

Abstract
The objective of the present study was to develop a population pharmacokinetic model for nelfinavir mesylate (NFV) and nelfinavir hydroxy-tert-butylamide (M8), the most abundant metabolite of NFV, in infants vertically infected with human immunodeficiency virus type 1 and participating in the Paediatric European Network for Treatment of AIDS 7 study. Plasma NFV concentrations were determined during repeated NFV administrations (two to three times a day). Eighteen infants younger that age 2 years participated in this study. The doses administered ranged from 71 to 203 mg/kg of body weight/day. Pharmacokinetic parameter estimates were obtained by a compartmental approach by using a kinetic model to simultaneously fit NFV and M8 (active metabolite) concentrations. M8 was shown to be formation rate limited and was characterized by first-order rate constants of formation and elimination. Body weight was found to be a more appropriate predictor than age of the changes in (i) the rate of metabolism, (ii) the elimination rate constant of NFV, and (iii) NFV clearance. Population parameters were computed to account for the relationship between the rate of metabolism and body weight. The estimated NFV and M8 elimination half-lives were 4.3 and 2.04 h, respectively. The estimated NFV clearance was 2.13 liters/h/kg. The M8 concentration-to-NFV concentration ratio was 0.64 ± 0.44. In conclusion, the population pharmacokinetic model describing the dispositions of NFV and M8 should facilitate the design of future studies to elucidate the relative contributions of the parent compound and M8 to the pharmacological and toxic effects of NFV therapy.
The introduction of protease inhibitors in clinical practice has caused an impressive decrease in the rates of morbidity and mortality among patients infected with human immunodeficiency virus (HIV) (5, 11). Although it is still not clear when antiretroviral treatment should be initiated in children, treatment in the first months of life is an important option to be considered when symptoms are present or the CD4-cell count is low (23). Nowadays, the standard of care for the treatment of HIV type 1 (HIV-1) infection includes two nucleoside reverse transcriptase inhibitors (NRTIs) in combination with one or two protease inhibitors or a nonnucleoside reverse transcriptase inhibitor (8, 10, 15).
Nelfinavir mesylate (NFV) is a nonpeptidic protease inhibitor (18) approved for use by the Food and Drug Administration in March 1997 for the treatment of HIV-1-infected children. Like other such agents, it prevents cleavage of the HIV-1 Gag and Gap-Pol precursor polyproteins by the protease, resulting in the production of immature, noninfectious virus particles. NFV is available in both tablet and powder formulations; after oral administration, the plasma NFV concentrations have been found to be similar for these two formulations (7). The powder formulation can feasibly be given to newborn infants. However, little is known about the safety and pharmacokinetics of NFV in this population (3, 4, 7, 9, 13, 16, 21, 22, 26, 27; R. C. Brundage, C. V. Fletcher, T. Fenton, W. D. Fiske, M. I. Becker, L. Purdue, J. McNamara, L. Mofenson, S. A. Spector, and S. E. Starr for the PACTG 382 Study Team, Abstr. 7th Conf. Retrovir. Opportunistic Infect., abstr. 719, 2000; E. Capparelli, J. Sullivan, L. Mofenson, E. Smith, B. Graham, P. Britto, M. I. Becker, and K. Luzuriaga, Abstr. 40th Intersci. Conf. Antimicrob. Agents Chemother., abstr. 1658, p. 338, 2000; E. V. Capparelli, S. Burchett, A. Kovacs, M. Khoury, V. Carey, E. Smith, L. Mofenson, J. Connor, B. Zimmer, and E. Hawkins, Abstr. 7th Conf. Retrovir. Opportunistic Infect., abstr. 661, 2000). Indeed, most pediatric studies performed to evaluate the pharmacokinetics of NFV included children older than 2 years of age but only a very small number of infants younger than 1 year of age (3, 7, 9, 16, 22, 27). Recently, Litalien et al. (13) reported the results of a pharmacokinetic study carried out with infants younger than 1 year of age, and Rongkavilit et al. (21) published the results of a dose-escalation pharmacokinetic study performed with neonates.
NFV is extensively metabolized by several cytochrome P-450 (CYP) isoenzymes (CYP3A4, CYP2C9, CYP2C19, and CYP2D6) in the body (at least four metabolites) (28; C. Merry, M. Barry, and M. Ryan, Abstr. 7th Eur. Conf. Clin. Aspects Treat. HIV Infect., abstr. 831, 1999). Moreover, it has been reported that NFV induces its own hepatic metabolism during the first 2 weeks of treatment (Merry et al., Abstr. 7th Eur. Conf. Clin. Aspects Treat. HIV Infect., 1999). The major metabolite, hydroxy-tert-butylamide (M8), is generated by hepatic CYP isoform 2C19 and constitutes a substantial portion of total drug in plasma. Both NFV and M8 demonstrate comparable antiretroviral activities in vitro (28). However, maturation of metabolic pathways takes place at different rates; the rate of metabolic clearance of drugs is very low at birth and then increases and reaches a maximum at about the age of 1 year, when it can exceed that in adults. In neonates, after 14 days of treatment (15, 30, and 45 mg/kg of body weight), the total clearance (CL/F) of NFV was 1.1 to 1.9 liters/h/kg and the elimination half-life was 3.3 to 5 h (21). In infants younger than 1 year of age (2.3 months to 1 year), CL/F was 2.7 to 4.2 liters/h/kg (4, 13) and the NFV and M8 elimination half-lives were 2.6 and 2.2 h, respectively (13). In older children, CL/F was 1 to 1.9 liters/h/kg, i.e., approximately twice the value reported for adults (12, 27; Capparelli et al., Abstr. 7th Conf. Retrovir. Opportunistic Infect., 2000).
The recommended NFV dose for adults is 2,250 to 2,500 mg/day, regardless of weight, in two or three divided doses, whereas the recommended pediatric dose is 60 to 90 mg/kg/day in three divided doses (1). The most appropriate dosage for children younger than 2 years of age is not well established but is higher than that for adults.
In the present study, NFV was administered in combination with didanosine (ddI) and stavudine (d4T) to infants less than or equal to 12 weeks of age at the time of enrollment. The objective was to develop a pharmacokinetic model that can be used to describe the dispositions of NFV and M8, in which metabolite formation is dependent on the disposition of the parent compound. Moreover, we examined the roles of age and weight in describing the variabilities in the estimated pharmacokinetic parameters for NFV and M8.
MATERIALS AND METHODS
Patient inclusion and exclusion criteria.
Eighteen vertically HIV-1-infected infants were included in the Paediatric European Network for Treatment of AIDS (PENTA) 7 study between September 1999 and February 2001. Infants were eligible if they were less than or equal to 12 weeks of age at the time of enrollment; had HIV-1 infection, as documented by HIV detection on two separate occasions (by HIV-1 DNA or RNA PCR, culture, or p24 antigen detection); and had not previously received protease inhibitors (except for prophylaxis to reduce maternofetal transmission). Exclusion criteria included in utero exposure to NFV, ddI, or d4T if such exposure was associated with resistance; abnormal hematological or biochemical parameters (grade 3 or more on the National Cancer Institute common toxicity criteria scale); the presence of vomiting, diarrhea, or malabsorption syndrome likely to interfere with drug intake or absorption; the need for therapy with drugs contraindicated for use with NFV; and any AIDS-defining event. Moreover, patients who were concurrently using medications known to be inhibitors or inducers of CYP3A were excluded from this study.
The study protocol was reviewed and approved by the institutional review board (Comité Consultatif de Protection des Personnes dans la Recherche Biomédicale Saint Louis, Paris, France). The study was performed in accordance with legal requirements and the Declaration of Helsinki and with present European Community and Food and Drug Administration guidelines for good clinical practice. Informed consent was obtained from the parents or legal guardians of the children.
Study design.
This study was a multiple-dose pharmacokinetic evaluation carried out with patients receiving NFV in combination with ddI and d4T. The initial dose of NFV was 40 mg/kg/dose three times a day (TID; 120 mg/kg/day). This dosage was chosen on the basis of unpublished data showing that doses of 20 to 30 mg/kg every 8 h fail to achieve therapeutic NFV levels in very young infants and on the basis of the findings of a recently published study (13). NFV was administered as tablets (250 mg) or as the pediatric formulation (50 mg of NFV per g of powder mixed with food prior to administration).
First pharmacokinetic evaluations were performed after at least 2 weeks of NFV treatment (PK1). As the first four patients enrolled had plasma NFV concentrations and areas under the plasma concentration-time curves well below those seen in older patients and adults, the dose of NFV was increased to 150 mg/kg/day, and at the same time the dosing interval was changed from TID to two times a day (BID) on the basis of recent data for adults that demonstrated comparable efficacies of the two dosing intervals (16). In addition, due to the unexpected results of the first pharmacokinetic study, additional pharmacokinetic evaluations were planned between 6 and 12 months of age (PK2) and between 16 and 24 months of age (PK3) in order to detect high levels of drug exposure and to reduce the risks of side effects. As there are significant changes in the rate of metabolic clearance of drugs (which is low at birth and which is higher than that in adults at age 1 year) and in renal function (which is low at birth and which is near that for adults at age 1 year), six children were considered new cases at the time of the second pharmacokinetic evaluation that was performed 12 to 28 months later. For each pharmacokinetic study, patients were admitted to the research unit for 12 h and extensive blood samples (2 ml each) were obtained just before administration of the NFV dose and at 1, 2, 4, 6, 8, and 12 h (BID dosing interval only) postdosing. In addition, from day 8 of treatment until the end of the study, blood samples (2 ml) were collected at 8- to 15-day intervals in heparinized tubes just before drug intake (to determine the minimum concentration of drug in serum; these samples represent about 20% of the total number of samples colleted during this study) for routine therapeutic drug monitoring; these samples were collected during a medical visit or when blood samples were taken for biochemical, hematological, and/or viral tests, according to the PENTA protocol. All samples were obtained during steady-state conditions (i.e., unchanged dosage for at least 8 days).
Strict instructions were given to the parents about the need to give NFV with food, as recommended by the manufacturer. The powder was diluted with a small amount of water, milk (or formula), or dietary supplements. For the tablet formulation, the doses were rounded to the nearest dose possible by using tablet or half-tablet sizes. The tablets were crushed and mixed with a small amount of water or milk.
Analytical method.
The drug analysis was undertaken at the Department of Pediatric Pharmacology and Pharmacogenetics of the Robert Debré Hospital. NFV, M8, and the internal standard [6,7-dimethyl-2,3-di(2-pyridyl)quinoxaline] were quantified in the plasma samples by using a liquid chromatography-electrospray ionization mass spectrometry method. The chromatographic conditions involved separation on a C30 high-pressure liquid chromatography column (250 by 2 mm [inner diameter]) packed with 5-μm particles. The mobile phase consisted of a mixture of acetonitrile-10 mM ammonium acetate buffer (pH 3.5) (70:30; vol/vol) and was split at a ratio of 1:5 at the entrance of the mass spectrometer. This method was validated according to good laboratory practice guidelines. Calibration curve standards were prepared at a concentration range of 0.02 to 7.5 μg/ml. The inter- and intra-assay precisions ranged from 0.57 to 6.8% for NFV and from 1.2 to 8.2% for M8. The lower limit of quantitation was 0.02 μg/ml for both analytes. Quality control samples were included in each analytical sequence to verify the stability of the study samples during storage and the accuracy and precision of the analysis.
Structural model development.
Pharmacokinetic model-building analysis was performed with NONMEM (version V.1) software (2) through the Visual-NM graphical interface (20). A total of 340 samples for pharmacokinetic analysis together with the individual full dosing history were included in the population pharmacokinetic database. The population characteristics of the pharmacokinetic parameters (fixed and random effects) were estimated by using the subroutine ADVAN-6 from the library of programs provided with the NONMEM-PREDPP package. Both first-order and first-order conditional estimation methods were used to estimate population pharmacokinetic parameters. The first-order method was used to fit the model because it markedly decreases the objective function. The plasma concentration-time curves of NFV and M8 were simultaneously fitted. We assumed that the time course of NFV kinetics was described by a one-compartment model with absorption from the gastrointestinal tract in the central compartment and a dual elimination process: the first one was associated with an excretion process and the second one was associated with metabolism (M8) represented by a complementary compartment (Fig. 1). The zero- or first-order input rate constant, as well as the presence or the absence of a lag time from the gastrointestinal tract, was evaluated. The best structural model was chosen on the basis of changes in the −2 log likelihood function and qualitative assessment of the diagnostic plots. The smaller objective function value was associated with the better model. A decrease in the objective function of at least 3.8 (P < 0.05 with 1 degree of freedom) was required for statistical significance to include the lag time in the model. A model with the first-order input rate and a lag time (Table 1; Fig. 1) best described the data and was therefore chosen as the most appropriate for the population analysis. The seven-dimensional vectors θ of the kinetic parameters considered in the population analysis were the volume of distribution of the two compartments (V1 and V2, respectively), the rate constant of metabolism (k12), the rate constant of absorption (ka), the elimination rate constants (k10 and k20), and the lag time. A bioavailability (F) of 1 was assumed in the absence of intravenous drug administration data.
FIG. 1.

Structural model used to fit NFV and M8 data. GI, gastrointestinal tract; 1 and 2, compartments of distribution; ka, first-order input rate; k12, first-order rate of metabolism; k10 and k20, first-order elimination rate constants; V1 and V2, estimated parameters defining the volumes of distribution of the two compartments, respectively.
TABLE 1.
Model-building steps
| Model | No. of parameters | Objective function values |
|---|---|---|
| Model 2, one-compartment model with first-order input rate and with a lag time | 7 | 4,534 |
| Model 3, one-compartment model with first-order input rate and without a lag time | 6 | 4,554 |
| Model 4, one-compartment model with zero-order input rate and with absorption duration estimated from the data | 6 | 4,970 |
The elimination rate constant (kel) of NFV is the sum of the rate constants associated with drug excretion and other routes of metabolism (k10) and M8 formation (k12). The following secondary pharmacokinetic parameters were calculated from the individual (Bayesian estimate) primary pharmacokinetic parameters: kel, NFV and M8 elimination half-lives, and total clearance (CL/F). Because the fraction of the NFV dose metabolized to M8 (Fm) was unknown for this patient population, the volume of distribution divided by Fm [V2/(F × Fm)] was estimated for M8.
Interindividual variability was assessed according to a proportional error model associated with each fixed-effect parameter; thus, for example, the volume of distribution (V1/F) of subject j was described by the relationship V1/Fj = V1/Fmean · exp(ηV1/F), where V1/Fmean is the population mean and ηV1/F is the difference between the population V1/Fmean value and the V1/Fj value in subject j; ηV1/F is assumed to be a Gaussian random variable with a mean of zero and a variance of ω2η. Various error models were also tested. A proportional (or constant coefficient of variation) error structure was chosen, with different errors assigned to NFV and M8.
Following selection of the basic structural and statistical models, the influences of covariates (weight, age, and gestational age) were assessed. First, the individual empirical pharmacokinetic estimates were plotted against all preselected potential covariates, and second, selected covariates were added to the model and tested for statistical significance (chi-square test).
The predicted concentrations in plasma (IPRED) were computed for each individual by using the empirical Bayes estimate of the pharmacokinetic parameters by using the POSTHOC option in the NONMEM program.
Statistical analysis.
At each step of the model building, qualitative assessment of diagnostic plots was performed by considering (i) the closeness and randomness around the line of identity (slope = 1 and intercept = 0) in the plot of the observed concentration (DV) versus the predicted concentration (PRED) and (ii) the randomness around the zero residual (RES) or weighted residual (WRES) line in the residual or weighted residual, respectively, versus the predicted concentration plot.
Relationships between covariates and estimates of CL/F, V1/F, k12, and kel for NFV, as well as V2/(F × Fm) and k20 for M8, were examined graphically. Due to the small number of infants enrolled in this study, it was not possible to properly evaluate the relationship between gender and pharmacokinetic parameters.
To evaluate the reliability of the post hoc oral clearance estimates, clearance values estimated by a Bayesian methodology (test values) were compared to the ones computed by noncompartmental analysis (reference values). For five patients, the number of blood samples available between two drug administrations did not allow computation of CL/F by simple noncompartmental analysis; thus, only 19 kinetic profiles were considered. Pk-fit software (version 2.0) (6) was used for this analysis. These comparisons were performed by using two criteria (24, 25). The bias or mean predictor error was calculated as
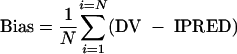 |
The precision or root mean square error was calculated as
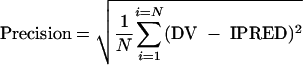 |
In these expressions the index i refers to the concentration number, and N is the sample size. The 95% confidence interval for bias was computed, and the t test was used to compare the bias to 0.
RESULTS
Patient characteristics.
A total of 24 pharmacokinetic studies were performed with data from 18 patients between 1 and 21 months of age. The baseline demographic characteristics of the 18 infants enrolled in this study are summarized in Table 2. The characteristics of the different pharmacokinetic studies (number of patients, dosage administration, and dosing schedule) are presented in Table 3. There was no evidence of hepatic dysfunction. None of the patients concurrently received a medication known to interfere with the metabolism of NFV (14).
TABLE 2.
Patient characteristics
| Characteristic | Value |
|---|---|
| No. of patients | 18 |
| Sex (no. of males; no. of females) | 8; 10 |
| Age at enrollment (days) | 81 ± 30 (26-137)a |
| Wt (kg) | 6.2 ± 2.4 (2.5-12.4) |
| Ethnicity (no. of patients) | |
| Black | 10 |
| White | 8 |
| Gestational age (wk) (n = 16)b | 36.7 ± 3.3 (29-40) |
| Mode of delivery (no. of patients) (n = 17)c | |
| Vaginal | 8 |
| Cesarean | 9 |
| Prophylactic regimens for vertical transmission (no. of patients) | |
| In utero | |
| Zidovudine | 5 |
| Two NRTIs + nevirapine | 6 |
| Two NRTIs + NFV | 1 |
| None | 7 |
| During labor | |
| Zidovudine | 6 |
| Zidovudine + nevirapine | 3 |
| None | 8 |
| Baseline plasma HIV RNA level (log10 copy no.) | 5.57 ± 0.82 (3.13-6.82) |
Values in parentheses are ranges.
Data are missing for two patients.
Data are missing for one patient.
TABLE 3.
Timing of pharmacokinetic studies and NFV administration
| Characteristic | PK1 | PK2 | PK3 |
|---|---|---|---|
| No. of patients | 10 | 10 | 4 |
| Age at the time of the study (mo) | 2.7 ± 0.8 | 7.8 ± 2.4 | 18.3 ± 2.2 |
| Daily dose (mg/kg/day) | 136 ± 20 | 138 ± 19 | 132 ± 20 |
| No. of patients dosed BID/no. of patients dosed TID | 6/4 | 10/0 | 4/0 |
| No. of patients who received powder/ no. of patients who received tablets | 4/6 | 1/9 | 0/4 |
Population pharmacokinetic parameters.
In six patients, eight NFV concentrations before dosing (about 30 ng/ml) were significantly lower than the concentration in the subsequent postdosing sample obtained at 12 h. These findings could be due to a problem of punctual compliance with treatment; indeed, the dose was given by the caregivers at home. Thus, a total of eight NFV concentrations were removed from the database. The M8 concentration-to-NFV concentration ratio was 0.64 ± 0.44. In a first step, population pharmacokinetic parameters were computed by assuming that no dependency existed between the pharmacokinetic parameters and the covariates. Then, the relationships between the pharmacokinetic parameters and the covariates were explored. Body weight and age were identified as sources of variability for k12, kel, and NFV CL/F. The best correlations were obtained between patient body weight and kel (slope = 0.012; intercept = 0.16; r = 0.60; P = 0.0024), between patient body weight and CL/F (slope = 1.84; intercept = −0.45; r = 0.70; P = 0.0002) (Fig. 2), and between patient body weight and the rate of metabolism (slope = 0.0122; intercept = 0.073; r = 0.60; P = 0.002) (Fig. 2). Finally, population parameters were computed to account for the relationship between k12 and body weight. The inclusion of this second-stage model significantly improved the relationship between the model-predicted and the observed concentrations (the objective function value dropped from 4,534 to 4,523) and the weighted residuals versus the model-predicted concentrations and reduced the interindividual variability in k12 from 22.1 to 19.7%. The adequacy of the fit of the model to the data is presented in Fig. 3 for NFV and in Fig. 4 for M8. Population pharmacokinetic parameters after covariate inclusion are displayed in Table 4. The precisions of the estimates of the rate of absorption and lag time were higher than those for the other parameters because few concentration data were available to properly estimate these parameters during the first hours after drug intake.
FIG. 2.

Relationships between patient body weight and NFV CL/F and between patient body weight and k12.
FIG. 3.

Model performance. (A) Model-predicted versus observed NFV concentrations based on population parameter estimates; (B) predicted NFV concentrations based on population parameter estimates versus time; (C) weighted residuals versus predicted NFV concentrations based on population parameter estimates. The dotted line represents the line of identity. The solid line represents the zero abscissa line.
FIG. 4.

Model performance. (A) Model-predicted versus observed M8 concentrations based on population parameter estimates; (B) predicted M8 concentrations based on population parameter estimates versus time; and (C) weighted residuals versus predicted M8 concentrations based on population parameter estimates. The dotted line represents the line of identity. The solid line represents the zero abscissa line.
TABLE 4.
Population pharmacokinetic parameters after covariate inclusiona
| Parameter | Population value
|
Interindividual variability (% CV) | Error of estimate (% CV) | |
|---|---|---|---|---|
| Mean | Error of estimate (% CV) | |||
| V1/F | 63.0 | 14.5 | 32.6 | 35.9 |
| V2/(F × Fm) (liters) | 17.3 | 16.1 | 37.0 | 35.3 |
| ka (h−1) | 0.666 | 40.3 | 43.2 | 57.9 |
| k10 (h−1) | 0.097 | 12.9 | 32.4 | 38.3 |
| k12 (h−1) | 0.083 | 10.5 | 19.7 | 43.1 |
| k20 (h−1) | 0.376 | 11.0 | 38.2 | 39.3 |
| ALAG (h) | 0.205 | 40.3 | 98.2 | 75.8 |
Residual intraindividual coefficients of variability were 7.6% for NFV and 24.1% for M8. Abbreviations: CV, coefficient of variability; ALAG, lag time. The other abbreviations are defined in the text.
The mean kel of NFV was 0.17 h−1 (i.e., elimination half-life, 4.3 h; range, 2.3 to 6.9 h), and the elimination half-life of M8 ranged from 1.0 to 4.3 h (mean, 2.04 h). The population CL/F was estimated to be 2.13 liters/h/kg (range, 1.0 to 4.0 liters/h/kg). For 19 kinetic profiles with intensive sampling times between two drug intakes, the CL/F values computed by use of a Bayesian approach (test approach) were compared to the ones estimated by use of the noncompartmental approach (reference approach). The ratio (test approach/reference approach) averaged 0.97. The bias (0.398 liter/h) was not statistically different from 0 (t test), and the 95% confidence interval included the 0 value (−0.21 to 1.01).
Typical posterior individual fittings are displayed in Fig. 5A for nelfinavir and Fig. 5B for the M8 metabolite. Simulations for a typical subject with the 95% confidence interval around the typical value are presented in Fig. 6.
FIG. 5.

Typical posterior individual fittings for a patient aged 6 months receiving NFV BID. (A) NFV; (B) M8. The open symbols represent the individual predicted plasma concentrations, and the filled symbols are the observed plasma concentrations.
FIG. 6.

Simulation for a typical patient. Heavy lines, population fitted model; thin lines, 95% confidence interval. The dose administered was 500 mg BID.
DISCUSSION
Adequate protease inhibitor concentrations have been shown to be critical for the overall success of antiretroviral therapy. From in vitro studies, a mean NFV concentration of 0.04 mg/liter (range, 0.005 to 0.09 mg/liter) (17) was reported to inhibit viral replication by 95%. In adult patients, a plasma NFV concentration of 0.4 mg/liter is the stated minimum effective concentration predicted on the basis of the 95% inhibitory concentration for several laboratory strains of HIV-1 and NFV protein binding in vivo (J. Montaner, personal communication, 1997). Nevertheless, no adult or pediatric studies have clearly established therapeutic NFV concentrations. Such pharmacodynamic studies are difficult to perform due to other variables, besides plasma NFV concentrations, that affect the optimal virologic response (19).
In the present study, children received a range of doses due to an interim analysis of the pharmacokinetic data. The mean initial dose of NFV administered ranged from 71 to 203 mg/kg/day. During the study period, this daily dose was increased by 13 to 20% in five children and by 23 to 30% in three children, due to low concentrations. Fourteen infants received NFV two times a day. Indeed, in the pediatric population, BID dosing is more practical and is expected to improve compliance. Four infants received dosages of 34 to 53 mg/kg TID. During the course of the study, the dose of NFV was increased by about 25%, and these infants were switched to BID dosing of NFV, while the same daily dose was maintained. Indeed, a crossover analysis with HIV-1-infected children who received NFV in both the TID and the BID regimens shown no significant differences in the pharmacokinetic parameters (22).
Little is known about the pharmacokinetics of NFV in infants and young children, an important age group requiring treatment for HIV infection from perinatal transmission. Thus, this study was initiated to investigate the steady-state pharmacokinetics of NFV and its metabolite, M8, in the plasma of children younger than age 1.5 years. For this purpose, a model that simultaneously fit NFV and M8 data was developed. An empirical Bayes methodology was used to estimate individual pharmacokinetic parameters. Such an approach allows estimation of the pharmacokinetic parameters for a patient with a limited number of blood samples. Despite the small number of patients included in this study, body weight was found to be a more appropriate predictor than age of the changes in (i) the rate of metabolism, (ii) the elimination rate constant of NFV, and (iii) the NFV CL/F. Indeed, in infants of about the same age, there were marked interpatient variations in weight (for example, in patients between 3 and 4 years of age, the body weight ranged from 2.5 to 6.7 kg). In the final model, the relationship between k12 (rate of metabolism) and weight was taken into account.
Marked interpatient variability in pharmacokinetic parameters was observed. This could be partially explained by the maturational changes in the CYP isoenzymes responsible for the metabolism of NFV. Table 5 compares the results of the present study with those previously reported by other workers (3, 4, 7, 12, 13, 16, 21, 22, 27; Capparelli et al., 40th ICAAC; Capparelli et al., Abstr. 7th Conf. Retrovir. Opportunistic Infect., 2000). In the present study, the NFV elimination half-life, 4.3 h, is close to the values reported by Litalien et al. (13), Rongkavilit et al. (21), and Van Heeswijk et al. (27). The apparent oral clearance (CL/F, 2.13 liters/h/kg) and the volume of distribution (12.3 liters/kg) of NFV found in our study are in agreement with previously reported values for children (21, 27). However, the values for these two parameters were about two times lower than those published by Litalien et al. (13). These findings could be explained by the differences in the patients' ages at the time of analysis of the kinetics: 2.3 to 7.7 months in the study of Litalien et al. (13) versus 1.5 to 16.2 months in the present study. We have found a relationship between post hoc oral clearance and body weight, but this result was not in accordance with the findings of Floren et al. (7). Indeed, those investigators reported that they detected no statistically significant correlation between weight and CL/F. These opposite results can be explained by the difference in the ages between the two populations of children (7.2 months to 16 years in the study of Floren et al. [7] versus 1.5 to 16 months in the present study). Indeed, there are marked interpatient variations in hepatic and renal functions, as well as gastrointestinal activity, during the first years of life.
TABLE 5.
Multiple-dose pharmacokinetic parameters for NFV in HIV-infected infants reported in the literature
| Reference | Age and wt | Dose (mg/kg/day) | No. of subjects | Elimination half-life (h) | CL/F (liters/h/kg) | V/F (liters/kg) | M8/NFV |
|---|---|---|---|---|---|---|---|
| Rongkavilit et al. (21) | 14 days | 30 BID | 6 | 3.3 | 1.05 | 5.5 | |
| 60 BID | 5 | 4.2 | 1.55 | 10.3 | |||
| 90 BID | 11 | 5.0 | 1.92 | 13.4 | |||
| 28 days | 30 BID | 6 | 4.7 | 1.73 | 10.5 | ||
| 60 BID | 5 | 4.2 | 1.90 | 14.7 | |||
| 90 BID | 11 | 4.8 | 2.43 | 18.7 | |||
| Litalien et al. (13) | 2.3-8.5 mo | 109.4-172.4 BID or TID | 14 | NFV, 2.6; M8, 2.2 | 4.2 | 22.3 | 0.12-0.89a |
| Capparelli et al. (4) | 15 days-2 yr | 75-110 BID or TID | 8 | 2.7 | 0.09b | ||
| Capparelli et al.c | 0.8-21.1 yr | 90 TID | 7 | 1.87 | |||
| Capparelli et al.d | ≤3 mo | 75 TID | 5 | 3.2 | |||
| Krogstad et al. (12) | 3.6-22 mo | 69 TID | 19 | 0.8-1.8 | |||
| Van Heeswijk et al. (27) | 2.1-10.8 yr | 90 BID or TID | 12 | 2.5-3.1 | 1-1.3 | 3.9-6 | |
| Pai and Nahata (16) | 2-7 yr | 60 TID | 8 | 1.25 | |||
| 7-13 yr | 60 TID | 7 | 2.08 | ||||
| Floren et al. (7) | 0.6-7 yr | 90 TID | 9 | 2.4 | 0.13-0.71a | ||
| 7.2-16 yr | 90 TID | 11 | 0.9 | ||||
| 3.4-11 yr | 110 BID | 6 | 1.2 | ||||
| Bergshoeff et al. (3) | <2 yr | 87 TID | 7 | 2.8 | 0.31a | ||
| ≥2 yr | 84 TID | 17 | 1.9 | 0.29a | |||
| Schuster et al. (22) | 2-14 yr | 18 | |||||
| <25 kg | 90-111 TID | 8 | 1.72 | ||||
| 90-110 BID | 1.54 | ||||||
| >25 kg | ≤90 TID | 10 | ≤2.30 | ||||
| ≤100 BID | ≤1.47 | ||||||
| This study | 1.5-16.2 mo | 81-203 BID and TID | 18 | NFV, 4.3; M8, 2.04 | 2.13 | 12.3 | 0.1-0.96a,e |
| 0.64b |
Area under the concentration-time curve ratio.
Concentration ratio.
Abstr. 7th Conf. Retrovir. Opportunistic Infect., 2000.
40th ICAAC.
Computed from 19 kinetic values by a simple noncompartmental approach between two drug administrations.
In conclusion, the population pharmacokinetic model describing the dispositions of NFV and M8 should facilitate the design of future studies to elucidate the relative contributions of the parent compound and M8 to the pharmacological and toxic effects of NFV therapy.
Acknowledgments
This study is presented on behalf of the PENTA 7 Executive Committee. PENTA is a concerted action supported by the European Commission (grant QLK2-2000-00150).
We thank those members of the PENTA Executive Committee (J.-P. Aboulker, A. Babiker, A. Compagnucci, J. Darbyshire, M. Debré, A. Faye, C. Giaquinto [chairperson], and D. M. Gibb) who contributed to the present study. We also thank the PENTA Steering Committee (J.-P. Aboulker, A. Babiker, S. Blanche, A. Britt-Bohlin, K. Butler, G. Castelli-Gattinara, P. Clayden, J. Darbyshire, M. Debré, A. Faye, C. Giaquinto [chairperson], D. M. Gibb, C. Griscelli, I. Grosch-Wörner, C. Kind, J. Levy, M. Mellado Pena, D. Nadal, C. Peckham, J. T. Ramos Amador, L. Rosado, C. Rudin, H. Scherpbier, M. Sharland, P. A. Tovo, G. Tudor-Williams, A. Volnay, V. Wahn, and U. Wintergerst) and the PENTA Pharmacology Committee (R. Hoetelmans, E. Jacqz-Aigrain, S. Khoo, and J.-M. Treluyer).
The national trials centers are the Medical Research Council Clinical Trials Unit, HIV Division, London, United Kingdom (A. Babiker, J. Darbyshire, D. M. Gibb, L. Harper, D. Johnson, P. Kelleher, L. McGee, and A. S. Walker) and INSERM SC10 Villejuif, France (J.-P. Aboulker, A. Compagnucci, M. Debré, V. Eliette, S. Girard, S. Leonardo, and Y. Saidi). The pharmacological work was performed at the Hôpital Robert Debré, Paris, France, by E. Jacqz-Aigrain.
REFERENCES
- 1.Bardsley-Elliot, A., and G. L. Plosker. 2000. Nelfinavir, an update of its use in HIV infection. Drugs 59:581-620. [DOI] [PubMed] [Google Scholar]
- 2.Beal, S. L., and L. B. Sheiner. 1994. NONMEM user's guide. University of California at San Francisco, San Francisco, Calif.
- 3.Bergshoeff, A. S., P. L. Fraaij, A. M. van Rossum, T. F. Wolfs, S. P. Geelen, R. de Groot, and D. M. Burger. 2003. Pharmacokinetics of nelfinavir in children: influencing factors and dose implications. Antivir. Ther. 8:215-222. [PubMed] [Google Scholar]
- 4.Capparelli, E. V., J. L. Sullivan, L. Mofenson, E. Smith, B. Graham, P. Britto, M. I. Becker, D. Holland, J. D. Connor, and K. Luzuriaga. 2001. Pharmacokinetics of nelfinavir in human immunodeficiency virus-infected infants. Pediatr. Infect. Dis. J. 20:746-751. [DOI] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention. 1998. Guidelines for the use of antiretroviral agents in pediatric HIV infection. Morb. Mortal. Wkly. Rep. 47:1-45. [Google Scholar]
- 6.Farenc, C., J. R. Fabreguette, and F. Bressolle. 2000. Pk-fit: a pharmacokinetic/pharmacodynamic and statistical data analysis software. Comput. Biomed. Res. 33:315-330. [DOI] [PubMed] [Google Scholar]
- 7.Floren, L. C., A. Wiznia, S. Hayashi, A. Jayewardene, K. Stanley, G. Johnson, S. Nachman, P. Krogstad, and F. T. Aweeka. 2003. Nelfinavir pharmacokinetics in stable human immunodeficiency virus-positive children: Pediatric AIDS Clinical Trials Group Protocol 377. Pediatrics 112:220-227. [DOI] [PubMed] [Google Scholar]
- 8.Funk, M. B., R. Linde, U. Wintergerst, G. Notheis, F. Hoffmann, T. Schuster, B. Kornhuber, P. Ahrens, and W. Kreuz. 1999. Preliminary experiences with triple therapy including nelfinavir and two reverse transcriptase inhibitors in previously untreated HIV-infected children. AIDS 13:1653-1658. [DOI] [PubMed] [Google Scholar]
- 9.Gatti, G., G. Castelli-Gattinara, M. Cruciani, S. Bernardi, C. R. De Pascalis, E. Pontali, L. Papa, F. Miletich, and D. Bassetti. 2003. Pharmacokinetics and pharmacodynamics of nelfinavir administered twice or thrice daily to human immunodeficiency virus type-1-infected children. Clin. Infect. Dis. 36:1476-1482. [DOI] [PubMed] [Google Scholar]
- 10.Gulik, R. M., J. H. Mellors, D. Havlir, J. J. Eron, C. Gonzalez, and D. McMahon. 1997. Treatment with indinavir, zidovudine and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N. Engl. J. Med. 337:734-739. [DOI] [PubMed] [Google Scholar]
- 11.Hammer, S. M., K. E. Squires, M. D. Hughes, J. M. Grimes, L. M. Demeter, J. S. Currier, J. J. Eron, Jr., J. E. Feinberg, H. H. Balfour, Jr., L. R. Deyton, J. A. Chodakewitz, M. A. Fischl, et al. 1997. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. N. Engl. J. Med. 337:725-733. [DOI] [PubMed] [Google Scholar]
- 12.Krogstad, P., A. Wiznia, K. Luzuriaga, W. Dankner, K. Nielsen, M. Gersten, B. Kerr, A. Hendricks, B. Boczany, M. Rosenberg, D. Jung, S. A. Spector, and Y. Bryson. 1999. Treatment of human immunodeficiency virus 1-infected infants and children with the protease inhibitor nelfinavir mesylate. Clin. Infect. Dis. 28:1109-1118. [DOI] [PubMed] [Google Scholar]
- 13.Litalien, C., A. Faye, A. Compagnucci, C. Giaquinto, L. Harper, D. M. Gibb, and E. Jacqz-Aigrain. 2003. Pharmacokinetics of nelfinavir and its active metabolite, hydroxy-tert-butylamide, in infants perinatally infected with human immunodeficiency virus type 1. Pediatr. Infect. Dis. J. 22:48-55. [DOI] [PubMed] [Google Scholar]
- 14.Malaty, L. I., and J. J. Kuper. 1999. Drug interactions of HIV protease inhibitors. Drug Safety 20:147-169. [DOI] [PubMed] [Google Scholar]
- 15.Montaner, J. S., P. Reiss, D. Cooper, S. Vella, M. Harris, B. Conway, M. A. Wainberg, D. Smith, P. Robinson, D. Hall, M. Myers, and J. M. Lange. 1998. A randomized, double-blind trial comparing combinations of nevirapine, didanosine, and zidovudine for HIV-infected patients: the INCAS Trial. Italy, The Netherlands, Canada and Australia study. JAMA 279:930-937. [DOI] [PubMed] [Google Scholar]
- 16.Pai, V. B., and M. C. Nahata. 1999. Nelfinavir mesylate: a protease inhibitor. Ann. Pharmacother. 33:325-339. [DOI] [PubMed] [Google Scholar]
- 17.Patick, A. K., H. Mo, M. Markowitz, K. Appelt, B. Wu, L. Musick, V. Kalish, S. Kaldor, S. Reich, D. Ho, and S. Webber. 1996. Design, synthesis, and evaluation of nonpeptidic inhibitors of human rhinovirus 3C protease. J. Med. Chem. 39:5072-5082. [DOI] [PubMed] [Google Scholar]
- 18.Perry, C. M., and P. Benfield. 1997. Nelfinavir. Drugs 54:81-87. [DOI] [PubMed] [Google Scholar]
- 19.Powderly, W. G., M. S. Saag, S. Chapman, G. Yu, B. Quart, and N. J. Clendeninn. 1999. Predictors of optimal virological response to potent antiretroviral therapy. AIDS 13:1873-1980. [DOI] [PubMed] [Google Scholar]
- 20.Research Development Population Pharmacokinetics. 1998. Visual-NM user's manual, version 5.1. Research Development Population Pharmacokinetics, Montpellier, France.
- 21.Rongkavilit, C., R. P. van Heeswijk, S. Limpongsanurak, P. Thaithumyanon, C. Boonrod, E. A. Hassink, A. Srigritsanapol, T. Chuenyam, S. Ubolyam, R. M. Hoetelmans, K. Ruxrungtham, J. M., Lange, D. A. Cooper, and P. Phanuphak. 2002. Dose-escalating study of the safety and pharmacokinetics of nelfinavir in HIV-exposed neonates. J. Acquir. Immune Defic. Syndr. 29:455-463. [DOI] [PubMed] [Google Scholar]
- 22.Schuster, T., R. Linde, U. Wintergerst, M. B. Funk, M. Kurowski, W. Kreuz, and D. Hofmann. 2000. Nelfinavir pharmacokinetics in HIV-infected children: a comparison of twice daily and three time daily dosing. AIDS 14:1466-1468. [DOI] [PubMed] [Google Scholar]
- 23.Shearer, W. T., T. C. Quinn, P. LaRussa, J. F. Lew, L. Mofenson, S. Almy, K. Rich, E. Handelsman, C. Diaz, M. Pagano, V. Smeriglio, L. A. Kalish, et al. 1997. Viral load and disease progression in infants infected with human immunodeficiency virus type 1. N. Engl. J. Med. 336:1337-1342. [DOI] [PubMed] [Google Scholar]
- 24.Sheiner, L. B., and S. L. Beal. 1981. Some suggestions for measuring predictive performance. J. Pharmacokinet. Biopharm. 9:503-512. [DOI] [PubMed] [Google Scholar]
- 25.Sheiner, L. B., and S. L. Beal. 1981. Evaluation of methods for estimating population pharmacokinetic parameters. II. Biexponential model and experimental pharmacokinetic data. J. Pharmacokinet. Biopharm. 9:635-651. [DOI] [PubMed] [Google Scholar]
- 26.Starr, S. E., C. V. Fletcher, S. A. Spector, F. H. Yong, T. Fenton, R. C. Brundage, D. Manion, N. M. Ruiz, M. Gersten, M. Becker, J. McNamara, and L. M. Mofenson for The Pediatric AIDS Clinical Trials Group 382 Team. 1999. Combination therapy with efavirenz, nelfinavir, and nucleoside reverse-transcriptase inhibitors in children infected with human immunodeficiency virus type 1. N. Engl. J. Med. 341:1874-1881. [DOI] [PubMed] [Google Scholar]
- 27.van Heeswijk, R. P., H. J. Scherpbier, L. A. de Koning, H. S. Heymans, J. M. Lange, J. H. Beijnen, and R. M. Hoetelmans. 2002. The pharmacokinetics of nelfinavir in HIV-1-infected children. Ther. Drug Monit. 24:487-491. [DOI] [PubMed] [Google Scholar]
- 28.Zhang, K. E., E. Wu, A. K. Patick, B. Kerr, M. Zorbas, A. Lankford, T. Kobayashi, Y. Maeda, B. Shetty, and S. Webber. 2001. Circulating metabolites of the human immunodeficiency virus protease inhibitor nelfinavir in humans: structural identification, levels in plasma, and antiviral activities. Antimicrob. Agents Chemother. 45:1086-1093. (Erratum, 45:2405.) [DOI] [PMC free article] [PubMed] [Google Scholar]