

Abstract
Penicillin-binding proteins (PBPs) catalyze the essential reactions in the biosynthesis of cell wall peptidoglycan from glycopeptide precursors. β-Lactam antibiotics normally interfere with this process by reacting covalently with the active site serine to form a stable acyl-enzyme. The design of novel β-lactams active against penicillin-susceptible and penicillin-resistant organisms will require a better understanding of the molecular details of this reaction. To that end, we compared the affinities of different β-lactam antibiotics to a modified soluble form of a resistant Enterococcus faecium PBP5 (Δ1-36 rPBP5). The soluble protein, Δ1-36 rPBP5, was expressed in Escherichia coli and purified, and the NH2-terminal protein sequence was verified by amino acid sequencing. Using β-lactams with different R1 side chains, we show that azlocillin has greater affinity for Δ1-36 rPBP5 than piperacillin and ampicillin (apparent Ki = 7 ± 0.3 μM, compared to 36 ± 3 and 51 ± 10 μM, respectively). Azlocillin also exhibits the most rapid acylation rate (apparent k2 = 15 ± 4 M−1 s−1). Meropenem demonstrates an affinity for Δ1-36 rPBP5 comparable to that of ampicillin (apparent Ki = 51 ± 15 μM) but is slower at acylating (apparent k2 = 0.14 ± 0.02 M−1 s−1). This characterization defines important structure-activity relationships for this clinically relevant type II transpeptidase, shows that the rate of formation of the acyl-enzyme is an essential factor determining the efficacy of a β-lactam, and suggests that the specific side chain interactions of β-lactams could be modified to improve inactivation of resistant PBPs.
Bacterial penicillin-binding proteins (PBPs) are members of the penicilloyl serine transferase family of enzymes (2, 3, 8, 11). PBPs catalyze the essential reactions in the biosynthesis of cell wall peptidoglycan from the glycolipid precursor, lipid II (2). These reactions are composed of two parts: transglycosylation and transpeptidation. Transglycosylation joins the disaccharides of lipid II monomers to the peptidoglycan, creating the N-acetylmuramic acid and N-acetylglucosamine backbone of the murein sacculus. Transpeptidation cross-links the glycosidic backbone into a network of interlocking units that confers exceptional tensile strength. There are three broad types of PBPs (types I to III). Type I PBPs are multifunctional, multidomain proteins catalyzing both peptidoglycan polymerization and cross-linking. Type II PBPs are monofunctional multidomain proteins catalyzing the peptidoglycan linkage (d,d-transpeptidases). Type III PBPs are monofunctional d,d-carboxypeptidases; these enzymes remove the terminal d-Ala to prevent further cross-linking (4).
β-Lactam antibiotics normally inactivate PBPs and interfere with the process of transpeptidation by forming an ester bond between the active site serine and the carbonyl carbon of the β-lactam. Hence, β-lactams mimic the acyl-d-Ala-d-Ala of the bacterial cell wall in their interactions with transpeptidases (13). This process is represented in equation 1, where L represents a β-lactam:
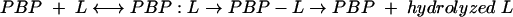 |
(1) |
Unfortunately, penicillin resistance mediated by PBPs that are, or have become, insensitive to the inhibition by β-lactams is found in many clinically important pathogens. Some of the more problematic pathogens are Staphylococcus aureus (methicillin-resistant S. aureus [MRSA]), coagulase-negative staphylococci (CNS), Neisseria gonorrhoeae, Streptococcus pneumoniae, Enterococcus faecium, and Enterococcus faecalis (6). The increasing prevalence and resistance of these pathogens are overcoming our best therapeutic efforts, since few antibiotics are available to treat these penicillin-resistant organisms.
One of the most pressing clinical problems in the chemotherapy of gram-positive infections is the treatment of high-level ampicillin-resistant E. faecium. Ampicillin-resistant E. faecium expresses variants of PBP5 that are very insensitive to attack by penicillin (herein designated rPBP5 for resistant PBP5) (14). Similar to PBP2a of MRSA, PBP5 of E. faecium acts as the principle cell wall-synthesizing enzyme when all the other PBPs have been inactivated by ampicillin or penicillin (9). The individual contribution of each amino acid mutation to penicillin resistance has been inferred from a direct comparison between penicillin-susceptible and penicillin-resistant enterococcal PBP5s (14). The recent crystal structure of E. faecium PBP5 has highlighted the relationships in the active site residues that impact upon the penicillin-resistant phenotype (12). Most notably, the increased rigidity of the penicillin-binding site due to the formation of a salt bridge between residues R464 and D481 and lower accessibility for the active site cleft as a result of a unique amino acid substitution (e.g., V465) defines the low affinity of benzylpenicillin for the active site.
Herein, we initiated investigations with a soluble form of rPBP5 of E. faecium to systematically characterize the affinities of different β-lactam antibiotics against this penicillin-resistant cell wall-synthesizing enzyme. Unlike other PBP5s, the PBP5 protein of E. faecium is not a d,d-peptidase (11). This is the first comparative study addressing structure-activity relationships of different β-lactams against enterococcal rPBP5. Comparing the effects of different R1 side chains, we demonstrate that the acylureido-penicillin, azlocillin, possesses the highest apparent affinity for the active site of Δ1-36 rPBP5. Using carbapenems as a means to explore the effects of the R2 side chains, we also show that meropenem possesses the same affinity as ampicillin for Δ1-36 rPBP5, but it is significantly slower at acylation. Taken together, these data explain why rPBP5 in E. faecium is resistant to many β-lactams and that the inactivation process exceeds the dividing time of enterococci.
MATERIALS AND METHODS
Bacterial strains and plasmids.
The clinical strain E. faecium vanB E45-7 was employed for isolation of chromosomal DNA used to construct Δ1-36 rPBP5 (Mensch et al., unpublished data). E. faecium DNA was digested with XbaI, and a 4.5-kb insert was identified containing the pbp5 gene, its promoter, and the napA gene. PCR primers were next designed based upon the DNA sequence of Enterococcus hirae PBP3r (EMBL accession number X69092) and used to generate a pbp5 amplification product. The amplicon was then cloned into the plasmid vector pUC19 (New England Biolabs). This was used in Southern hybridization experiments to screen a library of E. faecium DNA identifying the complete gene for rPBP5. The 2,037-bp open reading frame encoding a 679-amino-acid protein with an Mr of 73,780 was next identified. Plasmids pREP4, pDS56/RBS11, and pMC56 were used for cloning and overexpression of the rPBP5. The truncated protein (Δ1-36 rPBP5) was expressed in pMC56 and lacked the membrane-anchoring domain. A representation of Δ1-36 rPBP5 is shown in Fig. 1.
FIG. 1.

Ribbon representation of the structure of E. faecium Δ1-36 rPBP5.
Protein purification.
Purification of Δ1-36 rPBP5 was performed by using size exclusion chromatography, anion exchange with DEAE cellulose, and hydroxyapatite columns as described elsewhere (B. Mensch et al., unpublished). Protein concentrations were determined using a commercially available Bio-Rad protein assay reagent (Hercules, Calif.) and bovine serum albumin as a standard. Purity was assessed by resolution of Δ1-36 rPBP5 on sodium dodecyl sulfate-8% polyacrylamide gel electrophoresis (SDS-PAGE) gels.
N-Terminal amino acid sequencing.
Pulsed liquid-phase Edman degradation amino acid sequencing was performed on an Applied Biosystems Procise 494 protein sequencer (Applied Biosystems, Foster City, Calif.) to confirm truncation of Δ1-36 rPBP5 (Case School of Medicine Core Facility).
β-Lactam antibiotics.
BOCILLIN FL was obtained from Molecular Probes (Eugene, Oreg.). Benzylpenicillin was purchased from Fluka (Milwaukee, Wis.). Ampicillin, carbenicillin, azlocillin, and piperacillin were obtained from Sigma Chemical Co. (St. Louis, Mo.). Aztreonam and cefepime were purchased from Bristol-Myers Squibb (Princeton, N.J.). Ticarcillin was obtained from GlaxoSmithKline (Research Triangle Park, N.C.). Meropenem was purchased from Astra Zeneca (Wilmington, Del.), and imipenem-cilistatin and ertapenem were obtained from Merck Research Laboratories (Whitehouse Station, N.J.). The structure of each antibiotic is shown in Fig. 2. All antibiotics were dissolved in phosphate-buffered saline (PBS; 2 mM monobasic sodium phosphate, 8 mM dibasic sodium phosphate, 154 mM sodium chloride) at pH 7.4, at room temperature.
FIG. 2.

Chemical structures of β-lactams used in these studies.
Kinetics of β-lactam-Δ1-36 rPBP5 interaction.
All kinetic studies were performed in PBS at 37°C. A UV trans-illuminator with λ = 302 nm was used and fluorescence intensity (FI; binding of BOCILLIN FL to Δ1-36 rPBP5) was visualized with a Gel DOC 2000 imaging system (Quantity One software; Bio-Rad). Densitometry was performed using Scion Image, a Windows-compatible version of NIH Image (www.scioncorp.com), as previously described (12). In each assay, background fluorescence was subtracted. The maximum value of FI (FI max) defined complete saturation of 9.5 μM Δ 1-36 rPBP5.
Δ1-36 rPBP5, like other PBPs, is acylated by β-lactam antibiotics and deacylated according to the following scheme:
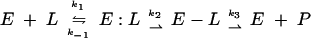 |
(2) |
where E represents the enzyme (Δ1-36 rPBP5), L is the β-lactam, E:L is the Michaelis complex, E-L is the acylated enzyme, and P is the hydrolyzed β-lactam product. In the analysis that follows, the ratio of k-1/k1 will be represented by the term K. In all experiments using BOCILLIN FL, the rate of formation of the hydrolyzed product P was very slow, which allowed us to neglect k3.
First, the apparent affinity of BOCILLIN FL for Δ1-36 rPBP5 (Km) was determined by reacting increasing concentrations (0 to 300 μM BOCILLIN FL) with 9.5 μM Δ1-36 rPBP5 for 4 h. FI was measured, graphed against the concentration of BOCILLIN FL, and fit to the working expression:
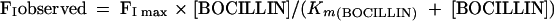 |
(3) |
Adapting the method of Golemi-Kotra et al. (5), the first-order rate constant for acylation (k2) of BOCILLIN FL to Δ1-36 rPBP5 was derived by measuring FI and kobs for increasing concentrations. BOCILLIN FL (0 to 500 μM) was mixed with 9.5 μM Δ1-36 rPBP5 for 0 to 360 min. Beginning with time zero (and at t = 10, 30, 60, 120, 180, 240, 300, and 360 min), a sample of the reaction mix was rapidly frozen to −80°C. Samples were thawed and resolved on SDS-8% PAGE gels. FI was measured, and the amount of enzyme acylated was determined by densitometry and graphed as a function of time. The data were fit to a first-order exponential rate equation:
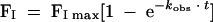 |
(4) |
and we derived values for kobs for each concentration. The measured kobs values were plotted against BOCILLIN FL concentration, and the slope of the line defined the apparent first-order rate constant (k2) for BOCILLIN FL, based on equation 5:
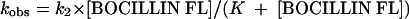 |
(5) |
Using BOCILLIN FL as the reporter substrate, we next derived the apparent first-order rate constant for acylation (k2) for unlabeled β-lactams in a competition reaction also adapted from the methods described by Golemi-Kotra et al. (5) and Frere et al. (1). This apparent first-order rate constant for acylation of the reaction with unlabeled β-lactams was determined using a fixed concentration (500 μM) of BOCILLIN FL. Here, BOCILLIN FL and concentrations of the unlabeled β-lactams that inhibited binding by approximately 50% were incubated with Δ1-36 rPBP5 at 37°C for 60 min, and the amount of BOCILLIN FL bound to Δ1-36 rPBP5 (amount acylated) was measured as described above. We derived the apparent k2 of the unlabeled β-lactam using equation 6:
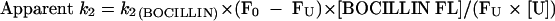 |
(6) |
Here, [U] represents the concentration of unlabeled β-lactam, and F0 and FU are the FIs of BOCILLIN FL and unlabeled β-lactam reacting with 9.5 μM Δ1-36 rPBP5, respectively.
To determine 50% inhibitory concentration (IC50) values, we performed direct competition assays. In brief, increasing concentrations of the unlabeled test β-lactam antibiotic were added to a solution of PBS containing 200 μM BOCILLIN FL. To these mixtures, 9.5 μM Δ1-36 rPBP5 was added. Samples were incubated for 4 h at 37°C and then rapidly frozen to −80°C. Aliquots were resolved on SDS-8% PAGE gels as described above. FI was measured, and the amount of acylation was estimated based upon the FI value obtained from a control sample run without competing test antibiotic.
To estimate the apparent inhibition constant (Ki) of each unlabeled β-lactam for Δ1-36 rPBP5, we reacted 9.5 μM Δ1-36 rPBP5, 200 μM BOCILLIN FL, and increasing concentrations of unlabeled β-lactam and then measured FI. The reciprocals of the densitometry values obtained were graphed versus concentration of unlabeled β-lactam, and the apparent Ki of each unlabeled β-lactam for Δ1-36 rPBP5 was determined from the slope and intercept of the reciprocal plot. We corrected each value for the affinity of BOCILLIN FL by using equation 7 (see also reference 7):
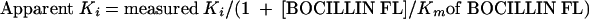 |
(7) |
Molecular representation.
Molecular modeling was performed using the program MOLOC (http://www.moloc.ch) and compared to the X-ray crystal structure of the PBP2a of MRSA complexed with benzylpenicillin and Δ1-36 rPBP5 (unpublished data). Azlocillin was docked manually in the active site of Δ1-36 rPBP5. Two-step energy minimizations were performed.
RESULTS AND DISCUSSION
Our investigations with Δ1-36 rPBP5 of E. faecium attempted to elucidate the structure-activity relationships of a series of β-lactam antibiotics against this highly resistant cell wall-synthesizing enzyme. This approach explored the effects of different R1 and R2 side chains in β-lactam binding.
Expression, purification, and characterization of E. faecium Δ1-36 rPBP5.
The truncated protein Δ1-36 rPBP5 was expressed in E. coli in soluble form and purified to homogeneity. To verify that the protein expressed did not possess the membrane-anchoring domain (Δ1-36), we performed N-terminal amino acid sequencing. Sequencing of the amino terminus NH2-MYQETQAVEAG demonstrated that our construct contained the correct N-terminal sequence. This form of the protein ensured solubility and facilitated purification, while preserving enzymatic activity (binding of β-lactams).
Kinetics of β-lactam-Δ1-36 rPBP5 interactions.
To study the affinity of the various β-lactams for Δ1-36 rPBP5, we first determined the binding of BOCILLIN FL to 14.5 μg (9.5 μM) of this protein with an excess of the fluorescent β-lactam. Our initial experiments demonstrated Δ1-36 rPBP5 reached a stable level of labeling within 4 h (FI max) (Fig. 3).
FIG. 3.

Determination of maximum FI (FI max) by reacting 200 μM BOCILLIN FL with Δ1-36 rPBP5 for 4 h.
We next determined the affinity of BOCILLIN FL for Δ1-36 rPBP5. As expected, a monophasic saturation curve resulted from the binding of BOCILLIN FL to Δ1-36 rPBP5. From this analysis, we calculated the affinity of BOCILLIN FL for Δ1-36 PBP5 to be 70 ± 27 μM.
Table 1 summarizes the IC50 and apparent inhibition constant (Ki) of unlabeled β-lactams when using 200 μM BOCILLIN FL as the competing β-lactam. These competition experiments were used as indicators of the sensitivity of the Δ1-36 PBP5 to inactivation by β-lactams. We tested 11 representative β-lactams (penams, penems, and cephems) under identical conditions. These included benzylpenicillin, one aminopenicillin (ampicillin), two carboxypenicillins (carbenicillin and ticarcillin), two ureidopenicillins (piperacillin and azlocillin), three carbapenems (meropenem, ertapenem, and imipenem), one monobactam (aztreonam), and one advanced-generation cephalosporin (cefepime). The penicillins chosen differed primarily in the composition of the R1 side chain and the length of the substituents from the N-8 position. The carbapenems differed primarily in the structure of the R2 side chains: the R1 side chain is uniform in each carbapenem (6-α-hydroxyethyl moiety).
TABLE 1.
Kinetics of different β-lactam substrates for Δ1-36 rPBP5a
| Substrate | IC50 (μM) | Apparent Ki (μM) | Apparent acylation rate (M−1 s−1) |
|---|---|---|---|
| Azlocillin | 25 | 7 ± 0.3 | 15 ± 4.0 |
| Piperacillin | 80 | 36 ± 3 | 2.2 ± 0.18 |
| Ampicillin | 125 | 51 ± 10 | 1.8 ± 0.01 |
| Meropenem | 200 | 51 ± 15 | 0.14 ± 0.02 |
| Benzylpenicillin | 300 | 118 ± 28 | NMc |
| Imipenem | 350 | 127 ± 9 | NM |
| Ertapenem | 500 | 138 ± 0.3 | 0.28 ± 0.02 |
| Carbenicillin | 4,000 | 1,166 ± 6 | NM |
| Ticarcillin | 4,000 | 1,223 ± 112 | NM |
| Cefepime | 10,000 | 3,247 ± 1,633 | NM |
| Aztreonam | >10,000 | NDb | NM |
Measured IC50s, apparent inhibition constants (Ki), and apparent acylation rates of different β-lactam substrates for Δ1-36 rPBP5, as determined in a direct competition assay with BOCILLIN FL. The affinity of BOCILLIN FL for Δ1-36 rPBP5 is 70 ± 27 μM, and k2 is 1.1 ± 0.1 M−1 s−1. IC50s were determined with 200 μM BOCILLIN FL for 4 h 37°C. Apparent acylation rates were determined with 500 μM BOCILLIN FL for 1 h at 37°C.
ND, not detected.
NM, not measured.
Azlocillin possessed the greatest apparent affinity for the active site of Δ1-36 rPBP5 (lowest Ki) (Fig. 4; Table 1). This result was unanticipated, since azlocillin was initially used as an antipseudomonal β-lactam and has not been explored as an agent effective against the enterococci. As expected, benzylpenicillin was less active than ampicillin or piperacillin. In contrast, the carboxypenicillins (carbenicillin and ticarcillin), cefepime, and aztreonam were the least-active β-lactams in competition with BOCILLIN FL.
FIG. 4.

Comparative binding studies (IC50 determinations) performed with 200 μM BOCILLIN. Azlocillin demonstrated the greatest affinity for Δ1-36 rPBP5 of E. faecium.
Among the penems, meropenem demonstrated a greater affinity (lower IC50) for Δ1-36 rPBP5 compared to imipenem or ertapenem (Table 1; Fig. 4). In fact, meropenem and ampicillin appear to have equal apparent affinity for Δ1-36 rPBP5. In contrast to ampicillin, benzylpenicillin, and piperacillin, the relative sizes and active site binding interactions of the carbapenems examined in this study suggest a role of the R2 side chains in β-lactam binding. Hence, this analysis led us to conclude that both R1 and R2 side chains influence substrate affinity for Δ1-36 rPBP5.
In order to understand the dynamic consequence of differing affinities of β-lactams in binding to Δ1-36 rPBP5, we determined the time dependence of the accumulation of BOCILLIN FL to Δ1-36 rPBP5 in the absence of a competing β-lactam. Based upon the reaction time courses of 50, 100, 150, 200, and 250 μM BOCILLIN binding, we calculated kobs for each concentration (0.11, 0.15, 0.21, 0.25, and 0.33 s−1, respectively). Graphing kobs versus BOCILLIN FL concentration, we derived a k2 of 1.1 ± 0.1 M−1 s−1 (r2 = 0.98). Using equation 6, we determined an apparent acylation rate for a series of other β-lactams by a competition method. The rank order corresponded in large part to the IC50 values (Table 1; Fig. 4 and 5). Again, we noted that the fastest acylation rate was observed with the acylureidopenicillin, azlocillin. Piperacillin and ampicillin acylated Δ1-36 rPBP5 at similar rates. Meropenem, a β-lactam with the same affinity as ampicillin, was significantly slower at acylating Δ1-36 rPBP5 than was ampicillin.
FIG. 5.

Determination of second-order rate constants for competing β-lactams. Each β-lactam was competed with 500 μM BOCILLIN for 1 h at 37°C.
This experimental approach shows that the properties of the side chains R1 and R2 are essential determinants in the binding of β-lactams to Δ1-36 rPBP5. It has not escaped our notice that R1 modifications of the penicillin structure can be used in drug synthesis strategies to find potent inhibitors of this bacterial type II transpeptidase. The modeling performed herein suggests that the additional ureido side chain of azlocillin penetrates deeper into the active site of Δ1-36 PBP5 than any of the other β-lactam side chains (Fig. 6). The importance of the additional noncovalent interactions remains to be proven.
FIG. 6.

(Left) Experimentally observed conformation of the benzylpenicillinoyl moiety of the acyl-enzyme complex formed with Δ1-36 rPBP5. (Right) Modeled conformation of the penicillinoyl moiety of the acyl-enzyme complex formed between Δ1-36 rPBP5 and azlocillin, based on the benzylpenicillin complex. The carbonyl oxygen of the ureido side chain offers H-bonding interactions not present with other β-lactams.
Preliminary attempts to use azlocillin in combination with another β-lactam, as a double β-lactam synergistic combination, in susceptibility tests were not very successful (data not shown). Screening a heterogeneous collection of ampicillin-resistant E. faecium isolates, we were not able to demonstrate, in a consistent manner, bacterial synergy using azlocillin (50 and 100 μg/ml) added to increasing concentrations of piperacillin. However, it is suspected from the data that despite its high affinity, the absolute rate of inactivation at clinically relevant concentrations was so low that there was no effective inhibition.
In conclusion, these experiments are a necessary first step in the study of the dynamic interactions of various β-lactams with a highly resistant target, Δ1-36 rPBP5. Studies such as this complement the existing investigations that have used a similar approach to analyze the S. pneumoniae PBP2x and the methicillin-resistant PBP2a of S. aureus (10). Like investigations with β-lactamase inhibitors and the inactivation of inhibitor-resistant SHV enzymes, these data shows that both the affinity and rate at which Δ1-36 rPBP is inactivated are significant factors in the evaluation of potent inhibitors of transpeptidation (7). A series of variants of Δ1-36 rPBP5 have been already constructed that are aimed at further exploring the role of individual amino acid substitutions on Δ1-36 rPBP5 binding of penicillin.
Acknowledgments
This work was supported by grants from the Veterans Affairs Medical Center Merit Review, Career Development, and VISN 10 programs (R.A.B., M.S.H., and L.B.R.). L.B.R. is also supported by the NIH (R01 AI 45626). J.D.B. acknowledges the continued support of the Robert A. Welch foundation.
REFERENCES
- 1.Frere, J. M., M. Nguyen-Disteche, J. Coyette, and B. Joris. 1992. Mode of action: interaction with the penicillin-binding proteins. Chapman and Hall, Glasgow, Scotland.
- 2.Ghuysen, J. M. 1994. Molecular structures of penicillin-binding proteins and beta-lactamases. Trends Microbiol. 2:372-380. [DOI] [PubMed] [Google Scholar]
- 3.Goffin, C., and J. M. Ghuysen. 2002. Biochemistry and comparative genomics of SxxK superfamily acyltransferases offer a clue to the mycobacterial paradox: presence of penicillin-susceptible target proteins versus lack of efficiency of penicillin as therapeutic agent. Microbiol. Mol. Biol. Rev. 66:702-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goffin, C., and J. M. Ghuysen. 1998. Multimodular penicillin-binding proteins: an enigmatic family of orthologs and paralogs. Microbiol. Mol. Biol. Rev. 62:1079-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Golemi-Kotra, D., J. Y. Cha, S. O. Meroueh, S. B. Vakulenko, and S. Mobashery. 2003. Resistance to β-lactam antibiotics and its mediation by the sensor domain of the transmembrane BlaR signaling pathway in Staphylococcus aureus. J. Biol. Chem. 278:18419-18425. [DOI] [PubMed] [Google Scholar]
- 6.Hakenbeck, R., and J. Coyette. 1998. Resistant penicillin-binding proteins. Cell Mol. Life Sci. 54:332-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Helfand, M. S., C. R. Bethel, A. M. Hujer, K. M. Hujer, V. E. Anderson, and R. A. Bonomo. 2003. Understanding resistance to beta-lactams and beta-lactamase inhibitors in the SHV β-lactamase: lessons from the mutagenesis of SER-130. J. Biol. Chem. 278:52724-52729. [DOI] [PubMed] [Google Scholar]
- 8.Joris, B., J. M. Ghuysen, G. Dive, A. Renard, O. Dideberg, P. Charlier, J. M. Frere, J. A. Kelly, J. C. Boyington, P. C. Moews, et al. 1988. The active-site-serine penicillin-recognizing enzymes as members of the Streptomyces R61 DD-peptidase family. Biochem. J. 250:313-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim, D., and N. C. Strynadka. 2002. Structural basis for the β-lactam resistance of PBP2a from methicillin-resistant Staphylococcus aureus. Nat. Struct. Biol. 9:870-876. [DOI] [PubMed] [Google Scholar]
- 10.Malouin, F., J. Blais, S. Chamberland, M. Hoang, C. Park, C. Chan, K. Mathias, S. Hakem, K. Dupree, E. Liu, T. Nguyen, and M. N. Dudley. 2003. RWJ-54428 (MC-02479), a new cephalosporin with high affinity for penicillin-binding proteins, including PBP 2a, and stability to staphylococcal β-lactamases. Antimicrob. Agents Chemother. 47:658-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Massova, I., and S. Mobashery. 1998. Kinship and diversification of bacterial penicillin-binding proteins and β-lactamases. Antimicrob. Agents Chemother. 42:1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sauvage, E., F. Kerff, E. Fonze, R. Herman, B. Schoot, J. P. Marquette, Y. Taburet, D. Prevost, J. Dumas, G. Leonard, P. Stefanic, J. Coyette, and P. Charlier. 2002. The 2.4-Å crystal structure of the penicillin-resistant penicillin-binding protein PBP5fm from Enterococcus faecium in complex with benzylpenicillin. Cell Mol. Life Sci. 59:1223-1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tipper, D. J., and J. L. Strominger. 1965. Mechanism of action of penicillins: a proposal based on their structural similarity to acyl-d-alanyl-d-alanine. Proc. Natl. Acad. Sci. USA 54:1133-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zorzi, W., X. Y. Zhou, O. Dardenne, J. Lamotte, D. Raze, J. Pierre, L. Gutmann, and J. Coyette. 1996. Structure of the low-affinity penicillin-binding protein 5 PBP5fm in wild-type and highly penicillin-resistant strains of Enterococcus faecium. J. Bacteriol. 178:4948-4957. [DOI] [PMC free article] [PubMed] [Google Scholar]