

Abstract
Background:
Postsynaptic density-95 (PSD-95) protein expression is dysregulated in schizophrenia in a variety of brain regions. We have designed experiments to examine PSD-95 mRNA splice variant expression in the dorsolateral prefrontal cortex from subjects with schizophrenia.
Methods:
We performed quantitative PCR and western blot analysis to measure PSD-95 expression in schizophrenia vs control subjects, rodent haloperidol treatment studies, rodent postmortem interval studies, and GluN1 knockdown (KD) mice vs controls.
Results:
We found decreased mRNA expression of beta (t = 4.506, df = 383, P < .0001) and truncated (t = 3.378, df = 383, P = .0008) isoforms of PSD-95, whereas alpha was unchanged. Additionally, we found decreased PSD-95 protein expression in schizophrenia (t = 2.746, df = 71, P = .0076). We found no correlation between PSD-95 protein and alpha, beta, or truncated mRNA isoforms in schizophrenia. PSD-95 beta transcript was increased (t = 3.346, df = 14, P < .05) in the GluN1 KD mouse model of schizophrenia. There was an increase in PSD-95 alpha mRNA expression (t = 2.905, df = 16, P < .05) in rats following long-term haloperidol administration.
Conclusions:
Our findings describe a unique pathophysiology of specific PSD-95 isoform dysregulation in schizophrenia, chronic neuroleptic treatment, and a genetic lesion mouse model of drastically reduced N-methyl-d-aspartate receptor (NMDAR) complex expression. These data indicate that regulation of PSD-95 is multifaceted, may be isoform specific, and biologically relevant for synaptic signaling function. Specifically, NMDAR-mediated synaptic remodeling, and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking and interaction may be impaired in schizophrenia by decreased PSD-95 beta and truncated expression (respectively). Further, increased PSD-95 beta transcript in the GluN1 KD mouse model poses a potential compensatory rescue of NMDAR-mediated function via increased postsynaptic throughput of the severely reduced GluN1 signal. Together, these data propose that disruption of excitatory signaling complexes through genetic (GluN1 KD), pharmacologic (antipsychotics), or disease (schizophrenia) mechanisms specifically dysregulates PSD-95 expression.
Keywords: isoform, mRNA, protein, schizophrenia, splice variant, postmortem, PSD-95
Introduction
Excitatory (glutamatergic) synapses facilitate interneuronal signaling with high fidelity by transducing extracellular information (ie, ligand binding induced ionotropic and metabotropic receptor activation) onto intracellular signaling cascades. Importantly, for accurate reproduction of signal intensity, receptors, ion channels, and signaling machinery need to be appropriately clustered within a specialized area of the postsynaptic neuron termed the postsynaptic density. Postsynaptic densities are nanodomains comprised of about 1000 unique proteins that interact with one another via specialized multipotent scaffolding molecules.1 Postsynaptic density (PSD-95) is the most abundant scaffolding protein within the postsynaptic density.2–4 PSD-95 links glutamate receptors, signaling molecules, and other structural proteins. More than 95% of PSD-95 expression is localized to excitatory synapses.5 Manipulating the expression of PSD-95 significantly changes synaptic function, mimicking long-term potentiation (LTP) and enhancing long-term depression (LTD). For example, PSD-95 overexpression increases the amplitude of excitatory postsynaptic currents (EPSCs) to the extent that additional stimulus is unable to further strengthen LTP.6–10 Reciprocally, PSD-95 knockdown (KD) suppresses EPSCs, indicating the importance of PSD-95 for the regulation of learning and memory.11,12
In humans, there are 3 distinct PSD-95 isoforms: alpha, beta, and truncated.13–15 The truncated isoform has identical N-terminal structures to the alpha form but stops prematurely. The beta isoform constitutes ~10% of total PSD-95 in human brain and has a unique N-terminus, consisting of 53 amino acids that differentiates it from the alpha isoform.13 The beta N-terminus is similar to that of SAP97 (another MAGUK family scaffolding protein), as both contain an L27 domain which is responsible for direct interaction with CASK. This L27 domain confers the ability to cluster PSD-95 beta at synaptic sites.13 For PSD-95 alpha, synaptic clustering is regulated by 2 N-terminal cysteines which may be palmitoylated and targeted to the plasma membrane. Rodents have alpha, beta, and gamma PSD-95 transcripts (all expressed in cortex), of which alpha and beta are homologous to the human isoforms.16
Along with findings of reduced dendritic spine density and synapse number, alterations in PSD-95 have been reported in schizophrenia in a variety of brain regions, including the anterior cingulate cortex (ACC), dorsolateral prefrontal cortex (DLPFC), and the occipital cortex, across multiple cohorts, suggesting alterations in synaptic clustering of proteins in excitatory synapses.17–25 In addition, a recent genetic analysis of PSD-95 in a Taiwanese population found 2 core region polymorphic markers associated with schizophrenia susceptibility.26 Another study found an association of PSD-95 single nucleotide polymorphism (SNP) rs17203281, which did not survive correction for multiple comparisons, but the authors did find a haplotype association involving 5 SNPs in both Japanese and Chinese populations.27 These studies suggest a genetic and functional link between alterations in PSD-95 protein expression and schizophrenia.
We have designed a series of experiments to examine alterations in PSD-95 mRNA splice variant expression in the DLPFC from human subjects with schizophrenia. We also examined PSD-95 protein levels in the DLPFC to explore the relationship between PSD-95 splice variants and protein levels, using an antibody that detects all isoforms. The DLPFC is a highly implicated region in schizophrenia with myriad studies detailing protein and mRNA alterations in this illness.18,28–32 The DLPFC was selected due to its importance in executive cognitive function and working memory, which are abnormal in subjects with schizophrenia.33,34 Our study adds to the growing literature of synaptic alterations in schizophrenia and tests the hypothesis that PSD-95 mRNA expression may be driven by isoform-specific changes in this critical element of synaptic plasticity.
Experimental Materials and Methods
Human Tissue Acquisition and Preparation
Postmortem brains were collected at the Human Brain Collection Core (HBCC), National Institute of Mental Health (NIMH), with informed consent from the legal next of kin according to the National Institutes of Health Institutional Review Board and ethical guidelines under NIMH protocol (90-M-0142). Clinical characterization, neuropathological screening, toxicological analyses, and dissection of the DLPFC were performed as previously described.35 Briefly, all patients met DSM-IV criteria for a lifetime axis I diagnosis of schizophrenia or schizoaffective disorder. Comparison subjects were defined as those individuals with no history of significant psychological problems or psychological care, psychiatric admissions, or drug detoxification and with no known history of psychiatric symptoms or substance abuse, as determined by both telephone screening and medical examiner documentation, as well as negative toxicology results. Brains were removed from the skull, wrapped in plastic, and transported on wet ice. The brains were hemisected, cut into 1.5 cm coronal slabs, rapidly frozen in a prechilled dry-ice/isopentane slurry bath (−40°C), and stored at −80°C. A portion of cerebellum was pulverized for pH measurement. For DLPFC dissections, grey matter tissue from the crown of the middle frontal gyrus was obtained from the coronal slab corresponding to the middle one-third immediately anterior to the genu of the corpus callosum. Subcortical white matter was carefully trimmed from the area immediately below the middle frontal gyrus. Blocks of DLPFC were sectioned at 14 µm and mounted on glass slides prior to protein studies, while pulverized tissues from DLPFC were used for mRNA measurements (table 1).
Table 1.
Subject Demographics
| mRNA | Protein | |||
|---|---|---|---|---|
| Dx | Ctl | Scz | Ctl | Scz |
| Age | 44.17 ± 15.5 | 50.22 ± 14.9 | 41.72 ± 14.7 | 58.63 ± 16.7 |
| pH | 6.55 ± 0.27 | 6.41 ± 0.25 | 6.54 ± 0.33 | 6.37 ± 0.35 |
| PMI (h) | 30.7 ± 14.5 | 38.5 ± 24.1 | 35 ± 15.7 | 42.2 ± 21.5 |
| Sex | 61f, 149m | 65f, 110m | 18f, 32m | 14f, 13m |
Note: Ctl, Control; Dx, diagnosis; PMI, postmortem interval; Scz, Schizophrenia. f, female; m, male.
Antipsychotic-Treated Rats
Rodent experiments were carried out according to University of Alabama guidelines and all procedures complied with IACUC regulations. Male Sprague-Dawley rats (250 g) were treated with haloperidol decanoate for 9 months. Rats were housed in pairs and injected intramuscularly every 3 weeks, for a total of 12 injections, with vehicle (sesame oil) or 28.5 mg/kg of haloperidol decanoate in sesame oil. Rats were decapitated and the brains were immediately removed, dissected on wet ice, and the left anterior cortex was then stored at −80°C. The tissue was then prepared for western blot and quantitative PCR (qPCR) analyses. Ten haloperidol decanoate treated and 10 control rats were used for each experiment.
GluN1 KD Mouse Study
Brains from 5 male transgenic GluN1 KD and 5 male littermate wild-type (WT) mice were provided by Dr Amy Ramsey (University of Toronto).36 The brains arrived on dry ice and were stored at −80°C until the time of the experiment. The frontal cortex was dissected from brain and processed for western blot and qPCR analyses.
mRNA Extraction and cDNA Production
Total RNA was extracted from ~100 mg of tissue using the RNeasy kit (Qiagen) according to the manufacturer’s protocol. The yield of total RNA was determined by spectrophotometry by measuring the absorbance at 260 nm. The RNA quality of human samples was assessed by high-resolution capillary electrophoresis on an Agilent Bioanalyzer 2100 (Agilent Technologies). Samples with RNA integrity number (RIN) <5 were excluded. cDNA was created from 4 µg total RNA using SuperScript First-Strand Synthesis System for real-time PCR (Invitrogen), according to the manufacturer’s protocol.
Human and Rodent Protein Extraction
Tissue sections (human samples) were scraped from each slide, and frontal cortex dissections (rat and mouse samples) were homogenized in 5 mM Tris–HCl, pH 7.4, 0.32 M sucrose, and HALT protease and phosphatase inhibitor. The homogenates were assayed for protein concentration using a BCA protein assay kit (Thermo Scientific) and stored at −80°C.
Western Blot Analysis
Human and rodent experiments were performed as follows. We tested the antibodies (PSD-95, 1:1000, Cell Signaling #D27E11; β-tubulin, 1:10000, Millipore #05-661) using varying concentrations of total protein of rat brain tissue homogenate. These control studies demonstrated that the assays were linear for the protein concentrations used in our western blot studies. Samples for western blot analysis were diluted with ultrapure (Milli-Q A10, Millipore) water and reducing buffer (6× solution: 4.5% sodium dodecyl sulfate (SDS), 15% β-mercaptoethanol, 0.018% Bromophenol blue, and 36% glycerol in 170 mM Tris–HCl, pH 6.8) to a concentration of 20 µg of protein per 12 µl and heated at 70°C for 10 minutes. Samples were then processed in duplicate by SDS–polyacrylamide gel electrophoresis (Invitrogen) using 4–12% gradient gels and transferred to polyvinylidene fluoride membranes via BioRad semi-dry transblotters. The membranes were blocked with LiCor blocking buffer for 1 hour at room temperature, then probed with primary antisera to PSD-95 (1:1000) diluted in 0.1% Tween LiCor blocking buffer. The membranes were washed twice for 10 minutes each in 0.1% Tween phosphate buffer solution (PBST) then probed with goat anti-mouse or goat anti-rabbit IR-Dye 670 or 800cw labeled secondary antisera (LiCor) in 0.1% Tween and 0.01% SDS LiCor blocking buffer for 1 hour at room temperature. Washes were repeated after secondary labeling, washing twice for 10 minutes in PBST then placed in water. Membranes were imaged using a LiCor Odyssey scanner. Boxes were manually placed around each band of interest which returned near-infrared fluorescent values of raw intensity with intra-lane background subtracted using Image Studio 4.0 analytical software (LiCor). The near-infrared fluorescence value for each protein of interest was normalized to the in-lane value of β-tubulin and this normalized ratio from duplicate lanes was averaged.37 We found no changes in raw intensity values for tubulin between the schizophrenia and comparison groups, consistent with previous reports.19,37,38
SYBR Green PCR
Rat SYBR green PCR experiments were performed as follows: primers were designed for the alpha, beta, and gamma isoforms of PSD-95 based on published studies, then confirmed by sequencing.16 The rodent alpha and beta isoforms are homologous to human alpha and beta, respectively. PCR assays for each target isoform were performed in duplicate on cDNA samples in 96-well optical plates on a StepOnePlus Real-Time PCR system (ThermoFisher). For every sample, an amplification plot was generated, showing the increase in the reporter dye fluorescence with each cycle of PCR. Peptidylprolyl isomerase A (PPIA, a.k.a. cyclophilin), beta-2-microglobulin (B2M), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were expressed in all samples and were used as standards. The relative expression levels of transcripts were calculated using the Relative Standard Curve Method as previously described.39,40 Standard curves were generated for each assay. The curve was generated from measurements of a mixture of serial dilutions of a pool of all experimental samples. Using the linear equations describing these standard curves, the relative amounts of each transcript were determined. The relative expression levels of target mRNAs were computed as the ratio of the target mRNA to the geometric mean of the 3 housekeeping assays.
Mouse SYBR green PCR experiments were performed almost identically to the rat qPCR experiments, with the exception of using alternative internal standards of glucuronidase beta (GUSB) and hypoxanthine phosphoribosyltransferase 1 (HPRT1), along with PPIA. GUSB and HPRT1 were used due to tamoxifen induced differences in B2M and GAPDH expression.
TaqMan PCR
Human experiments were performed as follows. Primers and probes used were standard proprietary assays designed by Applied Biosystems except for the truncated PSD-95 isoform. For the combined α and truncated analysis we used primer/probe #Hs01250877_m1, and #Hs01555378_m1 for the β isoform. We designed a custom primer/probe for the truncated only analysis using the following sequences: forward primer: TCCCTCTTCAGATATTCCCTATGAA, reverse primer: AATTGATTTTCCATCTCTTCTCCAA, probe: CCTCT GATCCTTGCTGTT. TaqMan gene expression assays were run on an ABI 7900HT with 10 µl of total reaction volume in 384-well format. Cycle threshold values were normalized to a standard curve as described above and then each reaction was normalized to the geometric mean of 3 housekeeping genes (actin #Hs99999903_m1, β2 microglobulin #Hs99999907_m1, and glucuronidase β #Hs99999908_m1).
Data Analysis
Data were analyzed using Statistica (Statsoft). All dependent measures were determined to have a Gaussian distribution. Correlation analyses were performed to determine associations between the dependent variables and pH, age, RIN, and postmortem interval (PMI). Sex and antipsychotic medication status were used as grouping variables for secondary analyses. One-way ANCOVA was used to analyze the data when significant correlations with potential covariates were found, otherwise unpaired t test was used. Data sets with differences in homogeneity of variance were analyzed by unpaired t test with Welch’s correction. No correlations between age, pH, RIN, or PMI and any dependent measures were detected.
Results
We found decreased mRNA expression of the beta (t = 4.506, df = 383, P < .0001) and truncated (t = 3.378, df = 383, P = .0008) forms of PSD-95 in the DLPFC of patients with schizophrenia (N = 175) as compared to control subjects (N = 210). The alpha isoform was unchanged (figure 1). No change was detected when mRNA isoform expression was analyzed post hoc by category of positive vs negative for anticonvulsants, antidepressants, or antipsychotics. PSD-95 protein expression analysis of subjects who tested positive vs negative for antipsychotics at time of death detected no change (protein cohort size did not permit the analysis of anticonvulsants or antidepressants).
Fig. 1.

Decreased isoform-specific postsynaptic density-95 (PSD-95) mRNA expression in the dorsolateral prefrontal cortex (DLPFC) in schizophrenia. Quantitative PCR analysis of mRNA isolated from control (ctl, N = 210) and schizophrenia (scz, N = 175) subjects revealed decreased isoform-specific expression in the DLPFC in schizophrenia. No change was found with an assay that detected both alpha (α) and truncated (trunc) isoforms, while schizophrenia samples had decreased beta (β) (t = 4.7, df = 346.7, P < .05) and truncated isoform expression (t = 3.5, df = 373.5, P < .05). *P < .05.
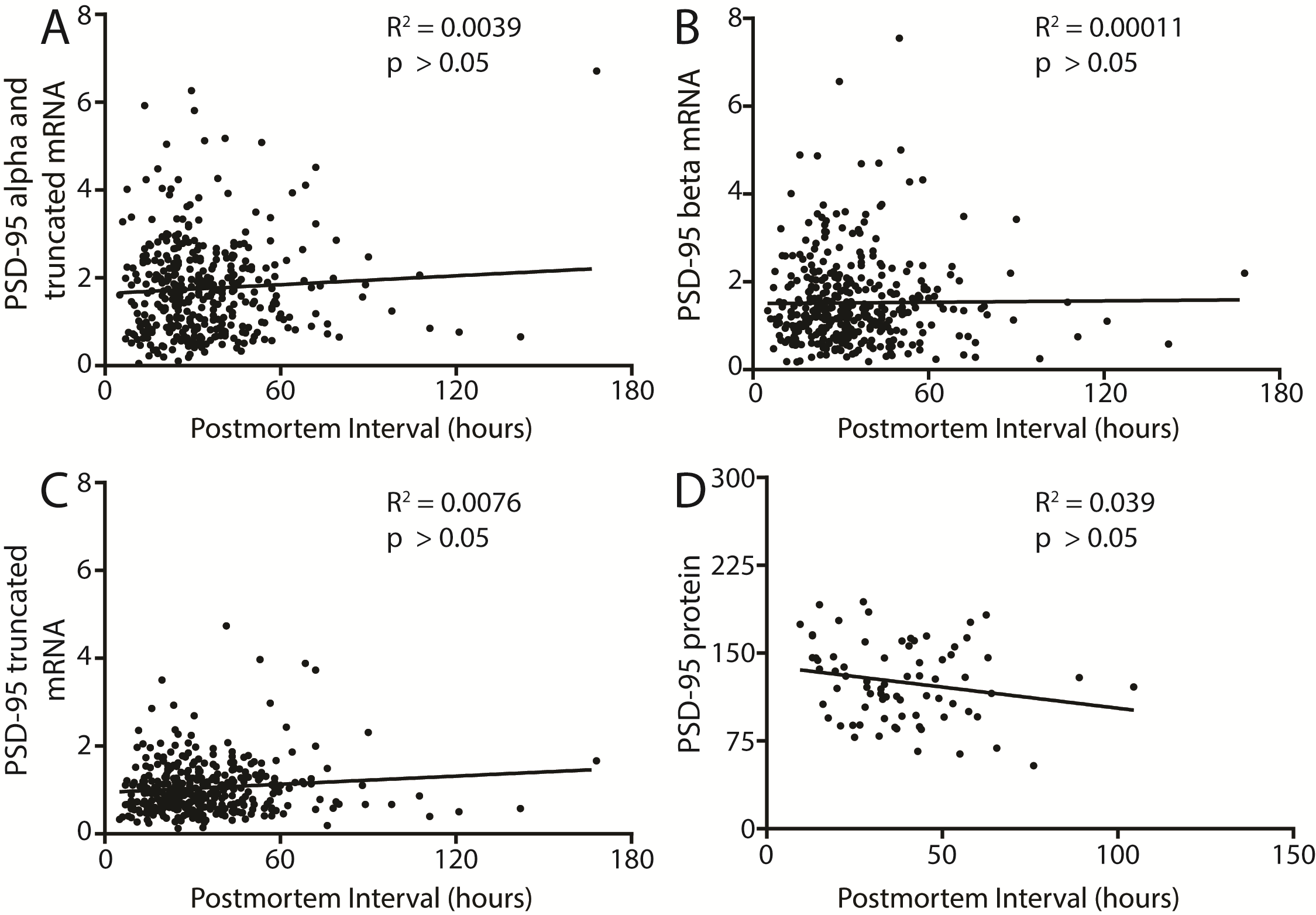
We also found decreased PSD-95 protein expression in the DLPFC in schizophrenia (N = 26) as compared to control subjects (N = 47; t = 2.746, df = 71, P = .0076) (figure 2). There were no significant correlations between PMI and any transcript isoform or PSD-95 protein expression in the human samples (supplementary figure S1). We also performed a rat PMI study to mimic the handling of human samples. We found no difference in PSD-95 protein expression between 0, 24, or 48 hours PMI in rat frontal cortex (supplementary figure S2).
Fig. 2.

Decreased postsynaptic density-95 (PSD-95) protein expression in the dorsolateral prefrontal cortex (DLPFC) in schizophrenia. Western blot analysis of protein isolated from control (C, N = 47) and schizophrenia (S, N = 26) subjects revealed decreased protein expression (t = 2.746, df = 71, P < .05) in the DLPFC from a subset of subjects used in the mRNA studies. (A) Graphed analysis of normalized PSD-95 protein expression. (B) Representative western blot, in duplicate for PSD-95 and β-tubulin. *P < .05.
We performed correlation analyses in the human DLPFC samples between total PSD-95 protein and mRNA isoforms. We detected no significant correlations between PSD-95 mRNA isoforms and total PSD-95 protein expression in this data set (data not shown). This may be due to the antibody detecting all forms of PSD-95 protein.
In haloperidol-treated rats, we found increased PSD-95 alpha isoform (PSD-95α) mRNA expression (t = 2.905, df = 16, P < .05) in frontal cortex vs vehicle control treated. No changes in the beta or gamma isoforms were detected (figure 3).
Fig. 3.

Haloperidol-treated rats have increased postsynaptic density-95 alpha (PSD-95α) mRNA expression in frontal cortex. Quantitative PCR analysis of mRNA isolated from control (ctl, N = 9) and haloperidol (hal, N = 9) treated rats revealed increased PSD-95α expression (t = 2.9, df = 16, P < .05) in the frontal cortex. We found no changes in PSD-95 beta or gamma isoforms. *P < .05.
In an analogous model of broken synapses (GluN1 KD mice),36 we measured PSD-95 transcript expression and found an increase in PSD-95 beta (t = 2.466, df = 7, P < .05) in the KD mice versus WT (figure 4). We then performed western blot analyses from these same samples and detected no difference between KD and WT (figure 4).
Fig. 4.

Analysis of the GluN1 NMDAR knockdown (KD) mouse model of schizophrenia. PSD-95 protein expression is unchanged in GluN1 KD mice (A, B). GluN1 KD (N = 5) mice have increased postsynaptic density-95 beta (PSD-95β) mRNA expression (t = 2.466, df = 7, P < .05) vs littermate wild-type (WT, N = 4) mice (C). *P < .05.
Discussion
In schizophrenia, substantial evidence suggests an abnormality of properly formed/functioning excitatory synapses, including decreased spine density and number.41,42 The experiments described here indicate alterations in trafficking and structural proteins that regulate synaptic protein-protein complexes important for cognition.43 Multiple reports indicate alterations of PSD-95 in the thalamus including, an increase, no change, and a decrease in mRNA measured by in situ, qPCR, and in situ (respectively) in schizophrenia.44–46 In cortical samples, PSD-95 transcript were unchanged in DLPFC, significantly decreased in BA9, unchanged in hippocampus, and increased in ACC samples from multiple different cohorts, including the Stanley Foundation and Bronx VA brain collections.47–49 Whereas, PSD-95 protein was significantly decreased in the ACC from the Bronx VA.18,19,49 These disparate findings may be a result of differences in technique, study cohort, or specificity of thalamic nuclei/cortical area/regional section used for the experiments. Additionally, many previous studies may have been underpowered (some with Ns ≤ 10 per group) to detect significant changes in transcript or protein. In the DLPFC, we show isoform-specific decreases in PSD-95 mRNA (figure 1), which suggests that the decreases in protein expression we find (figure 2) may be isoform specific. This is supported by the finding that PSD-95 isoforms have differential regulatory regions in the N-terminus and uniquely traffic to synaptic sites.13 There are many mechanisms whereby transcript and protein may be regulated independently of each other, and less than 30% of human genes show strong correlation between transcript and protein expression.50 Therefore, PSD-95 protein could be negatively regulated by a variety of posttranslational modifications to decrease expression, irrespective of mRNA levels. One possibility includes the ubiquitin proteasome pathway, whereby PSD-95 is ubiquitinated by Mdm2 and targeted to the proteasome for degradation.51 There is evidence indicating selective synaptic ubiquitination of PSD-95 by Mdm2 after a reduction in Cdk5 does not result in proteasome degradation.52 Thus, simple ubiquitin analyses of PSD-95 may not be informative to its fate. Interestingly, we have recently shown increased Cdk5 protein expression in the DLPFC from a different cohort, suggesting that PSD-95 may have less Mdm2-mediated ubiquitination in this region in schizophrenia.18 However, follow-up studies on PSD-95 ubiquitination and degradation would be necessary to inform and support this hypothesis.
To further explore the role of PSD-95 isoform expression on brain development, we measured transcripts and protein in the GluN1 KD mouse model of glutamatergic dysfunction.36 Unexpectedly, we found an increase in PSD-95β transcript expression (figure 4). These mice have been well characterized and show decreased expression of the disrupted in schizophrenia-1 (DISC1) gene.53 These data suggest that excitatory synapse composition is deleteriously remodeled when the GluN1 N-methyl-d-aspartate receptor (NMDAR) subunit is decreased (~90% less in GluN1 KD mice). Further, an increase in PSD-95β transcript could suggest a compensatory increase in NMDAR-mediated signaling, as PSD-95β is trafficked in response to NMDAR-mediated CaMKII activation, with subsequent downstream modulation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) localization.54 These data are interesting but should be considered cautiously when compared to schizophrenia, as schizophrenia is associated with polygenic risk vs a single lesion to the GluN1 subunit in this model.42 In these mice, we found no change in PSD-95 protein expression at the region level, which is not unexpected as PSD-95β is only thought to contribute ~10% to overall PSD-95 protein levels.13 Although our GluN1 KD study may have been underpowered (N = 5 per group) to detect a small change in protein expression, an increase in PSD-95β expression of even 50% would be insufficient to differentiate between isoforms at the protein level.
There are some caveats that should be noted related to the rodent and human PSD-95 isoform comparisons in this study. In rodents, there is no truncated version. Also, the rodent has a gamma isoform which is not present in human tissues. The alpha and beta isoforms of human and mice are homologous. Alpha PSD-95 comprises ~80–90% of human brain expression, with beta and truncated contributing another ~10–20%.13 Experimental data indicate that alternative forms of PSD-95 which lack their N-terminal domains block LTD.55 The N-terminal domain is necessary for AMPAR-mediated signaling but is insufficient to induce NMDAR-dependent LTP.9,55 This is likely due to the N-terminus playing an important role in the dimerization of PSD-95, and linkage to AMPARs but not NMDARs.55 Evidence for divergent roles between alpha and beta PSD-95 include shRNA KD of endogenous PSD-95α, which only alters AMPAR, but not NMDAR EPSCs.54 In the same study, rescue or overexpression of PSD-95α resulted in a 2- to 3-fold increase in EPSC over baseline vs recovery back to baseline with overexpression of PSD-95β. Additionally, the authors showed that PSD-95β-mediated trafficking of AMPARs requires activity-dependent NMDAR transmission. They went on to show that PSD-95β trafficking of AMPARs is CaMKII dependent. Therefore, isoform-specific decreases in PSD-95 protein levels could alter the ability of synapses to traffic AMPARs directly via PSD-95α and truncated or indirectly through NMDAR-mediated activation and trafficking of PSD-95β.
The N-terminus of PSD-95α contains palmitoylation sites important for trafficking, but other studies indicate an inability to reach synaptic sites in constructs without the C-terminus, suggesting the C-terminus is the primary mediator of synaptic localization.55 This was only uncovered after knockdown of endogenous PSD-95, due to the masking effect of truncated dimerization with full length endogenous PSD-95. Our findings suggest that lower PSD-95 protein expression may be due to decreased beta and truncated PSD-95 isoforms. Synaptic targeting of PSD-95β is dependent on Hrs, a vesicular transport protein, which binds to the L27 domain of PSD-95β.13 Functionally, PSD-95β couples with CASK (a Lin-2 homolog serine kinase) which is important for proper GluN2B localization. Therefore, if there were a selective reduction in PSD-95β and truncated isoforms, we would expect to find abnormal localization of the GluN2B NMDAR subunit (due to reduced beta interaction with CASK), and lower AMPAR-mediated currents (due to reduced dimerization of truncated with alpha isoforms). Indeed, there is evidence from postmortem studies in the DLPFC which show decreased CASK transcript and increased phosphorylation of Tyr1336 on GluN2B NMDAR subunit, suggesting increased extra-synaptic localization of GluN2B in schizophrenia.18 Additionally, we have shown evidence of abnormal trafficking of AMPAR subunits from postmortem schizophrenia.38 These data, coupled with recent reports that NMDARs are alternatively spliced in subjects with schizophrenia, with an increase in a particular isoform which exits from the endoplasmic reticulum at a faster rate due to a lack of the retention motif, suggest alterations of excitatory synaptic molecule trafficking and scaffolding structure that align with the data we present here.49,56
Although the PMI times were long, the measured RIN values of the RNA samples were well within acceptable ranges (Control = 8.25 ± 0.7, Schizophrenia = 7.84 ± 1). Moreover, we found no significant correlations between either PMI (supplementary figure S1) or RIN (data not shown) and the dependent measures.57 Additionally, we found no change in PSD-95 protein expression in rat cortex homogenate from brain samples subject to PMI treatment of 0, 24, or 48 hours as described in the methods (supplementary figure S2).
Antipsychotic medication may differentially influence expression of mRNA or protein. Therefore, we examined the effect of long-term haloperidol administration in rats on the expression of PSD-95 mRNA isoforms. These data indicate no change for rodent beta or gamma PSD-95, with a significant increase in the alpha variant. This is unexpected and may represent a functional change in PSD-95 whereby palmitoylation may play an exaggerated role in the trafficking of PSD-95 by typical antipsychotics. Future studies on the regulation of PSD-95 with atypical antipsychotics and a comprehensive analysis of PSD-95 posttranslational modifications after antipsychotic treatment would be of value. Unfortunately, there are currently no available antibodies that can distinguish between alpha and beta PSD-95. Although correlation analyses between protein and isoform-specific transcript did not indicate an effect, this is not unexpected, as we measured total protein rather than isoforms. It should be noted that related MAGUK family proteins, PSD-93 (DLG2) and SAP97 (DLG1), are genome-wide association studies (GWAS) schizophrenia risk genes, and mutations in PSD-93 or SAP97 may contribute to synaptic malformation or dysfunction, altering normal excitatory synaptic signaling and the postsynaptic landscape.58 Importantly, these genetic findings support the hypothesis that genetic disruption (SNP, isoform expression, deletion, or translocation) of synaptic structural, signaling, and trafficking proteins is a significant risk factor in the development of severe mental illness including schizophrenia.58
Accumulating evidence suggests that schizophrenia is a disorder of broken synapses, with convergence of genetic risk factors for synaptic proteins and other factors that modulate neuroplasticity.42,58–60 Our work supports and extends this hypothesis, identifying synaptic substrates associated with the pathophysiology of this often devastating illness. Taken together, our findings suggest that PSD-95 protein changes may be isoform specific. Biologically, this may represent abnormal regulation of differential trafficking of PSD-95, as alpha and beta isoforms are uniquely targeted to postsynaptic sites. This opens up the potential for future studies measuring isoform-specific PSD-95 protein expression in schizophrenia and other neurodevelopmental and degenerative diseases with PSD-95 alterations.
Supplementary Material
Supplementary data are available at Schizophrenia Bulletin online.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
The authors would like to thank the families of those with severe mental illness for their continued support of basic research to understand the cellular and molecular alterations in schizophrenia. We thank Jonathan Sirovatka and Vesna Imamovic for their technical expertise. We also thank the Offices of the Chief Medical Examiner of Washington, DC and of Northern Virginia-Northern District and Dr Maree Webster at the Stanley Medical Research Institute for the contribution of brain tissues. We also thank the families of the deceased for the donations of brain tissue and their time and effort devoted to the consent process and interviews. We would also like to thank the LIFE Foundation, the Lindsay Brinkmeyer Schizophrenia Research Fund, and UCNI for their financial support. This project was supported by NIMH grant numbers MH094445 and MH107916 and by the Intramural Research Program of the National Institute of Mental Health at the National Institutes of Health. The authors have declared that there are no conflicts of interest in relation to the subject of this study.
References
- 1. Bayés A, van de Lagemaat LN, Collins MO, et al. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat Neurosci. 2011;14:19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen X, Vinade L, Leapman RD, et al. Mass of the postsynaptic density and enumeration of three key molecules. Proc Nat Acad Sci USA. 2005;102:11551–11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cheng D, Hoogenraad CC, Rush J, et al. Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol Cell Proteomics. 2006;5:1158–1170. [DOI] [PubMed] [Google Scholar]
- 4. Sugiyama Y, Kawabata I, Sobue K, Okabe S. Determination of absolute protein numbers in single synapses by a GFP-based calibration technique. Nat Methods. 2005;2:677–684. [DOI] [PubMed] [Google Scholar]
- 5. Fortin DA, Tillo SE, Yang G, et al. Live imaging of endogenous PSD-95 using ENABLED: a conditional strategy to fluorescently label endogenous proteins. J Neurosci. 2014;34:16698–16712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Béïque JC, Andrade R. PSD-95 regulates synaptic transmission and plasticity in rat cerebral cortex. J Physiol. 2003;546:859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ehrlich I, Malinow R. Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci. 2004;24:916–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Elias GM, Elias LA, Apostolides PF, Kriegstein AR, Nicoll RA. Differential trafficking of AMPA and NMDA receptors by SAP102 and PSD-95 underlies synapse development. Proc Nat Acad Sci USA. 2008;105:20953–20958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Nat Acad Sci USA. 2002;99:13902–13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stein V, House DR, Bredt DS, Nicoll RA. Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J Neurosci. 2003;23:5503–5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ehrlich I, Klein M, Rumpel S, Malinow R. PSD-95 is required for activity-driven synapse stabilization. Proc Nat Acad Sci USA. 2007;104:4176–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA. Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron. 2006;52:307–320. [DOI] [PubMed] [Google Scholar]
- 13. Chetkovich DM, Bunn RC, Kuo SH, Kawasaki Y, Kohwi M, Bredt DS. Postsynaptic targeting of alternative postsynaptic density-95 isoforms by distinct mechanisms. J Neurosci. 2002;22:6415–6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stathakis DG, Hoover KB, You Z, Bryant PJ. Human postsynaptic density-95 (PSD95): location of the gene (DLG4) and possible function in nonneural as well as in neural tissues. Genomics. 1997;44:71–82. [DOI] [PubMed] [Google Scholar]
- 15. Rousset R, Fabre S, Desbois C, Bantignies F, Jalinot P. The C-terminus of the HTLV-1 Tax oncoprotein mediates interaction with the PDZ domain of cellular proteins. Oncogene. 1998;16:643–654. [DOI] [PubMed] [Google Scholar]
- 16. Bence M, Arbuckle MI, Dickson KS, Grant SG. Analyses of murine postsynaptic density-95 identify novel isoforms and potential translational control elements. Brain Res Mol Brain Res. 2005;133:143–152. [DOI] [PubMed] [Google Scholar]
- 17. de Bartolomeis A, Latte G, Tomasetti C, Iasevoli F. Glutamatergic postsynaptic density protein dysfunctions in synaptic plasticity and dendritic spines morphology: relevance to schizophrenia and other behavioral disorders pathophysiology, and implications for novel therapeutic approaches. Mol Neurobiol. 2014;49:484–511. [DOI] [PubMed] [Google Scholar]
- 18. Funk AJ, McCullumsmith RE, Haroutunian V, Meador-Woodruff JH. Abnormal activity of the MAPK- and cAMP-associated signaling pathways in frontal cortical areas in postmortem brain in schizophrenia. Neuropsychopharmacology. 2012;37:896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Funk AJ, Rumbaugh G, Harotunian V, McCullumsmith RE, Meador-Woodruff JH. Decreased expression of NMDA receptor-associated proteins in frontal cortex of elderly patients with schizophrenia. Neuroreport. 2009;20:1019–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kristiansen LV, Patel SA, Haroutunian V, Meador-Woodruff JH. Expression of the NR2B-NMDA receptor subunit and its Tbr-1/CINAP regulatory proteins in postmortem brain suggest altered receptor processing in schizophrenia. Synapse. 2010;64:495–502. [DOI] [PubMed] [Google Scholar]
- 21. Hahn CG, Wang HY, Cho DS, et al. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat Med. 2006;12:824–828. [DOI] [PubMed] [Google Scholar]
- 22. Kristiansen LV, Meador-Woodruff JH. Abnormal striatal expression of transcripts encoding NMDA interacting PSD proteins in schizophrenia, bipolar disorder and major depression. Schizophr Res. 2005;78:87–93. [DOI] [PubMed] [Google Scholar]
- 23. Dracheva S, Marras SA, Elhakem SL, Kramer FR, Davis KL, Haroutunian V. N-methyl-d-aspartic acid receptor expression in the dorsolateral prefrontal cortex of elderly patients with schizophrenia. Am J Psychiatry. 2001;158:1400–1410. [DOI] [PubMed] [Google Scholar]
- 24. Egbujo CN, Sinclair D, Borgmann-Winter KE, Arnold SE, Turetsky BI, Hahn CG. Molecular evidence for decreased synaptic efficacy in the postmortem olfactory bulb of individuals with schizophrenia. Schizophr Res. 2015;168:554–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dean B, Gibbons AS, Boer S, et al. Changes in cortical N-methyl-d-aspartate receptors and post-synaptic density protein 95 in schizophrenia, mood disorders and suicide. Aust NZ J Psychiatry. 2015;50: 275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cheng MC, Lu CL, Luu SU, et al. Genetic and functional analysis of the DLG4 gene encoding the post-synaptic density protein 95 in schizophrenia. PLoS One. 2010;5:e15107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Balan S, Yamada K, Hattori E, et al. Population-specific haplotype association of the postsynaptic density gene DLG4 with schizophrenia, in family-based association studies. PLoS One. 2013;8:e70302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lewis DA, González-Burgos G. Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology. 2008;33:141–165. [DOI] [PubMed] [Google Scholar]
- 29. Mueller HT, Meador-Woodruff JH. NR3A NMDA receptor subunit mRNA expression in schizophrenia, depression and bipolar disorder. Schizophr Res. 2004;71:361–370. [DOI] [PubMed] [Google Scholar]
- 30. Ritter LM, Meador-Woodruff JH, Dalack GW. Neurocognitive measures of prefrontal cortical dysfunction in schizophrenia. Schizophr Res. 2004;68:65–73. [DOI] [PubMed] [Google Scholar]
- 31. Beneyto M, Meador-Woodruff JH. Lamina-specific abnormalities of AMPA receptor trafficking and signaling molecule transcripts in the prefrontal cortex in schizophrenia. Synapse. 2006;60:585–598. [DOI] [PubMed] [Google Scholar]
- 32. Pinner AL, Haroutunian V, Meador-Woodruff JH. Alterations of the myristoylated, alanine-rich C kinase substrate (MARCKS) in prefrontal cortex in schizophrenia. Schizophr Res. 2014;154:36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zanello A, Curtis L, Badan Bâ M, Merlo MC. Working memory impairments in first-episode psychosis and chronic schizophrenia. Psychiatry Res. 2009;165:10–18. [DOI] [PubMed] [Google Scholar]
- 34. Potkin SG, Turner JA, Brown GG, et al. ; FBIRN. Working memory and DLPFC inefficiency in schizophrenia: the FBIRN study. Schizophr Bull. 2009;35:19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lipska BK, Deep-Soboslay A, Weickert CS, et al. Critical factors in gene expression in postmortem human brain: focus on studies in schizophrenia. Biol Psychiatry. 2006;60:650–658. [DOI] [PubMed] [Google Scholar]
- 36. Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98:427–436. [DOI] [PubMed] [Google Scholar]
- 37. Bauer D, Haroutunian V, McCullumsmith R, Meador-Woodruff J. Expression of four housekeeping proteins in elderly patients with schizophrenia. J Neural Transm. 2009;116:487–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hammond J, McCullumsmith R, Funk A, Haroutunian V, Meador-Woodruff J. Evidence for abnormal forward trafficking of AMPA receptors in frontal cortex of elderly patients with schizophrenia. Neuropsychopharmacology. 2010;35:2110–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Funk AJ, Haroutunian V, Meador-Woodruff JH, McCullumsmith RE. Increased G protein-coupled receptor kinase (GRK) expression in the anterior cingulate cortex in schizophrenia. Schizophrenia Res. 2014;159:130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. O’Donovan SM, Hasselfeld K, Bauer D, et al. Glutamate transporter splice variant expression in an enriched pyramidal cell population in schizophrenia. Trans Psychiatry. 2015;5:e579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Glantz LA, Lewis DA. Dendritic spine density in schizophrenia and depression. Arch Gen Psychiatry. 2001;58:203. [DOI] [PubMed] [Google Scholar]
- 42. Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hill WD, Davies G, van de Lagemaat LN, et al. Human cognitive ability is influenced by genetic variation in components of postsynaptic signalling complexes assembled by NMDA receptors and MAGUK proteins. Trans Psychiatry. 2014;4:e341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Clinton SM, Haroutunian V, Davis KL, Meador-Woodruff JH. Altered transcript expression of NMDA receptor-associated postsynaptic proteins in the thalamus of subjects with schizophrenia. Am J Psychiatry. 2003;160:1100–1109. [DOI] [PubMed] [Google Scholar]
- 45. Dracheva S, Byne W, Chin B, Haroutunian V. Ionotropic glutamate receptor mRNA expression in the human thalamus: absence of change in schizophrenia. Brain Res. 2008;1214:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Clinton SM, Meador-Woodruff JH. Abnormalities of the NMDA receptor and associated intracellular molecules in the thalamus in schizophrenia and bipolar disorder. Neuropsychopharmacology. 2004;29:1353–1362. [DOI] [PubMed] [Google Scholar]
- 47. Beneyto M, Meador-Woodruff JH. Lamina-specific abnormalities of NMDA receptor-associated postsynaptic protein transcripts in the prefrontal cortex in schizophrenia and bipolar disorder. Neuropsychopharmacology. 2008;33:2175–2186. [DOI] [PubMed] [Google Scholar]
- 48. Ohnuma T, Kato H, Arai H, Faull RL, McKenna PJ, Emson PC. Gene expression of PSD95 in prefrontal cortex and hippocampus in schizophrenia. Neuroreport. 2000;11:3133–3137. [DOI] [PubMed] [Google Scholar]
- 49. Kristiansen LV, Beneyto M, Haroutunian V, Meador-Woodruff JH. Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia. Mol Psychiatry. 2006;11:737–747, 705. [DOI] [PubMed] [Google Scholar]
- 50. Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genetics. 2012;13:227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Colledge M, Snyder E, Crozier R, et al. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bianchetta MJ, Lam TT, Jones SN, Morabito MA. Cyclin-dependent kinase 5 regulates PSD-95 ubiquitination in neurons. J Neurosci. 2011;31:12029–12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ramsey AJ, Milenkovic M, Oliveira AF, et al. Impaired NMDA receptor transmission alters striatal synapses and DISC1 protein in an age-dependent manner. Proc Nat Acad Sci USA. 2011;108:5795–5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schluter OM, Xu W, Malenka RC. Alternative N-terminal domains of PSD-95 and SAP97 govern activity-dependent regulation of synaptic AMPA receptor function. Neuron. 2006;51:99–111. [DOI] [PubMed] [Google Scholar]
- 55. Xu W, Schluter OM, Steiner P, Czervionke BL, Sabatini B, Malenka RC. Molecular dissociation of the role of PSD-95 in regulating synaptic strength and LTD. Neuron. 2008;57:248–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Standley S, Roche KW, McCallum J, Sans N, Wenthold RJ. PDZ domain suppression of an ER retention signal in NMDA receptor NR1 splice variants. Neuron. 2000;28:887–898. [DOI] [PubMed] [Google Scholar]
- 57. McCullumsmith RE, Meador-Woodruff JH. Novel approaches to the study of postmortem brain in psychiatric illness: old limitations and new challenges. Biol Psychiatry. 2011;69:127–133. [DOI] [PubMed] [Google Scholar]
- 58. Purcell SM, Moran JL, Fromer M, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fromer M, Pocklington AJ, Kavanagh DH, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ripke S, O’Dushlaine C, Chambert K, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nature Genetics. 2013;45:1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.