

Abstract
Two independent epigenome-wide association studies of Alzheimer’s disease cohorts have identified overlapping signals in four loci (ANK1, RPL13, RHBDF2 and CDH23), not previously associated with Alzheimer’s disease in any genetic study. These studies also suggest that epigenetic changes contribute more to Alzheimer’s disease than expected.
Alzheimer’s disease is a devastating condition, currently without effective cure, treatment or prevention, that robs people of their memories and abilities. With an estimated 26 million people living with the condition worldwide, and this figure expected to rise as populations age, it is one of the most burdesome threats to public health today.1 Despite this, and despite growing public attention and funding being directed into dementia research, the fundamental cause of Alzheimer’s disease remains elusive. While recent large, collaborative genome-wide association studies (GWAS) have been successful in identifying numerous genes associated with the condition,2 the variants and mechanisms underlying these associations are generally unclear. Additionally the proportion of the genetic heritability explained by common variants is only 3–4% for each locus. Therefore, it has proposed that other types of genetic variation, such as low frequency and rare variants or epigenetic changes, may help explain the condition.3 In this issue of Nature Neuroscience, De Jager et al. and Lunnon et al. present the results of the first two large-scale, epigenome-wide association studies (EWAS) in Alzheimer’s disease with results replicated in independent cohorts.4,5 Their results showcase one of the latest tools for dissecting the etiology of complex disorders and give new insights into the basis of Alzheimer’s disease.
Epigenetics refers to changes in gene regulation brought about through modifications to the DNA’s packaging proteins or the DNA molecules themselves without changing the underlying sequence.6 Such changes are thought to be a way in which the environment can influence genetics, bringing about potentially pathogenic alterations in the way genes are expressed. Many different types of epigenetic modifications exist; these can up- or downregulate gene expression. As there is no alteration of the actual DNA sequence, resequencing efforts looking for genetic variants influencing complex traits, many of which are still poorly understood, will overlook such changes entirely. It may be that this epigenetic interface between environmental and genetic risk factors holds the key to disentangling these etiologies, and this could bring huge leaps forward in risk prediction, diagnostics and therapeutics.7
Although epigenetic modifications, particularly methylation at CpG dinucleotides, which is often associated with downregulation or silencing of genes, have long been considered relevant to Alzheimer’s disease pathology, empirical evidence is hard to come by, and results of studies so far have often been conflicting and controversial.8 The methods for studying such modifications genome wide are still in their infancy, as are the accompanying analytical tools. Genetic methylation is thought to increase with age, complicating its study in diseases of old age such as late-onset Alzheimer’s disease9. A particular challenge in Alzheimer’s is that different populations of cells are known to have differences in epigenetic modifications; indeed, intra-individual differences between tissues far outstrip inter-individual differences in the same tissue type10. This is particularly problematic for Alzheimer’s because a typical brain biopsy will contain multiple cell types, including neurons, astrocytes and other glia cells all with different methylation patterns. In Alzheimer’s, where there is known to be a loss of neurons, establishing disease-related changes in methylation as opposed to differences in the proportion of cell types assayed is problematic.7
Whereas previous EWAS have struggled to control for such confounding factors, a likely reason for the lack of replication of early findings, the studies by De Jager et al.5 and Lunnun et al.4 provide compelling evidence for genuine, genome-wide significant associations between differentially methylated regions and Alzheimer’s disease, and suggest that the correlatory relationship could reflect a causative one, De Jager et al. adopted a three-stage study design, first screening cortical DNA for associations between methylation at 415,848 discrete CpG dinucleotides across the genome and neural plaque load, a key neuropathological feature of Alzheimer’s disease, in a large cohort. They found 71 discrete CpGs corresponding to 60 differentially methylated regions, including two known Alzheimer’s-associated loci identified by GWAS, ABCA7 and BIN1. Although the number of neurons sampled was not significantly different between cases and controls, the researchers included a variable to control for different proportions of cells present, combatting one of the potential sources of false positive findings mentioned above. To further strengthen the evidence, they then tested an independent replication cohort for association between the 71 significant CpGs from stage 1 and BRAAK staging, a post-mortem measure of Alzheimer’s neuropathological burden. Despite the considerably smaller sample size in this stage, they were able to replicate 12 of the CpGs (11 differentially methylated regions, as two CpGs fell close together near the RHBDF2 gene). Many of the loci that were not significant in this replication showed trends toward significance (including ABCA7 and BIN1), with similar effect sizes to those in the first cohort, suggesting that with more samples these would also validate. Finally, the authors validated the findings using mRNA from brain tissue by measuring expression levels of genes near significant CpGs in the replication stage. Out of 22 nearby genes, 9 showed differences in mRNA levels, linking a meaningful biological effect to the differential methylation observed.
Lunnon et al., meanwhile, adopted a similar strategy in their first stage, seeking associations between CpG methylation and BRAAK staging, this time in four different brain regions, three known to be affected by Alzheimer’s disease (enthorinal cortex, superior temporal gyrus and prefrontal cortex) and one largely left unaffected (cerebellum). Of the top ten differentially methylated CpGs in the enthorinal cortex, two fell within 91 bp of each other, close to the gene ANK1, which had also been identified by De Jager et al., providing further replication for this finding. These researchers too included a correction for estimated cellular heterogeneity in the samples used. The association of both these CpGs was confirmed in the other two Alzheimer’s-affected brain regions, but not in the cerebellum or pre-mortem whole blood samples. In a further two independent replication cohorts, the two CpGs showed a significant increase in methylation at the sites associated with BRAAK staging.
Together, these studies provide the first replicable and robust associations of differential methylation and Alzheimer’s pathology. It is likely that many more of the associations will replicate given sufficient sample sizes. However, despite the compelling evidence provided by each study’s robust and innovative designs, there remain limitations. Both groups used Illumina’s HumanMethylation450 beadset as the main quantification method for methylation. While this is the leading product for such studies on the market, it is far from perfect. The array only targets a small proportion of the CpG sites in the human genome (~2%), and while efforts have been made to capture the ones most relevant to disease investigations, current knowledge limits the effectiveness of the approach; the targeted CpG islands and promoter regions may not transpire to be the most significant players in this epigenetic regulation.10 Furthermore, the array cannot distinguish between DNA methylation and DNA hydroxymethylation, which have been reported to have opposing effects on gene regulation,11 and the analysis does not consider the many other forms of epigenetic modification that may affect disease phenotypes. Another intrinsic problem of the EWAS is that it is not clear what multiple-test correction should be used. In GWAS for common variants, the significant threshold has been set to 5.00 × 10−8, based on the number of independent single nucleotide polymorphisms. It is clear that EWAS significant threshold levels should account for multiple testing. Both Lunnon et al.4 and De Jager et al.5 used a Bonferroni correction based on the number of sites analyzed (~415,848; which yields a P < 1.20 × 10−7). However, DNA methylation levels at nearby (within 1–2 kb) CpG sites are correlated, which suggest that a Bonferroni correction is likely to be conservative. Conversely, not all CpG sites are tested in the most common methylation arrays; therefore, it is not clear how many independent CpG sites should be corrected for in a EWAS and what significance threshold should be adopted.
It also remains unclear whether the differences in methylation seen in Alzheimer’s are the cause of neurodegeneration or an early consequence of neuropathalogical changes. While the groups were able to demonstrate that the dysregulation of methylation is more pronounced in areas of the brain hit earlier by Alzheimer’s4,5, and that the changes can be seen in correlation with Alzheimer’s pathology in cognitively normal, presymptomatic individuals5 this does not establish causality. It remains to be seen how many of the other identified differentially methylated positions will replicate over time and hence whether the methodological and analytical practices in place are sufficient to address the numerous potential confounding influences.
In terms of the biology of Alzheimer’s disease, the genes identified largely fit with our prior knowledge of the condition. De Jager et al. were able to demonstrate that many of their differentially expressed mRNAs linked to identified CpGs connect with known Alzheimer’s disease susceptibility networks derived from protein:protein interactions, genes identified by GWAS and rare variant studies. Indeed, two of De Jager’s initial significant hits resided close to ABCA7 and BIN1, genes that were implicated in Alzheimer’s disease by GWAS.12,13 The association of the CpGs at these genes was shown to be independent of the GWAS variants, indicating that risk loci may harbor different types of genomic variation that can independently affect disease risk. Whether this will be the case for other loci in Alzheimer’s, and indeed in other complex disorders, remains to be established.
Notably, four new loci with differential methylation—RHBDF2, RPL13, C10orf54–CDH23 and ANK1—were independently identified in both studies, suggesting that these signals represent a real association with Alzheimer’s disease risk (Figure 1). RHDBF2 was found to be part of the same network of interacting proteins as the Alzheimer’s risk gene PTK2B, suggesting a potential role in microglia and macrophage activity. The same CpG site close to RPL13 was also differentially methylated on both studies. Networks analyses suggest that RPL13 interacts with PTK2B and APP, and it has been previously reported to be differentially expressed in Alzheimer’s diseases brains as compared to controls.14 Similarly, the same CpG site close to CDH23 and C10orf54 was found by two groups. CDH23 but not C10orf54 was found to be differentially expressed, suggesting that CDH23 drove the initial association. However, the roles of CDH23, RPL12 and RHDBF2 in Alzheimer’s disease risk are not yet clear. ANK1, while not previously linked to Alzheimer’s, is a known susceptibility gene for type 2 diabetes, the parallels between which and Alzheimer’s are receiving increasing attention.15 Lunnon et al. speculated that the brain-expressed ANK1 protein could be linked to Alzheimer’s pathology via its roles in compartmentalization of the plasma membrane and showed that the methylation differences observed were correlated with isoform-specific expression of the gene in the brain.
Figure 1. Loci showing consistent methylation changes in both studies.
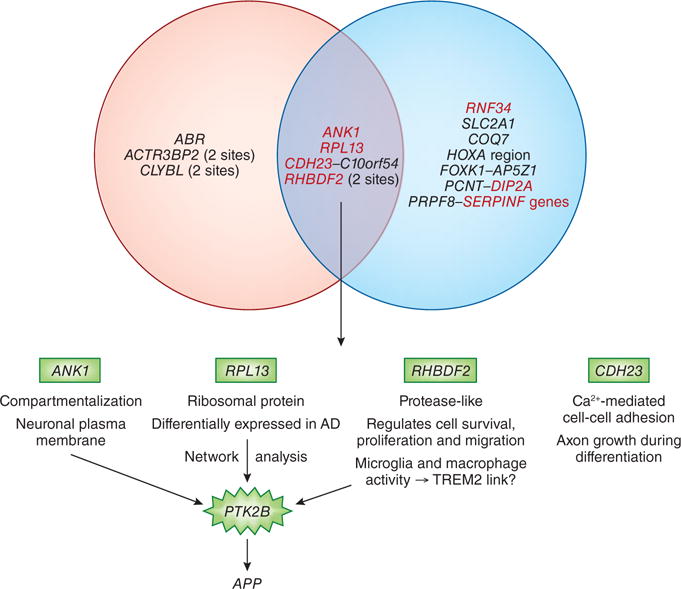
Four CpG sites close to ANK1, RHBDF2, RPL13, C10orf54–CDH23 were associated with Alzheimer’s disease pathology. Three of them (ANK1, RPL13 and RHBDF2) are biologically linked to PTK2B, a known AD gene. The fourth gene (CDH23) is involved in neuronal differentiation.
It is also worthy of note that De Jager et al., estimated the explanatory power of the significant CpGs from the first stage of their study and found that the differentially methylated CpGs together had a significantly higher explanatory power of neural plaque load than did all the known risk genes (APOE, and those identified by GWAS) for Alzheimer’s disease combined (13.9% for APOE and GWAS genes, 28.7% for the CpGs cumulatively). This suggests that epigenetic dysregulation of genes may have stronger effects on risk for Alzheimer’s and other complex diseases than variation within the genes themselves.
In summary, the two studies presented here provide the first robust and overlapping associations between Alzheimer’s disease and differential methylation patterns in brain regions known to be affected by the condition. The two studies provide independent replication for a number of findings both internally and collaboratively. Although causation has not been established, it is clear that these changes occur early in the disease process—indeed, before the onset of symptoms. The genes identified fit with our ever-increasing knowledge of Alzheimer’s etiology, suggesting potential avenues for future diagnostics, prediction and treatment strategies for the disorder. This is some of the most compelling evidence in epigenetic epidemiology so far, highlighting the importance of epigenetics to complex disorders. These well-designed studies pave the way for future investigations in the field, and while there are still limitations that need to be addressed, clearly we have the ability to detect disease associated methylation differences that are correlated with biologically relevant effects on gene expression in complex disorders.
Contributor Information
Jenny Lord, Department of Psychiatry, Washington University School of Medicine, St. Louis, Missouri, USA.
Carlos Cruchaga, Department of Psychiatry, Washington University School of Medicine, St. Louis, Missouri, USA; Hope Center Program on Protein Aggregation and Neurodegeneration, Washington University School of Medicine, St. Louis, Missouri, USA.
References
- 1.Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer’s disease. Alzheimers & Dementia. 2007;3:186–191. doi: 10.1016/j.jalz.2007.04.381. [DOI] [PubMed] [Google Scholar]
- 2.Lambert JC, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013 doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ertekin-Taner N. Genetics of Alzheimer disease in the pre- and post-GWAS era. Alzheimers Res Ther. 2010;2:3. doi: 10.1186/alzrt26. doi:alzrt26 [pii] 10.1186/alzrt26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lunnon K, Smith R, Volta M, Troakes C, Mill J. Cross-tissue methylomic profiling in Alzheimer’s disease implicates a role for cortex-specific deregulation of ANK1 in neuropathology. Nature Neuroscience. 2014 doi: 10.1038/nn.3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeJager P, et al. Alzheimer’s disease pathology is associated with early alterations in brain DNA 2 methylation at ANK1, BIN1 and other loci. Nature Neuroscience. 2014 doi: 10.1038/nn.3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mazzio EA, Soliman KF. Basic concepts of epigenetics: impact of environmental signals on gene expression. Epigenetics : official journal of the DNA Methylation Society. 2012;7:119–130. doi: 10.4161/epi.7.2.18764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mill J, Heijmans BT. From promises to practical strategies in epigenetic epidemiology. Nat Rev Genet. 2013;14:585–594. doi: 10.1038/nrg3405. [DOI] [PubMed] [Google Scholar]
- 8.Bennett DA, et al. Epigenomics of Alzheimer’s disease. Translational research : the journal of laboratory and clinical medicine. 2014 doi: 10.1016/j.trsl.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hernandez DG, et al. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum Mol Genet. 2011;20:1164–1172. doi: 10.1093/hmg/ddq561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davies MN, et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012;13:R43. doi: 10.1186/gb-2012-13-6-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coppieters N, et al. Global changes in DNA methylation and hydroxymethylation in Alzheimer’s disease human brain. Neurobiol Aging. 2014;35:1334–1344. doi: 10.1016/j.neurobiolaging.2013.11.031. [DOI] [PubMed] [Google Scholar]
- 12.Hollingworth P, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nature Genetics. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seshadri S, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. Jama. 2010;303:1832–1840. doi: 10.1001/jama.2010.574. doi:303/18/1832 [pii] 10.1001/jama.2010574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kong W, et al. Independent component analysis of Alzheimer’s DNA microarray gene expression data. Mol Neurodegener. 2009;4:5. doi: 10.1186/1750-1326-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jayaraman A, Pike CJ. Alzheimer’s disease and type 2 diabetes: multiple mechanisms contribute to interactions. Current diabetes reports. 2014;14:476. doi: 10.1007/s11892-014-0476-2. [DOI] [PMC free article] [PubMed] [Google Scholar]