

Abstract
The turnstile motion of two neighboring threonines sets up a dynamic side chain interplay that can accommodate both polar and apolar ligands in a small molecule allosteric protein binding site. A computational model based on SAR data and both X-ray and cryo-EM structures of the AAA ATPase p97 was used to analyze the effects of paired threonines at the inhibitor site. Specifically, the Thr side chain hydroxyl groups form a hydrogen bonding network that readily accommodates small, highly polar ligand substituents. Conversely, diametric rotation of the χ1 torsion by 150–180° orients the side chain β-methyl groups into the binding cleft, creating a hydrophobic pocket that can accommodate small, apolar substituents. This motif was found to be critical for rationalizing the affinities of a structurally focused set of inhibitors of p97 covering a >2,000-fold variation in potencies, with a preference for either small-highly polar or small-apolar groups. The threonine turnstile motif was further validated by a PDB search that identified analogous binding modes in ligand interactions in PKB, as well as by an analysis of NMR structures demonstrating additional gear-like interactions between adjacent Thr pairs. Combined, these data suggest that the threonine turnstile motif may be a general feature of interest in protein binding pockets.
Keywords: Protein homeostasis, ERAD, UPR, ubiquitin pathway inhibitor, autophagy, threonine turnstile, allosteric binding site, dynamics, docking
Graphical Abstract
The turnstile motion of two neighboring threonines accommodates both polar and apolar ligands in an allosteric binding site.
Introduction
The hexameric AAA+ (ATPases Associated with diverse cellular Activities) p97 serves as a major regulator of protein homeostasis. p97 assists in chromatin- and mitochondria-associated degradation, endoplasmic reticulum (ER)-associated degradation (ERAD), unfolded protein response (UPR), proteasome degradation, Golgi reassembly, endosomal tracking, protein aggregate processing, and autophagy.1 Key to p97’s function is a remarkable conformational mobility in its 6×3 multi-domain subunits. Binding to nucleotides during the ATPase cycle repositions these domains around the ring-shaped core, transducing phosphate hydrolysis into mechanical force exerted on protein substrates.2 The specific elements that control the coordinated ratchet motion within the D1, D2, and N domains in the 550 kDa hexameric complex are still under investigation.3 p97 has been recognized as a potential target for treating cancer as well as neurodegeneration,4,5 thus triggering drug discovery efforts in both industry and academia.6,7,8, 9,10
Results and discussion
In a recent series of indole-based allosteric inhibitors of p97, we were unable to completely rationalize the structure-activity relationship (SAR) of CF3-bioisosteres at the indole C-5 position (R1) of our lead structure (Figure 1).11 Specifically, the analysis of substituent effects revealed a remarkably divergent electronic trend since both the 5-nitro and the 5-methyl substituent showed superior binding affinities versus the corresponding pentafluorosulfanyl-, methoxy-, trifluoromethoxy-, and trifluoromethyl-substituted analogs.11 Even after the subsequent elucidation of a cryo-EM structure with a potent (IC50 = 55 nM) 5-fluoro-substituted inhibitor of the same structural series bound to p97 (PDB entry 5FTJ),3 the inhibitor binding data could not be fully explained by their steric, polar, or electronic properties. Therefore, in the present study, we computationally refined a model of this p97 allosteric site based on the SAR of additional 5-substituted indoles and select arene and heteroarene analogs, which together span a range of more than 3 orders of magnitude in a biochemical assay (Figure 1 and Table 1). Examination of these small molecule probes revealed an unprecedented turnstile motion of two neighboring but discontinuous threonine (Thr) residues, setting up a dynamic side chain - heterocyclic ligand interaction that can accommodate both polar and apolar moieties in the allosteric binding pocket.
Fig. 1.

Structures of inhibitors used for comparing substituent effects at the 5-position of the indole or analogous heterocyclic scaffolds. R1 see Table 1; R2 = H or Me. Additional indole replacements include 5- and 7-azaindoles, naphthalene, benzimidazole, and benzofuran.
Table 1.
Biochemical Activities of p97 Inhibitorsa
| Entry | Compound ID | R1 Indole Substitution (R2) | ADPGlo IC50 [μM] | ADPGlo Std. Dev. |
|---|---|---|---|---|
| 1 | UPCDC30283 (1) | 5-CN (H) | 0.044 | 0.045 |
| 2 | UPCDC30287 (2) | 5-NO2 (H) | 0.047 | 0.040 |
| 3 | UPCDC30346 (3) | 5-F (Me) | 0.050 | 0.044 |
| 4 | UPCDC30245 (4) | 5-F (H) | 0.055 | 0.087 |
| 5 | UPCDC30361 (5) | 5-CN (Me) | 0.087 | 0.047 |
| 6 | UPCDC30310 (6) | 5-CONH2 (H) | 0.10 | 0.032 |
| 7 | UPCDC30256 (7) | 5-OH (H) | 0.12 | 0.073 |
| 8 | UPCDC30341 (8) | 5-CO2Me (H) | 0.13 | 0.076 |
| 9 | UPCDC30083 (9) | 5-H (H) | 0.16 | 0.10 |
| 10 | UPCDC30317 (10) | 5-Cl (H) | 0.19 | 0.14 |
| 11 | UPCDC30288 (11) | 5-N3 (H) | 0.23 | 0.18 |
| 12 | UPCDC30318 (12) | 5-CH3 (H) | 0.24 | 0.11 |
| 13 | UPCDC30206 (13) | benzo[α]carbazole | 0.39 | 0.069 |
| 14 | UPCDC30367 (14) | 5-CONHMe (H) | 0.45 | 0.12 |
| 15 | UPCDC30238 (15) | 5-OCH3 (H) | 0.71 | 0.22 |
| 16 | UPCDC30257 (16) | 5-OCF3 (H) | 3.8 | 0.8 |
| 17 | UPCDC30345 (17) | 5-azaindole (H) | 4.0 | 1.7 |
| 18 | UPCDC30297 (18) | 5-CF3 (H) | 4.67 | 2.0 |
| 19 | UPCDC30381 (19) | 5-F,7-azaindole (H) | 4.74 | 1.3 |
| 20 | UPCDC30222 (20) | naphthalene (H) | 5.2 | 1.1 |
| 21 | UPCDC30221 (21) | benzofuran (H) | 20.1 | 4.60 |
| 22 | UPCDC30277 (22) | 5-SF5 (H) | 21.5 | 0.40 |
| 23 | UPCDC30368 (23) | 5-CONMe2 (H) | 31.4 | 1.60 |
| 24 | UPCDC30250 (24) | benzimidazole (H) | 38.5 | 6.60 |
| 25 | UPCDC30201 (25) | N-Me indole (H) | >50 | - |
Central to developing a protein binding model capable of rationalizing our p97 SAR was that all inhibitors considered for the refinement contained an identical tetramine side chain, thereby creating a structurally homologous set of compounds differing only in the substitution pattern in the indole region. Semiempirical calculations initially suggested a correlation between the assay data (Table 1) and surface area calculations of the 5-substituted indoles.13 Specifically, van der Waals surface areas of the phenyl indole moiety of compounds 1, 2, 4, 6–12, 14–16, 18, 22 and 23 formed a least-squares linear fit with the log IC50 data (R=0.72, df = 14, p <0.01). Upon selecting only examples with nonpolar 5-substitutions (9–12, 15, 16, 18 and 22), the linear fit slightly improved (R=0.91, df = 6, p <0.01). Conversely, the correlation for analogs with polar substitutions (1, 2, 4, 6–8, 14, and 23) dropped to R=0.81 (df = 6, p >0.01) (Figures S1–S3). Among several possible correlations of physicochemical parameters with activity, only the van der Waals surface areas provided R > 0.5. This suggested that for inhibitors with nonpolar indole 5-substituents, the main factor determining potent activity was the size of the substituent, with a preference for smaller groups. Polar substituents formed an independent cohort, and hydrogen bonding capabilities influenced the biological activity in addition to substituent size. Taken together, these data indicated that the protein binding subsite at the indole 5-position was sterically constrained and amphiphilic, accommodating hydrogen bond donors and acceptors, as well as small hydrophobic groups.
The cryo-EM co-structure of 5-F derivative 4 (UPCDC30245) bound to p97 showed how this specific inhibitor molecule engaged in multiple favorable contacts in the protein’s allosteric binding site, including a multipolar bond between the fluorine and a backbone carbonyl carbon (see Figure 2D in ref 3). However, the docking of other derivatives possessing polar and apolar 5-position substituents in the structure failed to provide a rationale for why substituents of divergent polarity should be equally active (Table 1). For example, the structure provided no explanation why a 5-NO2 substitution would result in greater affinity than a 5-amide substituent, or why a hydrophobic 5-CH3 substituent would be significantly more potent than a 5-OCF3 derivative (Figure S4). Therefore, to gain a better understanding of potential conformational variabilities in amino acid backbone and side chain arrangements surrounding this allosteric binding site in the highly dynamic protein, we examined all available hexameric p97 structures in the protein data bank (PDB), including both cryo-EM and X-ray structures (PDB entries 3CF1, 3CF2, 3CF3, 5C1A, 5C18, 5C19, 5FTJ, 5FTK, 5FTL, 5FTM, and 5FTN).3,14,15 Interestingly, we found that two separate loops (composed of residues 507–511 and 611–616), which border the site where the inhibitor indole 5-position is localized in the cryo-EM co-structure, displayed a high degree of conformational flexibility. In these different structures, the distances between the β-carbons of the amphiphilic sec-propanol side chains of residues Thr 509 and Thr 613 ranged from approx. 3.2 Å to 7.8 Å (Figure 2). Furthermore, we observed that at least one of the side chains of the Thr pair could form van der Waals contacts with substituents at the 5-position of the indole in some conformations of the canonical ensemble. Given the combined steric restriction and divergent SAR of the 5-position substituents, along with the conformational flexibility of these loops, we hypothesized that, upon inhibitor binding and concomitant hydrophobic collapse with the binding site loops, the amphiphilic Thr side chain pair could form a subsite in agreement with the sterically restricted, amphiphilic SAR of the indole series.
Fig. 2.

Variable orientations of loops 507–511 and 611–616 near the allosteric binding site of UPCDC30245 (4). Shown is the distance range between the side chain β-carbons of loop residues Thr 509 and Thr 613 observed in ADP-bound X-ray crystal structure 3CF3 (cyan) and ADP-bound cryo-EM structures 5FTJ (magenta, with inhibitor complex) and 5FTK (green), and ATPγS-bound cryo-EM structures 5FTL (yellow) and 5FTM (orange).
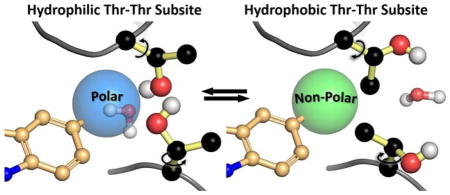
Using cryo-EM co-structure 5FTJ as a starting point, we employed constrained molecular mechanics and dynamics protocols to anneal the bis-Thr side chain pair to form a subsite for the 5-polar substitutions (e.g., the 5-OH of UPCDC30256, 7). This subsite is characterized by a water-coordinated, stabilized hydrogen bonding network with the polar inhibitors (Figure 3a). When the Thr side chain χ1 torsions are rotated in a turnstile mode (150–180°), a subsite formed by the Thr side chain methyl groups accommodates nonpolar 5-substitutions (e.g., 5-methyl indole inhibitor UPCDC30318, 12, Figure 3b). Moreover, χ rotations of the Thr pair are facilitated by the ATP binding cavity of the D2 domain, which is located on the opposite side of the loops bordering the allosteric site. Specifically, when the γ-hydroxyl groups rotate out of the binding pocket, the water molecule shown in Figure 3a is extruded into the ATP binding cavity (as shown in Figure 3b), and is accommodated, along with the γ-hydroxyls, by bulk solvent in the cavity (Figure 3b). Conversely, in the polar conformation, the γ-methyls of the Thr pair, which have very low solvent accessible surface areas, are oriented toward the ATP binding cavity, and the bulk solvent in the cavity is not significantly affected.
Fig. 3.

Upon inhibitor binding, the loops containing the Thr 509-Thr 613 pair collapse to form an amphiphilic binding site at the indole 5-position. (a) Polar subsite: the Thr γ-hydroxyls engage in a network of hydrogen bonds with polar inhibitor substituents. (b) Hydrophobic subsite: rotation of the Thr χ1 torsions by 150–180° results in γmethyl group interactions with small hydrophobic moieties on the indole 5-position. In this conformation, the bridging water molecule has been displaced from the binding pocket.
These alternate binding modes incorporate key inhibitor contacts observed in the cryo-EM co-structure (Figure 4), including the critical hydrogen bond between the backbone amide carbonyl of Val 493 and the inhibitor indole NH, π-stacking between the Phe 618 side chain phenyl and the inhibitor indole, favorable hydrophobic collapse between the inhibitor phenyl-indole and the side chains of residues Pro 510, Ala 537, Pro 571, and Lys 614 (i.e., the residue’s side chain methylenes), a favorable dipole-dipole interaction between the Cys 535 thiol and the inhibitor piperidine N, and a hydrogen bond between the carboxylate of Glu 534 and the piperidine-4-amino substituent. In total, these interactions result in a tightly bound phenyl indole scaffold that facilitates inhibitor tetramine side chain interactions with residues of helices 16 and 17 (Figure 4). Specifically, piperazine N1 is predicted to hydrogen bond with the side chain amide of Gln 494, while the substituent’s N4 hydrogen bonds with the carboxylate of Glu 534, and the isopropyl moiety desolvates via favorable hydrophobic collapse with the side chains of Trp 476 and Ile 531 (Figure 4).
Fig. 4.

Schematic of polar (top) and apolar (or hydrophobic) (bottom) inhibitor binding modes exemplified by 5-hydroxyl analog UPCDC30256 (7, top) and 5-methyl analog UPCDC30318 (12, bottom). In both binding modes, the yellow shaded region represents the amphiphilic bis-Thr subsite. Blue dashes indicate hydrogen bonds/dipole-dipole interactions, green dashes indicate hydrophobic contacts, and green lines depict the hydrophobic and steric continuity of the binding site. Pink shading indicates a critical hydrogen bond for optimal inhibitor activity, while green circles indicate π-stacking.
Molecular docking studies employing hydropathic scoring (Table S2 and Figure S5) 16, 17 with all of the inhibitors listed in Table 1 were used to further refine the allosteric site on p97. Several key SAR aspects were investigated. Binding modes for high affinity (IC50 <150 nM) polar 5-substitutions (entries 1–8; 5-CN, 5-NO2, 5-F, 5-CN, 5-CONH2, 5-OH, and 5-CO2Me) were distinguished by favorable hydrogen bonding networks similar to those shown in Figures 3a and 5a. For these derivatives, the polar 5-moieties had good complementary fits for the bis-Thr polar subsite that were characterized by either the formation of direct hydrogen bonds with a Thr side chain hydroxyl group, or via water mediation, or both. Similarly, good complementarity was achieved when docking potent (IC50 <500 nM) 5-substituted apolar moieties (entries 9–13; 5-H, 5-Cl, 5-N3, 5-CH3, and benzo[α]carbazole). These binding modes were characterized by favorable hydrophobic interactions with the side chain methyl groups of the bis-Thr, as depicted in Figure 3b.
Fig. 5.

Comparison of the binding modes of the 5-substituted amide series comprised of 6, 14, and 23, and for contrast, ester derivative 8. Yellow dashes = hydrogen bonds; red dashes = unfavorable contacts. (a) The potent binding affinity of UPCDC30310 (6, IC50 = 100 nM) is typical of the polar binding mode, with the two Thr side chain hydroxyl groups and the indole 5-position amide nitrogen coordinating a water molecule in a hydrogen bonding network. Additionally, the 5-position amide nitrogen forms a hydrogen bond with the backbone carbonyl carbon of Ser 511. (b) For UPCDC30367 (14), substitution of a single methyl group on the amide nitrogen necessitates the engagement of the Thr 613 methyl group to accommodate the hydrophobic addition, while the polar amide carbonyl group hydrogen bonds with the Thr 509 hydroxyl group. In this suboptimal binding mode (IC50 = 450 nM), the water-mediated hydrogen bond observed for 6 is lost, and the amide methyl moiety assumes an unfavorable, partially solvent-exposed orientation (red sphere). (c) Dimethylation of the amine in the C(5)-amide (UPCDC30368, 23) is sterically prohibited (red sphere), resulting in a near loss of all potency. (d) For comparison with methyl amide 14, the binding model of the more potent methyl ester UPCDC30341 (8, IC50 = 130 nM) indicates that this substituent’s methyl group, unlike the conformationally restricted N-methyl of 14, can avoid solvent exposure by orienting toward the bottom edge of the binding pocket, while the methoxy oxygen engages in a weak hydrogen bond with the coordinated water molecule.
Disruption of the indole NH to Val 493 hydrogen bond (Figure 4) by N-methylation in UPCDC30201 (25) or by replacement of the indole with a naphthalene isostere in UPCDC30222 (20) ablates or greatly diminishes activity, respectively. Introduction of a nitrogen atom at the indole 3-position in benzimidazole UPCDC30250 (24) reduces binding affinity by >2 orders of magnitude. The lone pair of the imine N of the benzimidazole presents an incompatible negative electrostatic potential near the hydrophobic α–, β–, and aromatic carbons of Phe 618. Furthermore, if this N atom is complexed to a water molecule (via hydrogen bonding) during the binding event, an even greater hydrophobic-polar incompatibility ensues.
The 5-azaindole nitrogen of UPCDC30345 (17) has an electrostatic potential similar to the benzimidazole N, and is also suboptimal for this binding pocket. Its negative electrostatic potential or coordination with water places it too close to the hydrophobic side chain components of Pro 510 and the hydrocarbon component of Lys 614. Similarly, other derivatives, such as 5-fluoro-7-azaindole UPCDC30381 (19) introduce a destabilizing polar mismatch between the 7-position N and the backbone carbonyl group of Leu 492.
Finally, some polar 5-substituents (14 and 23 (Figure 5b and c, respectively)) and hydrophobic substituents (15, 16, 18, and 22) are either sterically less favorable (e.g., 14) or simply too large to fit into either binding mode. For example, the increased steric bulk of dimethylamide 23 results in the loss of a water-mediated hydrogen bond, solvent exposure, and unfavorable steric contacts (i.e., versus potent amide analog 6) (Figure 5a–c). Interestingly, unlike methylamide 14 (IC50 = 450 nM), which is sterically less favorable than amide 6, methyl ester 8 is significantly more potent (IC50 = 130 nM), as the more flexible methoxy group can rotate slightly to avoid solvent exposure. This orientation also directs the methoxy oxygen of the ester to form a hydrogen bond with a water molecule (Figure 5d), thereby rationalizing its similar potency to C(5)-amide 6.
Based on the importance of the bis-Thr pair in rationalizing the amphiphilic SAR of our allosteric inhibitors of p97, we conducted a search of the PDB to examine the generality of this motif at small molecule ligand binding sites. All PDB entries with ligands having >200 residues in the longest chain and a resolution ≤2.5 Å were evaluated. Figure 6a illustrates the search criteria used for this study. Among the 19,881 entries meeting the criteria, 840 (4.2%) were found to possess discontinuous (>5 residues apart in loop sequence) Thr pairs for which the distance between the residues’ β-carbons was ≤5 Å (similar to our model), and where the Thr β-carbons were in the vicinity of a ligand (Figure 6a). Interestingly, we found an active site with a bis-Thr pair occurring on separate loops in protein kinase B (ATK1) (see Supporting Information and Figure S6 for additional details). Similar to our model, the Thr pair could also adopt conformations to accommodate either a polar or a hydrophobic contact in the same binding pose. In the co-crystal structure (PDB entry 2UVY) depicted in Figure 6b,18 the bis-Thr pair in the polar binding mode is positioned for hydrogen bonding directly to the 9H-nitrogen of the inhibitor, and when the 9H-nitrogen is replaced with a carbon (PDB entry 2X39, Figure 6c),19 the bis-Thr χ1 torsion changes to form an apolar conformation, with the Thr β-methyl groups closing off the hydrophobic pocket. The specific compounds from the indicated PDB entries possess different side chain substituents on the respective positions of their purine and pyrrolo-pyrimidine cores, and thus a comparison of their inhibitory activity is not straightforward. However, similar potencies are observed for analogous pairs with identical substitutions (see core pairs 14 and 15, 17 and 18, 20 and 21, 22 and 23, and 24 and 25 in Table 2 in reference 20).
Figure 6.

(a) Schematic of the criteria used to search the PDB for bis-Thr pairs in the vicinity of ligands. (b) Protein kinase B inhibitor with a 9H-purine component near the bis-Thr pair in the polar conformation. In this structure, the purine N9 engages in a hydrogen bond (yellow dash) with one of the Thr side chain hydroxyls. (c) Protein kinase B inhibitor with a 7H-pyrrolo[2,3-d]pyrimidine moiety near the bis-Thr pair in the apolar conformation. In this inhibitor, N9 is replaced with a carbon, which consequently engages in favorable hydrophobic contacts (orange dash) with a Thr methyl group.
To further broaden the scope of the PDB analysis, the initial search criteria were relaxed to allow for the identification of bis-Thr pairs (within 7 Å of a ligand) occurring in any secondary structural feature (i.e., helices, sheets, and loops). From this search we found additional X-ray co-crystal examples of ligands interacting with at least one Thr of a bis-Thr pair. Examples include heat shock protein 90 (HSP90), β-lactamases (CTX-M-9 and BEL-1), and AMPA glutamate receptor 2 (GluR2) (Figure S7). The HSP90 threonines both occur in β sheets, the β-lactamase threonines occur on a β sheet and a loop, and the GluR2 threonines occur on a β sheet and an α helix. While in these additional examples the bis-Thr pairs did not display the χ rotations observed in PKB inhibitor co-crystal structures, it is interesting that they support a consistently polar binding mode in the solid state (i.e., X-ray structures), as the interacting ligand moieties in these examples were always polar and interacted with the γ-hydroxyl of the Thr side chain. Consequently, we considered the possibility that the bis-Thr turnstile movement may not be readily observable in solid state structures. Therefore, we further expanded our search to examine solution NMR structures for paired Thr χ rotations, under the assumption that the cooperative motion between paired Thr residues’ side chains would be more readily detected in solution. Accordingly, 8,687 non-redundant NMR entries meeting the search criteria described above were identified. Of these, a subset of 271 structures contained Thr pairs where both side chain χ torsions rotated simultaneously by >50°. Although this subset did not contain ligands interacting with bis-Thr pairs, the finding that 271 structures did display full and simultaneous χ rotations occurring in Thr pairs signifies that any subsite created by these residues can be both polar and hydrophobic. It is noteworthy, yet not surprising, that such bis-Thr turnstile motion is more prominent in solution structures versus X-ray structures.
For example, Figure 7a shows the 15 conformers deposited with NMR PDB entry 2FIN. In Figures 7b and 7c, the Thr pair in the polar subsite conformation (conformer 12) and the hydrophobic subsite conformation (conformer 1) are shown, respectively. Movie S1, which was created directly from the 15 conformers deposited for the protein entry, clearly displays the residues simultaneously rotating in a turnstile manner to gear between polar and hydrophobic subsite orientations.
Fig. 7.

The bis-Thr pair in NMR structure, PDB entry, 2FIN. (a) The PDB entry contains 15 conformers, with a bis-Thr pair formed by residues Thr 78 and Thr 95. Over the 15 conformers of the protein, the two Thr residues display a range of χ torsion rotations. (b) An example of a polar bis-Thr conformation as seen in conformer 12 of the protein ensemble. In this example, the hydroxyl hydrogen atom of Thr 78 engages in a hydrogen bond (yellow dash) with the hydroxyl oxygen atom of Thr 95. (c) An example of a hydrophobic (or apolar) bis-Thr conformation as seen in conformer 1 of the protein ensemble. In this example, the methyl groups of Thr 78 and Thr 95 engage in a favorable hydrophobic contact (orange dash). Movie S1 shows the rotations observed for the 15 conformers in the PDB entry.
Conclusions
We utilized a series of focused small molecule probes and extensive structural information from the PDB to develop a unified computational model that is able to rationalize the divergent potency in a series of phenyl indole-based p97 inhibitors. The SAR of all 25 probe molecules can be comprehensively explained by an allosteric binding site model, with the dynamic turnstile nature of two amphiphilic Thr side chains providing a compelling rationalization for the observation that inhibitors possessing either polar or hydrophobic substituents at the same position are simultaneously accommodated in the binding site. Furthermore, the results from our PDB search identified a similar amphiphilic bis-Thr turnstile in ligand complexes of protein kinase B, suggesting this previously unrecognized feature may frequently occur in small molecule protein binding sites. In support of this hypothesis, an analysis of NMR/solution structures provided further evidence for the concept that side chains of Thr pairs are able to interact in a dynamic, gear-like manner in protein structures. We can speculate that the function of such Thr pairs is to position water molecules in protein allosteric sites, as well as other locations that are either functionally important, and/or structurally significant. For example, we suggest that, in the absence of an inhibitor, the function of the bis-Thr pair in the p97 allosteric site identified in the cryo-EM co-structure3 is to shuttle water molecules out of the structurally dynamic interface between the D1 and D2 domains. Future studies will investigate the functional role of this bis-Thr pair in p97 protein dynamics.
Experimental section
Materials and Methods
Synthetic summary schemes, computational methods and any associated references and additional discussion as well as a summary SAR table are available in the online version in the Supplementary Information of this paper.
Synthesis
General Synthetic Methods
All non-aqueous reactions were carried out under a nitrogen atmosphere in oven- or flame-dried glassware unless otherwise noted. Anhydrous tetrahydrofuran and diethyl ether were distilled from sodium benzophenone ketyl; anhydrous dichloromethane and toluene were distilled from CaH2; alternatively, the same solvents were obtained from a solvent purification system using alumina columns. All other solvents and reagents were used as obtained from commercial sources without further purification unless noted. Reactions were monitored via TLC using 250 μm pre-coated silica gel 60 F254 plates, which were visualized with 254 nm and/or 365 nm UV light and by staining with KMnO4 (1.5 g KMnO4, 10 g K2CO3, and 1.25 mL 10% NaOH in 200 mL water), cerium molybdate (0.5 g Ce(NH4)2(NO3)6, 12 g (NH4)6Mo7O24•4H2O, and 28 mL conc. H2SO4 in 235 mL water), or vanillin (6 g vanillin and 1.5 mL conc. H2SO4 in 100 mL EtOH). Flash chromatography was performed with SiliCycle silica gel 60 (230–400 mesh) or with ISCO MPLC. Microwave reactions were performed using a Biotage Initiator in glass microwave vials (cap sealed) with continuous magnetic stirring and an external surface temperature sensor. 1H and 31C NMR spectra were recorded on Bruker Avance 300, 400, or 500 MHz spectrometers, using the residual solvent as an internal standard. 19F NMR spectra were obtained using a proton-decoupled pulse sequence without internal standard. IR spectra were obtained on a Smiths IdentifyIR or PerkinElmer Spectrum 100. HRMS data were obtained on a Thermo Scientific Exactive HRMS coupled to a Thermo Scientific Accela HPLC system using a 2.1 × 50 mm 3.5 μm Waters XTerra C18 column eluting with MeCN/H2O containing 0.1% formic acid. Purity of compounds was assessed using the same HPLC system with either the PDA or an Agilent 385 ELSD. All final screening samples passed QC based on >95% purity by LC/MS/ELSD analysis.
Experimental Procedures
2-(3-Bromophenyl)-1H-indole-5-carbonitrile (26)
A solution of 3-bromo acetophenone (4.6 g, 23 mmol), 4-aminobenzonitrile (2.5 g, 21 mmol), and TsOH•H2O (36 mg, 0.20 mmol) in toluene (100 mL) was heated overnight under Dean-Stark conditions. The reaction mixture was cooled to room temperature, concentrated, and purified by chromatography on SiO2 (0 to 100% EtOAc/hexanes) to provide (E)-4-((1-(3-bromophenyl)ethylidene)amino)-benzonitrile (3.6 g, 12 mmol, 57%) as a yellowish oil that was used without further purification.
A solution of (E)-4-((1-(3-bromophenyl)ethylidene)amino)benzonitrile (3.0 g, 10 mmol), Pd(OAc)2 (225 mg, 1.0 mmol), and Cu(OAc)2 (5.58 g, 30 mmol) in DMSO (100 mL) was heated at 40 °C for 24 h, cooled to room temperature, diluted with EtOAc (150 mL), and filtered through Celite. The filtrate was washed with sat. NH4Cl and brine. The organic layer was dried (MgSO4), filtered, concentrated, and purified by chromatography on SiO2 (20% EtOAc/hexanes) followed by trituration with EtOAc and hexanes to give 2-(3-bromophenyl)-1H-indole-5-carbonitrile (26, 1.56 g, 5.25 mmol, 52%) as a yellow solid: IR (ATR) 3301, 2216, 1599, 1584, 1560, 1512, 1457, 1441, 1437, 1340, 1322, 1247, 1170, 876, 825, 800, 792, 774, 673 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.22 (s, 1 H), 8.13 (br s, 1 H), 8.09 (br s, 1 H), 7.91 (d, J = 7.6 Hz, 1 H), 7.56 (d, J = 8.4 Hz, 2 H), 7.48-7.43 (m, 2 H), 7.16 (s, 1 H); 31C NMR (100 MHz, DMSO-d6) δ 138.9, 138.5, 133.5, 131.1, 130.8, 128.1, 127.7, 125.8, 124.7, 124.4, 122.5, 120.5, 112.6, 101.7, 100.5; HRMS (ESI+) m/z calcd for C15H10BrN2 297.0022 (M+H), found 297.0022.
2-(3-(4-((2-(4-Isopropylpiperazin-1-yl)ethyl)amino)piperidin-1-yl)phenyl)-1H-indole-5-carbonitrile (1, UPCDC30283)
A solution of 2-(3-bromophenyl)-1H-indole-5-carbonitrile (26, 0.15 g, 0.50 mmol), LiHMDS (0.20 g, 1.2 mmol), Pd2(dba)3 (9.2 mg, 0.010 mmol), and CyJohnPhos (14.0 mg, 0.040 mmol) in anhydrous THF was treated with tert-butyl (2-(4-isopropylpiperazin-1-yl)ethyl)(piperidin-4-yl)carbamate3,11 (A, 0.213 g, 0.600 mmol). The reaction mixture was heated at 75 °C overnight, cooled to room temperature, diluted with sat. NaHCO3, and extracted with CH2Cl2 (3×). The combined organic layers were washed with brine, dried (Na2SO4), filtered, concentrated, and purified by chromatography on SiO2 (2% MeOH/CH2Cl2 with 1% TEA) followed by filtration through basic Al2O3 (CH2Cl2) to provide tert-butyl (1-(3-(5-cyano-1H-indol-2-yl)phenyl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)carbamate (0.16 g, 0.28 mmol, 56%) as a yellow foamy solid: IR (ATR) 2965, 2932, 2809, 2216, 1685, 1653, 1599, 1448, 1411, 1362, 1320, 1245, 1172, 1146, 1010, 971, 898, 805, 775 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.60 (br s, 1 H), 7.92 (s, 1 H), 7.44 (d, J = 8.4 Hz, 1 H), 7.34 (dd, J = 8.4, 1.6 Hz, 1 H), 7.29 (t, J = 8.0 Hz, 1 H), 7.20 (br s, 1 H), 7.15 (d, J = 7.6 Hz, 1 H), 6.88 (br d, J = 7.2 Hz, 1 H), 6.81 (s, 1 H), 4.05 (br s, 1 H), 3.74 (br s, 2 H), 3.22 (br s, 2 H), 2.66-2.44 (m, 13 H), 1.85-1.64 (m, 4 H), 1.48 (s, 9 H), 1.03 (d, J = 6.4 Hz, 6 H); 31C NMR (100 MHz, CDCl3) δ 155.6, 151.9, 141.2, 138.7, 132.4, 129.9, 129.1, 125.9, 124.9, 121.0, 116.91, 116.85, 113.8, 111.9, 103.0, 100.0, 80.1, 54.6, 53.90, 53.88, 49.5, 48.7, 30.14, 30.14, 28.6, 18.6; HRMS (ESI+) m/z calcd for C34H47O2N6 571.3755 (M+H), found 571.3759.
A solution of TFA (0.53 mL, 7.0 mmol) and triethylsilane (0.11 mL, 0.70 mmol) in CH2Cl2 (1 mL) was added to a solution of tert-butyl (1-(3-(5-cyano-1H-indol-2-yl)phenyl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)carbamate (40 mg, 0.07 mmol) in CH2Cl2 (0.5 mL). After 1 h, the reaction mixture was concentrated, diluted with sat. NaHCO3, and extracted with EtOAc (3×). The combined organic layer was washed with brine, dried (Na2SO4), filtered, concentrated, and purified by chromatography on SiO2 (8 to 10% MeOH/CH2Cl2 with 1% TEA) followed by filtration through basic Al2O3 (0 to 10% MeOH/CH2Cl2) to provide 2-(3-(4-((2-(4-isopropylpiperazin-1-yl)ethyl)amino)piperidin-1-yl)phenyl)-1H-indole-5-carbonitrile 1 (UPCDC30283, 21 mg, 0.045 mmol, 65%) as a yellow oil: IR (ATR) 3334, 2936, 2930, 2872, 2809, 2214, 1599, 1577, 1461, 1381, 1361, 1241, 1176, 1144, 1124, 982, 859, 803, 775, 734 cm−1; 1H NMR (500 MHz, acetone-d6) δ 11.28 (br s, 1 H), 8.00 (s, 1 H), 7.56 (d, J = 8.5 Hz, 1 H), 7.47 (br s, 1 H), 7.40 (dd, J = 8.3, 1.3 Hz, 1 H), 7.32-7.28 (m, 2 H), 7.02 (s, 1 H), 6.97 (d, J = 7.5 Hz, 1 H), 3.78-3.74 (m, 2 H), 2.90-2.84 (m, 2 H), 2.72 (t, J = 6.3 Hz, 2 H), 2.66-2.55 (m, 4 H), 2.46-2.40 (m, 8 H), 1.98-1.95 (m, 2 H), 1.48 (qd, J = 13.5, 3.5 Hz, 2 H), 0.97 (d, J = 7.0 Hz, 6 H); 31C NMR (125 MHz, acetone-d6) δ 153.2, 142.4, 139.8, 133.1, 130.5, 130.0, 126.2, 125.1, 121.2, 116.9, 116.8, 113.8, 113.1, 103.5, 100.2, 59.0, 55.6, 54.9, 54.6, 49.4, 48.6, 44.4, 33.2, 18.8; HRMS (ESI+) m/z calcd for C29H38N6 471.3231 (M+H), found 471.3231.
2-(5-Bromo-2-methylphenyl)-5-fluoro-1H-indole (27)
A solution of 1-(5-bromo-2-methylphenyl)ethanone21 (5.00 g, 23.5 mmol), 4-fluoroaniline (2.77 g, 24.6 mmol), and TsOH•H2O (91 mg, 0.47 mmol) in toluene (150 mL) was heated for 40 h under Dean-Stark conditions. The reaction mixture was cooled to room temperature, concentrated, and purified by chromatography on SiO2 (100% hexanes followed by 5% Et2O/hexanes with 1% TEA) to provide (E)-1-(5-bromo-2-methylphenyl)-N-(4-fluorophenyl)ethan-1-imine (7.03 g, 23.0 mmol, 98%) as a yellow oil that was used without further purification.
A solution of (E)-1-(5-bromo-2-methylphenyl)-N-(4-fluorophenyl)ethan-1-imine (4.03 g, 13.2 mmol), Pd(OAc)2 (0.296 g, 1.32 mmol), and Cu(OAc)2 (7.32 g, 39.5 mmol) in DMSO (130 mL) was heated at 40 °C for 45 h, cooled to room temperature, diluted with EtOAc (500 mL), and filtered through Celite. The filtrate was washed with water (3×) and brine. The organic layer was dried (Na2SO4), filtered, concentrated, and purified by chromatography on SiO2 (10% Et2O/hexanes) followed by trituration with Et2O and hexanes to give 2-(5-bromo-2-methylphenyl)-5-fluoro-1H-indole (27, 2.01 g, 6.61 mmol, 50%) as a white powder: IR (ATR) 3430, 2993, 2980, 1769, 1758, 1586, 1480, 1372, 1245, 1241, 1055 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 11.49 (s, 1 H), 7.73 (d, J = 2.1 Hz, 1 H), 7.47 (dd, J = 8.1, 2.1 Hz, 1 H), 7.39 (dd, J = 8.9, 4.7 Hz, 1 H), 7.34-7.30 (m, 2 H), 6.97 (td, J = 9.2, 2.4 Hz, 1 H), 6.66 (d, J = 1.5 Hz, 1 H), 2.44 (s, 3 H); 31C NMR (100 MHz, DMSO-d6) δ 157.1 (d, JCF = 231.0 Hz), 137.4, 134.9, 134.3, 133.2, 133.1, 130.9, 130.3, 128.4 (d, JCF = 10.0 Hz), 118.8, 112.3 (d, JCF = 10.0 Hz), 109.9 (d, JCF = 25.0 Hz), 104.6 (d, JCF = 23.0 Hz), 102.8 (d, JCF = 4.0 Hz), 20.6; 19F NMR (376 MHz, DMSO-d6) δ -124.6; HRMS (ESI+) m/z calcd for C15H12BrFN 304.0132 (M+H), found 304.0131.
1-(3-(5-Fluoro-1H-indol-2-yl)-4-methylphenyl)-N-(2-(4-isopropylpiperazin-1-yl)ethyl)piperidin-4-amine (3, UPCDC30346)
A solution of 2-(5-bromo-2-methylphenyl)-5-fluoro-1H-indole (27, 0.100 g, 0.409 mmol), tert-butyl (2-(4-isopropylpiperazin-1-yl)ethyl)(piperidin-4-yl)carbamate (A, 0.160 g, 0.450 mmol), Pd2(dba)3 (8 mg, 0.008 mmol), and CyJohnPhos (12 mg, 0.033 mmol) in anhydrous THF (0.5 mL) in a microwave vial was degassed by bubbling argon for 20–30 min. The reaction mixture was charged with LiHMDS (0.180 g, 1.02 mmol) and the vial was sealed and heated at 80 °C overnight. The reaction mixture was cooled to room temperature, diluted with water and extracted with EtOAc. The organic layer was washed with brine, dried (Na2SO4), filtered, concentrated, and purified by chromatography on SiO2 (5 to 15% MeOH/CH2Cl2) to provide tert-butyl (1-(3-(5-fluoro-1H-indol-2-yl)-4-methylphenyl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)carbamate (0.116 g, 0.201 mmol, 49%): 1H NMR (300 MHz, CDCl3) δ 8.23 (s, 1 H), 7.34-7.28 (m, 2 H), 7.18 (d, J = 8.4 Hz, 1 H), 7.01 (d, J = 2.1 Hz, 1 H), 6.97-6.86 (m, 2 H), 6.54 (d, J = 0.9 Hz, 1 H), 4.11 (br s, 1 H), 3.72 (d, J = 12.0 Hz, 2 H), 3.24 (br s, 2 H), 2.81-2.48 (m, 13 H), 2.38 (s, 3 H), 1.78 (br s, 4 H), 1.47 (s, 9 H), 1.11 (br s, 6 H); HRMS (ESI+) m/z calcd for C34H49FN5O2 578.3845 (M+H), found 578.3864.
To a solution of tert-butyl (1-(3-(5-fluoro-1H-indol-2-yl)-4-methylphenyl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)carbamate (0.100 g, 0.173 mmol) in CH2Cl2 (2 mL) at 0 °C was added trifluoroacetic acid (0.64 mL, 8.6 mmol). The reaction mixture was allowed to warm to room temperature and stirred for 3 h, treated with sat. NaHCO3, and extracted with EtOAc (3×). The combined organic layers were washed with brine, dried (MgSO4), filtered, concentrated, and purified by chromatography on SiO2 (2 to 5% MeOH/CH2Cl2 with 1% TEA) followed by filtration through basic Al2O3 (0 to 5% MeOH/CH2Cl2) to provide 1-(3-(5-fluoro-1H-indol-2-yl)-4-methylphenyl)-N-(2-(4-isopropylpiperazin-1-yl)ethyl)piperidin-4-amine 3 (UPCDC30346, 49 mg, 0.10 mmol, 60%) as an off-white solid: IR (ATR) 3157, 2930, 2807, 1607, 1491, 1448, 1379, 1178, 1123, 1107, 982, 848, 785, 760 cm−1; 1H NMR (400 MHz, CD3OD) δ 7.33 (dd, J = 8.8, 4.4 Hz, 1 H), 7.19 (dd, J = 10.0, 2.4 Hz, 1 H), 7.16 (d, J = 8.4 Hz, 1 H), 7.11 (d, J = 2.4, 1 H), 6.88 (dd, J = 8.4, 2.8 Hz, 1 H), 6.86 (td, J = 9.2, 2.4 Hz, 1 H), 6.48 (br s, 1 H), 3.69 (app d, J = 12.6 Hz, 2 H), 2.78-2.74 (m, 4 H), 2.65-2.50 (m, 12 H), 2.38 (s, 3 H), 2.00 (app d, J = 12.4 Hz, 2 H), 1.53 (qd, J = 11.9, 3.5 Hz, 2 H), 1.07 (d, J = 6.4 Hz, 6 H); 31C NMR (100 MHz, CD3OD) δ 159.2 (d, JCF = 230.3 Hz), 151.0, 141.5, 134.6, 134.4, 132.6, 130.5 (d, JCF = 10.3 Hz), 128.6, 118.9, 118.0, 112.6 (d, JCF = 9.8 Hz), 110.3 (d, JCF = 26.3 Hz), 105.3 (d, JCF = 23.3 Hz), 102.9 (d, JCF = 4.9 Hz), 58.4, 56.2, 55.9, 54.1, 50.5, 49.6, 43.8, 32.8, 20.4, 18.7; HRMS (ESI+) m/z calcd for C29H41FN5 478.3341 (M+H), found 478.3340.
2-(5-Bromo-2-methylphenyl)-1H-indole-5-carbonitrile (28)
Prepared by the same 2 step procedure as for the compound 27 using 1-(5-bromo-2-methylphenyl)ethanone18 (5.00 g, 23.5 mmol, 1.0 equiv) and 4-aminobenzonitrile (2.97 g, 24.6 mmol, 1.05 equiv). The crude residue was purified by chromatography on SiO2 (10 to 20% EtOAc/hexanes) followed by trituration with EtOAc and hexanes to give 2-(5-bromo-2-methylphenyl)-1H-indole-5-carbonitrile (28, 1.37 g, 4.39 mmol, step 1, 85% and step 2, 34%) as a yellow solid: IR (ATR) 3308, 2992, 2982, 2220, 1769, 1758, 1471, 1372, 1241, 1094, 1049 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.00 (s, 1 H), 8.10 (s, 1 H), 7.75 (d, J = 2.0 Hz, 1 H), 7.56 (d, J = 8.4 Hz, 1 H), 7.52-7.46 (m, 2 H), 7.32 (d, J = 8.4 Hz, 1 H), 6.81 (d, J = 1.2 Hz, 1 H), 2.43 (s, 3 H); 31C NMR (100 MHz, DMSO-d6) δ 138.13, 138.07, 135.2, 133.5, 133.2, 131.1, 130.7, 127.9, 125.8, 124.4, 120.7, 118.8, 112.5, 103.3, 101.4, 20.5; HRMS (ESI+) m/z calcd for C16H12BrN2 311.0178 (M+H), found 311.0176.
2-(5-(4-((2-(4-Isopropylpiperazin-1-yl)ethyl)amino)piperidin-1-yl)-2-methylphenyl)-1H-indole-5-carbonitrile (5, UPCDC30361)
A solution of 2-(5-bromo-2-methylphenyl)-1H-indole-5-carbonitrile (28, 0.100 g, 0.397 mmol), tert-butyl (2-(4-isopropylpiperazin-1-yl)ethyl)(piperidin-4-yl)carbamate (A, 0.123 g, 0.437 mmol), Pd2(dba)3 (6 mg, 0.006 mmol), and CyJohnPhos (9.0 mg, 0.025 mmol) in anhydrous THF (0.5 mL) in a microwave vial was degassed by bubbling argon for 20–30 min. The reaction mixture was charged with LiHMDS (0.180 g, 1.02 mmol) and the vial was sealed and heated at 80 °C for 12 h. The reaction mixture was cooled to room temperature, diluted with water and extracted with EtOAc (3×). The combined organic layers were washed with brine, dried (Na2SO4), filtered, concentrated, and purified by chromatography on SiO2 (0 to 10% MeOH/CH2Cl2) to provide tert-butyl (1-(3-(5-cyano-1H-indol-2-yl)-4-methylphenyl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)carbamate (0.093 g, 0.16 mmol, 51%) as a brown solid: IR (ATR) 3301, 2960, 2932, 2890, 2218, 1685, 1659, 1605, 1499, 1465, 1363, 1318, 1299, 1172, 1144, 1010, 898, 803, 749, 728 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.83 (br s, 1 H), 7.97 (s, 1 H), 7.47 (d, J = 8.0 Hz, 1 H), 7.42 (d, J = 8.5 Hz, 1 H), 7.18 (d, J = 8.0 Hz, 1 H), 7.00 (br s, 1 H), 6.90 (d, J = 8.0 Hz, 1 H), 6.63 (br s, 1 H), 4.08 (br s, 1 H), 3.71 (app d, J = 10.0 Hz, 2 H), 3.23 (br s, 2 H), 2.77-2.48 (m, 13 H), 2.37 (s, 3 H), 1.80-1.75 (m, 4 H), 1.47 (s, 9 H), 1.08 (d, J = 4.5 Hz, 6 H); 31C NMR (125 MHz, CDCl3) δ 155.5, 149.7, 140.6, 137.9, 132.2, 132.0, 128.7, 127.3, 126.0, 124.9, 121.0, 117.5, 117.4, 111.8, 103.17, 103.13, 80.1, 58.4, 55.0, 53.6, 53.1, 50.0, 48.6, 30.2, 28.7, 20.0, 18.5; HRMS (ESI+) m/z calcd for C35H49O2N6 585.3912 (M+H), found 585.3911.
To a solution of tert-butyl (1-(3-(5-cyano-1H-indol-2-yl)-4-methylphenyl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)carbamate (0.070 g, 0.112 mmol) in CH2Cl2 (2 mL) at 0 °C was added trifluoroacetic acid (0.36 mL, 4.8 mmol). The reaction mixture was allowed to warm to room temperature and stirred for 2 h, treated with sat. NaHCO3, and extracted with CH2Cl2 (3×). The combined organic layers were washed with brine, dried (MgSO4), filtered, concentrated, and purified by chromatography on SiO2 (0 to 10% MeOH/CH2Cl2 with 1% TEA) followed by filtration through basic Al2O3 (0 to 5% MeOH/CH2Cl2) to provide 2-(5-(4-((2-(4-isopropylpiperazin-1-yl)ethyl)amino)piperidin-1-yl)-2-methylphenyl)-1H-indole-5-carbonitrile 5 (UPCDC30361, 38 mg, 0.078 mmol, 66%) as a light yellow solid: IR (ATR) 3293, 3120, 2956, 2924, 2811, 2705, 2214, 1603, 1560, 1500, 1459, 1379, 1333, 1320, 1273, 1232, 1176, 1113, 982, 803 cm−1; 1H NMR (400 MHz, CD3OD) δ 7.98 (d, J = 0.8 Hz, 1 H), 7.52 (d, J = 8.8 Hz, 1 H), 7.38 (dd, J = 8.4, 1.6 Hz, 1 H), 7.18 (d, J = 8.8 Hz, 1 H), 7.11 (d, J = 2.8 Hz, 1 H), 6.94 (dd, J = 8.4, 2.8 Hz, 1 H), 6.64 (d, J = 0.8 Hz, 1 H), 3.71-3.68 (m, 2 H), 2.78-2.71 (m, 4 H), 2.66-2.50 (m, 12 H), 2.37 (s, 3 H), 2.00 (d, J = 10.8 Hz, 2 H), 1.52 (qd, J = 11.9, 3.6 Hz, 2 H), 1.07 (d, J = 6.4 Hz, 6 H); 31C NMR (100 MHz, CD3OD) δ 151.1, 142.4, 139.8, 133.5, 132.7, 130.1, 128.6, 126.6, 125.1, 121.9, 118.7, 118.4, 113.0, 103.4, 102.9, 58.2, 56.3, 56.0, 54.1, 50.3, 49.6, 43.7, 32.8, 20.3, 18.7; HRMS (ESI+) m/z calcd for C30H41N6 485.3387 (M+H), found 485.3388.
2-(3-(4-((2-(4-Isopropylpiperazin-1-yl)ethyl)amino)piperidin-1-yl)phenyl)-1H-indole-5-carboxamide (6, UPCDC30310)
To a solution of tert-butyl (1-(3-(5-cyano-1H-indol-2-yl)phenyl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)carbamate (62 mg, 0.11 mmol) in DMSO was added K2CO3 (15 mg, 0.11 mmol) in water (0.2 mL) at 0 °C then a solution of 30% w/w hydrogen peroxide (1.2 mL) was added dropwise to the mixture. The resulting mixture was stirred at 5 °C for 5 min then the ice bath was removed. After another 5 min, water (10 mL) was added to the reaction mixture and extracted with EtOAc (3×10mL). The combined organic layers were washed with brine, dried (MgSO4), filtered, concentrated, and purified by chromatography on SiO2 to provide tert-butyl (1-(3-(5-carbamoyl-1H-indol-2-yl)phenyl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)carbamate (35 mg, 0.059 mmol, 55%) as a yellow oil: IR (ATR) 3208, 2973, 2962, 1668, 1653, 1586, 1474, 1448, 1431, 1363, 1333, 1245, 1146, 1103, 1049, 1020, 757, 710, 689 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.55 (br s, 1 H), 8.15 (s, 1 H), 7.66 (d, J = 8.5 Hz, 1 H), 7.44 (d, J = 8.5 Hz, 1 H), 7.30 (t, J = 7.8 Hz, 2 H), 7.18 (d, J = 7.5 Hz, 1 H), 6.88 (d, J = 8.0 Hz, 1 H), 6.83 (s, 1 H), 6.36 (br s, 1 H), 5.74 (br s, 1 H), 4.11 (br s, 1 H), 3.81 (app d, J = 8.1 Hz, 2 H), 3.18 (br s, 2 H), 2.80-2.64 (m, 11 H), 2.45-2.44 (m, 2 H), 1.78-1.73 (m, 4 H), 1.47 (s, 9 H), 1.10 (d, J = 6.0 Hz, 6 H); HRMS (ESI+) m/z calcd for C34H48O3N6 589.3861 (M+H), found 589.3859.
A solution of TFA (0.45 mL, 5.9 mmol) and triethylsilane (0.10 mL, 0.59 mmol) in CH2Cl2 (1 mL) was added to a solution of tert-butyl (1-(3-(5-carbamoyl-1H-indol-2-yl)phenyl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)carbamate (35 mg, 0.059 mmol) in CH2Cl2 (0.5 mL). After 1 h, the reaction mixture was concentrated, diluted with sat. NaHCO3, and extracted with EtOAc (3×). The combined organic layer was washed with brine, dried (Na2SO4), filtered, concentrated, and purified by chromatography on SiO2 (8 to 10% MeOH/CH2Cl2 with 1% TEA) followed by filtration through basic Al2O3 (0 to 10% MeOH/CH2Cl2) to provide 2-(3-(4-((2-(4-isopropylpiperazin-1-yl)ethyl)amino)piperidin-1-yl)phenyl)-1H-indole-5-carboxamide 6 (UPCDC30310, 19 mg, 0.039 mmol, 65%) as a yellow foam: IR (ATR) 3302, 2943, 2829, 1637, 1603, 1540, 1474, 1435, 1384, 1338, 1250, 1176, 1146, 1020, 757, 738, 723, 710 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.71 (br s, 1 H), 8.10 (s, 1 H), 7.62 (d, J = 8.4 Hz, 1 H), 7.36 (d, J = 8.4 Hz, 1 H), 7.24 (d, J = 8.4 Hz, 2 H), 7.12 (d, J = 7.2 Hz, 1 H), 6.87 (d, J = 8.0 Hz, 1 H), 6.78 (s, 1 H), 6.39 (br s, 1 H), 5.98 (br s, 1 H), 3.69 (d, J = 12.0 Hz, 2 H), 2.80-2.70 (m, 4 H), 2.62-2.45 (m, 12 H), 1.94 (app d, J = 10.8 Hz, 2 H), 1.52-1.44 (m, 2 H), 1.02 (d, J = 6.4 Hz, 6 H); 31C NMR (100 MHz, CDCl3) δ 171.1, 152.2, 140.6, 139.1, 132.9, 129.9, 129.0, 125.2, 121.6, 120.8, 116.8, 116.4, 113.8, 111.2, 100.5, 58.1, 55.3, 54.6, 53.7, 48.9, 48.7, 43.5, 32.6, 18.9; HRMS (ESI+) m/z calcd for C29H41ON6 489.3336 (M+H), found 489.3335.
2-(3-(4-((2-(4-Isopropylpiperazin-1-yl)ethyl)amino)piperidin-1-yl)phenyl)-1H-indol-5-ol (7, UPCDC30256)
To a solution of N-(2-(4-isopropylpiperazin-1-yl)ethyl)-1-(3-(5-methoxy-1H-indol-2-yl)phenyl)piperidin-4-amine 151 (UPCDC30238, 60.0 mg, 0.124 mmol) in dry CH2Cl2 (15 mL) under nitrogen was added dropwise boron tribromide (1.0 M solution in CH2Cl2, 0.49 mL, 0.49 mmol) at room temperature and stirred for 1 h. The reaction was quenched with MeOH, concentrated and purified by chromatography on SiO2 (8 to 20% MeOH/CH2Cl2 with 1% TEA) followed by filtration through basic Al2O3 (0 to 10% MeOH/CH2Cl2) to provide 2-(3-(4-((2-(4-isopropylpiperazin-1-yl)ethyl)amino)piperidin-1-yl)phenyl)-1H-indol-5-ol 7 (UPCDC30256, 55.0 mg, 0.119 mmol, 96%) as a colorless oil: IR (ATR) 3260, 2960, 2932, 2818, 1705, 1623, 1599, 1584, 1489, 1452, 1420, 1381, 1363, 1312, 1295, 1273, 1253, 1221, 1200, 1178, 1146, 1118, 971 cm−1; 1H NMR (500 MHz, acetone-d6) δ 10.49 (s, 1 H), 7.42 (s, 1 H), 7.26-7.21 (m, 3 H), 6.97 (d, J = 2.5 Hz, 1 H), 6.88-6.86 (m, 1 H), 6.71-6.69 (m, 2 H), 3.76-3.72 (m, 2 H), 2.86-2.81 (m, 2 H), 2.73 (t, J = 6.3 Hz, 2 H), 2.65-2.55 (m, 2 H), 2.48-2.41 (m, 10 H), 1.97-1.94 (m, 2 H), 1.48 (qd, J = 14.0, 3.5 Hz, 2 H), 0.98 (d, J = 6.5 Hz, 6 H); 31C NMR (125 MHz, acetone-d6) δ 153.1, 152.2, 140.0, 134.4, 133.0, 131.0, 130.2, 116.6, 115.9, 113.5, 112.8, 112.3, 105.0, 99.1, 58.9, 55.7, 54.9, 54.5, 49.4, 48.8, 44.2, 33.2, 30.6, 18.8; HRMS (ESI+) m/z calcd for C28H40ON5 462.3227 (M+H), found 462.3227.
Methyl 2-(3-bromophenyl)-1H-indole-5-carboxylate (29)
A solution of 3-bromo acetophenone (8.3 g, 41 mmol), 4-aminobenzoate (5.7 g, 37 mmol), and 4 Å molecular sieves (24 g) in toluene (180 mL) was refluxed for 28 h under Dean-Stark conditions. A second portion of 4 Å molecular sieves (16 g) was added and refluxed for an additional 95 h. The reaction mixture was filtered through a pad of Celite, concentrated and purified by chromatography on SiO2 (5 to 10% Et2O/hexanes with 2% TEA) to give methyl (E)-4-((1-(5-bromo-2-methylphenyl)ethylidene)amino)-benzoate (2.40 g, ca 20%) as a yellowish-green solid.
A solution of methyl (E)-4-((1-(5-bromo-2-methylphenyl)ethylidene)amino)-benzoate (2.34 g, 7.04 mmol), Pd(OAc)2 (0.158 g, 0.704 mmol), and Cu(OAc)2 (3.84 g, 21.1 mmol) in DMSO (70 mL) was heated at 40 °C for 40 h, cooled to room temperature, diluted with water (280 mL), filtered through Celite, and extracted with EtOAc (4x). The combined organic layer was washed with brine, dried (Na2SO4), filtered, concentrated, and purified by chromatography on SiO2 (6 to 10% Et2O/hexanes) followed by trituration with EtOAc and hexanes to give methyl 2-(3-bromophenyl)-1H-indole-5-carboxylate (29, 1.6 g, 4.8 mmol, 69%) as a white solid: IR (ATR) 2993, 2911, 2227, 1433, 1404, 1308, 1042, 951, 925, 725, 697, 667 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.02 (s, 1 H), 8.25 (d, J = 1.2 Hz, 1 H), 8.11 (t, J = 1.8 Hz, 1 H), 7.91-7.88 (m, 1 H), 7.76 (dd, J = 8.6, 1.7 Hz, 1 H), 7.54 (ddd, J = 8.0, 1.9, 0.9 Hz, 1 H), 7.46 (dt, J = 15.0, 7.8 Hz, 2 H), 7.17 (s, 1 H), 3.85 (s, 3 H); 31C NMR (100 MHz, DMSO-d6) δ 167.1, 139.8, 137.7, 133.9, 131.1, 130.5, 128.0, 127.5, 124.2, 123.0, 122.8, 122.5, 121.1, 111.4, 101.1, 51.7; HRMS (ESI+) m/z calcd for C16H13BrO2N 330.0124 (M+H), found 330.0123.
Methyl 2-(3-(4-((2-(4-isopropylpiperazin-1-yl)ethyl)amino)piperidin-1-yl)phenyl)-1H-indole-5-carboxylate (8, UPCDC30341)
A solution of methyl 2-(3-bromophenyl)-1H-indole-5-carboxylate (29, 0.150 g, 0.445 mmol), tert-butyl (2-(4-isopropylpiperazin-1-yl)ethyl)(piperidin-4-yl)carbamate (A, 0.189 g, 0.534 mmol) and K3PO4 (0.146 g, 0.668 mmol) in deoxygenated dioxane (1 mL) in a 2–5 mL microwave vial was degassed by bubbling with argon for 20–30 min. The reaction mixture was charged with Pd2(dba)3 (8 mg, 0.009 mmol), and CyJohnPhos (13 mg, 0.036 mmol) and the vial was sealed and heated at 110 °C for 18 h. The reaction mixture was cooled to room temperature, diluted with sat. NaHCO3 and extracted with EtOAc (3×). The combined organic layer was washed with brine, dried (Na2SO4), filtered, concentrated, and purified by chromatography on SiO2 (0 to 15% MeOH/EtOAc) to provide methyl 2-(3-(4-((tert-butoxycarbonyl)(2-(4-isopropylpiperazin-1-yl)ethyl)amino)piperidin-1-yl)phenyl)-1H-indole-5-carboxylate (0.18 g, 0.30 mmol, 68%) as a brown solid: IR (ATR) 2958, 2813, 1707, 1685, 1601, 1577, 1446, 1435, 1308, 1247, 1165, 1144, 1124, 1089, 1008, 917, 768 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.87 (br s, 1 H), 8.38 (s, 1 H), 7.89 (dd, J = 8.6, 1.4 Hz, 1 H), 7.40 (d, J = 8.7 Hz, 1 H), 7.32 (t, J = 8.0 Hz, 1 H), 7.21 (s, 1 H), 7.14 (d, J = 7.5 Hz, 1 H), 6.91 (dd, J = 8.3, 1.4 Hz, 1 H), 6.86 (s, 1 H), 4.13 (br s, 1 H), 3.94 (s, 3 H), 3.81 (d, J = 12.3 Hz, 2 H), 3.25-3.24 (m, 2 H), 2.87-2.47 (m, 13 H), 1.85-1.77 (m, 4 H), 1.48 (s, 9 H), 1.06 (d, J = 6.6 Hz, 6 H); 31C NMR (75 MHz, CDCl3) δ 168.3, 155.5, 151.9, 140.1, 139.5, 133.0, 130.0, 129.0, 123.7, 123.6, 122.4, 116.8, 116.6, 113.7, 110.7, 101.0, 80.0, 54.7, 53.6, 52.0, 49.7, 48.5, 30.2, 28.7, 18.5; HRMS (ESI+) m/z calcd for C35H50O4N5 604.3857 (M+H), found 604.3856.
To a solution of methyl 2-(3-(4-((tert-butoxycarbonyl)(2-(4-isopropylpiperazin-1-yl)ethyl)amino)piperidin-1-yl)phenyl)-1H-indole-5-carboxylate (80 mg, 0.13 mmol) in CH2Cl2 (2 mL) at 0 °C was added TFA (0.40 mL, 5.3 mmol). The reaction mixture was allowed to warm to room temperature and stirred for 2 h, treated with sat. NaHCO3, and extracted with EtOAc (2x). The combined organic layer was washed with brine, dried (MgSO4), filtered, concentrated, and purified by chromatography on SiO2 (0 to 10% MeOH/CH2Cl2 with 1% TEA) followed by filtration through basic Al2O3 (0 to 5% MeOH/CH2Cl2) to provide methyl 2-(3-(4-((2-(4-isopropylpiperazin-1-yl)ethyl)amino)piperidin-1-yl)phenyl)-1H-indole-5-carboxylate 8 (UPCDC30341, 30 mg, 0.060 mmol, 45%): IR (ATR) 3327, 2958, 2930, 2809, 1707, 1691, 1599, 1577, 1545, 1433, 1379, 1344, 1310, 1249, 1169, 1122, 1088, 982, 768, 690 cm−1; 1H NMR (300 MHz, CD3OD) δ 8.29 (s, 1 H), 7.79 (dd, J = 8.4, 1.2 Hz, 1 H), 7.43 (app d, J = 9.0 Hz, 2 H), 7.32-7.26 (m, 2 H), 6.96-6.92 (m, 1 H), 6.90 (s, 1 H), 3.90 (s, 3 H), 3.79 (app d, J = 12.3 Hz, 2 H), 2.84-2.73 (m, 4 H), 2.66-2.49 (m, 12 H), 2.01 (app d, J = 11.7 Hz, 2 H), 1.53 (qd, J = 11.8, 3.1 Hz, 2 H), 1.07 (d, J = 6.6 Hz, 6 H); 31C NMR (75 MHz, CD3OD) δ 170.1, 153.4, 141.8, 141.5, 134.2, 130.7, 130.1, 124.1, 123.9, 122.4, 117.9, 117.5, 114.7, 111.8, 100.8, 58.4, 56.2, 55.9, 54.1, 52.3, 49.9, 43.8, 32.8, 18.7; HRMS (ESI+) m/z calcd for C30H41O2N5 504.3333 (M+H), found 504.3330.
8-(3-(1H-Indol-2-yl)phenyl)-1,4-dioxa-8-azaspiro[4.5]decane (30)
A suspension of 2-(3-bromophenyl)-1H-indole11 (0.40 g, 1.5 mmol), K3PO4 (0.48 g, 2.2 mmol), Pd2(dba)3 (32 mg, 0.034 mmol), CyJohnPhos (42 mg, 0.12 mmol) in dry, degassed dioxane (10 mL) was treated with 1,4-dioxa-8-azaspiro[4.5]decane (0.30 mL, 2.4 mmol). The flask was sealed and the reaction mixture was heated at 110 °C for 6 h under microwave irradiation. The reaction mixture was diluted with sat. NaHCO3 and extracted with EtOAc (3×). The combined organic layer was washed with brine, dried (Na2SO4), filtered, concentrated and purified by chromatography on SiO2 (30 to 40% EtOAc/hexanes) to provide 8-(3-(1H-indol-2-yl)phenyl)-1,4-dioxa-8-azaspiro[4.5]decane (30, 0.40 g, 1.2 mmol, 81%) as a pale yellow foam: Mp 142-143 °C; IR (ATR) 3348, 2957, 2927, 2886, 1600, 1484, 1354, 1219, 1096, 776, 746 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1 H), 7.64 (d, J = 7.6 Hz, 1 H), 7.40 (dd, J = 8.0, 0.4 Hz, 1 H), 7.32 (t, J = 8.0 Hz, 1 H), 7.27-7.26 (m, 1 H), 7.20 (td, J = 7.6, 1.2 Hz, 1 H), 7.16-7.12 (m, 2 H), 6.93 (dd, J = 8.4, 2.0 Hz, 1 H), 6.81 (app d, J = 1.2 Hz, 1 H), 4.02 (s, 4 H), 3.42 (t, J = 5.8 Hz, 4 H), 1.90 (t, J = 5.8 Hz, 4 H); 31C NMR (100 MHz, CDCl3) δ 151.5, 138.6, 136.8, 133.4, 129.9, 129.4, 122.3, 120.7, 120.3, 116.7, 116.2, 113.8, 111.0, 107.2, 100.0, 64.5, 48.0, 34.7; HRMS (ESI+) m/z calcd for C21H23O2N2 335.1754 (M+H), found 335.1758.
1-(3-(1H-Indol-2-yl)phenyl)-N-(2-(4-isopropylpiperazin-1-yl)ethyl)piperidin-4-amine (9, UPCDC30083)
A solution of 8-(3-(1H-indol-2-yl)phenyl)-1,4-dioxa-8-azaspiro[4.5]decane (30, 0.20 g, 0.60 mmol) in acetone (70 mL) and 3.5 M HCl (60 mL) was heated at 80 °C for 3.5 h. The reaction mixture was cooled to 0 °C, quenched with Na2CO3 (14 g) and extracted with EtOAc (3×). The combined organic layers were washed with brine, dried (Na2SO4), filtered, concentrated, and purified by chromatography on SiO2 (25 to 30% EtOAc/hexanes) to provide 1-(3-(1H-indol-2-yl)phenyl)piperidin-4-one (0.14 g, 0.48 mmol, 80%) as a pale yellow solid which is not stable under air and was used immediately: 1H NMR (300 MHz, CDCl3) δ 8.35 (s, 1 H), 7.63 (d, J = 7.8 Hz, 1 H), 7.42-7.35 (m, 2 H), 7.28-7.27 (m, 1 H), 7.23-7.10 (m, 3 H), 6.95 (app dd, J = 8.0, 2.3 Hz, 1 H), 6.82 (app d, J = 1.5 Hz, 1 H), 3.69 (t, J = 6.0 Hz, 4 H), 2.61 (t, J = 6.2 Hz, 4 H); HRMS (ESI+) m/z calcd for C19H19ON2 291.1492 (M+H), found 291.1489.
A solution of crude 1-(3-(1H-indol-2-yl)phenyl)piperidin-4-one (ca. 87 mg, 0.30 mmol) in 1,2-DCE (3 mL) was treated with 2-(4-isopropylpiperazin-1-yl)ethanamine22 (B, 0.077 g, 0.45 mmol) followed by Ti(OiPr)4 (0.10 mL, 0.32 mmol) and stirred at room temperature overnight. The reaction mixture was treated with NaBH(OAc)3 (130 mg, 0.60 mmol) in a single portion. After 2 h, the reaction mixture was diluted with sat. NaHCO3 and extracted with EtOAc (3×). The combined organic layers were washed with brine, dried (Na2SO4), filtered, concentrated and purified by chromatography on SiO2 (5 to 20% MeOH/CH2Cl2 with 1% TEA) followed by filtration through basic Al2O3 (0 to 7% MeOH/CH2Cl2) to provide 1-(3-(1H-indol-2-yl)phenyl)-N-(2-(4-isopropylpiperazin-1-yl)ethyl)piperidin-4-amine 9 (UPCDC0083, 95 mg, 0.21 mmol, 71% over 2-steps) as a pale yellow foam: IR (ATR) 3422, 3222, 2957, 2931, 2808, 1600, 1450, 1294, 1144, 776, 746 cm−1; 1H NMR (400 MHz, CD3OD) δ 7.52 (d, J = 8.0 Hz, 1 H), 7.40-7.37 (m, 2 H), 7.26-7.20 (m, 2 H), 7.11-7.07 (m, 1 H), 7.00 (app t, J = 7.4 Hz, 1 H), 6.82 (dt, J = 7.5, 1.9 Hz, 1 H), 6.77 (s, 1 H), 3.70-3.66 (m, 2 H), 2.66 (td, J = 12.2, 1.5 Hz, 2 H), 2.59-2.36 (m, 14 H), 1.87 (br d, J = 11.2 Hz, 2 H), 1.41 (qd, J = 11.8, 3.5 Hz, 2 H), 1.01 (d, J = 6.8 Hz, 6 H); 31C NMR (100 MHz, CD3OD) δ 153.3, 139.8, 138.7, 134.8, 130.6, 122.6, 121.2, 120.5, 117.8, 116.9, 114.5, 112.1, 99.7, 58.4, 56.0, 55.8, 54.0, 49.9, 49.5, 43.6, 32.9, 18.7; HRMS (ESI+) m/z calcd for C28H40N5 446.3278 (M+H), found 446.3277.
2-(3-Bromophenyl)-5-chloro-1H-indole (31)
A solution of 3-bromo acetophenone (4.0 g, 20 mmol), 4-chloroaniline (2.6 g, 20 mmol), and TsOH•H2O (35 mg, 0.20 mmol) in toluene (50 mL) was heated overnight under Dean-Stark conditions. The reaction mixture was cooled to room temperature, concentrated, and purified by chromatography on SiO2 (0 to 5% EtOAc/hexanes) to provide (E)-1-(3-bromophenyl)-N-(4-chlorophenyl)ethan-1-imine as a yellowish oil (4.6 g, 15 mmol, 74%) that was used without further purification.
A solution of (E)-1-(3-bromophenyl)-N-(4-chlorophenyl)ethan-1-imine (1.2 g, 3.9 mmol), Pd(OAc)2 (87 mg, 0.39 mmol), and Cu(OAc)2 (2.2 g, 12 mmol) in DMSO (100 mL) was heated at 40 °C for 24 h, cooled to room temperature, diluted with EtOAc (150 mL), and filtered through Celite. The filtrate was washed with sat. NH4Cl, and brine. The organic layer was dried (MgSO4), filtered, concentrated, and purified by chromatography on SiO2 (20% EtOAc/hexanes) followed by trituration with EtOAc and hexanes to give 2-(3-bromophenyl)-5-chloro-1H-indole (31, 0.76 g, 2.5 mmol, 63%) as a yellow solid: IR (ATR) 3431, 1683, 1564, 1452, 1439, 1305, 1281, 1249, 1057, 922, 865, 803, 772, 680 cm−1; 1H NMR (300 MHz, CD3OD) δ 7.94 (t, J = 1.7 Hz, 1 H), 7.72 (dt, J = 7.8, 1.4 Hz, 1 H), 7.50 (d, J = 1.8 Hz, 1 H), 7.43 (ddd, J = 8.0, 1.8, 1.1 Hz, 1 H), 7.35-7.29 (m, 2 H), 7.07 (dd, J = 8.7, 2.1 Hz, 1 H), 6.78 (s, 1 H); 31C NMR (75 MHz, CD3OD) δ 139.2, 137.3, 135.9, 131.7, 131.40, 131.38, 129.0, 126.3, 125.0, 124.0, 123.3, 120.6, 113.4, 100.4.
1-(3-(5-Chloro-1H-indol-2-yl)phenyl)-N-(2-(4-isopropylpiperazin-1-yl)ethyl)piperidin-4-amine (10, UPCDC30317)
A solution of 2-(3-bromophenyl)-5-chloro-1H-indole (31, 77 mg, 0.25 mmol), LiHMDS (0.10 g, 0.60 mmol), Pd2(dba)3 (4.6 mg, 0.005 mmol), and CyJohnPhos (7.0 mg, 0.020 mmol) in anhydrous THF was treated with tert-butyl (2-(4-isopropylpiperazin-1-yl)ethyl)(piperidin-4-yl)carbamate (A, 0.106 g, 0.300 mmol). The reaction mixture was heated at 75 °C overnight, cooled to room temperature, diluted with sat. NaHCO3, and extracted with CH2Cl2 (3×). The combined organic layer was washed with brine, dried (Na2SO4), concentrated, and purified by chromatography on SiO2 (2 to 10% MeOH/CH2Cl2) to give tert-butyl (1-(3-(5-chloro-1H-indol-2-yl)phenyl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)carbamate (68 mg, 0.12 mmol, 47%) as a foam: IR (ATR) 2962, 2956, 2949, 2932, 2926, 2807, 1685, 1653, 1599, 1575, 1463, 1446, 1411, 1381, 1363, 1329, 1303, 1269, 1245, 1172, 1144, 1103, 1059, 1048, 1010, 993, 982, 971, 915, 900, 861, 772, 755, 734, 718, 692 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.13 (br s, 1 H), 7.55 (d, J = 1.6 Hz, 1 H), 7.29-7.25 (m, 2 H), 7.17 (br s, 1 H), 7.13-7.08 (m, 2 H), 6.84 (br d, J = 7.2 Hz, 1 H), 6.69 (s, 1 H), 4.08 (br s, 1 H), 3.71-3.69 (m, 2 H), 3.21 (br s, 2 H), 2.69-2.44 (m, 13 H), 1.71 (s, 4 H), 1.49 (s, 9 H), 1.04 (d, J = 6.4 Hz, 6 H); 31C NMR (100 MHz, CDCl3) δ 155.6, 151.8, 140.1, 135.3, 133.0, 130.3, 129.8, 125.6, 122.3, 119.8, 118.2, 116.8, 116.5, 113.73, 113.72, 112.0, 99.3, 80.1, 58.4, 54.7, 53.8, 49.5, 48.6, 30.1, 28.6, 18.6; HRMS (ESI+) m/z calcd for C33H47ClO2N5 580.3413 (M+H), found 580.3411.
A solution of TFA (0.84 mL, 11.2 mmol) and triethylsilane (0.18 mL, 1.1 mmol) in CH2Cl2 (1 mL) was added to a solution of tert-butyl (2-(4-isopropylpiperazin-1-yl)ethyl)(1-(3-(5-(trifluoromethyl)-1H-indol-2-yl)phenyl)piperidin-4-yl)carbamate (65 mg, 0.11 mmol) in CH2Cl2 (0.5 mL). After 1 h, the reaction mixture was concentrated, diluted with sat. NaHCO3, and extracted with EtOAc (3×). The combined organic layer was washed with brine, dried (Na2SO4), filtered, concentrated, and purified by chromatography on SiO2 (8 to 10% MeOH/CH2Cl2 with 1% TEA) followed by filtration through basic Al2O3 (0 to 10% MeOH/CH2Cl2) to provide 1-(3-(5-chloro-1H-indol-2-yl)phenyl)-N-(2-(4-isopropylpiperazin-1-yl)ethyl)piperidin-4-amine 10 (UPCDC30317, 41 mg, 0.085 mmol, 76%) as a yellow foam: IR (ATR) 3181, 2960, 2932, 2926, 2814, 1599, 1577, 1461, 1448, 1381, 1359, 1344, 1310, 1294, 1273, 1217, 1176, 1146, 1118, 1059, 917, 861, 790, 775, 755, 738, 690 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.75 (s, 1 H), 7.56 (d, J = 2.0 Hz, 1 H), 7.29 (t, J = 8.0 Hz, 2 H), 7.19 (t, J = 2.0 Hz, 1 H), 7.12-7.07 (m, 2 H), 6.90 (dd, J = 8.4, 2.0 Hz, 1 H), 6.70 (d, J = 1.6 Hz, 1 H), 3.73-3.70 (m, 2 H), 2.85-2.75 (m, 4 H), 2.66-2.48 (m, 13 H), 2.01-1.98 (m, 2 H), 1.52 (qd, J = 11.6, 3.0 Hz, 2 H), 1.04 (d, J = 6.8 Hz, 6 H); 31C NMR (100 MHz, CDCl3) δ 152.1, 140.2, 135.2, 132.9, 130.4, 129.8, 125.7, 122.4, 119.9, 116.4, 116.3, 113.6, 111.9, 99.4, 58.1, 55.1, 54.6, 53.6, 48.8, 48.6, 43.5, 32.6, 18.8; HRMS (ESI+) m/z calcd for C28H39ClN5 480.2889 (M+H), found 480.2887.
tert-Butyl (1-(3-(5-amino-1H-indol-2-yl)phenyl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)carbamate (32)
A solution of tert-butyl (2-(4-isopropylpiperazin-1-yl)ethyl)(1-(3-(5-nitro-1H-indol-2-yl)phenyl)piperidin-4-yl)carbamate11 (0.102 g, 0.173 mmol) in MeOH (5 mL) was evacuated, flushed with argon (2x) and treated with 10% Pd/C (0.019 g, 0.17 mmol). The reaction mixture was evacuated and subjected to H2 (1 atm - balloon). After 2 h, the solution was filtered through a plug of Celite, rinsed with MeOH and concentrated to provide tert-butyl (1-(3-(5-amino-1H-indol-2-yl)phenyl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)carbamate (32, 0.070 g, 0.15 mmol, 72%) as an orange oil that was used without further purification: 1H NMR (400 MHz, CD3OD) δ 7.36 (br s, 1 H), 7.26-7.20 (m, 3 H), 6.94 (br s, 1 H), 6.89 (br d, J = 5.2 Hz, 1 H), 6.69 (br d, J = 8.4 Hz, 1 H), 6.61 (br s, 1 H), 4.09-3.81 (m, 3 H), 3.35-3.25 (m, 2 H), 2.85-2.50 (m, 13 H), 2.01-1.74 (m, 4 H), 1.47 (s, 9 H), 1.11 (br s, 6 H); HRMS (ESI+) m/z calcd for C33H49O2N6 561.3912 (M+H), found 561.3912.
tert-Butyl (1-(3-(5-azido-1H-indol-2-yl)phenyl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)carbamate (33)
Into a dried flask under nitrogen was added tert-butyl (1-(3-(5-amino-1H-indol-2-yl)phenyl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)carbamate (32, 0.085 g, 0.15 mmol) in AcOH (4 mL). The reaction mixture was cooled to 0 °C and protected from light (foil wrap) and treated with a solution of NaNO2 (0.012 g, 0.17 mmol) in H2O (0.4 mL). After 10 min, a solution of NaN3 (0.010 g, 0167 mmol) in H2O (0.4 mL) was added dropwise. After 45 min, the mixture was slowly poured into H2O (10 mL) and sat. Na2CO3 was added (8 mL) until neutral pH. The mixture was extracted with EtOAc (3×), washed with brine, dried (Na2SO4), filtered, and concentrated. The crude product was purified by chromatography on SiO2 (0 to 10 % MeOH/CH2Cl2) to provide tert-butyl (1-(3-(5-azido-1H-indol-2-yl)phenyl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)carbamate (33, 0.053 g, 0.090 mmol, 60%) as an orange foam: 1H NMR (400 MHz, acetone-d6) δ 10.92 (br s, 1 H), 7.47-7.43 (m, 2 H), 7.29-7.26 (m, 3 H), 6.95-6.92 (m, 1 H), 6.87 (br s, 1 H), 6.83 (dd, J = 8.6, 2.2 Hz, 1 H), 4.47 (br s, 1 H), 3.91 (br d, J = 11.6 Hz 2 H), 3.24 (br s, 2 H), 2.80 (t, J = 12.0 Hz, 2 H), 2.67 (dt, J = 13.1, 6.5 Hz, 1 H), 2.53 (br s, 8 H), 2.44 (t, J = 7.4 Hz, 3 H), 1.93 (br s, 2 H), 1.75 (br s, 2 H), 1.45 (s, 9 H), 1.00 (d, J = 6.4 Hz, 6 H); HRMS (ESI+) m/z calcd for C33H47O2N8 587.3816 (M+H), found 587.3815.
1-(3-(5-Azido-1H-indol-2-yl)phenyl)-N-(2-(4-isopropylpiperazin-1-yl)ethyl)piperidin-4-amine (11, UPCDC30288)
To a cooled solution of tert-butyl (1-(3-(5-azido-1H-indol-2-yl)phenyl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)carbamate (33, 0.050 g, 0.085 mmol) and lutidine (0.020 mL, 0.17 mmol) in dry CH2Cl2 (5 mL) was added dropwise a solution of TMSOTf (0.023 mL, 0.127 mmol) in CH2Cl2 (5 mL) at 0 °C. After 2 h, the reaction mixture was quenched with sat. NaHCO3 and extracted with EtOAc (3×). The combined organic layer was washed with water followed by brine, dried (Na2SO4), filtered, concentrated, and purified by chromatography on SiO2 (8 to 20% MeOH/CH2Cl2 with 1% TEA) followed by filtration through basic Al2O3 (10% MeOH/CH2Cl2) to provide 1-(3-(5-azido-1H-indol-2-yl)phenyl)-N-(2-(4-isopropylpiperazin-1-yl)ethyl)piperidin-4-amine 11 (UPCDC30288, 0.025 g, 0.051 mmol, 60%) as an orange foam: IR (ATR) 2960, 2937, 2931, 2876, 2811, 1599, 1582, 1577, 1476, 1458, 1452, 1381, 1359, 1342, 1331, 1305, 1266, 1217, 1176, 1146, 1117, 852, 777, 772, 751 cm−1; 1H NMR (400 MHz, acetone-d6) δ 10.84 (br s, 1 H), 7.44-7.42 (m, 2 H), 7.30-7.24 (m, 3 H), 6.94-6.91 (m, 1 H), 6.87-6.86 (m, 1 H), 6.83 (dd, J = 8.6, 2.2 Hz, 1 H), 3.74 (dt, J = 12.5, 3.4 Hz, 2 H), 2.86 (td, J = 11.9, 2.3 Hz, 2 H), 2.71 (t, J = 6.2 Hz, 2 H), 2.66-2.56 (m, 2 H), 2.47-2.38 (m, 10 H), 1.99-1.94 (m, 2 H), 1.50-1.40 (m, 2 H), 0.98 (d, J = 6.4 Hz, 6 H); 31C NMR (100 MHz, acetone-d6) δ 153.2, 141.4, 141.3, 136.0, 135.9, 133.72, 133.68, 132.4, 131.1, 131.0, 130.4, 116.7, 116.4, 114.3, 113.6, 113.23, 113.18, 110.3, 99.5, 99.4, 59.0, 55.6, 54.9, 54.6, 49.4, 48.7, 44.3, 33.3, 18.8; HRMS (ESI+) m/z calcd for C28H39N8 487.3292 (M+H), found 487.3293.
2-Bromo-6,11-dihydro-5H-benzo[a]carbazole (34) 23
A mixture of phenylhydrazine (0.506 g, 4.44 mmol) and 7-bromo-1-tetralone (1.02 g, 4.44 mmol) in EtOH (10 mL) and conc. HCl (0.5 mL) was heated at reflux for 4 h. The reaction mixture was concentrated and the resulting solid was suspended in hexanes/CH2Cl2 (21 mL, 20:1) and stirred for 30 min. The light red solid (34, 0.84 g, 2.8 mmol, 63%) was collected by filtration and dried: 1H NMR (300 MHz, CDCl3) δ 8.11 (br s, 1 H), 7.56 (d, J = 7.5 Hz, 1 H), 7.44-7.43 (m, 1 H), 7.38 (d, J = 8.1 Hz, 1 H), 7.29-7.28 (m, 1 H), 7.23-7.22 (m, 1 H), 7.20-7.12 (m, 1 H), 3.02-2.95 (m, 4 H); HRMS (ESI+) m/z calcd for C16H13NBr 298.0220 (M+H), found 298.0224.
1-(6,11-Dihydro-5H-benzo[a]carbazol-2-yl)-N-(2-(4-isopropylpiperazin-1-yl)ethyl)piperidin-4-amine (13, UPCDC30206)
A suspension of 34 (0.079 g, 0.26 mmol), K3PO4 (0.087 g, 0.40 mmol), Pd2(dba)3 (0.005 g, 0.005 mmol) and CyJohnPhos (0.0076 g, 0.021 mmol) in dry degassed dioxane (1 mL) was treated with A (0.11 g, 0.32 mmol) in dioxane (2.5 mL). The reaction mixture was degassed by bubbling argon for 15 min, sealed and heated at 120 °C for 48 h. The reaction mixture was cooled to room temperature, diluted with sat. NaHCO3 and extracted with CH2Cl2 (3×). The combined organic layer was washed with brine, dried (Na2SO4) and concentrated. The crude residue was purified by chromatography on SiO2 (2% MeOH/CH2Cl2 with 1% TEA) followed by filtration through basic Al2O3 (CH2Cl2) to afford tert-butyl (1-(6,11-dihydro-5H-benzo[a]carbazol-2-yl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)carbamate (0.050 g, 0.087 mmol, 33%) as a pale yellow amorphous solid: 1H NMR (300 MHz, CDCl3) δ 8.28 (br s, 1 H), 7.54 (d, J = 7.5 Hz, 1 H), 7.37 (d, J = 8.1 Hz, 1 H), 7.20-7.09 (m, 3 H), 6.94 (d, J =1.5 Hz, 1 H), 6.75 (dd, J = 1.8, 8.1 Hz, 1 H), 3.75-3.72 (m, 2 H), 3.28-3.24 (m, 2 H), 2.96-2.80 (br s, 4 H), 2.83-2.76 (m, 2 H), 2.56-2.31 (m, 11 H), 1.81-1.72 (m, 6 H), 1.48 (s, 9 H), 1.04 (d, J = 6.3 Hz, 6 H); HRMS (ESI+) m/z calcd for C35H50O2N5 572.3959 (M+H), found 572.3956.
A solution of trifluoroacetic acid (1.0 mL, 13.3 mmol) and triethylsilane (0.046 mL, 0.29 mmol) in CH2Cl2 (1.0 mL) was added to a solution of tert-butyl (1-(6,11- dihydro-5H-benzo[a]carbazol-2-yl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)carbamate (0.033 g, 0.058 mmol) in CH2Cl2 (1.0 mL). After 2 h, the reaction mixture was concentrated, diluted with sat. NaHCO3, and extracted with EtOAc (3×). The combined organic layer was washed with brine, dried (Na2SO4) and concentrated. The crude residue was purified by chromatography on SiO2 (5 to 10% MeOH/CH2Cl2 with 1% TEA) followed by filtration through basic Al2O3 (0 to 2% MeOH/CH2Cl2) to afford 13 (UPCDC30206, 0.015 g, 0.032 mmol, 57%) as a pale yellow oil: IR (ATR) 3218, 2924, 2839, 2181, 1728, 1609, 1465, 1381, 1195, 740 cm−1; 1H NMR (500 MHz, acetone-d6) δ 10.65 (br s, 1 H), 7.49 (d, J = 8.0 Hz, 1 H), 7.36 (d, J = 8.0 Hz, 1 H), 7.31 (d, J = 1.5 Hz, 1 H), 7.11-7.06 (m, 2 H), 7.03-6.99 (m, 1 H), 6.73 (dd, J = 1.5, 8.0 Hz, 1 H), 3.66 (d, J = 12.5 Hz, 2 H), 2.96-2.91 (m, 4 H), 2.89-2.76 (m, 2 H), 2.72-2.69 (m, 2 H), 2.61-2.56 (m, 3 H), 2.46-2.39 (m, 10 H), 1.96-1.93 (m, 2 H), 1.44 (qd, J = 13.5, 3.5 Hz, 2 H) 0.97 (d, J = 6.5 Hz, 6 H); 31C NMR (100 MHz, acetone-d6) δ 151.8, 138.3, 134.8, 130.5, 129.5, 128.3, 127.5, 122.4, 119.9, 119.1, 115.1, 112.3, 111.9, 110.2, 59.0, 55.7, 54.9, 54.6, 49.4, 49.2, 44.4, 33.4, 20.7, 18.8; HRMS (ESI+) m/z calcd for C30H42N5 472.3440 (M+H), found 472.3433.
2-(3-(4-((tert-Butoxycarbonyl)(2-(4-isopropylpiperazin-1-yl)ethyl)amino)piperidin-1- yl)phenyl)-1H-indole-5-carboxylic acid (35)
To a solution of methyl 2-(3-(4-((tert-butoxycarbonyl)(2-(4-isopropylpiperazin-1-yl)ethyl)amino)piperidin-1-yl)phenyl)-1H-indole-5-carboxylate (0.650 g, 1.08 mmol) in a mixture of THF (10 mL) and H2O (5 mL) was added LiOH (0.079 g, 3.23 mmol, portionwise), and the mixture was stirred at 60 °C for 24 h. The reaction mixture was cooled to room temperature and diluted with EtOAc and the phases were separated. The basic aqueous layer was acidified with 1N HCl and extracted with EtOAc (3×). The combined organic layer was washed with brine, dried (Na2SO4), filtered, concentrated to provide crude 2-(3-(4-((tert-butoxycarbonyl)(2-(4-isopropylpiperazin-1-yl)ethyl)amino)piperidin-1- yl)phenyl)-1H-indole-5-carboxylic acid (35, 0.443 g, 0.751 mmol, 70%) as a brown solid, which was used without further purification: HRMS (ESI+) m/z calcd for C34H48O4N5 590.3701 (M+H), found 590.3701.
2-(3-(4-((2-(4-Isopropylpiperazin-1-yl)ethyl)amino)piperidin-1-yl)phenyl)-N-methyl-1H-indole-5-carboxamide (14, UPCDC30367)
To a solution of 2-(3-(4-((tert-butoxycarbonyl)(2-(4-isopropylpiperazin-1-yl)ethyl)amino)piperidin-1- yl)phenyl)-1H-indole-5-carboxylic acid (35, 0.100 g, 0.169 mmol), methylamine hydrochloride (17.5 mg, 0.254 mmol) and TEA (0.191 mL, 1.36 mmol) in dry acetonitrile (0.5 mL) was slowly added T3P (0.107 g, 0.144 mmol; 50% EtOAc solution) at 0 °C. After 1 h, another portion of T3P (0.107 g, 0.144 mmol; 50% EtOAc solution) was added and the reaction mixture was allowed to warm to room temperature overnight. The reaction mixture was quenched with slow addition of 0.5M NaOH and the mixture was stirred at room temperature for 1 h. The crude product was extracted with EtOAc (3×) and the combined organic layer was washed with water followed by brine, dried (Na2SO4), filtered and concentrated to provide crude tert-butyl (2-(4-isopropylpiperazin-1-yl)ethyl)(1-(3-(5-(methylcarbamoyl)-1H-indol-2-yl)phenyl)piperidin-4-yl)carbamate (77.5 mg, 0.129 mmol, 76%), which was used without further purification: 1H NMR (500 MHz, CD3OD) δ 8.07 (s, 1 H), 7.60 (d, J = 8.5 Hz, 1 H), 7.43 (d, J = 9.0 Hz, 2 H), 7.32-7.27 (m, 2 H), 6.96 (d, J = 7.5 Hz, 1 H), 6.89 (s, 1 H), 4.04 (br s, 1 H), 3.93-3.91 (m, 2 H), 3.27 (br s, 2 H), 2.95 (s, 3 H), 2.86 (t, J = 12.3 Hz, 2 H), 2.69-2.50 (m, 11 H), 1.98-1.93 (m, 2 H), 1.77-1.76 (m, 2 H), 1.47 (s, 9 H), 1.10 (d, J = 6.0 Hz, 6 H); HRMS (ESI+) m/z calcd for C35H51O3N6 603.4017 (M+H), found 603.4013.
To a solution of tert-butyl (2-(4-isopropylpiperazin-1-yl)ethyl)(1-(3-(5-(methylcarbamoyl)-1H-indol-2-yl)phenyl)piperidin-4-yl)carbamate (72.5 mg, 0.120 mmol) in CH2Cl2 (2 mL) at 0 °C was added trifluoroacetic acid (0.36 mL, 4.8 mmol). The reaction mixture was allowed to warm to room temperature and stirred for 2 h, treated with sat. NaHCO3 and extracted with CH2Cl2 (3×). The combined organic layers were washed with brine, dried (Na2SO4), filtered, concentrated, and purified by chromatography on SiO2 (0 to 5% MeOH/CH2Cl2 to 10% MeOH/CH2Cl2 with 1% TEA) followed by filtration through basic Al2O3 (0 to 5% MeOH/CH2Cl2) to provide 2-(3-(4-((2-(4-Isopropylpiperazin-1-yl)ethyl)amino)piperidin-1-yl)phenyl)-N-methyl-1H-indole-5-carboxamide 14 (UPCDC30367, 30 mg, 0.059 mmol, 49%): IR (ATR) 3275, 2923, 2813, 1627, 1599, 1547, 1458, 1407, 1176, 1148, 1117, 980, 805, 777, 764 cm−1; 1H NMR (500 MHz, CD3OD) δ 8.07 (s, 1 H), 7.60 (d, J = 8.5 Hz, 1 H), 7.43 (d, J = 8.5 Hz, 2 H), 7.32-7.27 (m, 2 H), 6.96 (app d, J = 7.0 Hz, 1 H), 6.88 (s, 1 H), 3.82 (app d, J = 12.5 Hz, 2 H), 2.95 (s, 3 H), 2.85-2.79 (m, 4 H), 2.72-2.53 (m, 12 H), 2.05 (app d, J = 11.5 Hz, 2 H), 1.56 (qd, J = 11.8, 3.3 Hz, 2 H), 1.09 (d, J = 6.5 Hz, 6 H); 31C NMR (125 MHz, CD3OD) δ 172.3, 153.4, 141.6, 140.6, 134.4, 130.6, 130.1, 126.8, 121.8, 121.0, 118.0, 117.4, 114.7, 111.8, 100.6, 58.3, 56.3, 55.9, 54.1, 50.0, 43.8, 32.7, 27.0, 18.7; HRMS (ESI+) m/z calcd for C30H43ON6 503.3493 (M+H), found 503.3492.
((3-Bromophenyl)ethynyl)trimethylsilane (36) 24
To a solution of 1-bromo-3-iodobenzene (2.00 g, 6.72 mmol) in dry, degassed acetonitrile (20.0 mL) was added PdCl2(PPh3)2 (0.24 g, 0.34 mmol) and CuI (0.065 g, 0.34 mmol). The mixture was degassed and backfilled with argon and charged with Et3N (0.95 mL, 6.7 mmol) and trimethylsilylacetylene (1.94 mL, 13.4 mmol). The mixture was stirred at room temperature for 1 h. The reaction mixture was concentrated and the crude product was extracted with EtOAc, washed with water, brine, dried (Na2SO4), filtered and concentrated. The residue was purified by chromatography on SiO2 (100% hexanes) to obtain 36 as an orange oil (1.7 g, 6.6 mmol, 99%): IR (ATR) 2956, 2160, 1582, 1588, 1470, 1450, 1260, 1245, 1079 cm−1; 1H NMR (500 MHz, CDCl3) 7.62 (t, J = 2.0 Hz, 1 H), 7.45-7.43 (m, 1 H), 7.16 (app t, J = 7.5 Hz, 1 H), 0.25 (s, 9 H); 31C NMR (CDCl3, 125 MHz) 134.8, 131.7, 130.6, 129.7, 125.3 122.2, 103.4, 96.0, 0.00.
8-(3-((Trimethylsilyl)ethynyl)phenyl)-1,4-dioxa-8-azaspiro[4.5]decane (37)
A solution of 36 (200 mg, 0.790 mmol), 1,4-dioxa-8-azaspiro[4.5]decane (127 mg, 0.87 mmol), Pd2(dba)3 (36 mg, 0.039 mmol), DavePhos (16 mg, 0.039 mmol) in dry THF (5.0 mL) was degassed by bubbling argon and backfilled with argon three times. To this solution was added LiHMDS (340 mg, 1.97 mmol). The reaction mixture was degassed and the reaction vial was sealed and heated at 70 °C for 3 h. The reaction mixture was cooled to room temperature, diluted with sat. NaHCO3 and extracted with EtOAc (3×). The combined organic layer was washed with brine, dried (Na2SO4), filtered and concentrated. The residue was purified by chromatography on SiO2 (10 to 30% EtOAc/hexanes) to provide 37 (0.17 g, 0.54 mmol, 68%) as an off-white sticky solid: IR (ATR) 2957, 2879, 2829, 2143, 1589, 1478, 1246, 1101, 833 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.16 (t, J = 7.8 Hz, 1 H), 7.03 (br s, 1 H), 6.94-6.88 (m, 2 H), 3.99 (s, 4 H), 3.32 (t, J = 5.7 Hz, 4 H), 1.82 (t, J = 5.7 Hz, 4 H), 0.24 (s, 9 H); HRMS (ESI+) m/z calcd for C18H26NO2Si 316.1733 (M+H), found 316.1725.
8-(3-Ethynylphenyl)-1,4-dioxa-8-azaspiro[4.5]decane (38)
A solution of 37 (820 mg, 2.47 mmol) in THF (10.0 mL) was treated with TBAF (0.95 mL, 75 wt % H2O) at 0 °C. The reaction mixture was warmed to room temperature. After 20 min, the reaction mixture was extracted with EtOAc (2x), washed with H2O, brine, dried (Na2SO4), filtered and concentrated. The residue was purified by chromatography on SiO2 (10 to 30% EtOAc/hexanes) to give 38 (0.508 g, 2.08 mmol, 85%) as a yellow oil: IR (ATR) 3282, 2956, 2926, 2881, 1664, 1591, 1569, 1235, 1097 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.18 (t, J = 8.0 Hz, 1 H), 7.06 (dd, J = 1.6, 2.4 Hz, 1 H), 6.96-6.92 (m, 2 H), 3.99 (s, 4 H), 3.33 (t, J = 5.6 Hz, 4 H), 3.02 (s, 1 H), 1.82 (t, J = 5.6 Hz, 4 H); 31C NMR (100 MHz, CDCl3) 150.7, 129.2, 123.1, 122.8, 119.9, 117.3, 107.2, 84.4, 76.5, 64.5, 47.5, 34.5; HRMS (ESI+) m/z calcd for C15H18O2N 244.1332 (M+H), found 244.1331.
3-Iodopyridin-4-amine (39) 25
To a refluxing solution of 4-aminopyridine (2.00 g, 21.3 mmol) and Na2CO3 (1.35 g, 12.8 mmol) in H2O (7.6 mL) was slowly added a solution of KI (3.99 g, 23.8 mmol) and I2 (4.09 g, 15.9 mmol) in H2O (16.8 mL) and the mixture was heated at reflux for 22 h. The reaction mixture was cooled to room temperature and extracted with EtOAc (2x). The combined organic layer was washed with satd Na2S2O3, followed by brine, dried (MgSO4), filtered and concentrated. The residue was purified by chromatography on SiO2 (50 to 75% EtOAc/hexanes) to give 39 as an off-white solid (1.66 g, 7.54 mmol, 36%): 1H NMR (300 MHz, CDCl3) δ 8.55 (s, 1 H), 8.08 (d, J = 5.4 Hz, 1 H), 6.57 (d, J = 5.4 Hz, 1 H), 4.72 (br s, 2 H).
N-(3-Iodopyridin-4-yl)acetamide (40) 26
A solution of 39 (1.65 g, 7.50 mmol) in CH2Cl2 (15 mL) was treated with acetic anhydride (0.71 mL, 7.5 mmol) and TEA (1.58 mL, 11.3 mmol) at room temperature. After 17 h, the solution was concentrated. The residue was diluted with THF/H2O (6.0 mL, 1/1) and treated with LiOH (0.20 g, 1.1 mmol) at room temperature. After 30 min, the reaction mixture was extracted with EtOAc (3×). The combined organic layer was washed with brine, dried (MgSO4), filtered and concentrated to give 40 (1.68 g, 6.40 mmol, 85%, containing approx. 5% residual solvent) as a light orange solid: 1H NMR (400 MHz, CDCl3) δ 8.81 (s, 1 H), 8.39 (d, J = 5.6 Hz, 1 H), 8.32 (d, J = 5.6 Hz, 1 H), 7.61 (br s, 1 H), 2.28 (s, 3 H).
8-(3-(1H-Pyrrolo[3,2-c]pyridin-2-yl)phenyl)-1,4-dioxa-8-azaspiro[4.5]decane (41)
To a microwave vial equipped with a magnetic stir bar was added 38 (200 mg, 0.822 mmol), 40 (323 mg, 1.23 mmol), PdCl2(PPh3)2 (29 mg, 0.041 mmol), CuI (8.0 mg, 0.04 mmol), 1,1,3,3-tetra-methylguanidine (0.31 mL, 2.5 mmol) and DMF (1.0 mL). The mixture was degassed by bubbling argon for 15 min, sealed, and then heated at 80 °C for 1 h under microwave irradiation. The reaction mixture was diluted with H2O and extracted with EtOAc (2x). The combined organic layer was washed with H2O, brine, dried (Na2SO4), filtered and concentrated. The solid was precipitated from a solution of MeOH/CH2Cl2/hexanes to afford 41 as a white powder (89 mg, 0.27 mmol, 33%): IR (ATR) 3092, 2956, 2829, 2726, 1595, 1573, 1541, 1494, 1494, 1224, 1095 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 11.88 (s, 1 H), 8.83 (br s, 1 H), 8.19 (br s, 1 H), 7.46 (s, 1 H), 7.38 (br s, 1 H), 7.29-7.28 (m, 2 H), 7.02 (s, 1 H), 6.94 (s, 1 H), 3.93 (s, 4 H), 3.39 (t, J = 5.4 Hz, 4 H), 1.75 (t, J = 5.1 Hz, 4 H); 31C NMR (75 MHz, DMSO-d6) δ 150.8, 140.3, 140.1, 139.5, 132.0, 129.6, 115.8, 115.3, 112.5, 106.4, 97.3, 63.7, 46.6, 33.9; HRMS (ESI+) m/z calcd for C20H22N3O2 336.1707 (M+H), found 336.1704.
1-(3-(1H-Pyrrolo[3,2-c]pyridin-2-yl)phenyl)-N-(2-(4-isopropylpiperazin-1-yl)ethyl)piperidin-4-amine (17, UPCDC30345)
To a solution of 41 (0.080 g, 0.24 mmol) in acetone (15 mL) was added 3M HCl (10 mL) and the reaction mixture was heated at 80 °C for 6 h. The solution was cooled to room temperature and kept at this temperature overnight. The solution was cooled to 0 °C, neutralized with solid Na2CO3 and extracted with EtOAc (2x). The combined organic layer was washed with brine, dried (Na2SO4), filtered and concentrated to afford 1-(3-(1H-pyrrolo[3,2-c]pyridin-2-yl)phenyl)piperidin-4-one as a yellow solid that was used without further purification: HRMS (ESI+) m/z calcd for C18H18ON3 292.1444 (M+H), found 292.1443.
To a suspension of 1-(3-(1H-pyrrolo[3,2-c]pyridin-2-yl)phenyl)piperidin-4-one (0.070 g, 0.24 mmol), B (0.045 g, 0.26 mmol) in a mixture of 1,2-DCE and THF (2:1, 1.5 mL) was added Ti(OiPr)4 (0.080 mL, 0.26 mmol). After 1.5 h, the reaction mixture was treated with NaBH(OAc)3 (0.031 g, 0.14 mmol). After 2 h, a second portion of NaBH(OAc)3 (0.025 g, 0.012 mmol) was added. After 1 h, the solution was treated with 0.5 M NaOH (aq) and the resulting suspension was extracted with EtOAc (2x). The combined organic layer was washed with brine, dried (Na2SO4), filtered and concentrated. The residue was purified by chromatography on SiO2 (0 to 10% MeOH/CH2Cl2 with 1% TEA) followed by filtration through basic Al2O3 (0 to 5% MeOH/CH2Cl2) to give 17 as an off-white solid (UPCDC30345, 47 mg, 0.11 mmol, 42% - 2 steps): IR (ATR) 3081, 2943, 2924, 2808, 1772, 1601, 1575, 1541, 1496 cm−1; 1H NMR (400 MHz, CD3OD) δ 8.75 (s, 1 H), 8.11 (d, J = 5.6 Hz, 1 H), 7.43-7.41 (m, 2 H), 7.34-7.28 (m, 2 H), 7.00 (d, J = 7.2 Hz, 1 H), 6.96 (s, 1 H), 3.82 (d, J = 12.8 Hz, 2 H), 2.86-2.78 (m, 4 H), 2.70-2.52 (m, 12 H), 2.05-2.01 (m, 2 H), 1.54 (qd, J = 12.0, 3.5 Hz, 2 H), 1.08 (d, J = 6.4 Hz, 6 H); 31C NMR (100 MHz, CD3OD) δ 153.4, 143.3, 142.5, 142.2, 140.3, 133.6, 130.8, 127.7, 118.1, 117.8, 114.8, 107.9, 98.8, 58.4, 56.3, 55.9, 54.1, 49.9, 49.5, 43.8, 32.8, 18.7; HRMS (ESI+) m/z calcd for C27H39N6 447.3231 (M+H), found 447.3229.
5-Fluoro-3-iodopyridin-2-amine (42) 27
To a stirring solution of 5-fluoropyridin-2-amine (1.5 g, 13.4 mmol) in H2SO4 (2M, 20.0 mL) was slowly added KIO3 (1.4 g, 6.7 mmol). The reaction mixture was heated at 100 °C and a solution of KI (2.24 g, 13.4 mmol) in H2O (10.0 mL) was added. After 30 min, the reaction mixture was cooled to room temperature and the solution was treated with NaHCO3 until pH 8–9. The reaction mixture was extracted with EtOAc (3×). The combined organic layers were washed with sat. NaHSO3, H2O, brine, dried (Na2SO4), filtered and concentrated to afford 42 (2.29 g, 9.62 mmol, 72%) that was used without further purification.
N-(5-Fluoro-3-iodopyridin-2-yl)acetamide (43)
A solution of 42 (1.20 g, 5.04 mmol) in AcOH (10.0 mL) was treated with acetic anhydride (0.47 mL, 5.0 mmol) and the mixture was heated at 100 °C for 4 h. The reaction mixture was cooled to room temperature and carefully treated with sat. K2CO3. The reaction mixture was extracted with EtOAc (3×). The combined organic layer was washed with H2O, brine, dried (Na2SO4), filtered and concentrated. The product was purified by chromatography on SiO2 (0 to 70% EtOAc/hexanes) to obtain 43 as a white powder (926 mg, 3.31 mmol, 66%): M.p. 148–150 °C; IR (ATR) 3223, 3189, 3029, 3001, 1688, 1655, 1569, 1515, 1431, 1262 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.27 (d, J = 2.4 Hz, 1 H), 7.89 (dd, J = 7.2, 2.8 Hz, 1 H), 7.66 (br s, 1 H), 2.33 (s, 3 H); 31C NMR (100 MHz, CDCl3) δ 170.1, 155.6 (d, JCF = 259 Hz), 147.6, 135.6 (d, JCF = 24 Hz), 135.1 (d, JCF = 21 Hz), 24.0; HRMS (ESI+) m/z calcd for C7H7FIN2O 280.9587 (M+H), found 280.9579.
8-(3-(5-Fluoro-1H-pyrrolo[2,3-b]pyridin-2-yl)phenyl)-1,4-dioxa-8-azaspiro[4.5]decane (44) 28
To a microwave vial equipped with a magnetic stir bar was added 38 (300 mg, 1.23 mmol), 43 (432 mg, 1.47 mmol), PdCl2(PPh3)2 (44 mg, 0.062 mmol), CuI (19.0 mg, 0.098 mmol), 1,1,3,3-tetra-methylguanidine (0.47 mL, 3.7 mmol) and DMF (1.0 mL). The reaction mixture was degassed for 10 min. by argon bubbling and the vial was sealed and heated at 80 °C under microwave irradiation. The reaction mixture was diluted with H2O and extracted with EtOAc (2x). The combined organic layer was washed with H2O, brine, dried (Na2SO4), filtered and concentrated. The residue was purified by chromatography on SiO2 (30 to 50% EtOAc/hexanes) to obtain 44 as a light yellow solid (145 mg, 0.41 mmol, 33%): IR (ATR) 3211, 2872, 2821, 1614, 1576, 1489, 1446, 1287, 1196, 1131, 1110, 876, 758 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 12.22 (s, 1 H), 8.17 (s, 1 H), 7.78 (d, J = 9.5 Hz, 1 H), 7.52 (s, 1 H), 7.33-7.27 (m, 2 H), 6.95 (d, J = 8.0 Hz, 1 H), 6.91 (s, 1 H), 3.93 (s, 4 H), 3.39 (t, J = 5.0 Hz, 4 H), 1.74 (t, J = 5.0 Hz, 4 H).
1-(3-(5-Fluoro-1H-pyrrolo[2,3-b]pyridin-2-yl)phenyl)piperidin-4-one (45)
To a solution of 44 (0.135 g, 0.382 mmol) in acetone (20 mL) was added 3M HCl (15 mL). The reaction mixture was heated at 80 °C for 6 h. The solution was cooled to 0 °C, neutralized with Na2CO3, and extracted with EtOAc (2x). The combined organic layer was washed with brine, dried (Na2SO4), filtered and concentrated. The solid that formed upon concentration was collected by filtration, washed with hexanes and dried in vacuo to give 45 as a brown solid (82 mg, 0.26 mmol, 69%, approx. 90% purity): 1H NMR (300 MHz, CDCl3) δ 11.51 (s, 1 H), 8.19 (s, 1 H), 7.64 (dd, J = 2.7, 9.0 Hz, 1 H), 7.46 (app t, J = 7.8 Hz, 1 H), 7.40 (s, 1 H), 7.32 (d, J = 7.5 Hz, 1 H), 7.03 (dd, J = 2.1, 8.1 Hz, 1 H), 6.75 (d, J = 2.1 Hz, 1 H), 3.69 (t, J = 6.3 Hz, 4 H), 2.62 (t, J = 6.0 Hz, 4 H); HRMS (ESI+) m/z calcd for C18H17FN3O 310.1356 (M+H), found 310.1348.
1-(3-(5-Fluoro-1H-pyrrolo[2,3-b]pyridin-2-yl)phenyl)-N-(2-(4-isopropylpiperazin-1-yl)ethyl)piperidin-4-amine (19, UPCDC30381)
To a suspension of 45 (0.070 g, 0.23 mmol) and B (0.046 g, 0.27 mmol) in THF/1,2-dichloroethane (1/2, 1.5 mL) was added Ti(OiPr)4 (0.083 mL, 0.27 mmol) at room temperature. After 30 min, NaBH(OAc)3 (0.030 g, 0.13 mmol) was added. After 2 h, a second portion of NaBH(OAc)3 (0.031 g, 0.14 mmol) was added. After 1 h, additional NaBH(OAc)3 (0.030 g, 0.13 mmol) was added and the reaction mixture was stirred overnight. The reaction mixture was diluted with sat. NaHCO3 and EtOAc/MeOH. The suspension was filtered through Celite and the filtrate was concentrated. The residue was extracted with EtOAc, washed with H2O, brine, dried (Na2SO4), filtered and concentrated. The residue was purified by chromatography on SiO2 (5 to 10% MeOH/CH2Cl2 with 1% TEA) followed by filtration through basic Al2O3 (5% MeOH/CH2Cl2) to give 19 as an off-white foamy solid (UPCDC30381, 0.051 g, 0.11 mmol, 49%): IR (ATR) 3170, 2930, 2807, 1610, 1586, 1489, 1446, 1343, 1176, 980, 758 cm−1; 1H NMR (500 MHz, CD3OD) δ 8.06 (s, 1 H), 7.68 (dd, J = 2.5, 9.5 Hz, 1 H), 7.45 (s, 1 H), 7.34-7.31 (m, 2 H), 7.01-6.99 (m, 1 H), 6.81 (s, 1 H), 3.85 (d, J = 12.5 Hz, 2 H), 2.90 (t, J = 6.5 Hz, 2 H), 2.87-2.82 (m, 3 H), 2.73-2.57 (m, 11 H), 2.09 (d, J= 12.0 Hz, 2 H), 1.62 (qd, J = 11.5, 3.0 Hz, 2 H), 1.10 (d, J = 6.5 Hz, 6 H); 31C NMR (125 MHz, CD3OD) δ 157.2 (d, JCF = 238 Hz), 153.2, 147.6, 143.4, 133.7, 131.3 (d, JCF = 30 Hz), 130.7, 123.5 (d, JCF = 8 Hz), 118.0 (d, JCF = 39 Hz), 114.9, 114.8, 114.6, 98.3 (d, JCF = 4 Hz), 57.5, 56.4, 56.1, 53.8, 49.7, 43.5, 31.9, 18.6; 19F NMR (376 MHz, CD3OD) δ -141.4; HRMS (ESI+) m/z calcd for C27H38FN6 465.3136 (M+H), found 465.3133.
2-(3-Bromophenyl)naphthalene (46)29
To a flask containing 1-bromo-3-iodobenzene (0.28 mL, 2.2 mmol), 2-naphthalene boronic acid (0.26 g, 1.5 mmol), Pd(PPh3)2Cl2 (0.023 g, 0.033 mmol) and K2CO3 (0.405 g, 2.93 mmol) was added degassed DMF (4.0 mL). The reaction mixture was heated at 80 °C overnight. The reaction mixture was cooled to room temperature, diluted with H2O, and extracted with EtOAc (3×). The combined organic layer was washed with brine, dried (Na2SO4), filtered and concentrated. The residue was purified by chromatography on SiO2 (0 to 10% EtOAc/hexanes) to give 46 (0.265 g, 0.94 mmol, 64%) as an off-white solid: 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 1.2 Hz, 1 H), 7.94-7.90 (m, 2 H), 7.89-7.86 (m, 2 H), 7.70 (dd, J = 2.0, 8.4 Hz, 1 H), 7.66-7.63 (m, 1 H), 7.55-7.48 (m, 3 H), 7.35 (app t, J = 8.0 Hz, 1 H); 31C NMR (100 MHz, CDCl3) δ 143.5, 130.6, 130.5, 130.4, 128.8, 128.4, 127.8, 126.6, 126.4, 126.2, 126.1, 125.4; HRMS (ESI+) m/z calcd for C16H11Br 282.0044 (M+), found 282.0062.
N-(2-(4-Isopropylpiperazin-1-yl)ethyl)-1-(3-(naphthalen-2-yl)phenyl)piperidin-4-amine (20, UPCDC30222)
A suspension of 46 (0.100 g, 0.353 mmol), A (0.150 g, 0.424 mmol), K3PO4 (0.116 g, 0.529 mmol), Pd2(dba)3 (0.006 g, 0.007 mmol) and CyJohnPhos (0.010 g, 0.028 mmol) in dry degassed dioxane (5 mL) was heated at 110 °C overnight. The reaction mixture was cooled to room temperature, diluted with sat. NaHCO3 and extracted with EtOAc (3×). The combined organic layer was washed with brine, dried (Na2SO4) and concentrated. The crude residue was purified by chromatography on SiO2 (10 to 30% EtOAc/CH2Cl2) to give tert-butyl (2-(4-isopropylpiperazin-1-yl)ethyl)(1-(3-(naphthalen-2-yl)phenyl)piperidin-4-yl)carbamate (0.101 g, 0.18 mmol, 51%) as a light yellow oil: IR (ATR) 2959, 2928, 2807, 1679, 1592, 1448, 1410, 1383, 1362, 1178, 1146, 1010, 982, 907, 767 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 0.8 Hz, 1 H), 7.91-7.85 (m, 3 H), 7.72 (dd, J = 2.0, 8.8 Hz, 1 H), 7.52-7.45 (m, 2 H), 7.36 (app t, J = 8.0 Hz, 1 H), 7.25-7.23 (m, 1 H), 7.19 (d, J = 7.6 Hz, 1 H), 6.95 (dd, J = 2.0, 8.0 Hz, 1 H), 4.17 (br s, 1 H), 3.85 (d, J = 12.4 Hz, 2 H), 3.25 (br s, 2 H), 2.88-2.84 (m, 2 H), 2.69-2.47 (m, 11 H), 1.86-1.79 (m, 4 H), 1.47 (s, 9 H), 1.05 (d, J = 6.4 Hz, 6 H); 31C NMR (100 MHz, CDCl3) δ 155.4, 151.8, 142.3, 139.3, 133.7, 132.7, 129.7, 128.4, 128.3, 127.8, 126.4, 126.0, 125.92, 125.91, 119.1, 116.1, 115.8, 79.9, 54.7, 53.9, 49.9, 48.7, 30.2, 28.7, 18.7; HRMS (ESI+) m/z calcd for C35H49N4O2 557.3850 (M+H), found 557.3849.
A solution of trifluoroacetic acid (0.83 mL, 11.0 mmol) and triethylsilane (0.09 mL, 0.52 mmol) in CH2Cl2 (1.5 mL) was added to a solution of tert-butyl (2-(4-isopropylpiperazin-1-yl)ethyl)(1-(3-(naphthalen-2-yl)phenyl)piperidin-4-yl)carbamate (0.055 g, 0.099 mmol) in CH2Cl2 (1.5 mL). After 1 h, the reaction mixture was concentrated, diluted with sat. NaHCO3, and extracted with EtOAc (3×). The combined organic layer was washed with brine, dried (Na2SO4) and concentrated. The crude residue was purified by chromatography on SiO2 (8 to 20% MeOH/CH2Cl2 with 1% TEA) followed by filtration through basic Al2O3 (0 to 10% MeOH/CH2Cl2) to afford 20 (UPCDC30222, 0.030 g, 0.066 mmol, 67%, containing approx. 5% residual solvents) as a light yellow oil: IR (ATR) 2957, 2932, 2805, 1702, 1593, 1461, 1379, 1232, 1176, 1129, 982, 854, 779 cm−1; 1H NMR (500 MHz, acetone-d6) δ 8.15 (d, J = 1.0 Hz, 1 H), 7.98-7.96 (m, 2 H), 7.91 (d, J = 8.0 Hz, 1 H), 7.82 (dd, J = 2.0, 8.5 Hz, 1 H), 7.54-7.48 (m, 2 H), 7.35-7.32 (m, 2 H), 7.19 (dd, J = 0.5, 7.5 Hz, 1 H), 6.99 (dd, J = 2.5, 8.5 Hz, 1 H), 3.77 (dt, J = 3.0, 12.5 Hz, 2 H), 2.90-2.85 (m, 2 H), 2.72 (t, J = 6.0 Hz, 2 H), 2.65-2.55 (m, 3 H), 2.47-2.40 (m, 10 H), 1.98-1.95 (m, 2 H), 1.47 (qd, J = 13.0, 4.0, 2 H), 0.98 (d, J = 6.5 Hz, 6 H); 31C NMR (125 MHz, acetone-d6) δ 153.3, 142.5, 140.1, 134.7, 133.6, 130.3, 129.1, 129.0, 128.4, 127.1, 126.7, 126.4, 126.3, 118.6, 116.2, 115.9, 59.0, 55.7, 54.9, 54.6, 49.4, 48.9, 44.4, 33.4, 18.8; HRMS (ESI+) m/z calcd for C30H41N4 457.3331 (M+H), found 457.3322.
2-(3-Bromophenyl)benzofuran (47)30
Benzofuran-2-ylboronic acid (0.408 g, 2.47 mmol), 1-bromo-3-iodobenzene (0.482 mL, 3.70 mmol), PdCl2(PPh3)2 (0.088 g, 0.12 mmol), K2CO3 (0.682 g, 4.94 mmol) was treated with DMF (4.0 mL) and heated at 80 °C overnight. The reaction mixture was diluted with H2O and extracted with EtOAc (3×). The combined organic layer was washed with brine, dried (Na2SO4), filtered and concentrated. The residue was purified by chromatography on SiO2 (0 to 10% EtOAc/hexanes) to give 47 as an off-white solid (0.55 g, 2.0 mmol, 82%): 1H NMR (400 MHz, CDCl3) δ 8.02 (t, J = 1.2 Hz, 1 H), 7.80-7.77 (m, 1 H), 7.61-7.58 (m, 1 H), 7.54-7.51 (m, 1 H), 7.49-7.46 (m, 1 H), 7.34-7.29 (m, 2 H), 7.27-7.23 (m, 1 H), 7.05 (d, J = 0.8 Hz, 1 H); 31C NMR (100 MHz, CDCl3) δ 155.1, 154.3, 132.6, 131.5, 130.5, 129.0, 128.0, 124.9, 123.6, 123.3, 123.1, 121.3, 111.4, 102.6; HRMS (ESI+) m/z calcd for C14H9OBr 271.9837 (M+), found 271.9858.
1-(3-(Benzofuran-2-yl)phenyl)-N-(2-(4-isopropylpiperazin-1-yl)ethyl)piperidin-4-amine (21, UPCDC30221)
A suspension of A (187 mg, 0.527 mmol), 2-(3-bromophenyl)benzofuran (47, 120 mg, 0.44 mmol) and K3PO4 (144 mg, 0.66 mmol) in dry degassed dioxane (1.5 mL) was degassed for 5 min with argon. To this mixture was added Pd2(dba)3 (8 mg, 0.009 mmol) and CyJohnPhos (12 mg, 0.035 mmol). The reaction vial was sealed and the mixture was heated at 110 °C for 12 h. The reaction mixture was diluted with sat. NaHCO3 and extracted with EtOAc (3×). The combined organic layer was washed with brine, dried (Na2SO4), filtered and concentrated. The residue was purified by chromatography on SiO2 (10 to 30% EtOAc/CH2Cl2) to give tert-butyl (1-(3-(benzofuran-2-yl)phenyl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)-carbamate (0.19 g, 0.36 mmol, 82%) as a light yellow oil: IR (ATR) 2962, 2930, 2807, 1685, 1601, 1450, 162, 1143, 1008, 775, 749 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.50 (dd, J = 0.8, 7.6 Hz, 1 H), 7.46 (d, J = 8.0 Hz, 1 H), 7.38 (s, 1 H), 7.28-7.14 (m, 4 H), 6.93 (d, J = 0.8 Hz, 1 H), 6.65 (dt, J = 7.2, 1.6 Hz, 1 H), 4.11 (br s, 1 H), 3.76 (d, J = 12.0 Hz, 2 H), 3.19 (br s, 2 H), 2.86-2.75 (m, 2 H), 2.63-2.42 (m, 11 H), 1.80-1.76 (m, 4 H), 1.43 (s, 9 H), 0.99 (d, J = 6.8 Hz, 6 H); 31C NMR (100 MHz, CDCl3) δ 156.3, 155.4, 154.8, 151.5, 131.2, 129.5, 129.3, 124.2, 122.9, 120.8, 116.8, 116.4, 112.9, 111.1, 101.3, 79.8, 58.4, 54.6, 53.8, 53.0, 49.6, 48.6, 39.9, 30.1, 28.5, 18.6; HRMS (ESI+) m/z calcd for C33H47O3N4 547.3643 (M+H), found 547.3643.
A solution of tert-butyl (1-(3-(benzofuran-2-yl)phenyl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)-carbamate (0.100 g, 0.18 mmol) in CH2Cl2 (1.5 mL) was treated with trifluoroacetic acid (1.5 mL) and triethylsilane (0.15 mL, 0.95 mmol) in CH2Cl2 (1.5 mL). After 1 h, the solution was concentrated, diluted with sat. NaHCO3 and extracted with EtOAc (3×). The combined organic layer was washed with brine, dried (Na2SO4) and concentrated. The crude residue was purified by chromatography on SiO2 (0 to 10% MeOH/CH2Cl2) to afford 21 as a yellow foam (UPCDC30221, 0.060 g, 0.13 mmol, 73%): IR (ATR) 2957, 2928, 2807, 1599, 1567, 1489, 1450, 1254, 1220, 1176, 1144, 1122, 982, 747 cm−1; 1H NMR (500 MHz, acetone-d6) δ 7.62 (d, J = 7.5 Hz, 1 H), 7.55 (d, J = 8.0 Hz, 1 H), 7.52-7.51 (m, 1 H), 7.35-7.28 (m, 3 H), 7.26-7.22 (m, 1 H), 7.00 (dt, J = 7.5, 2.0 Hz, 1 H), 3.75 (dt, J = 12.5, 3.0 Hz, 2 H), 2.92-2.87 (m, 3 H), 2.73 (app t, J = 6.0 Hz, 3 H), 2.67-2.56 (m, 3 H), 2.48-2.41 (m, 9 H), 2.00-1.96 (m, 2 H); 1.46 (qd, J = 13.5, 4.0 Hz, 2 H), 0.98 (d, J = 6.5 Hz, 6 H); 31C NMR (125 MHz, acetone-d6) δ 157.4, 155.6, 153.0, 131.8, 130.3, 130.2, 125.1, 123.9, 121.8, 117.4, 116.1, 112.9, 111.8, 102.2, 59.0, 55.6, 54.9, 54.6, 49.4, 48.6, 44.4, 32.3, 18.8; HRMS (ESI+) m/z calcd for C28H39N4O 447.3118 (M+H), found 447.3118.
2-(3-(4-((2-(4-Isopropylpiperazin-1-yl)ethyl)amino)piperidin-1-yl)phenyl)-N,N-dimethyl-1H-indole-5-carboxamide (23, UPCDC30368)
Prepared according to the procedure for 14 using dimethylamine hydrochloride instead of methylamine hydrochloride to provide crude tert-butyl (1-(3-(5-(dimethylcarbamoyl)-1H-indol-2-yl)phenyl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)carbamate (77 mg, 0.13 mmol, 74%), which was used for the next reaction without further purification: 1H NMR (500 MHz, CD3OD) δ 7.65 (s, 1 H), 7.45 (d, J = 8.0 Hz, 1 H), 7.42 (br s, 1 H), 7.32-7.28 (m, 2 H), 7.19 (d, J = 8.5 Hz, 1 H), 6.96 (app d, J = 7.0 Hz, 1 H), 6.87 (s, 1 H), 4.05 (br s, 1 H), 3.93-3.91 (m, 2 H), 3.28 (br s, 2 H), 3.11 (s, 6 H), 2.87 (t, J = 12.0 Hz, 2 H), 2.71-2.51 (m, 11 H), 1.97-1.91 (m, 2 H), 1.78-1.76 (m, 2 H), 1.48 (s, 9 H), 1.08 (d, J = 6.5 Hz, 6 H); HRMS (ESI+) m/z calcd for C36H53O3N6 617.4174 (M+H), found 617.4172.
To a solution of tert-butyl (1-(3-(5-(dimethylcarbamoyl)-1H-indol-2-yl)phenyl)piperidin-4-yl)(2-(4-isopropylpiperazin-1-yl)ethyl)carbamate (82.3 mg, 0.133 mmol) in CH2Cl2 (2 mL) at 0 °C was added trifluoroacetic acid (0.39 mL, 5.3 mmol). The reaction mixture was allowed to warm to room temperature and stirred for 2 h, treated with sat. NaHCO3, and extracted with CH2Cl2 (3×). The combined organic layers were washed with brine, dried (Na2SO4), filtered, concentrated, and purified by chromatography on SiO2 (0 to 10% MeOH/CH2Cl2 with 1% TEA) followed by filtration through basic Al2O3 (0 to 5 % MeOH/CH2Cl2) to provide 2-(3-(4-((2-(4-isopropylpiperazin-1-yl)ethyl)amino)piperidin-1-yl)phenyl)-N,N-dimethyl-1H-indole-5-carboxamide 23 (UPCDC30368, 36 mg, 0.069 mmol, 52%): IR (ATR) 3225, 2930, 2808, 1599, 1541, 1499, 1444, 1383, 1318, 1176, 1146, 1071, 982, 803, 779, 764 cm−1; 1H NMR (400 MHz, CD3OD) δ 7.65 (s, 1 H), 7.45 (d, J = 8.4 Hz, 1 H), 7.42 (br s, 1 H), 7.32-7.27 (m, 2 H), 7.19 (dd, J = 8.2, 1.4 Hz, 1 H), 6.96-6.94 (m, 1 H), 6.86 (s, 1 H), 3.81 (app d, J = 12.4 Hz, 2 H), 3.11 (s, 6 H), 2.85-2.78 (m, 4 H), 2.70-2.52 (m, 12 H), 2.04 (app d, J = 12.0 Hz, 2 H), 1.55 (qd, J = 11.9, 3.2 Hz, 2 H), 1.08 (d, J = 6.4 Hz, 6 H); 31C NMR (100 MHz, CD3OD) δ 175.6, 153.4, 141.5, 139.4, 134.4, 130.6, 129.9, 128.0, 121.8, 120.7, 118.2, 117.4, 114.7, 111.9, 100.2, 58.4, 56.3, 55.9, 54.1, 50.0, 43.8, 32.8, 18.7; HRMS (ESI+) m/z calcd for C31H45ON6 517.3649 (M+H), found 517.3649.
1-Benzhydryl-2-(3-bromophenyl)-1H-benzo[d]imidazole (48)
A mixture of 3-bromobenzaldehyde (0.216 g, 2.00 mmol), o-phenylenediamine (0.388, 2.10 mmol), boric acid (0.006 g, 0.09 mmol) and glycerin (1 drop) in H2O (3 mL) was heated at 80 °C until disappearance of starting material. The water was decanted off and MeOH (ca 10 mL) was added. The mixture was stirred for 2 h at room temperature. The formed precipitate was collected by filtration and washed with cold MeOH to afford 2-(3-bromophenyl)-1H-benzo[d]imidazole31 as a white solid (0.32 g, 1.2 mmol, 59%): Mp. 264–265 °C; IR (ATR) 3038, 2957, 2799, 1562, 1436, 1398, 1357, 742, 727 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 13.04 (s, 1 H), 8.37 (t, J = 2.0 Hz, 1 H), 8.20-8.18 (m, 1 H), 7.69 (ddd, J = 8.0, 1.0, 1.0 Hz, 2 H), 7.56-7.50 (m, 2 H), 7.27-7.19 (m, 2 H); 31C NMR (125 MHz, DMSO-d6) δ 150.1, 144.1, 135.5, 132.9, 132.8, 131.6, 129.3, 125.8, 123.4, 122.7, 122.4, 119.5, 112.0; HRMS (ESI+) m/z calcd for C13H10N2Br 273.0027 (M+H), found 273.0023. To a solution of 2-(3-bromophenyl)-1H-benzo[d]imidazole (0.273 g, 0.99 mmol) in anhydrous THF (8 mL) was added NaH (0.048 g, 1.2 mmol, 60% dispersion). The reaction mixture was stirred at room temperature for 20 min then diphenylmethyl chloride (0.41 g, 1.9 mmol) was added. The mixture was stirred at room temperature for 30 min and then heated to 70 °C. After 24 h, the reaction mixture was cooled to room temperature, diluted with sat. NaHCO3, extracted with EtOAc, dried (Na2SO4), and concentrated. The crude residue was purified by chromatography on SiO2 (10 to 20% EtOAc/hexanes) to afford 48 as a white foam (0.50 g, 1.1 mmol, 89%): 1H NMR (500 MHz, CDCl3) δ 7.85-7.82 (m, 2 H), 7.66-7.64 (m, 1 H), 7.51 (d, J = 7.5 Hz, 1 H), 7.36 (app q, J = 3.5 Hz, 7 H), 7.33 (t, J = 8.0 Hz, 2 H), 7.27-7.24 (m, 2 H), 7.18-7.15 (m, 5 H), 7.06-7.03 (m, 1 H), 6.96 (s, 1 H), 6.82 (d, J = 8.5 Hz, 1 H).
1-(3-(1H-Benzo[d]imidazol-2-yl)phenyl)-N-(2-(4-isopropylpiperazin-1-yl)ethyl)piperidin-4-amine (24, UPCDC30250)
A suspension of A (87 mg, 0.24 mmol), 1-benzhydryl-2-(3-bromophenyl)-1H-benzo[d]imidazole (48, 90 mg, 0.21 mmol) and K3PO4 (67 mg, 0.31 mmol) in dry degassed dioxane (1.5 mL) was degassed for 20 min with argon. To this mixture was added Pd2(dba)3 (4 mg, 0.004 mmol) and CyJohnPhos (6 mg, 0.02 mmol). The reaction vial was sealed and the mixture was heated at 110 °C for 12 h. The reaction mixture was diluted with sat. NaHCO3 and extracted with EtOAc (3×). The combined organic layer was washed with brine, dried (Na2SO4), concentrated and purified by chromatography on SiO2 (10 to 20% MeOH/CH2Cl2) to afford tert-butyl (1-(3-(1-benzhydryl-1H-benzo[d]imidazol-2-yl)phenyl)piperidin-4-yl)(2-(4-isopropyl-piperazin-1-yl)ethyl)carbamate as a pale yellow solid (89 mg, 0.12 mmol, 61%): 1H NMR (500 MHz, CDCl3) δ 7.82 (d, J = 8.0 Hz, 1 H), 7.32 (app t, J = 3.5 Hz, 6 H), 7.21 (t, J = 7.5 Hz, 2 H), 7.16 (app t, J = 3.5 Hz, 4 H), 7.10 (d, J = 7.5 Hz, 1 H), 7.04-6.97 (m, 4 H), 6.82 (d, J = 8.5 Hz, 1 H), 4.10 (br s, 1 H), 3.57 (d, J = 11.5 Hz, 2 H), 3.28-3.20 (m, 2 H), 2.67-2.47 (m, 13 H), 1.69 (br s, 5 H), 1.49 (s, 9 H), 1.10 (d, J = 6.5 Hz, 6 H); HRMS (ESI+) m/z calcd for C45H57N6O2 713.4538 (M+H), found 713.4531.
A solution of tert-butyl (1-(3-(1-benzhydryl-1H-benzo[d]imidazol-2-yl)phenyl)piperidin-4-yl)(2-(4-isopropyl-piperazin-1-yl)ethyl)carbamate (0.089 g, 0.13 mmol) in CH2Cl2 (2 mL) was treated with trifluoroacetic acid (2.0 mL) followed by triethylsilane (0.40 mL, 2.5 mmol). The reaction was stirred at room temperature overnight. The solution was then heated at 70 °C overnight, concentrated, diluted with sat. NaHCO3 and extracted with EtOAc (3×). The combined organic layer was washed with brine, dried (Na2SO4) and concentrated. The crude residue was purified by chromatography on SiO2 (10 to 25% MeOH/CH2Cl2) to afford a pale yellow foam which was filtered through basic Al2O3 (0 to 10% MeOH/CH2Cl2) to afford 24 as a pale yellow foam (UPCDC30250, 0.023 g, 0.051 mmol, 41%): IR (ATR) 3069, 2938, 2912, 2811, 1600, 1450, 1357, 1176, 1115, 776, 736 cm−1; 1H NMR (500 MHz, CD3OD) δ 7.74 (t, J = 2.0 Hz, 1 H), 7.61 (dd, J = 6.0, 3.5 Hz, 2 H), 7.53 (d, J = 7.5 Hz, 1 H), 7.38 (t, J = 8.0 Hz, 1 H), 7.27 (dd, J = 6.0, 3.5 Hz, 2 H), 7.11 (dd, J = 8.5, 2.5 Hz, 1 H), 3.85 (d, J = 12.5 Hz, 2 H), 2.84 (t, J = 12.0 Hz, 2 H), 2.77 (t, J = 7.0 Hz, 2 H), 2.68-2.51 (m, 11 H), 2.02 (d, J = 11.5 Hz, 2 H), 1.53 (qd, J = 12.0, 3.5 Hz, 2 H), 1.09 (d, J = 6.5 Hz, 6 H); 31C NMR (125 MHz, CD3OD, several signals missing) δ 152.5, 151.9, 130.2, 129.4, 122.5, 118.1, 117.1, 114.2, 57.0, 54.8, 54.5, 52.7, 48.2, 48.1, 42.4, 31.3, 17.3; HRMS (ESI+) m/z calcd for C27H39N6 447.3231 (M+H), found 447.3230.
2-(3-Bromophenyl)-1-methyl-1H-indole (49)
To a solution of 2-(3-bromophenyl)-1H-indole11 (0.499 g, 1.83 mmol) in DMF (9 mL) was added NaH (0.110 g, 2.75 mmol, 60% dispersion) at 0 °C. The reaction mixture was warmed to room temperature. After 45 min, the reaction mixture was cooled to 0 °C and treated with iodomethane (0.12 mL, 1.9 mmol). The reaction mixture was warmed to room temperature and stirred overnight. The reaction mixture was diluted with H2O and extracted with EtOAc (3×). The combined organic layer was washed with brine, dried (Na2SO4), filtered and concentrated. The residue was purified by chromatography on SiO2 (40% hexanes/CH2Cl2) to give 49 (0.52 g, 1.8 mmol, 99%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.68 (s, 1 H), 7.65 (d, J = 8.0 Hz, 1 H), 7.54 (d, J = 8.0 Hz, 1 H), 7.44 (d, J = 8.0 Hz, 1 H), 7.38 (d, J = 8.8 Hz, 1 H), 7.33 (d, J = 8.0 Hz, 1 H), 7.28 (d, J = 7.6 Hz, 1 H), 7.16 (t, J = 7.6 Hz, 1 H), 6.59 (s, 1 H), 3.75 (s, 3 H); HRMS (ESI+) m/z calcd for C15H13NBr 286.0226 (M+H), found 286.0225.
N-(2-(4-Isopropylpiperazin-1-yl)ethyl)-1-(3-(1-methyl-1H-indol-2-yl)phenyl)piperidin-4-amine (25, UPCDC30201)
A suspension of A (105 mg, 0.297 mmol), 2-(3-bromophenyl)-1-methyl-1H-indole (49, 71 mg, 0.25 mmol) and K3PO4 (81 mg, 0.37 mmol) in dry degassed dioxane (2.0 mL) was treated with Pd2(dba)3 (5 mg, 0.005 mmol) and CyJohnPhos (7 mg, 0.02 mmol). The reaction vial was sealed and the mixture was heated at 110 °C for 11 h under microwave irradiation conditions. The reaction mixture was diluted with sat. NaHCO3 and extracted with EtOAc (3×). The combined organic layers were washed with brine, dried (Na2SO4), concentrated and purified by chromatography on SiO2 (10 to 20%, MeOH/CH2Cl2) to afford tert-butyl(2-(4-isopropylpiperazin-1-yl)ethyl)(1-(3-(1-methyl-1H-indol-2-yl)phenyl)-piperidin-4-yl)carbamate as a pale yellow oil (100 mg, 0.17 mmol, 72%): 1H NMR (500 MHz, CDCl3) δ 7.63 (d, J = 7.5 Hz, 1 H), 7.36-7.32 (m, 2 H), 7.26-7.22 (m, 1 H), 7.15-7.12 (m, 1 H), 7.05 (d, J = 1.5 Hz, 1 H), 6.96 (d, J = 8.5 Hz, 2 H), 6.55 (s, 1 H), 4.15 (br s, 1 H), 3.81 (d, J = 12.5 Hz, 2 H), 3.75 (s, 3 H), 3.25 (br s, 2 H), 2.84-2.82 (m, 2 H), 2.71-2.48 (m, 11 H), 1.85 (br s, 4 H), 1.48 (s, 9 H), 1.06 (d, J = 6.5 Hz, 6 H); 31C NMR (125 MHz, CDCl3) δ 155.4, 151.2, 142.1, 138.3, 133.7, 129.2, 128.0, 121.6, 120.5, 120.4, 119.8, 117.6, 116.0, 109.6, 101.4, 79.8, 58.5, 54.6, 53.8, 53.1, 49.5, 48.6, 40.0, 31.2, 30.1, 28.6, 18.6; HRMS (ESI+) m/z calcd for C34H50O2N5 560.3965 (M+H), found 560.3959.
A solution of trifluoroacetic acid (1.5 mL) and triethylsilane (0.15 mL, 0.93 mmol) in CH2Cl2 (1.5 mL) was added to a solution of tert-butyl(2-(4-isopropylpiperazin-1-yl)ethyl)(1-(3-(1-methyl-1H-indol-2-yl)phenyl)-piperidin-4-yl)carbamate (0.10 g, 0.18 mmol) in CH2Cl2 (1.5 mL). The reaction mixture was stirred at room temperature for 1 h, concentrated, diluted with sat. NaHCO3 and extracted with EtOAc (3×). The combined organic layers were washed with brine, dried (Na2SO4) and concentrated. The crude residue was purified by chromatography on SiO2 (8 to 20% CH2Cl2/MeOH with 1% TEA) followed by filtration through basic Al2O3 (0 to 10% MeOH/CH2Cl2) to afford 25 as a pale yellow solid (UPCDC30201, 0.061 g, 0.13 mmol, 74%): IR (ATR) 2981, 2808, 1596, 1462, 1339, 1178, 1145, 984, 776 cm−1; 1H NMR (400 MHz, acetone-d6) δ 7.56 (d, J = 7.6 Hz, 1 H), 7.40 (dd, J = 8.4, 0.8 Hz, 1 H), 7.34 (t, J = 8.0 Hz, 1 H), 7.19 (td, J = 8.0, 0.8 Hz, 1 H), 7.11-7.07 (m, 2 H), 7.03 (dd, J = 8.0, 2.0 Hz, 1 H), 6.94 (d, J = 7.6 Hz, 1 H), 6.52 (d, J = 0.8 Hz, 1 H), 3.75-3.71 (m, 5 H), 2.86 (td, J = 12.4, 2.8 Hz, 2 H), 2.71 (t, J = 6.0 Hz, 2 H), 2.69-2.62 (m, 2 H), 2.61-2.39 (m, 10 H), 1.96-1.92 (m, 2 H), 1.45 (qd, J = 13.5, 4.0 Hz, 2 H), 0.98 (d, J = 6.5 Hz, 6 H); 31C NMR (100 MHz, acetone-d6) δ 152.6, 143.0, 139.3, 134.3, 129.9, 129.0, 122.1, 120.9, 120.3, 120.2, 117.6, 116.4, 110.5, 101.8, 59.0, 55.6, 54.9, 54.6, 49.4, 48.6, 44.4, 33.2, 31.5, 18.8; HRMS (ESI+) m/z calcd for C29H42N5 460.3440 (M+H), found 460.3443.
Supplementary Material
Acknowledgments
This project was funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Chemical Biology Consortium Contract No. HHSN261200800001E Agreement No. 29XS127TO15. The authors thank Mr. Yongzhao Yan and Dr. Zhizhou Yue for synthetic contributions and Ms. Mary Liang and Taber Lewis for analytical and administrative support. The authors also greatly appreciate the stimulating discussions and valuable suggestions from all members of the CBC p97 team, particularly, Drs. Eric Baldwin (Leidos Biomedical Research), Tsui-Fen Chou (UCLA), Raymond Deshaies (Caltech), Andrew Flint (Leidos Biomedical Research), Gunda Georg (U. Minnesota), Neal Green (Leidos Biomedical Research), William J. Moore (Leidos Biomedical Research), Barbara Mroczkowski (NIH/NCI), R. Jeffrey Neitz (UCSF), Shizuko Sei (Leidos Biomedical Research), Gordon Stott (Leidos Biomedical Research), Sriram Subramaniam (NIH/NCI), and Michael Walters (U. Minnesota).
Footnotes
Author contributions. R.G., D.M.H. and P.W. devised the experiments, J.C.B., L.P.S., M.K., R.C., C.F.M., A.R.H, B.D.P., S.L.B., C.L., M.G.L. and M.R.A. conducted the theoretical and experimental investigations, and everybody interpreted results and contributed to the preparation of parts of this manuscript.
Competing financial interests. The authors declare no competing financial interests.
Additional information. Supporting Information, including copies of 1H and 31C NMR spectra, reprints and permissions information is available at www.xxx.xxx. The authors declare no competing financial interests.
Notes and References
- 1.Xia D, Tang WK, Ye Y. Gene. 2016;583:64–77. doi: 10.1016/j.gene.2016.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li G, Huang C, Zhao G, Lennarz WJ. Proc Nat Acad Sci USA. 2012;109:3737–3741. doi: 10.1073/pnas.1200255109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banerjee S, Bartesaghi A, Merk A, Rao P, Bulfer SL, Yan Y, Green N, Mroczkowski B, Neitz RJ, Wipf P, Falconieri V, Deshaies RJ, Milne JLS, Huryn D, Arkin M, Subramaniam S. Science. 2016;351:871–875. doi: 10.1126/science.aad7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou H-J, Wang J, Yao B, Wong S, Djakovic S, Kumar B, Rice J, Valle E, Soriano F, Menon M-K, Madriaga A, Kiss von Soly S, Kumar A, Parlati F, Yakes FM, Shawver L, Le Moigne R, Anderson DJ, Rolfe M, Wustrow D. J Med Chem. 2015;58:9480–9497. doi: 10.1021/acs.jmedchem.5b01346. [DOI] [PubMed] [Google Scholar]
- 5.Halawani D, Tessier S, Anzellotti D, Bennett DA, Latterich M, Leblanc AC. J Neurosci. 2010;30:6132–6142. doi: 10.1523/JNEUROSCI.5874-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chapman E, Maksim N, De La Cruz F, La Clair JJ. Molecules. 2015;20:3027–3049. doi: 10.3390/molecules20023027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alverez C, Bulfer SL, Chakrasali R, Chimenti MS, Deshaies RJ, Green N, Kelly M, LaPorte MG, Lewis TS, Liang M, Moore WJ, Neitz RJ, Peshkov VA, Walters MA, Zhang F, Arkin MR, Wipf P, Huryn DM. ACS Med Chem Lett. 2016;7:182–187. doi: 10.1021/acsmedchemlett.5b00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gui L, Zhang X, Li K, Frankowski KJ, Li S, Wong DE, Moen DR, Porubsky PR, Lin HJ, Schoenen FJ, Chou TF. ChemMedChem. 2016;11:953–957. doi: 10.1002/cmdc.201600036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tillotson J, Bashyal BP, Kang M, Shi T, De La Cruz F, Gunatilaka AAL, Chapman E. Org Biomol Chem. 2016;14:5918–5921. doi: 10.1039/c6ob00560h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Her N-G, Toth JI, Ma C-T, Wei Y, Motamedchaboki K, Sergienko E, Petroski MD. Cell Chem Biol. 2016;23:517–528. doi: 10.1016/j.chembiol.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alverez C, Arkin MR, Bulfer SL, Colombo R, Kovaliov M, LaPorte MG, Lim C, Liang M, Moore WJ, Neitz RJ, Yan Y, Yue Z, Huryn DM, Wipf P. ACS Med Chem Lett. 2015;6:1225–1230. doi: 10.1021/acsmedchemlett.5b00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chou T-F, Bulfer SL, Weihl CC, Li K, Lis LG, Walters MA, Schoenen FJ, Lin HJ, Deshaies RJ, Arkin MR. J Mol Biol. 2014;426:2886–2899. doi: 10.1016/j.jmb.2014.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.See SI for graphs.
- 14.Davies JM, Brunger AT, Weis WI. Structure. 2008;16:715–726. doi: 10.1016/j.str.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 15.Hänzelmann P, Schindelin H. Structure. 2016;24:127–139. doi: 10.1016/j.str.2015.10.026. [DOI] [PubMed] [Google Scholar]
- 16.Burnett JC, Ruthel G, Stegmann CM, Panchal RG, Nguyen TL, Hermone AR, Stafford RG, Lane DJ, Kenny TA, McGrath CF, Wipf P, Stahl AM, Schmidt JJ, Gussio R, Brunger AT, Bavari S. J Biol Chem. 2007;282:5004–5014. doi: 10.1074/jbc.M608166200. [DOI] [PubMed] [Google Scholar]
- 17.Nguyen TL, McGrath C, Hermone AR, Burnett JC, Zaharevitz DW, Day BW, Wipf P, Hamel E, Gussio R. J Med Chem. 2005;48:6107–6116. doi: 10.1021/jm050502t. [DOI] [PubMed] [Google Scholar]
- 18.Donald A, McHardy T, Rowlands MG, Hunter LJK, Davies TG, Berdini V, Boyle RG, Aherne GW, Garrett MD, Collins I. J Med Chem. 2007;50:2289–2292. doi: 10.1021/jm0700924. [DOI] [PubMed] [Google Scholar]
- 19.McHardy T, Caldwell JJ, Cheung K-M, Hunter LJ, Taylor K, Rowlands M, Ruddle R, Henley A, de Haven Brandon A, Valenti M, Davies TG, Fazal L, Seavers L, Raynaud FI, Eccles SA, Aherne GW, Garrett MD, Collins I. J Med Chem. 2010;53:2239–2249. doi: 10.1021/jm901788j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caldwell JJ, Davies TG, Donald A, McHardy T, Rowlands MG, Aherne GW, Hunter LK, Taylor K, Ruddle R, Raynaud FI, Verdonk M, Workman P, Garrett MD, Collins I. J Med Chem. 2008;51:2147–2157. doi: 10.1021/jm701437d. [DOI] [PubMed] [Google Scholar]
- 21.Good JAD, Wang F, Rath O, Kaan HYK, Talapatra SK, Podgórski D, MacKay SP, Kozielski F. J Med Chem. 2013;56:1878–1893. doi: 10.1021/jm3014597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tapia I, Alonso-Cires L, López-Tudanca PL, Mosquera R, Labeaga L, Innerárity A, Orjales A. J Med Chem. 1999;42:2870–2880. doi: 10.1021/jm981098j. [DOI] [PubMed] [Google Scholar]
- 23.Buu-Hoï NP, Hoán N, Khôi NH, Xuong ND. J Org Chem. 1951;16:309–314. [Google Scholar]
- 24.Malamas MS, Barnes K, Johnson M, Hui Y, Zhou P, Turner J, Hu Y, Wagner E, Fan K, Chopra R, Olland A, Bard J, Pangalos M, Reinhart P, Robichaud AJ. Bioorg Med Chem. 2010;18:630–639. doi: 10.1016/j.bmc.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 25.Livecchi M, Calvet G, Schmidt F. J Org Chem. 2012;77:5006–5016. doi: 10.1021/jo300481s. [DOI] [PubMed] [Google Scholar]
- 26.Calvet G, Livecchi M, Schmidt F. J Org Chem. 2011;76:4734–4740. doi: 10.1021/jo200480h. [DOI] [PubMed] [Google Scholar]
- 27.Dyke HJ, Gazzard LJ, Williams K. WO2011/073263 1,7-Diazacarbazoles as chk1 inhibitors, their preparation, pharmaceutical compositions, and use as anticancer agents and their use in the treatment of cancer. 2011
- 28.Hopkins CR, Collar N. Tetrahedron Lett. 2004;45:8631–8633. [Google Scholar]
- 29.Wagner AM, Hickman AJ, Sanford MS. J Am Chem Soc. 2013;135:15710–15713. doi: 10.1021/ja408112j. [DOI] [PubMed] [Google Scholar]
- 30.Ruan L, Shi M, Mao S, Yu L, Yang F, Tang J. Tetrahedron. 2014;70:1065–1070. [Google Scholar]
- 31.Mukhopadhyay C, Tapaswi PK, Butcher RJ. Aust J Chem. 2009;62:140–144. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.