Briefly, spleens were harvested, crushed and red-blood cell depleted mononuclear suspensions were made by ammonium chloride lysis. Cells were enumerated using an automated cell counter (Cellometer Auto T4, Nexcelom Bioscience LLC, Lawrence, MA, USA), resuspended in phosphate buffered saline containing bovine serum albumin and stained with antibodies for flow cytometry. Thymic mononuclear cells were prepared in the same manner barring the ammonium chloride lysis step by harvesting thymus (38).
Assessing in vitro proliferative response to staphylococcal enterotoxin B
Red blood cell-depleted splenic mononuclear cells were prepared as above. Cells were cultured in HEPES-buffered RPMI-1640 containing 5% horse serum, serum supplement, streptomycin, and penicillin, at a concentration of 10 × 105 cells/well in 100 μl volumes in 96-well round-bottomed tissue culture plates. Cells were incubated with medium alone or SEB (1 μg/ml) for a total of 48 hours. During the last 18 hours, tritiated thymidine (1 μCi) was added to each well. The next day, the cells were harvested using a semi-automated harvester (Brandel, Gaithersburg, MD, USA) and the incorporated radioactivity was determined using an automated scintillation counter (MicroBeta® TriLux, Perkin Elmer, Waltham, MA, USA).
Direct comparison of in vivo proliferation of T cells from WT and DKO HLA-DR3 mice by adoptive transfer
CD3+ T cells from WT and DKO HLA-DR3 mice were isolated using enrichment columns (R&D systems, Minneapolis, MN), labeled with Carboxyfluorescein succinimidyl ester (CFSE, Invitrogen, Thermo Fisher Scientific, MA, USA) and mixed in equal numbers. CD3- fraction of the DR3 WT splenocytes obtained from the above process was collected, irradiated and mixed with CFSE-labeled CD3+ T cell fraction as antigen presenting cells. Ten million cells were injected intravenously into 8-week-old NOD.SCID recipient mice. Recipient mice were injected with either PBS or SEB (50 μg) through intraperitoneal route 24-hours after adoptive transfer. Animals were killed 72-hours after SEB challenge, spleens harvested and stained with PerCp conjugated anti-CD3 antibodies and APC conjugated anti-CD4 and anti-CD8 antibodies and analyzed by flow cytometry. CD3-gated cells were further divided into CD4/CD8+ population and CD4/CD8- population. CFSE staining pattern within these gated populations was determined.
Acute challenge with staphylococcal enterotoxin B
Each experimental mouse received 50 μg of SEB dissolved in 200 μl of PBS by intraperitoneal route while control mice received 200 μl of PBS alone. Mice were closely monitored for symptoms of shock such as hunched posture, ruffled fur, diarrhea, hypothermia and reduced activity.
Implantation of miniosmotic pump
Miniosmotic pumps (Alzet Corporation, Cupertino, CA, USA) capable of continuous constant delivery of liquids over 7-days were loaded with either PBS or SEB (50 μg, dissolved in PBS) as per supplier's protocol. PBS- or SEB-filled pumps were implanted subcutaneously into surgically prepared experimental mice of either sex (8 to 12-weeks old) under anesthesia as per standard procedure. Surgical incisions were closed with stainless steel wound clips.
Serum cytokine quantification and histopathology
Age-matched WT and DKO HLA-DR3 were challenged intraperitoneally with either 50μg SEB or PBS. Animals were bled four hours later and blood was collected from the mice into serum separation tubes (BD Biosciences, San Jose, California). The serum was separated and then stored frozen in aliquots at –80°C. Concentration of cytokines was determined in the serum samples by using a multiplex bead assay using the manufacturer's protocol and their software and hardware (Bio-Plex; Bio-Rad, Hercules, CA, USA). Tissues collected in buffered formalin were paraffin embedded, cut, and stained with H&E per standard procedure for histopathologic analysis. To numerically quantify the extent of inflammation, H&E stained sections were objectively evaluated in a blinded fashion as described previously (39). A score of 0 to 3 were given to each slide based on the extent of tissue involvement. Score 0 – No inflammation, Score 1 – less than 25% of the tissue shows inflammatory changes with patchy aggregates of inflammatory cells, Score 2 – 25 to 50% of the tissue shows inflammatory changes with the presence of more intense aggregates of inflammatory cells and Score 3 – More than 50% of the tissue shows inflammatory changes with large prominent inflammatory cell aggregates. Representative sections were photographed using an Olympus AX70 research microscope fitted with an Olympus DP70 camera (Olympus America, Center Valley, PA, USA).
Immunofluorescence
Immediately following sacrifice as discussed above, tissues from mice were also collected in optimal cutting compound (OCT, Sakura Finetek Tissue-Tek) and stored frozen at -80°C. Five μm sections were cut using a cryostat, fixed in cold acetone and stained with fluorochrome-conjugated anti-CD3 antibodies as per standard techniques. Sections were mounted using the Slowfade Gold antifade reagent with DAPI (Invitrogen) and were analyzed using an Olympus AX70 research microscope (Olympus America Inc., Center Valley, PA, USA). Images were acquired using an Olympus DP70 camera.
Detection and quantification of autoantibodies
Antinuclear antibodies were determined using HEp-2 cells as per manufacturer's protocol (Bio-Rad, Hercules, CA) at 1 in 50 dilution. Binding of mouse autoantibodies was detected using a FITC-conjugated goat anti-mouse antibody (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA). Titers of antinuclear antibodies were quantitatively determined using mouse Anti-Nuclear Antigens (ANA/ENA) total Ig kit (Alpha Diagnostic Intl. Inc., San Antonio, Texas, USA) following manufacturer's recommendation. Sera were tested at a dilution of 1:20.
Statistics
All analyses were performed using GraphPad Prism (version 6.0a; San Diego, CA). Parametric testing between two unmatched groups was performed by unpaired t test. P values below 0.05 were considered significant.
Results
T cell development in thymus in mice lacking CD4 and CD8 coreceptors
To study the impact of deficiency of both CD4 and CD8 coreceptors on early T cell development, thymocytes extracted from 3-week-old WT and DKO HLA-DR3 mice were enumerated and analyzed by flow cytometry. As shown in Fig 1A, the total thymic cellularity was comparable between WT and DKO HLA-DR3 mice at a young age. However, the numbers of CD3+ thymocytes and TCR αβ+ thymocytes were dramatically reduced within the thymus of DKO HLA-DR3 mice. On the other hand, the numbers of thymocytes bearing γδ TCR were comparable between WT and DKO HLA-DR3 mice (Fig 1B). The expression patterns of CD44 and CD25 molecules, which are traditionally used to identify various thymocyte developmental stages, were comparable between WT and DKO mice (Supplemental Fig 1). The frequency of NKT cells in the thymus, as determined using CD1d tetramer staining, was comparable between WT and DKO mice (Data not shown). However, DKO had significantly reduced numbers of FoxP3+ T cells (0.58 ± 0.12% versus 0.046 ± 0.008% in DR3 WT and DR3.DKO mice, respectively (Supplemental Fig 2). We also analyzed the thymocyte numbers at 16-weeks of age. As expected, DR3 WT mice had fewer thymocytes compared at 3-weeks of age due to age-dependent thymic atrophy. Even though HLA-DR3 DKO mice had consistently higher total thymocyte count than WT mice at 16-weeks of age, this difference was not statistically significant (14.4±3.3 × 106 and 20.2±6.0 × 106 for WT and DKO thymii, respectively). Nonetheless, the numbers of CD3+ thymocytes and TCR β+ thymocytes still remained severely reduced even at 16-weeks of age in DKO mice (Not shown). Overall, while the total thymocyte numbers were not affected in DKO mice, lack of co-receptors significantly reduced the number of thymocytes expressing CD3 and TCRβ, indicating defects in maturation of T cells expressing TCR αβ.
Figure 1. The impact of concurrent targeted disruption of CD4 and CD8 genes on T cell development in the thymus in HLA-DR3 transgenic mice.

Thymocytes extracted from 3-week-old WT and DKO HLA-DR3 mice of either sex were enumerated and analyzed by flow cytometry following staining with indicated antibodies. Panel A represents total thymocyte count and panel B depicts distribution of various thymocyte subsets in thymus. Each bar represents mean±SE values from 6-8 mice/group. * p<0.05 and NS – Not significant.
Splenocyte compartment in mice lacking CD4 and CD8 coreceptors
We next investigated the presence of DNT cell population in the spleens. The total splenocyte numbers were significantly reduced in HLA-DR3 DKO mice at 6- to 8-weeks of age (Fig 2A). Further breakdown of various immune cell subsets in the spleens revealed a dramatic reduction in the total CD3+ T cells as well as T cells expressing TCR β (Fig 2B and C). Even though the percentage of B cells was not significantly different between WT and DKO mice, when converted to absolute numbers, DKO mice had significantly fewer B220+ cells and the same observations applied to CD11b+ cells (Fig 2B and C). While the percentage of T cells bearing γδ TCR was increased significantly in DKO mice, when converted to absolute numbers, this significance did not stand (Fig 2B and C).
Figure 2. Splenic immune compartment in HLA-DR3 transgenic mice lacking both CD4 and CD8 coreceptors.

Splenic mononuclear cells collected from 6- to 8-week-old WT and DKO HLA-DR3 mice of either sex were counted and analyzed by flow cytometry following staining with indicated antibodies. Panel A represents total splenocyte counts in WT and DKO mice. Panels B and C depict the percentage and absolute numbers of various splenocyte subsets, respectively. The percentage of T cells expressing TCR Vβ6 and Vβ8 within the total lymphocyte gate (panel D) and specifically within the CD3 gate (panel E) are also given. Each bar represents mean±SE values from 6-8 mice/group. * p<0.05 and NS – Not significant.
We also determined the TCR repertoire in DKO mice using antibodies to various TCR Vα and Vβ families. Splenic CD3+ gated DNT cells from DKO mice expressed a wide repertoire of TCR comparable to WT mice. However, the number of cells expressing each TCR family was reduced in DKO mice because the total numbers of CD3+ T cells were lower in these mice (Data not shown). Fig 2D, 2E, and Supp. Fig 3 represent the percentage of T cells expressing selected TCR Vβ families in WT and DKO mice. Splenic and liver NKT cell numbers, as determined using CD1d tetramers, were comparable between both the groups (Data not shown). Of interest, while our HLA-DR3 WT mice expressed both DX5 and NK1.1 markers, DKO mice did not express the NK1.1 marker but expressed only DX5, possibly due to the fact that the CD4 and CD8 knockout mice were generated on a BL/D2 (H-2b/d) genetic background (40). Nonetheless, the percentages of CD3+DX5+ cells were comparable between WT and DKO mice (Data not shown), whereas the numbers of FoxP3+ cells were reduced by 2-folds in DKO mice (Supp. Fig 4). It is interesting to note that while the FoxP3+ cells were reduced by 10-folds in the thymii of DKO mice, in the spleens, they were reduced only by 3-5-folds, suggesting expansion of Tregs either due to de novo differentiation of FoxP3- cells to FoxP3+ cells or proliferation of FoxP3+ cells in the periphery [reviewed in (41)]. Even at 16-weeks of age, DKO harbored significantly fewer splenocytes (55±7.8 × 106 and 31.5±6.6 ×106, for HLA-DR3 WT and HLA-DR3 DKO mice, respectively) and as a result the numbers of various splenocyte subsets were also reduced (Data not shown). Analysis of expression profiles of CD44 and CD62L on CD3+ splenocytes from adult DKO mice showed a more activated phenotype, with higher levels of expression of CD44 (CD44hi) and lower level of CD62L expression (CD62Llo) (Supp. Fig 4). Overall, these results indicated that T cells bearing the αβ TCR are still present in significantly higher numbers (approximately 90% of CD3+ T cells) in the periphery of mice lacking both the CD4 and CD8 T cell coreceptors compared to only 2-5% of CD3+ T cells from HLA-DR3 WT mice which are DNT cells (Supp. Fig 4) as shown in other studies (42). Hence, the DKO mice provided us an opportunity to investigate the biology of DNT cells independently of CD4+ and CD8+ T cells.
DNT cells respond efficiently to staphylococcal superantigen
Having demonstrated the presence of DNT cells in the periphery of DKO mice, we next investigated their functionality. Staphylococcal superantigens (SSAg) activate T cells in a coreceptor independent manner (30). Therefore, measuring their in vitro and in vivo responses to SSAg is an ideal way to study the functionality of T cells lacking CD4 or CD8 coreceptors. We have demonstrated previously that HLA-DR3/HLA-DQ8 mice mount a robust response to the (SSAg) compared to conventional laboratory mice expressing endogenous MHC class II molecules due to superior presentation of SSAg by HLA class II molecules compared to mouse MHC class II molecules (31). Therefore, in the first set of experiments splenic mononuclear cells from WT or DKO mice were stimulated in vitro with SEB. As shown in Fig 3, splenocytes from DKO proliferated robustly to SEB. As DKO HLA-DR3 mice have fewer CD3+ T cells than WT HLA-DR3 mice, lower thymidine incorporation in DKO mice is expected. We next performed a series of experiments to study the responses of DNT cells in vivo.
Figure 3. In vitro responsiveness of splenocytes from HLA-DR3 DKO mice to staphylococcal enterotoxin B in comparison to WT HLA-DR3 mice.

Splenic mononuclear cells collected from 8-week-old WT and DKO HLA-DR3 mice belonging to either sex were stimulated with SEB. The extent of cell proliferation was determined by thymidine incorporation assay. Each bar represents Mean±SE values from triplicate wells. Representative data from 3 similar experiments is shown. NS – Not significant.
In the first set of in vivo experiments, CD3+ T cells were isolated from WT and DKO mice, labeled with CFSE separately, mixed and adoptively transferred into recipient NOD-SCID mice along with irradiated CD3-depleted splenocytes from WT HLA-DR3 mice (as antigen presenting cells) as described in methods section. A day later, recipient NOD-SCID mice were acutely challenged with PBS or SEB and all mice were killed 72 hours after SEB injection. Splenocytes were harvested, stained with antibodies and analyzed by flow cytometry. As shown in Fig 4, CD3+ cells from both WT and DKO showed CFSE dilution to a certain extent even in control (PBS challenged) NOD-SCID recipient mice, likely due to homeostatic proliferation as recipient mice lack T and B cells. However, CFSE dilution was more pronounced in both WT and DKO T cells collected from recipient mice that were challenged with SEB. Histogram overlays (Fig 4) show comparable proliferative potential of DKO T cells compared to WT T cells. However, a careful analysis of CFSE peaks revealed a somewhat sluggish proliferation of DKO T cells compared to WT T cells because fewer T cells from DKO mice had diluted their CFSE to the level seen in WT T cells (peaks 5 and 6 in histograms, Fig 4C).
Figure 4. Determining the efficacy of proliferation of splenic CD3+ T cells from HLA-DR3 DKO mice to staphylococcal enterotoxin B in comparison to WT HLA-DR3 mice in an adoptive transfer model.

CD3+ splenocytes isolated from 8-week-old WT and DKO HLA-DR3 mice of either sex were labeled with CFSE, mixed and adoptively transferred into NOD.SCID recipients along with CD3-depleted splenocytes harvested from HLA-DR3 WT mice as APCs. Recipient mice were challenged with PBS or SEB. Mice were killed 72-hours later and the CFSE levels in CD4/CD8+ and CD4/CD8- populations was determined by flow cytometry. (A) Representative dot plots of splenocytes obtained from PBS- or SEB-challenged recipient mice showing expression patterns of CFSE and CD4/CD8. (B) Representative histogram overlays depicting CFSE expression in splenocytes obtained from PBS- or SEB-challenged recipient mice gated on CD4/CD8 expression. (C) Representative histogram overlays depicting CFSE expression in splenocytes obtained from WT (left) or DKO (right) mice challenged with PBS or SEB. (D) Table represents % of cells within each peak. Representative data from 2 similar experiments is shown.
In the next set of in vivo experiments, HLA-DR3 WT and DKO mice were acutely challenged with SEB, bled 4 hours later and the concentrations of a panel of Th1, Th2, Th17 cytokines and chemokines were determined by multiplex assay. As shown in Fig 5, sera from naïve WT and DKO mice had very low levels of various biomarkers tested as expected. However, following SEB challenge, the serum levels of all the cytokines and chemokines tested increased rapidly as we have demonstrated in our prior studies (31, 43). Surprisingly, even though HLA-DR3 DKO mice had significantly fewer CD3+ T cells, the serum levels of most of the cytokines/chemokines were highly elevated in SEB-challenged HLA-DR3 DKO mice compared to control HLA-DR3 DKO mice. Serum concentrations of a few cytokines/chemokines were significantly lower in SEB-challenged HLA-DR3 DKO mice compared to SEB-challenged WT HLA-DR3 mice. Nonetheless, it should be noted that they were still present at significantly high levels compared to control HLA-DR3 DKO mice. While DKO mice had lower serum IFN-γ levels compared to WT mice, this did not attain statistical significance due to a wider range of IFN-γ levels in WT HLA-DR3 mice.
Figure 5. Staphylococcal enterotoxin B induced systemic cytokine/chemokine response in WT and DKO HLA-DR3 mice lacking CD4, CD8 coreceptors.

Eight-week-old WT and DKO HLA-DR3 mice of either sex were challenged with either PBS or SEB (50 μg/mouse). Animals were bled 4 hours later and the concentrations of various cytokines and chemokines in the sera was determined using multiplex bead arrays. Each bar represents mean±SE values from 4-6 mice/group. Solid black bar – WT HLA-DR3 challenged with PBS, bar with slanting lines – WT HLA-DR3 challenged with SEB, Solid white bar – DKO HLA-DR3 challenged with PBS, bar with vertical lines - DKO HLA-DR3 challenged with SEB. * p<0.05, ** p<0.005 and NS – Not significant.
In addition to causing systemic cytokine/chemokine storm, acute challenge with SEB causes an expansion of splenic CD3+ T cells bearing TCR Vβ8, deletion of immature CD4CD8 DN thymocytes and inflammation in various organs which peak at day 3 post-SEB challenge. Therefore, we investigated these parameters. As shown in Fig 6A, systemic challenge with SEB caused a robust increase in the numbers of T cells bearing TCR Vβ8 but not TCR Vβ6 in WT HLA-DR3 mice. Comparable increase in the numbers of TCR Vβ8-bearing T cells was seen in DKO mice. As the baseline numbers of TCR Vβ8+ T cells were lower in the DKO mice compared to WT mice, the total numbers of TCR Vβ8+ cells in SEB challenged DKO mice were low. However, when represented as fold-increase, the extent of expansion of TCR Vβ8+ T cells were even slightly higher in DKO mice (Fig 6B). In vivo challenge with SAg causes massive deletion of DN thymocytes whereas the mature CD4 and CD8 single positive thymocytes remain intact. As DN thymocytes form a major proportion of thymocytes, the total thymocyte count is significantly reduced following SEB challenge. As shown in Fig 6C, the total numbers of thymocytes were significantly reduced in both WT and DKO mice following SEB challenge. However, due to lack of CD4 and CD8 coreceptors, we could not establish that reduction in thymocyte was in fact due to the deletion of CD4CD8 DN thymocyte. As the numbers of TCR Vβ8+ and TCR Vβ6+ thymocytes remained comparable between naïve and SEB-challenged mice, we can safely conclude that the DN thymocytes underwent deletion.
Figure 6. Staphylococcal enterotoxin B induced expansion of splenocytes and deletion of thymocytes in WT and DKO HLA-DR3 mice lacking CD4, CD8 coreceptors.

Eight-week-old WT and DKO HLA-DR3 mice of either sex were challenged with either PBS or SEB (50 μg/mouse). Animals were killed 3 days later. Panel A represents the number of CD3+ splenocytes expressing indicated TCR Vβ families as determined by flow cytometry. Panel B represents fold increase in the absolute numbers of CD3+ T cells expressing TCR Vβ6 and 8 in SEB challenged mice compared to naïve mice. Panel C represents thymocytes gated on the indicated markers. Each bar represents mean±SE values from 4 mice/group. * p<0.05 when compared to respective naive mice and # p<0.05 when compared to SEB-challenged WT DR3 mice.
Acute or chronic activation of DNT by SEB causes immunopathology
We next examined the organs from WT and DKO HLA-DR3 mice that were acutely challenged with SEB to study the extent of immunopathology. As shown in Fig 7, lungs and livers from WT mice challenged with SEB showed extensive perivascular infiltration as we have shown in previous studies (31, 43). Interestingly, lungs and livers from DKO mice also showed extensive inflammation concurring with the elevated serum cytokine/chemokine levels in DKO mice similar to WT mice. As in WT mice challenged with SEB, even in DKO mice challenged with SEB, the inflammatory infiltrates were primarily seen around blood vessels in the lungs and predominantly comprised of mononuclear cells. In the livers, extensive lymphocytic infiltrations were seen predominantly around the central vein. Organs from control, PBS-treated mice were normal. Semi-quantitative evaluation and scoring of hisotpatholgical changes confirmed comparable immunopathology in SEB-challenged HLA-DR3 WT and DKO mice (Fig 7I).
Figure 7. The impact of deficiency of CD4 and CD8 coreceptors on SEB-induced acute immunopathology in the lungs and livers of HLA-DR transgenic mice.
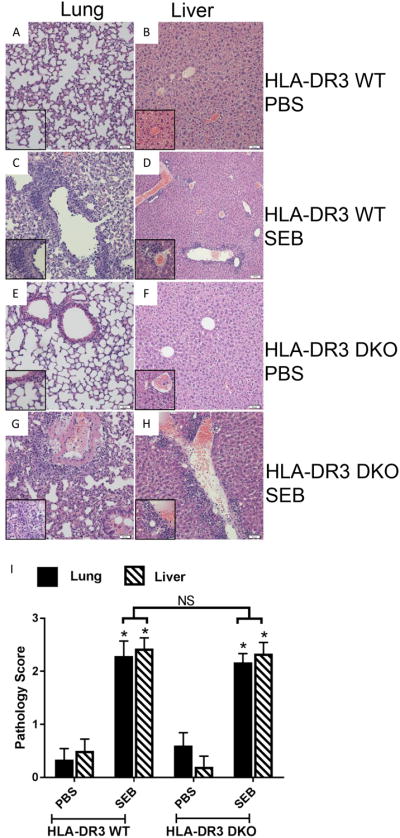
WT and DKO HLA-DR3 transgenic mice of either sex (8-10 weeks-old) were acutely challenged with PBS or SEB. Three days later, mice were euthanized, organs were collected, formalin fixed and paraffin embedded. H&E stained sections prepared from these tissue blocks were evaluated by light microscopy. Representative images are shown. Insets within each panel show higher magnifications. Panels A and B – Respective lung and liver sections from WT HLA-DR3 transgenic mice challenged with PBS. Panels C and D – Respective lung and liver sections from WT HLA-DR3 transgenic mice challenged with SEB. Panels E and F – Respective lung and liver sections from DKO HLA-DR3 transgenic mice challenged with PBS. Panels G and H – Respective lung and liver sections from DKO HLA-DR3 transgenic mice challenged with SEB. Panel I shows the histopathology scores which were determined as described in methods section. Each bar represents mean±SE values from 4-6 mice/group. * p<0.05 when compared to respective PBS-treated mice and NS – Not significant.
Having examined the acute responses of DNT cells, we next investigated how DNT cells would respond to recurrent activation. We have recently demonstrated that chronic activation of CD4+ and CD8+ T cells with SSAg induces a systemic inflammatory disease in HLA-DQ8 transgenic mice (35). Therefore, to investigate the responses of DNT cells to chronic activation with SEB, we generated DKO mice on the HLA-DQ8 background. Careful analyses of thymocyte and splenocyte subsets showed similar defects in thymus and in spleen in HLA-DQ8 DKO mice as in HLA-DR3 DKO (Data not shown). These results confirm that development of DNT cells can occur across different MHC class II alleles (Note. Both HLA-DR3 and HLA-DQ8 transgenic mice express murine endogenous MHC class I molecule of the H-2b haplotype). Subsequently, WT and DKO HLA-DQ8 transgenic mice were implanted with miniosmotic pumps delivering SEB or PBS over 7 days. Lungs, livers and kidneys from mice implanted with SEB pumps showed significant inflammatory changes both in WT and DKO HLA-DQ8 transgenic mice (Fig 8 i). The lymphocytic infiltration was mainly perivascular in these organs. Immunostaining with anti-CD3 antibodies showed the presence of CD3+ cells among the inflammatory infiltrates confirming that DNT cells are infiltrating the organs (Fig 8 ii). Semi-quantitative evaluation of hisotpatholgical changes showed the presence of comparable histopathological changes in lungs, liver and kidneys from WT and DKO HLA-DQ8 transgenic mice implanted with pumps delivering SEB (Fig 8 iii). Even though lungs from SEB-treated WT and DKO HLA-DQ8 transgenic mice showed distinct perivascular infiltration with mononuclear cells compared to mice implanted with PBS pumps, this did not attain statistical significance in our semi-quantitative scoring system. However, livers and kidneys from SEB-treated WT and DKO HLA-DQ8 transgenic mice had significantly higher pathology scores compared to PBS-treated mice.
Figure 8. Chronic exposure to SEB causes multiple organ immunopathology and CD3+ T lymphocyte infiltration in liver.

Ten to twelve weeks-old WT and DKO HLA-DQ8 transgenic mice of either sex subcutaneously implanted with 7-day mini osmotic pumps delivering either PBS or SEB (50 μg) were killed 7 days after implantation. (i) Formalin-fixed, paraffin-embedded tissue sections from mice treated as above were stained with H&E and evaluated microscopically. Representative images are shown. Panels A, B and C – Respective lung, kidney and liver sections from WT HLA-DQ8 transgenic mice challenged with PBS. Panels D, E and F – Respective lung, kidney and liver sections from WT HLA-DQ8 transgenic mice challenged with SEB. Panels G, H and I – Respective lung, kidney and liver sections from DKO HLA-DQ8 transgenic mice challenged with PBS. Panels J, K and L – Respective lung, kidney and liver sections from DKO HLA-DQ8 transgenic mice challenged with SEB. (ii) Frozen liver sections from WT HLA-DQ8 transgenic mice challenged with PBS (panel M) or SEB (panel N) and liver sections from DKO HLA-DQ8 transgenic mice challenged with PBS (panel M) or SEB (panel N) were stained with FITC-conjugated anti-CD3 antibodies. Representative images from PBS and SEB treated mice are shown. (iii) Mean organ histopathology scores from WT and DKO HLA-DQ8 transgenic mice challenged with PBS or SEB was determined as described in methods section. Mean data from 4-5 mice/group. * p<0.05 when compared to respective PBS-treated mice and NS – Not significant.
This was accompanied by expansion of CD3+ T cells bearing TCR Vβ8+ in the spleens as shown in our previous chronic SEB activation study (35) (Fig 9). We have also shown that chronic activation of T cells with SEB results in production of antinuclear antibodies (ANA) along with systemic multi-organ inflammatory response (35). While sera from WT and DKO mice implanted with PBS pumps showed no reactivity to HEp-2 cells, sera from both WT and DKO HLA-DQ8 mice implanted with SEB pumps showed strong positivity suggesting the presence of ANA, which was confirmed by quantitative ELISA (Fig 10). Overall, our investigation confirms that DNT cells can efficiently develop in mice lacking CD4 and CD8 coreceptors, and that they can respond robustly to SSAgs.
Figure 9. Chronic exposure to SAg causes expansion of DNT cells.

Ten to twelve weeks-old male and female WT and DKO HLA-DQ8 transgenic mice of were implanted with 7-day mini osmotic pumps delivering either PBS or SEB (50 μg). Spleens were collected at the time of sacrifice and distribution of CD3+ T cells bearing TCR Vβ6 and TCR Vβ8 was determined by flow cytometry. Each bar represents mean±SEM from 4-6 mice per group. * p<0.05 when compared to respective PBS-treated mice.
Figure 10. Chronic stimulation with SEB elicits autoantibody production and immune cell infiltration of multiple organs in CD4CD8 DKO mice.

Ten to twelve weeks-old male and female WT and DKO HLA-DQ8 transgenic mice subcutaneously implanted with 7-day mini osmotic pumps delivering either PBS or SEB (50 μg) were killed 7 days after implantation. Sera were tested for antinuclear antibodies using Hep-2 cells. Panels A and B represent the staining pattern with negative control (NC) and positive control (PC) sera provided along with the kit. Representative staining patterns with sera obtained from WT (panel C) and DKO (panel E) HLA-DQ8 transgenic mice implanted with PBS pumps. Representative staining patterns with sera obtained from WT (panel D) and DKO (panel F) HLA-DQ8 transgenic mice implanted with PBS pumps. The sera from WT and DKO HLA-DQ8 transgenic mice implanted with PBS or SEB pumps were also tested for the presence of antinuclear antibodies by ELISA. Each bar represents mean±SEM from 3-4 mice per group. * p<0.05 and NS – Not significant.
Discussion
Double negative T (DNT) cells lacking CD4 and CD8 co-receptors and expressing αβ TCR are present in the periphery of healthy individuals. However, very little is known regarding their development and function in vivo. DNT cells have been shown to play a beneficial role in certain infections (9, 10), type 1 diabetes and transplantation (11-15); and on the contrary a pathogenic role in certain autoinflammatory diseases such as ALPS, lupus (17, 18) and in HIV infections (44). Therefore, to better understand the biology and functions of DNT cells in vivo, we generated CD4-CD8 double knockout mice on HLA-DR3 and HLA-DQ8 backgrounds with the hypothesis that the concomitant absence of both CD4 and CD8 coreceptors might facilitate development of DNT cells in these mice.
T cell development and maturation within the thymus consists of a series of well-orchestrated events (45). The first step involves recruitment of thymocyte progenitors into the thymus. Within the thymus, the immature thymocytes lacking CD4, CD8 and TCR/CD3 complex go on to express both CD4 and CD8 coreceptors to become double positive (DP) thymocytes. Subsequently, the DP thymocytes undergo a series of developmental stages, express CD3, rearranged TCR β chain and then the rearranged TCR α chain, to become DP cells expressing functional TCR αβ/CD3 complexes. Depending on the MHC ligand (class I or class II) with which their TCR optimally interact with, these DP thymocytes then lose the expression of one of the coreceptors and become single positive (SP) Thymocytes (CD8 or CD4, respectively). The SP thymocytes undergo further rounds of selection processes wherein cells expressing TCR αβ that bind with very high or very low affinities to self MHC-peptide complexes are deleted and only those cells expressing the TCR αβ that bind with optimal affinity to self MHC-peptide complexes survive and exit the thymus as mature TCR αβ-bearing T cells expressing either CD4 or CD8 coreceptors (46).
Comparable numbers of total thymocytes in WT and DKO mice indicated that CD4 and CD8 co-receptors are not required for recruitment of thymocyte progenitors into the thymus, consistent with the current understanding of thymocyte development (45). However, the numbers of thymocytes expressing CD3 and TCR β chain were significantly reduced in DKO mice, further confirming the well-established roles for CD4 and CD8 in thymocyte development. Absence of CD4 or CD8 coreceptors likely prevented the T cells from binding with optimal affinity to MHC class II or class I molecules and receiving the necessary survival signals (47). Therefore, a majority of the thymocytes failed to survive and unable to advance to the next developmental stage. Hence, DKO mice had far fewer cells expressing TCR αβ and CD3. Nevertheless, there were still some CD3+ cells in the thymus expressing diverse rearranged TCR Vα and Vβ families suggesting that CD4 and CD8 coreceptors are not absolutely required for development of mature thymocytes (48). About 1/5th of TCR+ thymocytes (compared to WT mice) ultimately became fully mature thymocytes even in the absence of CD4 and CD8 coreceptors and entered the peripheral lymphoid organs. As expected the development of NKT cells and γδ T cells was not affected as these cells do not rely on CD4 or CD8 coreceptors for their development (49, 50). While the development of FoxP3+ T regulatory cells was compromised likely due to the deficiency of CD4 coreceptors, they were still present in DKO mice as described in other studies [reviewed in (11)].
The DNT cells in the spleens responded robustly to SSAg because of the unique mechanisms by which SSAg cause T cell activation. Unlike conventional exogenous antigens, SSAg do not undergo any intracellular antigen processing. Conversely, they bind directly to MHC class II molecules, either the α chain, β chain or both, outside of the peptide-binding groove formed by the MHC α and β chains. MHC class II bound SSAg subsequently bind only to certain TCR Vβ families (or TCR Vα families for the superantigen staphylococcal enterotoxin H) expressed by both CD4+ and CD8+ T cells irrespective of their antigen specificities, crosslink their TCR resulting in robust T cell activation without involving the CD4 or CD8 coreceptors (30, 51). Therefore, it is well known that even though SSAg bind to MHC class II molecules, they can activate both CD4+ and CD8+ T cells. In line with these observations, SEB was able to robustly activate DNT cells in vivo causing a significant elevation in systemic cytokine/chemokine levels and other associated responses such as expansion of mature T cells expressing certain TCR Vβ families, deletion of immature thymocytes and immunopathology in multiple organs (31). Surprisingly, even though DKO mice had fewer T cells, the magnitude of cytokine/chemokine storm and organ pathology was comparable to WT mice following acute SSAg challenge. Even chronic activation of DNT cells with SSAg elicited robust inflammatory responses in HLA-DQ8 transgenic mice. In the chronic SSAg exposure model, when the HLA class II-rich autoreactive B cells repeatedly present SSAg to T cells (either WT or DKO), such self-reactive B cells would receive the costimulatory signals and other factors from SSAg-activated T cells (collectively called “T cell help”) resulting in the expansion of self-reactive B cells and their differentiation to autoantibody secreting plasma cells. The reason behind the profound systemic immunopathology in DKO mice mimicking systemic lupus-like autoimmune disease following chronic activation with SSAg in spite of the presence of far fewer T cells in them is not clear. However, we put forth the following explanation based on the study by Van Laethem, et al., done on quad knockout mice lacking CD4 and CD8 coreceptors as well as MHC class I and class II molecules (48).
As TCRs inherently lack signaling ability, the Lck molecules associated with CD4 and CD8 coreceptors contribute to the signaling process. Lck associated with CD4 and CD8 molecules get activated only when these coreceptors interact with MHC class II or class I molecules. Therefore, binding to MHC class I or class II by CD8 or CD4 molecules, respectively, not only imprints coreceptor restriction to DP thymocytes, but also gives the survival signals (48). In the DKO mice, due to the absence of the coreceptors (and the coreceptor associated Lck signaling), there is insufficient signaling in the thymocytes through the TCR. As a result most of the thymocytes fail to survive and poorly differentiate into mature T cells. Hence, even though the total number of thymocytes were normal, there was a significant reduction in mature CD3+ and TCR β+ thymocytes. Nevertheless, some TCR might still bind with sufficiently high affinity to the self MHC-peptide complexes in DKO mice (which would normally cause deletion of T cells in WT mice in the presence of coreceptors) such that they can now receive survival signals through TCR without requiring CD4- or CD8-associated Lck molecules (48). Such cells would undergo maturation in thymus and reach peripheral lymphoid organs. Since these DNT cells in coreceptor-deficient mice react with higher affinity to self-MHC-peptide complexes, recurrent activation of these T cells in the periphery by SSAg causes a more severe autoimmune-like syndrome even though the total T cells are significantly lower in DKO mice. While we did not see any spontaneous autoimmune disease in these DKO mice at 16-weeks of age, a much longer follow-up may be needed to demonstrate immune dysregulation as shown in Quad-deficient mice lacking both coreceptors as well as both MHC (class I and class II) molecules (48). Nonetheless, chronic activation of DNT cells in HLA-DQ8 transgenic mice with SSAg accelerated the onset of the systemic inflammatory disease in our DKO mice. Given that DNT cells are readily demonstrable in systemic inflammatory diseases such as lupus (20, 52), that there is a significant association between S. aureus carriage and lupus in humans (53-56) and that a lupus-like disease could be induced in DKO mice with SSAg, SSAg may play an important role in the immunopathogenesis of lupus (35). Further studies are warranted to gain an in-depth understanding of the role of DNT cells in autoimmune diseases.
Supplementary Material
Acknowledgments
We thank Dr. Tak Mak for CD4 and CD8 single knockout mice. We thank Dr. Virginia Shapiro, Department of Immunology, Mayo Clinic, Rochester, MN for help with CD1d tetramer staining of NKT cells. We thank Julie Hanson for mouse husbandry and Michele Smart for help with genotyping animals.
Footnotes
This study was supported by NIH R01 to GR and CSD.
References
- 1.Egawa T. Chapter One - Regulation of CD4 and CD8 Coreceptor Expression and CD4 Versus CD8 Lineage Decisions. In: Frederick WA, editor. Advances in Immunology. Academic Press; 2015. pp. 1–40. [DOI] [PubMed] [Google Scholar]
- 2.Singer A, Bosselut R. Advances in Immunology. Academic Press; 2004. CD4/CD8 Coreceptors in Thymocyte Development, Selection, and Lineage Commitment: Analysis of the CD4/CD8 Lineage Decision; pp. 91–131. [DOI] [PubMed] [Google Scholar]
- 3.Alberts B, Johnson A, Lewis J, et al. Helper T Cells and Lymphocyte Activation 2002 [Google Scholar]
- 4.Andersen MH, Schrama D, thor Straten P, Becker JC. Cytotoxic T Cells. Journal of Investigative Dermatology. 2006;126:32–41. doi: 10.1038/sj.jid.5700001. [DOI] [PubMed] [Google Scholar]
- 5.Fischer K, Voelkl S, Heymann J, Przybylski GK, Mondal K, Laumer M, Kunz-Schughart L, Schmidt CA, Andreesen R, Mackensen A. Isolation and characterization of human antigen-specific TCRαβ+CD4-CD8- double-negative regulatory T cells. Blood. 2005;105:2828. doi: 10.1182/blood-2004-07-2583. [DOI] [PubMed] [Google Scholar]
- 6.Kusunoki Y, Hirai Y, Kyoizumi S, Akiyama M. Evidence for in vivo clonal proliferation of unique population of blood CD4-/CD8- T cells bearing T-cell receptor alpha and beta chains in two normal men. Blood. 1992;79:2965. [PubMed] [Google Scholar]
- 7.Reimann J. Double-Negative (CD4-CD8-), TCRαβ-Expressing, Peripheral T Cells. Scandinavian Journal of Immunology. 1991;34:679–688. doi: 10.1111/j.1365-3083.1991.tb01592.x. [DOI] [PubMed] [Google Scholar]
- 8.D'Acquisto F, Crompton T. CD3+CD4−CD8− (double negative) T cells: Saviours or villains of the immune response? Biochemical Pharmacology. 2011;82:333–340. doi: 10.1016/j.bcp.2011.05.019. [DOI] [PubMed] [Google Scholar]
- 9.Cowley SC, Hamilton E, Frelinger JA, Su J, Forman J, Elkins KL. CD4−CD8− T cells control intracellular bacterial infections both in vitro and in vivo. The Journal of Experimental Medicine. 2005;202:309–319. doi: 10.1084/jem.20050569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riol-Blanco L, Lazarevic V, Awasthi A, Mitsdoerffer M, Wilson BS, Croxford A, Waisman A, Kuchroo VK, Glimcher LH, Oukka M. IL-23 receptor regulates unconventional IL-17-producing T cells that control infection. Journal of immunology (Baltimore, Md : 1950) 2010;184:1710–1720. doi: 10.4049/jimmunol.0902796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ligocki AJ, Niederkorn JY. Advances on Non-CD4+Foxp3+ T regulatory cells: CD8+, Tr1, and double negative T regulatory cells in organ transplantation. Transplantation. 2015;99:1553–1559. doi: 10.1097/TP.0000000000000813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Juvet SC, Zhang L. Double negative regulatory T cells in transplantation and autoimmunity: recent progress and future directions. Journal of Molecular Cell Biology. 2012;4:48–58. doi: 10.1093/jmcb/mjr043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu T, Cong M, Sun G, Wang P, Tian Y, Shi W, Li X, You H, Zhang D. Combination of double negative T cells and anti-thymocyte serum reverses type 1 diabetes in NOD mice. Journal of Translational Medicine. 2016;14:57. doi: 10.1186/s12967-016-0815-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomson CW, Lee BPL, Zhang L. Double-negative regulatory T cells. Immunologic Research. 2006;35:163–177. doi: 10.1385/IR:35:1:163. [DOI] [PubMed] [Google Scholar]
- 15.Cong M, Liu T, Tian D, Guo H, Wang P, Liu K, Lin J, Tian Y, Shi W, You H, Jia J, Zhang D. Interleukin-2 Enhances the Regulatory Functions of CD4+T Cell-Derived CD4−CD8− Double Negative T Cells. Journal of Interferon & Cytokine Research. 2016 doi: 10.1089/jir.2015.0093. [DOI] [PubMed] [Google Scholar]
- 16.Sherlock JP, Joyce-Shaikh B, Turner SP, Chao CC, Sathe M, Grein J, Gorman DM, Bowman EP, McClanahan TK, Yearley JH, Eberl G, Buckley CD, Kastelein RA, Pierce RH, LaFace DM, Cua DJ. IL-23 induces spondyloarthropathy by acting on ROR-[gamma]t+ CD3+CD4-CD8- entheseal resident T cells. Nat Med. 2012;18:1069–1076. doi: 10.1038/nm.2817. [DOI] [PubMed] [Google Scholar]
- 17.Tarbox JA, Keppel MP, Topcagic N, Mackin C, Abdallah MB, Baszis KW, White AJ, French AR, Cooper MA. Elevated Double Negative T Cells in Pediatric Autoimmunity. Journal of clinical immunology. 2014;34:594–599. doi: 10.1007/s10875-014-0038-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lev A, Simon AJ, Amariglio N, Rechavi G, Somech R. Thymic functions and gene expression profile distinct double-negative cells from single positive cells in the autoimmune lymphoproliferative syndrome. Autoimmunity Reviews. 2012;11:723–730. doi: 10.1016/j.autrev.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 19.Russell TB, Kurre P. Double-negative T cells are non–ALPS-specific markers of immune dysregulation found in patients with aplastic anemia. Blood. 2010;116:5072. doi: 10.1182/blood-2010-09-306910. [DOI] [PubMed] [Google Scholar]
- 20.Crispín JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, Kyttaris VC, Juang YT, Tsokos GC. Expanded Double Negative T Cells in Patients with Systemic Lupus Erythematosus Produce IL-17 and Infiltrate the Kidneys. Journal of immunology (Baltimore, Md : 1950) 2008;181:8761–8766. doi: 10.4049/jimmunol.181.12.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roths JB, Murphy ED, Eicher EM. A new mutation, gld, that produces lymphoproliferation and autoimmunity in C3H/HeJ mice. The Journal of Experimental Medicine. 1984;159:1–20. doi: 10.1084/jem.159.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–317. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi T, Tanaka M, Brannan CI, Jenkins NA, Copeland NG, Suda T, Nagata S. Generalized lymphoproliferative disease in mice, caused by a point mutation in the fas ligand. Cell. 76:969–976. doi: 10.1016/0092-8674(94)90375-1. [DOI] [PubMed] [Google Scholar]
- 24.Cohen PL, Eisenberg RA. Lpr and gld: Single Gene Models of Systemic Autoimmunity and Lymphoproliferative Disease. Annual Review of Immunology. 1991;9:243–269. doi: 10.1146/annurev.iy.09.040191.001331. [DOI] [PubMed] [Google Scholar]
- 25.Matsumoto K, Yoshikai Y, Asano T, Himeno K, Iwasaki A, Nomoto K. Defect in negative selection in lpr donor-derived T cells differentiating in non-lpr host thymus. The Journal of Experimental Medicine. 1991;173:127–136. doi: 10.1084/jem.173.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kishimoto H, Surh CD, Sprent J. A Role for Fas in Negative Selection of Thymocytes In Vivo. The Journal of Experimental Medicine. 1998;187:1427–1438. doi: 10.1084/jem.187.9.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singer PA, Theofilopoulos AN. Novel origin of lpr and gld cells and possible implications in autoimmunity. Journal of Autoimmunity. 1990;3:123–135. doi: 10.1016/0896-8411(90)90136-g. [DOI] [PubMed] [Google Scholar]
- 28.Morse HC, Davidson WF, Yetter RA, Murphy ED, Roths JB, Coffman RL. Abnormalities induced by the mutant gene Ipr: expansion of a unique lymphocyte subset. The Journal of Immunology. 1982;129:2612–2615. [PubMed] [Google Scholar]
- 29.Martina MN, Noel S, Saxena A, Rabb H, Hamad ARA. Double Negative (DN) αβ T Cells: misperception and overdue recognition. Immunology and cell biology. 2015;93:305–310. doi: 10.1038/icb.2014.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fraser DJ, Proft T. The bacterial superantigen and superantigen-like proteins. Immunological Reviews. 2008;225:226–243. doi: 10.1111/j.1600-065X.2008.00681.x. [DOI] [PubMed] [Google Scholar]
- 31.Tilahun AY, Marietta EV, Wu TT, Patel R, David CS, Rajagopalan G. Human leukocyte antigen class II transgenic mouse model unmasks the significant extrahepatic pathology in toxic shock syndrome. Am J Pathol. 2011;178:2760–2773. doi: 10.1016/j.ajpath.2011.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernando MMA, Stevens CR, Walsh EC, De Jager PL, Goyette P, Plenge RM, Vyse TJ, Rioux JD. Defining the Role of the MHC in Autoimmunity: A Review and Pooled Analysis. PLoS Genetics. 2008;4:e1000024. doi: 10.1371/journal.pgen.1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mangalam AK, Taneja V, David CS. HLA Class II Molecules Influence Susceptibility versus Protection in Inflammatory Diseases by Determining the Cytokine Profile. The Journal of Immunology. 2013;190:513–519. doi: 10.4049/jimmunol.1201891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mangalam AK, Rajagopalan G, Taneja V, David CS. HLA class II transgenic mice mimic human inflammatory diseases. Adv Immunol. 2008;97:65–147. doi: 10.1016/S0065-2776(08)00002-3. [DOI] [PubMed] [Google Scholar]
- 35.Chowdhary VR, Tilahun AY, Clark CR, Grande JP, Rajagopalan G. Chronic Exposure to Staphylococcal Superantigen Elicits a Systemic Inflammatory Disease Mimicking Lupus. The Journal of Immunology. 2012;189:2054–2062. doi: 10.4049/jimmunol.1201097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tilahun AY, Chowdhary VR, David CS, Rajagopalan G. Systemic Inflammatory Response Elicited by Superantigen Destabilizes T Regulatory Cells, Rendering Them Ineffective during Toxic Shock Syndrome. The Journal of Immunology. 2014;193:2919–2930. doi: 10.4049/jimmunol.1400980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rajagopalan G, Sen MM, Singh M, Murali NS, Nath KA, Iijima K, Kita H, Leontovich AA, Gopinathan U, Patel R, David CS. Intranasal exposure to staphylococcal enterotoxin B elicits an acute systemic inflammatory response. Shock. 2006;25:647–656. doi: 10.1097/01.shk.0000209565.92445.7d. [DOI] [PubMed] [Google Scholar]
- 38.Rajagopalan G, Smart MK, Krco CJ, David CS. Expression and function of transgenic HLA-DQ molecules and lymphocyte development in mice lacking invariant chain. J Immunol. 2002;169:1774–1783. doi: 10.4049/jimmunol.169.4.1774. [DOI] [PubMed] [Google Scholar]
- 39.Krogman A, Tilahun A, David CS, Chowdhary VR, Alexander MP, Rajagopalan G. HLA-DR polymorphisms influence in vivo responses to staphylococcal toxic shock syndrome toxin-1 in a transgenic mouse model. HLA. 2017;89:20–28. doi: 10.1111/tan.12930. [DOI] [PubMed] [Google Scholar]
- 40.Penninger JM, Schilham MW, Timms E, Wallace VA, Mak TW. T cell repertoire and clonal deletion of Mtv superantigen-reactive T cells in mice lacking CD4 and CD8 molecules. European Journal of Immunology. 1995;25:2115–2118. doi: 10.1002/eji.1830250748. [DOI] [PubMed] [Google Scholar]
- 41.Akbar AN, Taams LS, Salmon M, Vukmanovic-Stejic M. The peripheral generation of CD4(+) CD25(+) regulatory T cells. Immunology. 2003;109:319–325. doi: 10.1046/j.1365-2567.2003.01678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ford MS, Zhang ZX, Chen W, Zhang L. Double-Negative T Regulatory Cells Can Develop Outside the Thymus and Do Not Mature from CD8+ T Cell Precursors. The Journal of Immunology. 2006;177:2803–2809. doi: 10.4049/jimmunol.177.5.2803. [DOI] [PubMed] [Google Scholar]
- 43.Tilahun AY, Holz M, Wu TT, David CS, Rajagopalan G. Interferon gamma-dependent intestinal pathology contributes to the lethality in bacterial superantigen-Induced toxic shock syndrome. PLoS ONE. 2011;6:e16764. doi: 10.1371/journal.pone.0016764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marodon G, Warren D, Filomio MC, Posnett DN. Productive infection of double-negative T cells with HIV in vivo. Proceedings of the National Academy of Sciences. 1999;96:11958–11963. doi: 10.1073/pnas.96.21.11958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shah DK, Zúñiga-Pflücker JC. An Overview of the Intrathymic Intricacies of T Cell Development. The Journal of Immunology. 2014;192:4017–4023. doi: 10.4049/jimmunol.1302259. [DOI] [PubMed] [Google Scholar]
- 46.Gascoigne NRJ, Rybakin V, Acuto O, Brzostek J. TCR Signal Strength and T Cell Development. Annual Review of Cell and Developmental Biology. 2016;32:327–348. doi: 10.1146/annurev-cellbio-111315-125324. [DOI] [PubMed] [Google Scholar]
- 47.Artyomov MN, Lis M, Devadas S, Davis MM, Chakraborty AK. CD4 and CD8 binding to MHC molecules primarily acts to enhance Lck delivery. Proceedings of the National Academy of Sciences. 2010;107:16916–16921. doi: 10.1073/pnas.1010568107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van Laethem F, Sarafova SD, Park JH, Tai X, Pobezinsky L, Guinter Terry I, Adoro S, Adams A, Sharrow SO, Feigenbaum L, Singer A. Deletion of CD4 and CD8 Coreceptors Permits Generation of αβT Cells that Recognize Antigens Independently of the MHC. Immunity. 2007;27:735–750. doi: 10.1016/j.immuni.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 49.Xiong N, Raulet DH. Development and selection of γδ T cells. Immunological Reviews. 2007;215:15–31. doi: 10.1111/j.1600-065X.2006.00478.x. [DOI] [PubMed] [Google Scholar]
- 50.Bendelac A, Killeen N, Littman DR, Schwartz RH. A subset of CD4+ thymocytes selected by MHC class I molecules. Science. 1994;263:1774. doi: 10.1126/science.7907820. [DOI] [PubMed] [Google Scholar]
- 51.Li H, Llera A, Malchiodi EL, Mariuzza RA. The structural basis of T cell activation by superantigens. Annu Rev Immunol. 1999;17:435–466. doi: 10.1146/annurev.immunol.17.1.435. [DOI] [PubMed] [Google Scholar]
- 52.Anand A, Dean GS, Quereshi K, Isenberg DA, Lydyard PM. Characterization of CD3+ CD4- CD8- (double negative) T cells in patients with systemic lupus erythematosus: activation markers. Lupus. 2002;11:493–500. doi: 10.1191/0961203302lu235oa. [DOI] [PubMed] [Google Scholar]
- 53.Hajialilo M, Ghorbanihaghjo A, Khabbazi A, Valizadeh H, Raeisi S, Hasani A, Varshochi M, kolahi S, Nakhjavani MR. Nasal carriage rate of Staphylococcus aureus among patients with systemic lupus erythematosus and its correlation with disease relapse. The Egyptian Rheumatologist. 2015;37:81–84. [Google Scholar]
- 54.Conti F, Ceccarelli F, Iaiani G, Perricone C, Giordano A, Amori L, Miranda F, Massaro L, Pacucci VA, Truglia S, Girelli G, Fakeri A, Taliani G, Temperoni C, Spinelli FR, Alessandri C, Valesini G. Association between Staphylococcus aureus nasal carriage and disease phenotype in patients affected by systemic lupus erythematosus. Arthritis Research & Therapy. 2016;18:177. doi: 10.1186/s13075-016-1079-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ceccarelli F, Iaiani G, Miranda F, Giordano A, Spinelli FR, Perricone C, Truglia S, Alessandri C, Conti F, Valesini G. AB0512 Nasal Carriage of Staphylococcus Aureus in Patients with Systemic Lupus Erythematosus: Cause or Effect? Annals of the Rheumatic Diseases. 2014;73:975–976. [Google Scholar]
- 56.Cavalcante EG, Guissa VR, Jesus AA, Campos LM, Sallum AM, Aikawa NE, Silva CA. Stevens-Johnson syndrome in a juvenile systemic lupus erythematosus patient. Lupus. 2011 doi: 10.1177/0961203311408377. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.