

SUMMARY
The p- and m-isothiocyanate adenosine derivatives N6-[4-[[[4-[[[[2-[[[(p-(m)-isothiocyanatophenyl)amino]thiocarbonyl]amino]-ethyl]amino]carbonyl]methyl]anilino]carbonyl]methyl]phenyl]-adenosine (p- and m-DITC-ADAC) were examined for irreversible agonist effects at the A1-adenosine receptor (A1-AdoR) in DDT1 MF-2 (DDT) cells and a functional A1-AdoR response in the guinea pig isolated heart. The p- and m-DITC-ADAC inhibited (−)-isoproterenol stimulated cAMP accumulation in DDT cells in the low nanomolar range, and the maximal responses elicited by both compounds were similar to that for N6-cyclopentyladenosine. Once established, the p-DITC-ADAC-mediated inhibition of cAMP accumulation in DDT cells was not affected by the addition of the AdoR antagonist 8-cy-clopentyl-1,3-dipropylxanthine (CPX). Pretreatment of DDT cells with p-DITC-ADAC (1 μM), followed by washing, reduced [3H]CPX binding to the A1-AdoR by 44% without altering the Kd value for the radioligand to the remaining receptors. The relationship between irreversible A1-AdoR occupancy by p-DITC-ADAC and inhibition of cAMP accumulation revealed a relatively large receptor reserve (64%) for the maximal response. In guinea pig isolated hearts, m-DITC-ADAC (5 μM) prolonged the stimulus to His bundle (SH) interval by 2.1-fold; this response could be prevented by the antagonist 8-cyclopentyltheophylline (5 μM). However, after the SH interval prolongation was established, extensive washout or the addition of 8-cyclopentyltheophylline had little reversal effect on the m-DITC-ADAC response. Binding of [3H]CPX to the guinea pig ventricular membranes after m-DITC-ADAC treatment and washing was reduced by 35%. The A1-AdoR occupancy response relationship for m-DITC-ADAC to prolong the SH interval indicated a small (10–20%) receptor reserve. Both p -and m-DITC-ADAC seem to be irreversible full agonists at the A1-AdoR and may prove to be useful probes to further investigate A1-AdoR structure-function relationships.
Adenosine is a ubiquitous modulator of physiological processes that acts to both increase tissue oxygen supply and reduce metabolic demand. The actions of adenosine are mediated by cell surface receptors and at least four subtypes, designated A1, A2A, A2B, and A3, have been identified, with each subtype mediating cellular effects by coupling to G proteins (1–4). The AdoR regulates the activity of several cellular components, including inhibition of adenylyl cyclase activity, stimulation of phospholipases A2 and C, and guanylyl cyclase, and regulates the ion permeability of calcium and potassium channels (2–4). The A2-AdoRs, on the other hand, mediate stimulation of adenylyl cyclase activity (1, 2). Although less is known about the A3-AdoR, it has been reported that this AdoR subtype mediates an inhibition of adenylyl cyclase and a stimulation of phospholipase C, resulting in an elevation of inositol phosphates (5, 6). The characterization, function, and regulation of the AdoRs have been greatly facilitated by the development of potent and selective agonists and antagonists (1, 7, 8).
Chemoreactive ligands are useful pharmacological probes in structure and function studies of receptors (9). These ligands are usually composed of two components: a pharmacophore and a reactive moiety. The pharmacophore is usually derived from known agonists or antagonists, which provide affinity and selectivity for the receptor. The reactive moiety allows covalent incorporation of the ligand into the receptor, which prevents ligand dissociation (9). These ligands have been used in a variety of studies, including determination of receptor reserve (10, 11), receptor subtype discrimination (12, 13), basal receptor turnover (14, 15), and ligand binding site mapping (16, 17).
Jacobson et al. (18) have synthesized a series of potential irreversible ligands for the adenosine receptor by modifying the high affinity agonist pharmacophore ADAC with substituents containing reactive electrophilic groups. Coupling of ADAC with 1,3- or 1,4-phenylene DITC produced p- and m-DITC-ADAC, which were shown by radioligand binding assays to be relatively selective and irreversible ligands for the rat brain A1-AdoR. In the current study, the agonist properties of p- and m-DITC-ADAC for the A1-AdoR were determined and contrasted with an established agonist. In addition, as potential irreversible agonists, under the appropriate experimental conditions these compounds occupy and activate a fixed fraction of the total receptor population. From the fixed fraction receptor occupancy-response relationship, the receptor reserve for the irreversible agonist can be determined. Toward these goals, the p- and m-DITC-ADAC binding properties and cAMP accumulation effects mediated by the A1-AdoR in DDT cells and the A1-AdoR-induced slowing of AV nodal conduction in the guinea pig isolated heart were investigated.
Experimental Procedures
Materials
The radioligands [3H]cAMP (28.4 Ci/mmol) and [3H]CPX (88.2–120 Ci/mmol) were purchased from New England Nuclear (Boston, MA). CPX and CPT were from Research Biochemicals (Natick, MA). Dulbecco’s modified Eagle’s medium, HBSS, horse serum, and fetal bovine serum were from GIBCO (Grand Island, NY). DDT cells were purchased from American Type Culture Collection (Rockville, MD). All other reagents were from Sigma Chemical (St. Louis, MO) or Fisher Scientific (Orlando, FL). Rolipram was a gift of Berlex Laboratories (Cedar Knolls, NJ).
Drug preparations
The synthesis and chemical characterization of p- and m-DITC-ADAC have been previously reported (18). The structures of these compounds are shown in Fig. 1. Stock solutions of p- and m-DITC-ADAC, CPA, and CPT (10 mM) were prepared in dimethylsulfoxide, stored at −20°, and diluted with incubation buffer immediately before use. Stock solutions of NECA were made in 5 mM HCl, stored at −20°, and diluted with buffer before use.
Fig. 1.
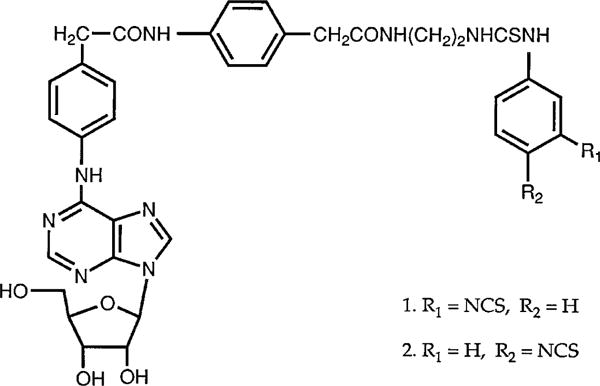
Structure of p- and m-DITC-ADAC.
Cell culture
DDT cells were grown as monolayers in 150-mm culture dishes with Dulbecco’s modified Eagle’s medium containing 5% fetal bovine serum, 100 units/ml penicillin G, 0.1 mg/ml streptomycin, and 2.5 μg/ml amphotericin B in a water-humidified 5% CO2/95% air mixture at 37°. Cells were seeded at 0.2–1 × 104 cells/cm2 and subcultured twice weekly after detachment using 1 mM EDTA in HBSS without the divalent cations. Experiments were performed on cells 1 day before confluency unless otherwise indicated.
Determination of cAMP accumulation
DDT cells were detached using a cell lifter in 5 ml of HBSS and pelleted by centrifugation at 500 × g for 5 min. Cells (0.2–0.3 mg of protein) were then incubated in microfuge tubes with 500 μl of HBSS containing 100 μM rolipram and the indicated concentrations of drugs. Incubations were performed at 37° for 10 min unless otherwise indicated. Reactions were terminated by placing the tubes in a boiling water bath for 5 min. After cooling to room temperature, the tubes were centrifuged for 2 min at 10,000 × g, and the supernatants were saved for assays.
The cAMP content was determined by a modified competitive protein binding assay (19). Briefly, an aliquot of the supernatant (50 μl) was incubated in a total volume of 0.2 ml of 25 mM Tris-HCl buffer, pH 7.0, containing 8 mM theophylline, 0.8 pmol of [3H]cAMP, and 24 μg of bovine heart cAMP-dependent protein kinase at 4° for 60 min. At the end of the incubation, 70 μl of 50% (v/v) hydroxyapatite/water suspension was added, followed by 4 ml of ice-cold 10 mM Tris-HCl buffer, pH 7.0. The suspension was then filtered through Whatman GF/B glass-fiber filters under reduced pressure using a Brandel Cell Harvester (Montreal, Quebec, Canada); the filters were washed twice with 3 ml of ice-cold 10 mM Tris-HCl, pH 7.0, and placed in a scintillation vial with 3 ml of Scinti-Verse BD. The radioactivity retained on the filters was determined in a liquid scintillation counter. The amount of cAMP in the assay was calculated from a standard curve determined with known concentrations of unlabeled cAMP.
Membrane preparation
Monolayers of DDT cells were washed two times with 10 ml of ice-cold HBSS, and the cells were scraped free in 5 ml of ice-cold 50 mM Tris-HCl buffer, pH 7.4, containing 5 mM MgCl2. The suspensions were then homogenized with a SDT-100EN homogenizer (Tekmar, Cincinnati, OH) at the lowest speed for 5 sec, diluted to 35 ml with homogenization buffer, and centrifuged at 27,000 × g for 10 min in a Sorvall RC-5B centrifuge. The supernatants were discarded, and the pellets were washed two additional times by resuspension and centrifugation as above. The final pellets were resuspended in one volume of homogenization buffer for assays.
Guinea pig ventricular membranes were prepared as previously described (20). Briefly, guinea pig hearts were perfused for 10 min with oxygenated Krebs-Henseleit solution to remove blood. Ventricles were then isolated, minced, and homogenized in 10 volumes of ice-cold 50 mM Tris-HCl buffer, pH 7.4. The homogenate was centrifuged at 48,000 × g for 15 min to pellet the membranes. The pellet was resuspended in 30 ml of homogenization buffer and centrifuged as above, and the membranes were washed twice more by resuspension and centrifugation. The final pellet was resuspended in one volume of appropriate buffer for assays. The protein content of membrane preparations was determine according to Bradford (21) method, with a BioRad (Richmond, CA) assay and bovine serum albumin as standard.
Radioligand binding assays
A1-AdoRs in DDT cell and cardiac membranes were determined according to the specific binding of [3H]CPX as described by Scammells et al. (22) and Belardinelli et al. (20). Briefly, membrane protein (0.1–0.2 mg) was incubated in a volume of 0.25 ml containing 50 mM Tris·HCl buffer, pH 7.4, 5 mM MgCl2, 2 units/ml adenosine deaminase, and 0.06–4 nM [3H]CPX for 90 min at 25°. Nonspecific binding was determined using CPX (10 μM) to displace the specific binding. At the end of the incubation, the suspensions were diluted with 3 ml of ice-cold incubation buffer, and the membranes with bound radioligand were collected by filtration through GF/B glass-fiber filters under reduced pressure. The filters were washed with an additional 6 ml of ice-cold buffer and placed in scintillation vials with 3 ml of Scinti-Verse BD, and the radioactivity was determined using a liquid scintillation counter. Specific [3H]CPX binding to the A1-AdoR was calculated as the difference between total binding in the absence of CPX and the nonspecific binding determined in the presence of CPX.
The functional A1 response in guinea pig isolated hearts
Guinea pig isolated hearts were perfused at a constant flow (10 ml/min) and instruments for measurement of AV nodal conduction time were attached as described previously (23). The hearts were perfused with oxygenated Krebs-Henseleit solution (pH 7.4, 35°) containing the drugs as indicated. The atrium of the heart was stimulated at a constant cycle length of 300 msec throughout the experiment. The A1-AdoR-mediated prolongation of the SH interval, an index of the delay in AV nodal conduction caused by adenosine and other compounds, was measured on a beat-by-beat throughout the experiment.
Data analysis
The receptor concentrations (Bmax) and dissociation constants (Kd) for the radiolabeled ligands were determined from nonlinear regression analysis of Rosenthal (24) plots. The concentration of drugs that inhibited cAMP accumulation by 50% (IC50) was determined using a concentration-effect analysis with a nonlinear regression algorithm (Marquardt-Levenberg). The occupancy-response relationship was fitted to a sigmoid equation using nonlinear regression (Table Curve V2.0; Jandel Scientific, San Rafel, CA) to estimate the receptor reserve for the maximal response defined as >90%. Statistical analysis of the experimental data was performed using the Student’s t test and were considered significant if p < 0.05.
Results
Effect of p- and m-DITC-ADAC on A1-AdoRs in DDT cells
The effect of p- and m-DITC-ADAC to decrease (A1-AdoR effect) cAMP accumulation was examined using DDT cells. For comparison, the well established A1-AdoR agonist CPA was also tested. Fig. 2 illustrates the effects of p- and m-DITC-ADAC and CPA to attenuate (−)-isoproterenol-stimulated cAMP accumulation in DDT cells. (−)-Isoproter-enol (5 μM) alone increased cAMP from 5 to 60 pmol/mg/min, a 12-fold increase above basal. This increase in cellular cAMP accumulation was inhibited by both m- and p-DITC-ADAC and CPA in a concentration-dependent manner with the same maximal attenuation (85%). Under the incubation conditions used (10 min, 37°), the IC50 values for p- and m-DITC-ADAC and CPA to inhibit (−)-isoproterenol-stimulated cAMP accumulation were 19.4 ± 0.2, 11.0 ± 2.0, and 1.4 ± 0.1 nM, respectively. The inhibitory effect of the three agonists at 0.1 μM was prevented by coincubation with the selective A1 antagonist CPX (10 μM, data not shown), indicating that this inhibitory response is mediated by the A1-AdoR.
Fig. 2.
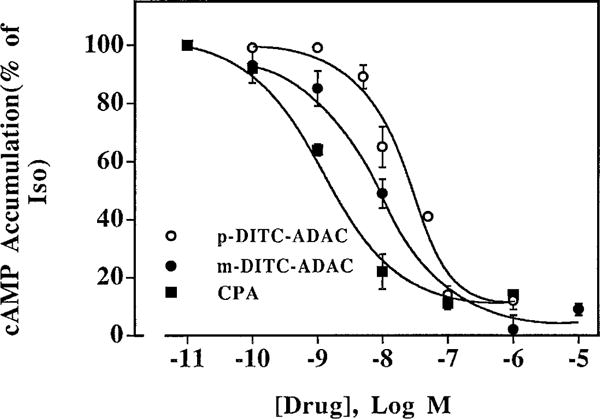
Effects of p- and m-DITC-ADAC on inhibition of (−)-isoproterenol-stimulated cAMP accumulation in DDT cells. Intact DDT cells were incubated with 1 μM (−)-isoproterenol, 50 μM rolipram, and the indicated concentrations of CPA or p- or m- DITC-ADAC for 10 min at 37°. At the end of the incubation, the cAMP accumulated was determined as described in Experimental Procedures. Values are the mean ± standard error of four separate experiments, each performed in duplicate. The basal cAMP was 5.0 ± 1.7 pmol/mg of protein/min, and the cAMP accumulated in the presence of 5 μM (−)-isoproterenol alone was 60.0 ± 9.0 pmol/mg of protein/min.
To examine the reversibility of the p-DITC-ADAC and CPA effects in DDT cells, the selective A1 antagonist CPX was used to attempt to displace the agonists bound at the A1-AdoR. As shown in Fig. 3, (−)-isoproterenol (1 μM) increased cAMP accumulation in a time-dependent manner from 20 (basal) to 605 pmol/mg of protein over a 12-min incubation period. This increase of cAMP accumulation was attenuated by 70% and 80% after a 5-min incubation in the presence of 1 μM p-DITC-ADAC or 1 μM CPA, respectively, and by 80% and 81%, respectively, after 12 min of incubation. When CPX (1 μM) was added after incubation with p-DITC-ADAC (1 μM) and (−)-isoproterenol (1 μM), no reversal of the p-DITC-ADAC-mediated attenuation of (−)-isoproterenol-stimulated cAMP accumulation was observed. This demonstrated that once receptor activation had been established, the inhibition by p-DITC-ADAC was insensitive to CPX, a finding consistent with an irreversible interaction between p-DITC-ADAC and the A1-AdoR. In contrast, the addition of CPX after a 4-min incubation with (−)-isoproterenol and CPA resulted in a time-dependent increase in cAMP accumulation such that at the end of the 12-min incubation period, the attenuation of cAMP accumulation by CPA was reduced from 81% in the absence of CPX to 36% in its presence. This indicates that CPA-mediated attenuation of cAMP accumulation and the interaction of CPA with the A1-AdoR are reversible.
Fig. 3.
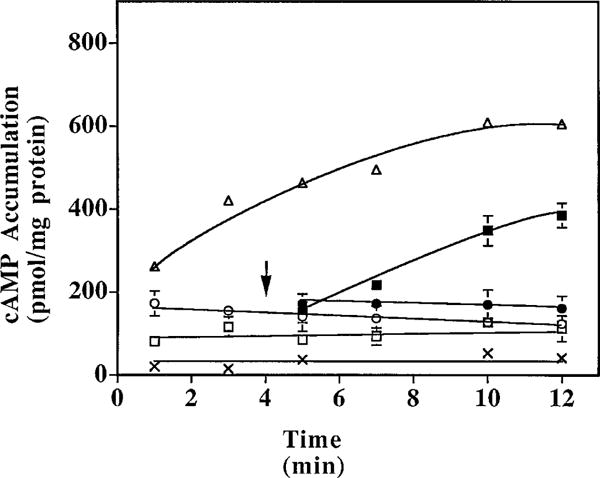
Time course effect of p-DITC-ADAC on the inhibition of (−)-isoproterenol-stimulated cAMP accumulation in DDT cells. DDT cells were incubated with 50 μM rolipram and 1 μM (−)-isoproterenol and without (Δ) or with 1 μM p-DITC-ADAC (○) or 1 μM CPA (□) at 37°. After 4 min of incubation with p-DITC-ADAC (●) or CPA (■), 1 μM CPX was added, and the accumulated cAMP was determined at the indicated time points. X, Basal cAMP levels. Points, mean ± standard error of three separate determinations assayed in duplicate; arrow, addition of CPX.
To further investigate the interaction of p-DITC-ADAC with the A1-AdoR, radioligand binding experiments were performed. DDT cells were incubated with 1 μM p-DITC-ADAC at 37° for 1 hr and washed six times with HBSS, and the Bmax and Kd values for [3H]CPX binding to cell membranes were determined on the basis of Rosenthal (24) plots. As shown in Table 1, preincubation of DDT cells with p-DITC-ADAC (1 μM) followed by extensive cell washing reduced the Bmax value of [3H]CPX binding to the A1-AdoR by 44%, with no change in the Kd value for the remaining receptors. In contrast, pretreatment with CPA (1 μM), followed by cell washing, had no effect on the Bmax or Kd value of [3H]CPX binding. A representative Rosenthal plot of [3H]CPA binding to DDT cell membranes is shown in Fig. 4. These results further indicate that the interaction of p-DITC-ADAC with the A1-AdoR of DDT cells is irreversible. Similar to p-DITC-ADAC, pretreatment of DDT cells with 1 μM m-DITC-ADAC resulted in a reduction of A1-AdoRs (data not shown). As illustrated in Fig. 5, the p-DITC-ADAC-induced loss of [3H]CPX binding sites (i.e., A1-AdoRs) in DDT cells was time dependent. After incubation of DDT cells with 1 μM p-DITC-ADAC for 5, 15, 30, and 60 min, there was a 16%, 29%, 39%, and 47% reduction, respectively, of specific [3H]CPX binding to cell membranes.
TABLE 1. Effects of pretreatment of DDT cells with p-DITC-ADAC on radioligand binding to cell membranes.
DDT cells were incubated with 1 μM p-DITC-ADAC or 1 μM CPA for 1 hr at 37° followed by six washes with HBSS. Cell membranes were then prepared, and the A1-AdoRs were determined by [3H]CPX (0.125–4 nM) binding to DDT cell membranes. The Kd and Bmax values were derived from Rosenthal plots. Data are the mean ± standard error of four or five separate determinations.
| [3H]CPX binding | Bmax | Kd |
|---|---|---|
| fmol/mg of protein | nM | |
| Pretreatment | ||
| Control | 275 ± 14 | 0.57 ± 0.06 |
| CPA | 262 ± 16 | 0.57 ± 0.02 |
| p-DITC-ADAC | 153 ± 11a | 0.56 ± 0.02 |
p < 0.0005 versus control.
Fig. 4.
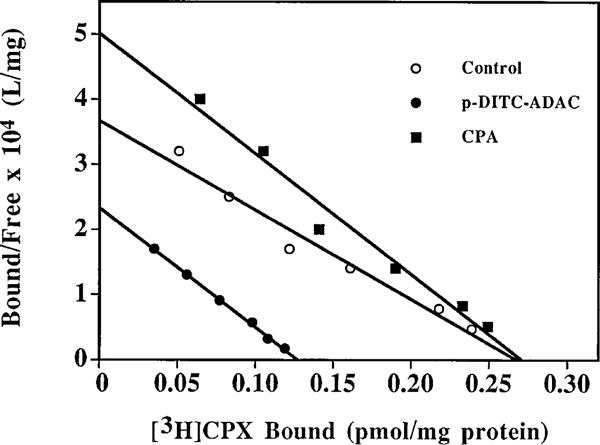
Representative Rosenthal plots of [3H]CPX binding to DDT cell membranes after pretreatment of intact cells with p-DITC-ADAC. DDT cells were incubated with 1 μM p-DITC-ADAC or 1 μM CPA at 37° for 1 hr followed by six washes with HBSS. Cell membranes were then prepared, and the A1-AdoRs were determined by [3H]CPX (0.06–4 nM) binding in DDT cell membranes. Points, mean of triplicate determinations.
Fig. 5.
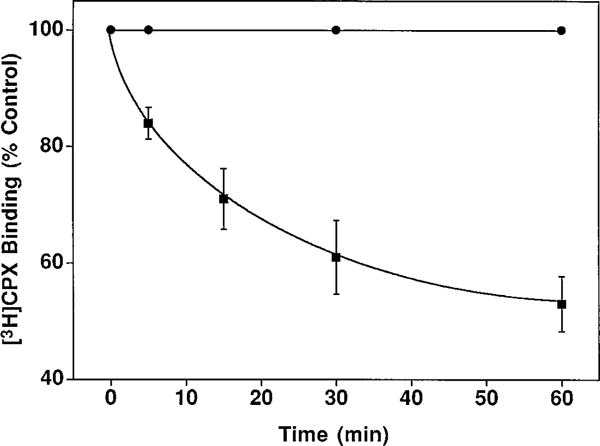
Time course of p-DITC-ADAC-induced loss of specific [3H]CPX binding to DDT cells. DDT cells were incubated without (F) or with (f) 1 μM p-DITC-ADAC for the indicated times followed by six washes with HBSS. The cells were then homogenized in 50 mM Tris-HCl buffer, pH 7.4, containing 5 mM MgCl2, and the suspension was centrifuged at 48,000 × g for 15 min. The membranes were resuspended in buffer, and the A1-AdoR content was determined with 4 nM [3H]CPX as described in Experimental Prcedures. Points, mean ± standard error of three separate experiments assayed in triplicate. The control specific [3H]CPX binding was 212 ± 30 fmol/mg of protein.
The relationship between A1-AdoR occupancy by p-DITC-ADAC and responsiveness in DDT cells
The relationship between p-DITC-ADAC-mediated inhibition of (−)-isoproterenol-stimulated cAMP accumulation and A1-AdoR occupancy in DDT cells is shown in Fig. 6. After preincubation of cells with increasing concentrations of p-DITC-ADAC (0.1–2 μM) at 37°C for 1 hr followed by six cell washes, (−)-isoproterenol (5 μM)-stimulated cAMP accumulation was determined in the presence of 1 μM CPX. This A1-AdoR antagonist was added to prevent any response from residual p-DITC-ADAC present in the cells after washing. In parallel samples, the A1-AdoR occupancy by p-DITC-ADAC was determined by the loss of specific [3H]CPX binding. At receptor occupancy levels of 16%, 29%, 34%, and 46%, there was a 32%, 58%, 73%, and 78% inhibition of (−)-isoproterenol-stimulated cAMP accumulation, respectively (Fig. 6). Using nonlinear regression analysis, the calculated receptor reserve for p-DITC-ADAC in DDT cells was ~64% because the maximal inhibition of (−)-isoproterenol-stimulated cAMP accumulation (defined as >90%) was obtained at 36% receptor occupancy. This level of receptor reserve for p-DITC-ADAC is based on the assumption that all A1-AdoRs that were measured are coupled to the inhibition of cAMP accumulation.
Fig. 6.
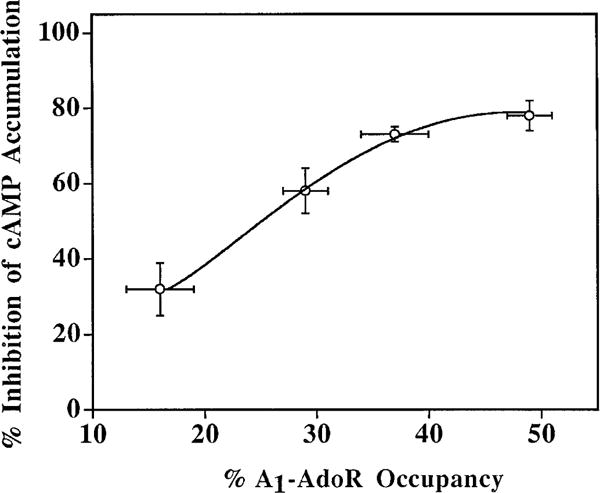
The relationship between the inhibition of (−)-isoproterenol-stimulated cAMP accumulation and receptor occupancy by p-DITC-ADAC in DDT cells. Cells were pretreated with 0.1, 0.5, 1.0, or 2.0 μM p-DITC-ADAC for 1 hr at 37° followed by six washes with HBSS. The inhibition of cAMP accumulation by the A1-AdoRs occupied by pretreatment with p-DITC-ADAC was determined by incubating the cells with 50 μM rolipram, 1 μM CPX, and 5 μM (−)-isoproterenol at 37° for 10 min. The accumulated cAMP was determined as described in Experimental Procedures. The A1-AdoR occupancy by p-DITC-ADAC was determined as the percent loss of specific [3H]CPX (5 nM) binding to cell membranes compared with untreated controls. Values are mean ± standard error of three or four separate experiments, each performed in duplicate or triplicate. The control (−)-isoproterenol (5 μM)-stimulated cAMP formation was 50 ± 5 pmol/mg of protein/min, and the control-specific [3H]CPX binding was 195 ± 4 fmol/mg of protein. The maximal inhibition of (−)-isoproterenol-stimulated cAMP accumulation in the presence of 5 μM p-DITC-ADAC was 82 ± 3% (three experiments).
Effect of m-DITC-ADAC on A1-AdoR-mediated functional response in guinea pig isolated hearts
As shown in Fig. 7A, CPA (50 nM) prolonged the SH interval of isolated guinea pig hearts from 35 to 70 msec. This effect of CPA was rapidly and nearly completely reversed on washout or the addition of the antagonist CPT (5 μM). Similar to CPA, m-DITC-ADAC (5 μM) prolonged the SH interval from 37 to 79 msec, which was sustained during a 40-min washout (Fig. 7B). Furthermore, perfusion of the heart with a supramaximal concentration of CPT (5 μM) caused only a small attenuation of the m-DITC-ADAC effect. In contrast, when the hearts were pretreated with the antagonist CPT (5 μM) before exposure to m-DITC-ADAC, the negative dromotropic effect (i.e., SH interval prolongation) of m-DITC-ADAC could no longer be elicited (Fig. 8). On washout of CPT but still in the presence of 5 μM m-DITC-ADAC, the SH interval rapidly lengthened from 40 to 82 msec and remained prolonged during a subsequent 45-min washout period. Thus, the functional effect of m-DITC-ADAC could be prevented by preincubation with the antagonist CPT, but once the effect was established, it persisted and could not be reversed.
Fig. 7.
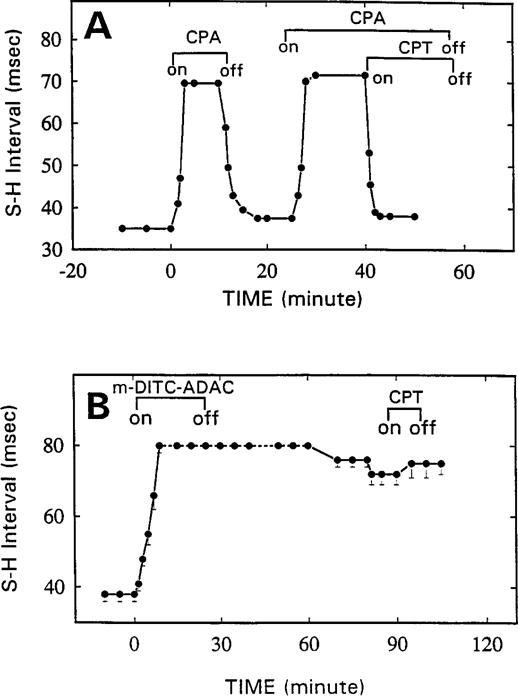
Time course of the effects of CPA, m-DITC-ADAC, and A1-AdoR antagonist CPT on SH interval of guinea pig isolated, perfused hearts. Hearts were paced at an atrial cycle length of 300 msec throughout an experiment. A, CPA (50 nM) maximally and reversibly prolonged the SH interval. This effect was rapidly and nearly completely reversed by its washout or by the antagonist CPT (5 μM). Points, averages of SH interval determinations from two hearts. B, Lack of reversibility of m-DITC-ADAC (5 μM, 20 min)-induced SH interval prolongation. m-DITC-ADAC (5 μM) added to the perfusate increased the SH interval and caused second-degree AV block. After a 40-min washout, 1:1 AV conduction resumed, but the SH interval remained prolonged (79 ± 2 msec). A supramaximal concentration of CPT (5 μM) caused only a small (p > 0.05) reversal of the effect of m-DITC-ADAC. Points, mean ± standard error of responses of four hearts.
Fig. 8.
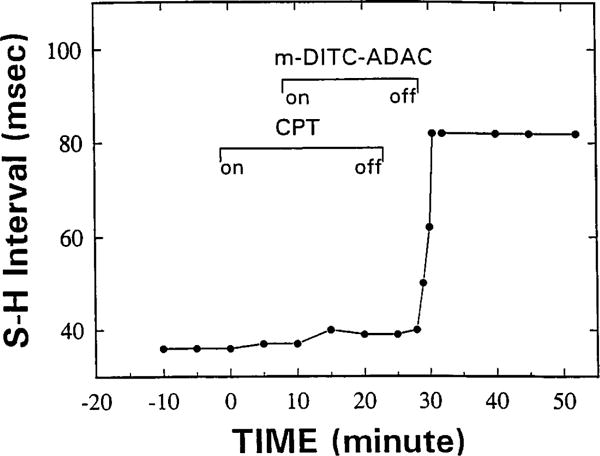
Ado-R blockade prevents the negative dromotropic effect of m-DITC-ADAC on the guinea pig isolated, perfused heart. The heart was paced at a constant atrial cycle length of 300 msec. CPT (5 μM) prevented the prolongation by m-DITC-ADAC (5 μM) of the SH interval. After washout of CPT in the presence of m-DITC-ADAC, the SH interval rapidly lengthened and remained prolonged despite washout of m-DITC-ADAC. Identical results were obtained in two additional hearts.
The effect of m-DITC-ADAC on [3H]CPX binding to the A1-AdoR in guinea pig ventricle membranes is illustrated in Fig. 9. In control membranes, maximal [3H]CPX binding was 38.5 ± 3.9 fmol/mg of protein with a Kd value of 4.3 ± 0.5 nM. Preincubation of the membranes with m-DITC-ADAC (1 μM) for 20 min followed by four wash cycles caused a 35% reduction in the Bmax value with a Kd value of 1.4 ± 0.1 nM for [3H]CPX binding to the remaining receptors.
Fig. 9.
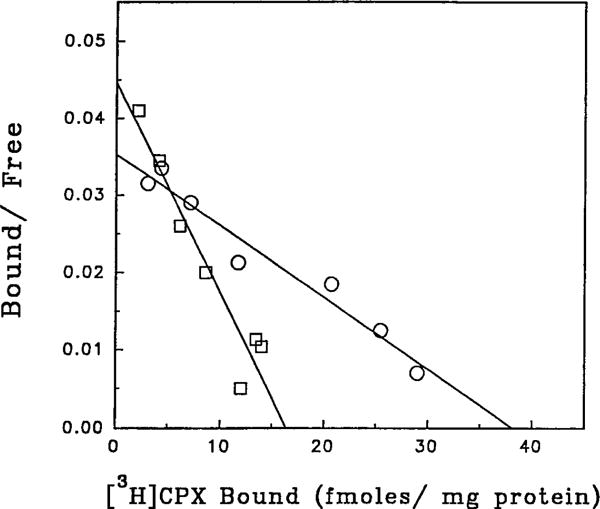
Rosenthal plot of specific [3H]CPX binding to guinea pig ventricular membranes after pretreatment with m-DITC-ADAC. Membranes were incubated with 1 μM m-DITC-ADAC for 20 min and then washed four times by centrifugation and resuspension. The A1-AdoR density was then determined by the specific binding of [3H]CPX (0.6–10 nM) as described in Experimental Procedures. Points, mean of four determinations assayed in triplicate. The Kd values are 4.3 ± 0.5 and 1.4 ± 0.1 nM for control and m-DITC-ADAC-treated membranes, respectively.
The relationship between the SH interval prolongation and A1-AdoR occupancy with m-DITC-ADAC in guinea pig isolated, perfused hearts is shown in Fig. 10. After a 5-, 10-, 15-, or 30-min perfusion with 5 μM m-DITC-ADAC, the hearts were perfused with 5 μM CPT, and the SH response was recorded. In the same perfused hearts, the A1-AdoR occupancy at each time point was determined by [3H]CPX binding after preparation of ventricular membranes. A nearly linear relationship between receptor occupancy by m-DITC-ADAC and prolongation of the SH interval was observed, with the maximal response requiring ~80–90% receptor occupancy. With the assumption that all of the A1-AdoRs are coupled to the prolongation of the SH interval, the receptor reserve for m-DITC-ADAC to produce the maximal response is 10–20%.
Fig. 10.
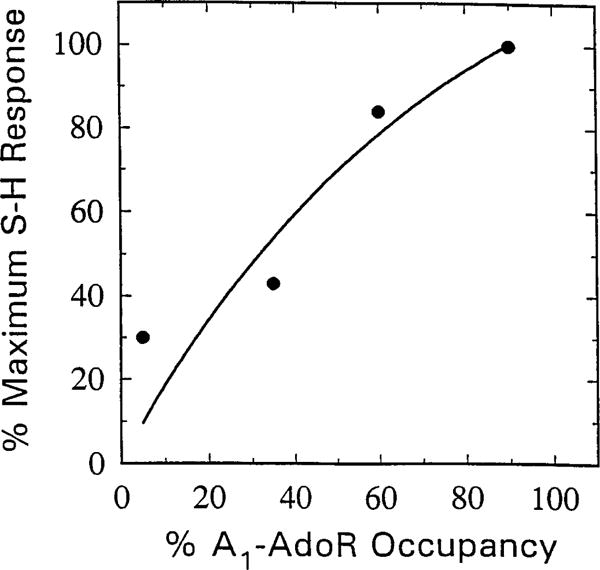
Correlation between SH interval prolongation and A1-AdoR occupancy by m-DITC-ADAC in guinea pig isolated, perfused hearts. Hearts paced at a constant atrial cycle length of 300 msec were perfused with Krebs-Henseleit solution containing 5 μM m-DITC-ADAC for 5, 10, 15, and 30 min. At the end of each perfusion period, the tissue was perfused with 5 μM CPT, and the irreversible component of SH interval prolongation was recorded. After washout for 1 hr, the ventricles were harvested, and membranes were prepared for determination of the number (Bmax) and affinity (Kd) of A1-AdoRs by measuring specific binding of [3H]CPX. The irreversible component of SH interval prolongation caused by m-DITC-ADAC is plotted as a function of percent of total A1-AdoRs occupied [Bmax (fmol/mg of protein of untreated control)/Bmax of m-DITC-ADAC-treated hearts × 100). The Bmax and Kd values of four untreated hearts were 33 ± 4 fmol/mg of protein and 3.2 ± 0.2 nM, respectively. There was no significant difference between the Kd values of [3H]CPX binding to membranes of untreated and m-DITC-ADAC-treated hearts.
Discussion
Agonist effects and irreversible binding of p- and m-DITC-ADAC to the A1-AdoR
The results from the current study show that p- and m-DITC-ADAC are potent and full agonists at the A1-AdoR in DDT cells. Under the study incubation conditions, the IC50 values for both compounds to inhibit (−)-isoproterenol-stimulated cAMP accumulation were in the low nanomolar range, whereas the maximal inhibition (85%) was the same as for the full A1-AdoR agonist CPA. As discussed below, p- and m-DITC-ADAC are irreversible ligands for the A1-AdoR; therefore, the IC50 values obtained will depend on the incubation time. The observation that p- and m-DITC-ADAC- and CPA-induced inhibition of cAMP accumulation was prevented by coincubation with the A1-AdoR-selective antagonist CPX indicates that the inhibitory effect elicited by all three agonists was mediated by the A1-AdoR.
Several criteria using radioligand binding assays are commonly used to determine whether a chemoreactive ligand binds in an irreversible manner to a receptor: (i) a decrease in radioligand binding capacity, (ii) lack of receptor recovery after extensive washing to remove the free chemoreactive ligand, and (iii) a time-dependent loss of the receptor (9). In the current study, incubation of intact DDT cells with p- or m-DITC-ADAC or of guinea pig ventricular membranes with m-DITC-ADAC reduced the Bmax value for [3H]CPX binding to the A1-AdoR with little or no change in the Kd value for this radioligand to the receptors remaining. The p- and m-DITC-ADAC-induced decreases in A1-AdoRs in DDT cells and guinea pig isolated heart were time dependent and not reversed by extensive washing to remove the free ligand. These findings are consistent with the two agonists binding to the A1-AdoR in an irreversible manner and confirm the findings with these two compounds as reported by Jacobson et al. (18), who used rat brain membranes as the receptor source. Because the preincubation protocol involved intact DDT cells, the p- and m-DITC-ADAC-induced A1-AdoR decreases could be explained by a combination of receptor acylation and agonist-induced receptor down-regulation. However, the contribution of agonist-induced down-regulation is likely to be minor because pretreatment of the cells with the higher potency reversible agonist CPA for the same time period (1 hr) did not result in a decrease in specific [3H]CPX binding to the A1-AdoR. Furthermore, the half-life of agonist-induced down-regulation of the A1-AdoR in DDT cells has been reported to be ~8 hr (25). This relatively slow rate of agonist-induced down-regulation suggests that only a small fraction of the receptors would have been lost during the 1-hr preincubation protocol of the current study.
The apparent irreversible binding of p- and m-DITC-ADAC to the A1-AdoR is likely due to an addition reaction leading to covalent bond formation between the electrophilic isothiocyanate moiety of the compounds and a nucleophilic amino acid located within or in close proximity to the ligand binding site of the receptor. Whether both p- and m-isomers of DITC-ADAC react with the same or different nucleophiles in the A1-AdoR remains to be determined.
In several experiments, the interaction of p-DITC-ADAC with the A2A-AdoR in PC12 cells was investigated using [3H]NECA, a radioligand previously used to label A2A-AdoRs in PC12 cells (26, 27). Pretreatment of PC12 cells with p-DITC-ADAC (1 μM) for 60 min, conditions that reduced the A1-AdoR content of DDT cells, did not affect [3H]NECA binding to the A2A-AdoR (data not shown). This suggests that p-DITC-ADAC did not irreversibly bind to the A2A-AdoR. However, an irreversible interaction of p-DITC-ADAC with the A2A-AdoR may be possible if higher concentrations of the chemoreactive ligand or longer incubation periods are used.
Sustained A1-AdoR activation by p- and m-DITC-ADAC
The results from the time course of cAMP accumulation in DDT cells and prolongation of the SH interval in guinea pig isolated hearts show that p- and m-DITC-ADAC elicit sustained, antagonist-insensitive A1-AdoR-mediated responses. This was shown by the observation that the addition of a 20-fold excess of CPX after a 4-min incubation with p-DITC-ADAC did not reverse the inhibition of (−)-isoproterenol-stimulated cAMP accumulation in DDT cells (Fig. 3). As expected, when CPX was added after a 4-min incubation with CPA alone, the inhibition of cAMP accumulation was largely reversed, which is consistent with CPA being a reversible A1-AdoR agonist. Similar to the effects in DDT cells, the m-DITC-ADAC-induced prolongation of the SH interval in the guinea pig isolated heart, an A1-AdoR-mediated response (4), was not reversed by the addition of an excess of the antagonist CPT or by extensive washout of m-DITC-ADAC (Fig. 7). This contrasts with the predicable behavior of the reversible agonist CPA; that is, the CPA-induced prolongation of the SH interval was completely reversed by washout of this agonist or addition of the antagonist CPT. Thus, the antagonist-insensitive effects of p- and m-DITC-ADAC to mediate cell and tissue responses are consistent with the radioligand binding data indicating these compounds bind irreversibly to the A1-AdoR. Irreversible activation of the A1-AdoR by m-DITC-ADAC would explain the sustained decrease in heart rate caused by this agonist in the rabbit isolated heart (28). Other irreversible agonists for AdoRs have been reported. Lohse et al. (29) showed that the photoaffinity ligand (R)-2-azido-N6-p-hydroxyphenylisopropyl-adenosine produced irreversible activation of the A1-AdoR in fat cells. This compound, however, caused an irreversible blockade of the A2-AdoR in human platelets (30). More recently, Niiya et al. (31) studied an arylisothiocyanate derivative of a functionalized congener of adenosine, 2-[(2-amino-ethyl-aminocarbonylethyl)phenylethylamino]-5′-N-ethyl-carboxamidoadenosine, and this compound produced sustained, antagonist insensitive coronary vasodilation in the guinea pig heart, indicating irreversible activation of the A2A-AdoR. It is not known whether this latter compound acts as an irreversible agonist at the A1-AdoR.
Receptor reserve for p- and m-DITC-ADAC
The concept of receptor reserve, in which a maximal response can be achieved at submaximal receptor occupancy, was introduced by Stephenson in 1956 (32). Typically, the receptor reserve is estimated with the use of irreversible antagonists (9–11, 33, 34). The persistent activation of the A1-AdoR by p- and m-DITC-ADAC allowed us to determine the relationship between fixed fraction receptor occupancy and response and hence the receptor reserve for these irreversible agonists. In DDT cells, the maximal inhibition of (−)-isoproterenol-stimulated cAMP accumulation was achieved when ~36% of the A1-AdoRs were irreversibly activated by p-DITC-ADAC (Fig. 6), which indicates that there is a ~64% receptor reserve for the maximal response produced by p-DITC-ADAC in DDT cells. The presence of a relatively high receptor reserve for the A1-AdoR-mediated inhibition of isoproterenol-stimulated cAMP accumulation in fat cells has also been reported for a photoaffinity A1 adenosine agonist (29). It should be pointed out that the magnitude of the receptor reserve, as determined in the current study, may be affected by several cell-dependent factors, including the presence of internalized receptors and the possibility that not all of the receptors measured are coupled to G proteins. Furthermore, in addition to the inhibition of cAMP accumulation, the A1-AdoR in DDT cells has been shown to mediate inositol phosphate accumulation (35). Thus, the receptor reserve for an agonist eliciting more than one response in a cell by the same receptor may be different, as has been shown for the muscarinic receptor in chick heart cells (36).
In contrast to DDT cells, the occupancy-response relationship for m-DITC-ADAC-mediated prolongation of the SH interval showed that 80–90% of the A1-AdoRs must be irreversibly activated to achieve the maximal response, indicating a 10–20% receptor reserve (Fig. 10). This small receptor reserve is similar to that determined, with an irreversible antagonist, for A1-AdoR activation of an inwardly rectifying K+ current by adenosine in guinea pig atrial myocytes (37). On the other hand, Dennis et al. (11), using an irreversible antagonist in combination with the method developed by Furchgott (38), calculated a 54% receptor reserve for the maximal CPA-mediated increase in the SH interval of the guinea pig heart. Because the receptor reserve is dependent on both the tissue and agonist (36), the difference in receptor reserve for m-DITC-ADAC and CPA to maximally increase the SH interval may in part be due to CPA having a higher intrinsic efficacy than m-DITC-ADAC. Alternatively, the difference in receptor reserve for these two agonists may be related to CPA being a reversible agonist, whereas m-DITC-ADAC irreversibly activates the A1-AdoR.
In summary, the data from the current study show that p-and m-DITC-ADAC are relatively potent, full agonists at the A1-AdoR. The irreversible binding and persistent agonist properties of these compounds suggest that they or related derivatives may be useful probes for further studies on the mechanism of receptor activation and effector coupling, desensitization and, in radiolabeled form, for elucidation of the structure of the A1-AdoR agonist binding site.
Acknowledgments
We thank Dr. Malgorzata Deyrup for helpful discussions during preparation of the manuscript.
This work was supported by a Grant-in-Aid from the American Heart Association, Florida Affiliate, and National Institutes for Health Grant HL35272.
ABBREVIATIONS
- AdoR
adenosine receptor
- AV
atrioventricular
- CPA
N6-cyclopentyladenosine
- CPT
8-cyclopentyltheophylline
- CPX
8-cy-clopentyl-1,3-dipropylxanthine
- DITC
diisothiocyanate
- HBSS
Hanks’ balanced salt solution
- NECA
5′-N-ethylcarboamidoadenosine
- SH
stimulus-to-His bundle
- ADAC
N6-[4-[[[4-[[[(2-aminoethyl)-amino]carbonyl]methyl]anilino]carbonyl]methyl]phenyl]adenosine
References
- 1.van Galen PJM, Stiles GL, Michaels G, Jacobson KA. Adenosine A1 and A2 receptors: structure-function relationships. Med Res Rev. 1992;12:423–471. doi: 10.1002/med.2610120502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collis MG, Hourani SMO. Adenosine receptor subtypes. Trends Pharmacol Sci. 1993;14:360–366. doi: 10.1016/0165-6147(93)90094-z. [DOI] [PubMed] [Google Scholar]
- 3.Linden J. Cloned adenosine A3 receptors: pharmacological properties, species differences and receptor functions. Trends Pharmacol Sci. 1994;15:298–306. doi: 10.1016/0165-6147(94)90011-6. [DOI] [PubMed] [Google Scholar]
- 4.Belardinelli L, Shryock JC, Wang SD, Srinivas M. Ionic basis of the electrophysiological actions of adenosine on cardiomyocytes. FASEB J. 1995;9:369–365. doi: 10.1096/fasebj.9.5.7896004. [DOI] [PubMed] [Google Scholar]
- 5.Zhou Q-Y, Li C, Olah ME, Johnson RA, Stiles GL, Civelli O. Molecular cloning and characterization of an adenosine receptor: the A3 adenosine receptor. Proc Natl Acad Sci USA. 1992;89:7432–7436. doi: 10.1073/pnas.89.16.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abbracchio MP, Brambilla R, Ceruti S, Kim HO, von Lubitz DKJE, Jacobson KA, Cattabeni F. G protein-dependent activation of phospholipase C by adenosine A3 receptors in rat brain. Mol Pharmacol. 1995;48:1038–1045. [PubMed] [Google Scholar]
- 7.Jacobson KA, van Galen PJM, Williams M. Adenosine receptors: pharmacology, structure-activity relationships, and therapeutic potential. J Med Chem. 1992;35:407–422. doi: 10.1021/jm00081a001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Galen PJM, Van Bergen AH, Gallo-Rodriguez C, Melman N, Olah ME, IJzerman ADP, Stiles GL, Jacobson KA. A binding site model and structure-activity relationships for the rat A3 adenosine receptor. Mol Pharmacol. 1994;45:1101–1111. [PMC free article] [PubMed] [Google Scholar]
- 9.Baker SP, Deyrup MD. Development of novel irreversible ligands. Neuroprotocols. 1994;4:66–75. [Google Scholar]
- 10.Jasper JR, Motulsky HJ, Insel PA. Characterization of a bromoacetylated derivative of pindolol as a high affinity, irreversible beta adrenergic antagonist in cultured cells. J Pharmacol Exp Ther. 1988;244:820–824. [PubMed] [Google Scholar]
- 11.Dennis D, Jacobson K, Belardinelli L. Evidence of spare A1-adenosine receptors in guinea pig atrioventricular node. Am J Physiol. 1992;262:H661–H671. doi: 10.1152/ajpheart.1992.262.3.H661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han C, Abel PW, Minneman KP. Heterogeneity of μ1-adrenergic receptors revealed by chlorethylclonidine. Mol Pharmacol. 1987;32:505–510. [PubMed] [Google Scholar]
- 13.Salles J, Wallace MA, Fain JN. Differential effects of alkylating agents on the multiple muscarinic receptor subtypes linked to activation of phospholipase C by carbachol in rat brain cortical membranes. J Pharmacol Exp Ther. 1993;264:521–529. [PubMed] [Google Scholar]
- 14.Hunter DD, Nathanson NM. Biochemical and physical analyses of newly synthesized muscarinic acetylcholine receptors in cultured embryonic chicken cardiac cells. J Neurosci. 1986;6:3739–3748. doi: 10.1523/JNEUROSCI.06-12-03739.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mahan LC, McKernan RM, Insel PA. Metabolism of alpha- and beta-adrenergic receptors in vitro and in vivo. Annu Rev Pharmacol Toxicol. 1987;27:215–235. doi: 10.1146/annurev.pa.27.040187.001243. [DOI] [PubMed] [Google Scholar]
- 16.Dohlman HG, Caron MG, Strader CD, Amlaiky N, Lefkowitz RJ. Identification and sequence of a binding site peptide of the μ2-adrenergic receptor. Biochemistry. 1988;27:1813–1817. doi: 10.1021/bi00406a002. [DOI] [PubMed] [Google Scholar]
- 17.Curtis CAM, Wheatley M, Bansal S, Birdsall NJM, Eveleigh P, Pedder EK, Poyner D, Hulme EC. Propylbenzilycholine mustard labels an acidic residue in transmembrane helix 3 of the muscarinic receptor. J Biol Chem. 1989;264:489–495. [PubMed] [Google Scholar]
- 18.Jacobson KA, Barone S, Kammula U, Stiles GL. Electrophilic derivatives of purines as irreversible inhibitors of A1 adenosine receptors. J Med Chem. 1989;32:1043–1051. doi: 10.1021/jm00125a019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Standifer KA, Pitha J, Baker SP. Carbostyril-based beta-adrenergic agonists: evidence for long lasting or apparent irreversible receptor binding and activation of adenylate cyclase in vitro. Naunyn-Schmiedebergs Arch Pharmacol. 1989;339:129–137. doi: 10.1007/BF00165134. [DOI] [PubMed] [Google Scholar]
- 20.Belardinelli L, Shryock JC, Zhang Y, Scammells PJ, Olsson R, Dennis D, Milner P, Pfister J, Baker SP. 1,3-Dipropyl-8-[2-(5,6-epoxy)norbornyl]-xanthine, a potent, specific and selective A1 adenosine receptor antagonist in the guinea pig heart and in DDT1MF-2 cells. J Pharmacol Exp Ther. 1995;275:1167–1176. [PubMed] [Google Scholar]
- 21.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 22.Scammells PJ, Baker SP, Belardinelli L, Olsson RA. Substituted 1, 3-dipropylxanthines as irreversible antagonists of A1 adenosine receptors. J Med Chem. 1994;37:2704–2712. doi: 10.1021/jm00043a010. [DOI] [PubMed] [Google Scholar]
- 23.Jenkins JR, Belardinelli L. Atrioventricular nodal accommodation in isolated guinea pig hearts: physiological significance and role of adenosine. Circ Res. 1988;63:97–116. doi: 10.1161/01.res.63.1.97. [DOI] [PubMed] [Google Scholar]
- 24.Rosenthal HE. A graphic method for the determination and presentation of binding parameters in a complex system. Anal Biochem. 1967;20:525–532. doi: 10.1016/0003-2697(67)90297-7. [DOI] [PubMed] [Google Scholar]
- 25.Ramkumar V, Olah ME, Jacobson KA, Stiles GA. Distinct pathways of desensitization of A1- and A2-adenosine receptors in DDT1 MF-2 cells. Mol Pharmacol. 1991;40:639–647. [PMC free article] [PubMed] [Google Scholar]
- 26.Williams M, Abreu M, Jarvis MF, Noronha-Blob L. Characterization of adenosine receptors in the PC12 pheochromocytoma cell line using radioligand binding: evidence for A-2 selectivity. J Neurochem. 1987;48:498–502. doi: 10.1111/j.1471-4159.1987.tb04120.x. [DOI] [PubMed] [Google Scholar]
- 27.Hide I, Padgett WL, Jacobson KA, Daly JW. A2A adenosine receptors from rat striatum and rat pheochromocytoma PC12 cells: characterization with radioligand binding and by activation of adenylate cyclase. Mol Pharmacol. 1992;41:352–359. [PMC free article] [PubMed] [Google Scholar]
- 28.Liu GS, Jacobson KA, Downey JM. An irreversible A1-selective adenosine agonist preconditions rabbit heart. Can J Cardiol. 1996;12:517–521. [PMC free article] [PubMed] [Google Scholar]
- 29.Lohse MJ, Klotz KN, Schwabe U. Agonist photoaffinity labeling of A1 adenosine receptors: persistent activation reveals spare receptors. Mol Pharmacol. 1986;30:403–409. [PubMed] [Google Scholar]
- 30.Lohse MJ, Klotz KN, Schwabe U. Mechanism of A2 adenosine receptor activation. I. Blockade of A2 adenosine receptors by photoaffinity labeling. Mol Pharmacol. 1991;39:517–523. [PubMed] [Google Scholar]
- 31.Niiya K, Jacobson KA, Silvia SK, Olsson RA. Covalent binding of a selective agonist irreversibly activates guinea pig coronary artery A2 adenosine receptors. Naunyn-Schmiedebergs Arch Pharmacol. 1993;347:521–526. doi: 10.1007/BF00166745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stephenson RP. A modification of receptor theory. Br J Pharmacol. 1956;11:379–393. doi: 10.1111/j.1476-5381.1956.tb00006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kenakin TP. The classification of drugs and drug receptors in isolated tissues. Pharmacol Rev. 1984;36:165–222. [PubMed] [Google Scholar]
- 34.Kenakin TP. Receptor reserve as a tissue misnomer. Trends Pharmacol Sci. 1986;7:93–95. [Google Scholar]
- 35.White TE, Dickenson JM, Alexander SPH, Hill SJ. Adenosine A1-receptor stimulation of inositol phospholipid hydrolysis and calcium mobilization in DDT1 MF-2 cells. Br J Pharmacol. 1992;106:215–221. doi: 10.1111/j.1476-5381.1992.tb14317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown JH, Goldstein D. Differences in muscarinic receptor reserve for inhibition of adenylate cyclase and stimulation of phosphoinositide hydrolysis in chick heart cells. Mol Pharmacol. 1986;30:566–570. [PubMed] [Google Scholar]
- 37.Srinivas M, Shryock J, Dennis D, Belardinelli L. Agonist affinity of adenosine and the absence of receptor reserve for activation of IKAdo in guinea pig atria. Circulation. 1996;94(Suppl. I):I-47–I-48. [Google Scholar]
- 38.Furchgott RF. The use of haloalkylamines in the differentiation of receptors and the determination of dissociation constants of receptor-agonist complexes. Adv Drug Res. 1966;3:21–55. [Google Scholar]