

Abstract
Frontotemporal Dementia (FTD) is a heterogeneous disorder with distinct clinical phenotypes associated with multiple neuropathologic entities. Presently, the term FTD encompasses clinical disorders that include changes in behavior, language, executive control and often motor symptoms. The core FTD spectrum disorders include: behavioral variant FTD (bvFTD), nonfluent/agrammatic variant primary progressive aphasia (nfvPPA), and semantic variant PPA (svPPA). Related FTD disorders include frontotemporal dementia with motor neuron disease (FTD-MND), progressive supranuclear palsy syndrome (PSP-S) and corticobasal syndrome (CBS). In this chapter we will discuss the clinic presentation, diagnostic criteria, neuropathology, genetics and treatments of these disorders.
Keywords: Frontotemporal Dementia (FTD), Primary Progressive Aphasia, nonfluent PPA, semantic PPA, motor neuron disease, progressive supranuclear palsy (PSP), corticobasal syndrome (CBS)
Introduction
Frontotemporal dementia (FTD) has undergone numerous changes in nomenclature and categorization schemes since it was first described by Pick in 1892. Presently, FTD encompasses clinical disorders that include changes in behavior, language, executive control and motor symptoms. Here, we use the term to characterize the core FTD spectrum disorders: behavioral variant FTD (bvFTD), nonfluent/agrammatic variant primary progressive aphasia (nfvPPA), and semantic variant PPA (svPPA). Related FTD disorders will be discussed including frontotemporal dementia with motor neuron disease (FTD-MND), progressive supranuclear palsy syndrome (PSP-S) and corticobasal syndrome (CBS). The term Frontotemporal Lobar Degeneration (FTLD) is used for pathological conditions that cause degeneration of frontal and temporal lobes. FTD is a heterogeneous disorder with distinct clinical phenotypes associated with multiple neuropathologic substrates.
A brief history of Frontotemporal Dementia
In 1892, Pick, a Czech neurologist, provided the first known description of a patient with FTD(Pick, 1892). He depicted a patient with progressive deterioration of language associated with left temporal lobe atrophy, a process that would presently be classified as svPPA (Gorno-Tempini et al., 2011). Histologic analysis of Pick’s clinical cases, performed by Alois Alzheimer in 1911, showed silver staining argyrophilic cytoplasmic inclusions within neurons (Alzheimer, 1911). In 1923, Gans described “Pick’s atrophy” to characterize unique cases with atrophy in the frontal and temporal lobes (Thibodeau & Miller, 2013). By 1926, Pick’s students Onari and Spatz expanded on Alzheimer’s pathologic description by delineating Pick’s bodies from Pick’s cells identifying “Pick’s disease” as a neuropathological entity (Rosen, Lengenfelder, & Miller, 2000; Thibodeau & Miller, 2013).
There was then a dearth of research into dementia until the 1970’s. During this period, the majority of clinical, anatomical and pathological patterns gleaned by these early pioneers was largely overlooked (Bruce L. Miller, 2014). Until the late 1950’s to early 1960’s, a vascular etiology was the accepted cause of “senility,” purportedly emanating from decreased cerebral blood flow and miniature infarctions and deemed “arteriosclerotic dementia” (Jellinger, 2007; Roman, 2003). Only a few groups continued Pick’s work during this time, using accurate clinical descriptions correlated with neuroanatomical and pathological analysis. The perseverance of these researchers would not be appreciated until decades afterwards. Marginal progress in frontotemporal dementia research was made until Delay, Brion and Escourolle, a French group of researchers, published their seminal paper emphasizing the clinical and neuropathological differences between Alzheimer’s disease and Pick’s disease. Pick’s disease was described to feature frontotemporal atrophy with sparing of the posterior lobes with histology revealing ballooned cells and cortical-sub-cortical gliosis (Thibodeau & Miller, 2013). The clinical syndrome of Pick’s disease showed increased behavioral alterations, lack of insight, and relative freedom from apraxia and agnosia (Thibodeau & Miller, 2013). In contrast, Alzheimer’s disease featured more diffuse cerebral atrophy and on histology showed neurofibrillary tangles and senile plaques. Clinically, Alzheimer’s patients had symptoms of agnosia, apraxia and problems with spatial orientation. In 1974, Constantinidis divided Pick’s disease into three subtypes. Only one of the three subtypes had classic Pick’s bodies suggesting that Pick’s bodies were not required for a diagnosis of Pick’s disease (Constantinidis, Richard, & Tissot, 1974).
By the 1970’s, there was a major shift in reasoning: arteriosclerotic dementia was no longer considered the underlying pathology for senility and the concept of dementia became associated with Alzheimer’s neuropathology (Roman, 2003; Ryan, Rossor, & Fox, 2015). In 1976 Katzman wrote about an Alzheimer’s epidemic suggesting that in the United States 880,000–1,200,000 people over the age of 65 may have Alzheimer’s disease (Katzman, 1976). During this period, the phrase, “don’t pick Pick’s disease” was often repeated to young researchers looking for careers in neurology, based on the misconception that Pick’s disease was both very rare and indistinguishable from Alzheimer’s disease (AD) during life (Bruce L. Miller, 2014). While the majority of dementia research in the United States was focused on Alzheimer’s disease, two groups in Europe started following large cohorts of persons with non-Alzheimer’s dementias. In Lund, Sweden, Gustafson, Ingvar and Brun, found clinical correlation of frontal lobe atrophy with hypoperfusion in the frontal lobes and only 20% of cases had classic Pick bodies on autopsy (Brun, 1993; Ingvar & Gustafson, 1970). In Manchester, England, Neary, Snowden and Mann described a large cohort of patients with dementia of the frontal type and found clinical correlations with neuroimaging (SPECT), neuropsychiatric testing and neuropathology (Rosen et al., 2000). Around the same time, Mesulam described patients with non-fluent and fluent aphasia without Alzheimer’s pathology (M. M. Mesulam, 1982; Rosen et al., 2000). Mesulam eventually coined the term primary progressive aphasia (PPA)(M. M. Mesulam, 2001). In 1989, Snowden suggested the term “semantic dementia” to describe the patient with predominant left temporal atrophy and aphasia that Pick originally described (J.S. Snowden, Goulding, & Neary, 1989), while predominant right temporal lobe atrophy was not associated with behavioral disturbances and less language involvement until later (Edwards-Lee et al., 1997). Collaboration between the two groups in Manchester, England and Lund, Sweden led to the first research criteria for FTD (“Clinical and neuropathological criteria for frontotemporal dementia. The Lund and Manchester Groups,” 1994). The clinical diagnostic criteria were revised in the late 1990s, when the FTD spectrum was divided into a behavioral variant, a nonfluent aphasia variant and a semantic dementia variant (Neary et al., 1998). With modernized neuroimaging techniques and neuropsychological evaluations, these groups proved that FTD was distinct from AD during life and that within FTD specific subtypes could better characterize patients.
Further advances in neuroimaging, genetics and neuropathology have been incorporated in the most recent revision of the clinical research criteria (Gorno-Tempini et al., 2011; Rascovsky et al., 2011). This chapter will focus on providing the most up to date information on the FTD clinical spectrum and its neuropathologic and genetic substrates.
Epidemiology
The incidence of FTD is estimated to be 1.61 to 4.1 cases per one hundred thousand people annually (Coyle-Gilchrist et al., 2016; Knopman & Roberts, 2011). One study showed the prevalence of FTD, PSP and CBS was 10.8 per 100,000 with peak prevalence at the age range of 65 to 69 years (Coyle-Gilchrist et al., 2016). There is an estimated twenty to thirty thousand people in the United States with FTD at one time (Knopman & Roberts, 2011). FTD accounts is the second most common dementia in persons under the age of 65, and is thought to be less frequent that Alzheimer’s disease (AD) (Hodges, Davies, Xuereb, Kril, & Halliday, 2003; Knopman & Roberts, 2011). The average age of onset is between 45 and 65 but there have been documented cases younger than age 30 and in the elderly (J. S. Snowden, Neary, & Mann, 2002). In a systematic review of 26 studies on FTD prevalence, men and women were found to be equally affected, and the diagnosis of bvFTD was four times more prevalent than the PPA diagnoses (Hogan et al., 2016). BvFTD accounts for roughly 60% of FTD cases and the other 40% are language variants of FTD (Onyike & Diehl-Schmid, 2013). FTD is likely underdiagnosed amongst non-neurologists due to the lack of recognition and the overlap with a multitude of psychiatric disorders (Knopman & Roberts, 2011; Lanata & Miller, 2016).
Behavioral Variant Frontotemporal Dementia
Behavioral variant frontotemporal dementia (bvFTD) presents with early changes in behavior, personality, emotion and executive control (Rascovsky et al., 2011). These changes can include new behavioral symptoms such as disinhibition, new compulsions, dietary changes or symptoms like apathy and lack of empathy. Many of these initial symptoms are easily mistaken for a psychiatric illness making bvFTD patients at high risk for misdiagnosis (Woolley, Khan, Murthy, Miller, & Rankin, 2011). In order to meet clinical criteria for a diagnosis of bvFTD, there needs to be a constellation of at least three symptoms fitting into the six categories which include: disinhibition, apathy, lack of empathy, compulsions, hyperorality and executive dysfunction (Rascovsky et al., 2011) (Table 1: Diagnostic Criteria for behavioral variant FTD). These will be discussed in further detail below.
Table 1. Diagnostic Criteria for behavioral variant FTD.
(From Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011)
|
I. Neurodegenerative disease The following symptom must be present to meet criteria for bvFTD
|
|
II. Possible bvFTD Three of the following behavioral/cognitive symptoms (A–F) must be present to meet criteria. Ascertainment requires that symptoms be persistent or recurrent, rather than single or rare events.
|
|
III. Probable bvFTD All of the following symptoms (A–C) must be present to meet criteria.
|
|
IV. Behavioral variant FTD with definite FTLD Pathology Criterion A and either criterion B or C must be present to meet criteria.
|
|
V. Exclusionary criteria for bvFTD Criteria A and B must be answered negatively for any bvFTD diagnosis. Criterion C can be positive for possible bvFTD but must be negative for probable bvFTD.
|
The behavioral symptoms of bvFTD correlate with dysfunction in the paralimbic areas including medial frontal, orbital frontal, anterior cingulate and frontoinsular cortices (Rosen et al., 2005; Seeley et al., 2008). Right hemisphere atrophy is associated more with behavior changes (Rosen et al., 2005). Von Economo neurons (VENs), present primarily in large brained, socially complex animals (Seeley et al., 2006) are found in the pregenual anterior cingulate cortex, frontoinsular and orbital frontal cortex (Seeley, 2008; Seeley et al., 2006). Seeley has shown that VENs are selectively vulnerable in bvFTD but not AD. These VEN-containing areas within the salience network are selectively vulnerable and represent the epicenter of bvFTD pathology, before there is spread to other areas within the network (Schroeter, Raczka, Neumann, & von Cramon, 2008; Seeley et al., 2007; Seeley, Zhou, & Kim, 2012). Neurodegeneration is thought to spread within the network with toxic protein aggregates moving from cell to cell in a prion-like manner (Kfoury, Holmes, Jiang, Holtzman, & Diamond, 2012; Walker & Jucker, 2015).
It is imperative to take an excellent and thorough clinical history with the goal of identifying the brain region where the disease begins. New symptoms developing through time can localize the spread of neuropathologic changes with disease progression. There are heterogeneous clinical phenotypes with bvFTD based upon the regional spread in the individual patient. Executive control is lost once the neuropathology involves the dorsal lateral prefrontal cortex that interacts with bilateral parietal lobes (Kramer et al., 2003; Seeley et al., 2007).
Disinhibition includes socially inappropriate behavior such as invading interpersonal space, inappropriate touching or over-familiarity with strangers. There can be impulsive or careless actions like new onset gambling, stealing, poor decision-making without regards to the consequences (e.g. buying matching motor cycles for themselves and their 7 year old son). Disinhibition is linked to right orbital frontal cortex degeneration (Tekin & Cummings, 2002; Tranel, Bechara, & Denburg, 2002). New criminal behaviors are seen in 37%–54% of bvFTD patients (Diehl-Schmid, Perneczky, Koch, Nedopil, & Kurz, 2013; Liljegren et al., 2015). Loss of social decorum such as telling off color jokes, using crude language, and rudeness with lack of embarrassment is common in bvFTD. There can be dramatic change in self-awareness, with lack of insight into one’s disease (Rankin, Baldwin, Pace-Savitsky, Kramer, & Miller, 2005).
Apathy can present in many ways (Chow et al., 2009). Affective apathy presents as indifference or not caring. Motor apathy manifests as decreased drive to move and less movement overall (Merrilees, Hubbard, Mastick, Miller, & Dowling, 2009) whereas cognitive apathy is a loss of desire to engage in goal oriented activities (Chow et al., 2009). Symptoms of apathy may also include social withdrawal in work activities, family functions or hobbies. Patients often need prompting to stay engaged in conversation, do chores or even to move. Apathy is easily misinterpreted as depression. Atrophy in the medial prefrontal lobes and anterior cingulate have been correlated with apathy in bvFTD (Holroyd & Yeung, 2012; Rosen et al., 2005; Rosen, Gorno-Tempini, et al., 2002).
Lack of empathy or sympathy is common. In one scenario a bvFTD patient has an improper response to a family member being diagnosed with a serious medical condition. Other responses that fit this category include insensitivity and lack of interest towards others or making cruel comments towards others. Another symptom included within this category is a patient’s indifference towards their own diagnosis of bvFTD and the impact it may have on others, which has been called “frontal anosodiaphoria” (Mendez & Shapira, 2011). Lack of empathy has been more strongly correlated with atrophy in the right temporal lobe in right svPPA patients and the subcallosal gyrus in bvFTD patients (Rankin et al., 2006).
Perseverative, stereotyped or compulsive behaviors often ritualistic in quality can occur in bvFTD (Ames, Cummings, Wirshing, Quinn, & Mahler, 1994; Josephs, Whitwell, & Jack, 2008; Perry et al., 2012; Rosso et al., 2001). Simple repetitive motor behaviors include tapping, clapping, rubbing, picking, and lip smacking. More complex behaviors included in this category are collecting cigarette butts, counting rituals, walking fixed routes or repetitive trips to the bathroom. Speech can also become stereotyped with specific repetitive patterns. Stereotyped and compulsive behaviors have been associated with several brain areas. Aberrant motor behavior has been affiliated with atrophy in the right dorsal anterior cingulate and left pre motor cortex (Rosen et al., 2005). Atrophy in the striatum has been associated with stereotypies (tapping, rocking, protruding ones tongue) (Josephs et al., 2008). Complex compulsions have been associated with asymmetric temporal lobe atrophy (Rosso et al., 2001). Obsessive compulsive behaviors in bvFTD have been correlated with loss of grey matter in bilateral globus pallidus, left putamen, and lateral temporal pole (left middle and inferior temporal gyri) (Perry et al., 2012).
Hyperorality and major changes in dietary habits can also manifest in bvFTD. Changes in food preference, particularly an inclination towards sweets or carbohydrates, are common and lead to weight gain (B. L. Miller, Darby, Swartz, Yener, & Mena, 1995). Patients may overeat, stuffing food in their mouth and in such cases satiety will be an insufficient cue to stop. One study showed that bvFTD patients continued to eat sweet sandwiches despite claiming to be full (Woolley et al., 2007). As patients become more disinhibited they may grab food off other people’s plates. Later in the course of bvFTD, hyperorality can occur, with oral exploration or eating inedible objects. Changes in eating behavior have been associated with atrophy in the orbital frontal cortex, right insular cortex and the striatum (Whitwell et al., 2007; Woolley et al., 2007). Some suggest involvement of the hypothalamus (Piguet, 2011).
Neuropsychological testing of patients with bvFTD should test multiple domains and be anatomically oriented to target specific brain structures (Bruce L. Miller, 2014). Notation of abnormal behaviors and impaired emotional processing should be described by the examiner if present. Formal neuropsychological testing can be normal in early stages of the disease (Gregory, Serra-Mestres, & Hodges, 1999). As pathology involves the dorsal lateral prefrontal cortex, executive function will emerge on neuropsychological testing and can help differentiate bvFTD patients from those with Alzheimer’s disease (Kramer et al., 2003).
There is subset of patients that exhibit a clinical presentation called “FTD-phenocopy” or slowly progressive bvFTD, that meet clinical criteria for possible bvFTD. In some instances, these patients do not suffer from a neurodegenerative condition (Kipps, Hodges, & Hornberger, 2010). However, in one study, two of four patients identified as slowly progressive bvFTD, carried the pathogenic repeat in the C9ORF72 gene (Khan et al., 2012). Recently, at UCSF, a new sub group was identified with slow progression associated with minimal cortical atrophy but with subcortical atrophy, of whom 88% carry the C9ORF72 repeat (Ranasinghe et al., 2016). This suggests that patients with slowly progressive bvFTD, or the “FTD-phenocopy” presentation should be tested for the C9ORF72 repeat as a possible etiology for their neurodegeneration.
Frontal and/or anterior temporal lobe atrophy on structural (MRI or CT) neuroimaging or hypometabolism on PET or SPECT can further support a diagnosis of bvFTD (Figure 1: bvFTD MRI) (Rascovsky et al., 2011). Neurologists are encouraged to interpret their patients’ MRI images, as radiologists may underestimate atrophy patterns that are suggestive of bvFTD (Suarez et al., 2009).
Figure 1. bvFTD MRI.
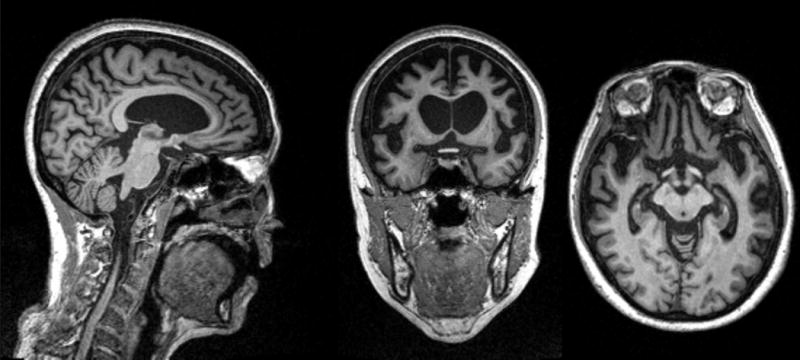
MRI of a patient with bvFTD showing severe frontal and temporal lobe atrophy. The patient is a 60 year old female who developed symptoms of withdrawal, not taking care of her farm animals, 8 years prior. Her symptoms progressed to impaired hygiene, lack of empathy, and disobeying the rules of the road. In the last 2 years she developed repetitive behaviors, hyperorality, incontinence and a non-fluent aphasia.
Primary Progressive Aphasias
The PPAs are neurodegenerative syndromes, where language is the primary impairment in the first two years of symptom onset, that Mesulam initially described as a “slowly progressive aphasia” and later renamed primary progressive aphasia (M.-M. Mesulam, 1987; M. M. Mesulam, 1982). For approximately 2 decades, PPAs were divided into semantic dementia and progressive non fluent aphasia, although several PPA cases did not fit into this binary categorization (Gorno-Tempini et al., 2011). A third type of variant that was fluent with syntactically simple sentences but frequent word finding pauses was described by Gorno-Tempini et al and called logopenic primary progressive aphasia (lvPPA) (Gorno-Tempini et al., 2004; Gorno-Tempini et al., 2011). The most recent clinical research criteria for PPAs include semantic variant of PPA (svPPA), nonfluent/agramamtic variant of PPA (nfvPPA) and logopenic variant PPA (Gorno-Tempini et al., 2011). The logopenic variant of PPA has been correlated predominantly with AD pathology so will not be discussed in detail in this chapter (M. Mesulam et al., 2008; Rabinovici et al., 2008). The other two types of PPA, svPPA and nfvPPA, are considered part of the FTD spectrum and are predominantly associated with FTD neuropathology.
Semantic Variant Primary Progressive Aphasia
The study of temporal lobe variants of FTD has furthered our understanding of language and behavior. If the left temporal lobe is involved, as in left svPPA, symptoms are predominantly language-based with a slow loss of semantic knowledge. When the right temporal lobe is primarily involved (right svPPA), behavioral symptoms predominate. As time progresses both temporal lobes become involved and symptoms begin to overlap (Seeley et al., 2005). By five to seven years, patients get more frontal lobe structures involved and develop symptoms of bvFTD with disinhibition, change in food preference and weight gain (Seeley et al., 2005). Patients with svPPA, tend to have slow progression and can live a decade or more after symptom onset (Hodges et al., 2010). Of the core clinical phenotypes in FTD, svPPA is the least likely to have a genetic cause underlying the etiology (Flanagan et al., 2015; Goldman et al., 2005).
The initial features of classic left temporal svPPA are anomia and single word comprehension deficits (Gorno-Tempini et al., 2011). The earliest symptom is often poor comprehension of single low frequency words such as “giraffe” with initial preservation of higher frequency words like “dog”. Neuropsychogical testing must include semantic screening for low frequency words, with a test like the Boston Naming Test (BNT), as the 2 words (pen, watch) in the MMSE are not sensitive enough to detect early svPPA (Kramer et al., 2003; Bruce L. Miller, 2014). As symptoms progress, patients also lose semantic knowledge about objects (Gorno-Tempini et al., 2011), so if a patient does not know the word “giraffe,” they will not improve with phonemic cues or semantic hints like “animal with a long neck.” When svPPA patients are asked to read irregularly spelled words (yacht, gnat), they will sound them out, attempting phonetic “regularization”, having lost the knowledge or their unconventional sound (i.e. y-act, ga-nat), a phenomenon known as surface dyslexia (Gorno-Tempini et al., 2011). In svPPA, repetition is spared, speech apraxia is not present, and syntax and grammar remain impressively intact. If the mesial temporal lobes are involved, memory can be affected but executive function and visuospatial skills typically are preserved (Hodges et al., 1999). Clinical research criteria for language predominant (left) svPPA must include the core features of impaired confrontational naming and impaired single word comprehension. There also must be three of the following four criteria: impaired object knowledge, surface dyslexia, spared repetition, and spared speech production (Gorno-Tempini et al., 2011) (Table 2: Diagnostic Criteria for semantic variant PPA).
Table 2. Diagnostic Criteria for the semantic variant PPA.
(From Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011)
|
I. Clinical diagnosis of semantic variant PPA Both of the following core features must be present:
|
At least three of the following other diagnostic features must be present:
|
|
II. Imaging-supported semantic variant PPA diagnosis Both of the following criteria must be present:
|
|
III. Semantic variant PPA with definite pathology Clinical diagnosis (criterion A below) and either criterion B or C must be present:
|
Right temporal svPPA patients present with prominent behavioral changes that include emotional distance, irritability, social isolation, bizarre alterations in dress, compulsions, disruption of sleep, appetite and libido (Edwards-Lee et al., 1997; B. L. Miller, Chang, Mena, Boone, & Lesser, 1993; Seeley et al., 2005). Patients with right temporal svPPA can often lack social pragmatics. They can be viscous, with telling long-winded stories, interrupting loved ones and not picking up on normal social cues that they may be acting inappropriately. It is thought that this inability to pick up social cues has to do with an inability to read the emotions on faces. Worsening degrees of right amygdala atrophy has been correlated with impairment in processing facial emotions (Rosen, Perry, et al., 2002).
Patients with either left or right temporal svPPA can have new compulsions. Left svPPA patients have been shown to have compulsions directed towards visual or non-verbal stimuli where objects are devoid of verbal information like coins or pictures. Right svPPA have compulsions around games with words and symbols (Seeley et al., 2005).
An interesting phenomenon is the development of new artistic abilities that has been observed in svPPA. Left svPPA patients have developed new visual abilities in painting, drawing, music, and gardening (B. L. Miller, Boone, Cummings, Read, & Mishkin, 2000; B. L. Miller et al., 1998; B. L. Miller, Ponton, Benson, Cummings, & Mena, 1996). Right svPPA patients can have emergent abilities in writing despite their progressive neurodegenerative condition (Bruce L. Miller, 2014). One patient with bilateral, right worse than left, svPPA was studied in depth at UCSF. As the right temporal lobe became more involved, he showed decline in emotional perception displayed through his new skill of painting (Liu et al., 2009). The topics of the painting would show couples and landscapes, but as his disease progressed the facial expressions became less genuine. Painted couples would stand next to each other, not holding hands, with bizarre smiles showing teeth (Liu et al., 2009). One could theorize that this case illustrates the difficulty the patient had understanding emotions in others as the right temporal lobe involvement progressed (Figure 2: Paintings of a right svPPA patient).
Figure 2. Paintings.

Paintings made by a 53 year old male with FTLD that had both language and behavioral symptoms. He developed compuslions for painting as one of his early symptoms and it is postulated that as his right temporal lobe become more involved that the faces in his work became less detailed and began to have a more generic smile with teeth.
Neuroimaging can further support a diagnosis of svPPA, with anterior temporal pole atrophy on structural imaging or hypoperfusion on functional imaging (Gorno-Tempini et al., 2011) (Figure 3: Left svPPA MRI, Figure 4: Right svPPA MRI). Traditionally, early symptoms originate from anterior and inferior temporal lobes involving the amygdala, with later symptoms deriving from pathologic spread into the contralateral temporal lobe and then into the ventromedial frontal cortex and insula (Rohrer & Rosen, 2013; Rosen, Gorno-Tempini, et al., 2002; Seeley et al., 2005).
Figure 3. Left svPPA MRI.
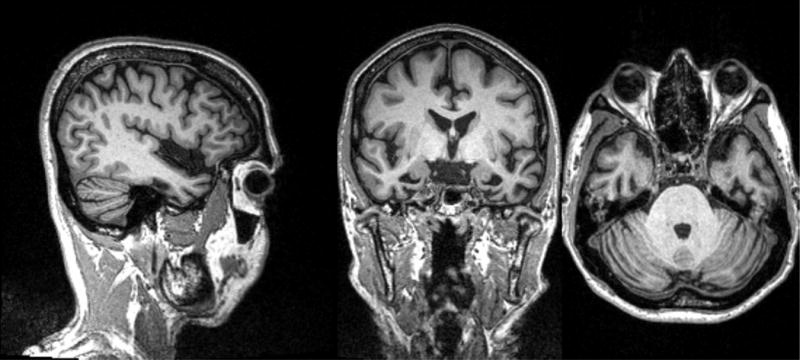
MRI showing left temporal lobe atrophy in sagittal, coronal and axial cuts.
The patient is a 61 year old man, whose first symptom was 6 years prior with the inability to name apples in a fruit bowl. His problems with semantics and naming progressed and 3 years prior he had difficulty recognizing faces of neighbors. More recent symptoms include the urge to lick people and things (hyperorality).
Figure 4. Right svPPA MRI.
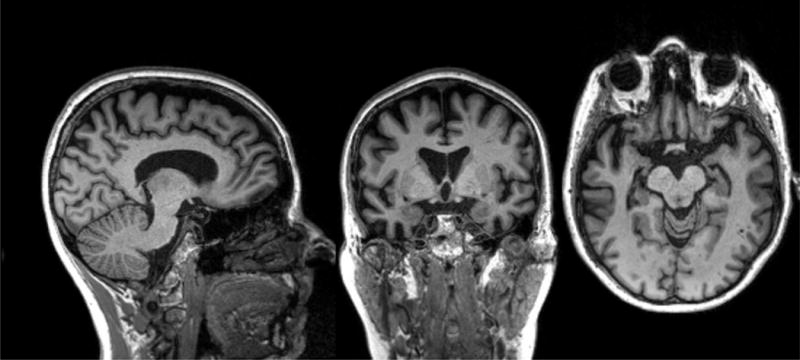
MRI shows bitemporal atrophy right greater than left. The patient is a 64 year old woman, whose first symptom was 10 years prior, telling repetitive stories that slowly became less appropriate and embarrassed her friends and family. Six years prior, she developed obsessions of taking specific walking routes to collect cigarette butts. More recently she has shown lack of empathy towards her family and dog.
Nonfluent/agrammatic Primary Progressive Aphasia
Patients with nonfluent/agrammatic primary progressive aphasias (nfvPPA) first present with effortful speech and often endorse word-finding problems. Over time, the speech becomes labored, slow, slurred, and choppy with disrupted prosody. The patient begins making inconsistent speech sound errors without awareness of this deficit. There can be inconsistent insertions, deletions, distortions, and substitutions in speech sounds. If a patient is asked to repeat a complexly structured word like “catastrophe” five times in a row, he or she will demonstrate a motor speech apraxia by repeating this word differently each time (Ogar et al., 2006). Improper use of grammar is frequently present in nfvPPA, but can be subtle, or even absent in the early stages of the illness. Patients can often have an aphemia, but can still communicate correctly with written language. Eventually agrammatism will involve both verbal and written language. Effortful speech often occurs before clear apraxia of speech or agrammatism (Gorno-Tempini et al., 2011). The neuroanatomical correlate for the symptoms of nfvPPA are Broadman’s Area 44, 45 (Broca’s area) in the left inferior frontal gyrus and the anterior insula (Gorno-Tempini et al., 2004) As time progresses, patients will have decreased verbal output and eventually become non-verbal. Mutism in nfvPPA is correlated to a larger lesion expanding beyond the typical inferior frontal and insular regions involved early in nfvPPA (Gorno-Tempini et al., 2006).
On neuropsychological testing, patients with nfvPPA eventually show deficits on the Boston Naming Test (BNT) and will still show retained semantic knowledge for these pictures (Gorno-Tempini et al., 2004). Usually, at least during the early stages, word-finding difficulty is common, but frank anomia is rare. Comprehension typically remains intact, except for longer complex sentences. Executive function can show subtle dysfunction but memory and visuospatial skills remain relatively spared early in the disease course (Gorno-Tempini et al., 2004).
For a patient to meet clinical criteria for nfvPPA, they must have effortful speech with motor speech apraxia or agrammatism, as well as supporting features of nfvPPA (Table 3: Diagnostic Criteria for nonfluent agrammatic variant PPA) (Gorno-Tempini et al., 2011). Neuroimaging support for nfvPPA consists of atrophy on MRI or hypometabolism on PET or SPECT scan in the left inferior frontal gyrus, near the perisylvian area and involving Broca’s area (Gorno-Tempini et al., 2004) (Figure 5: nfvPPA MRI).
Table 3. Diagnostic criteria for nonfluent/agrammatic variant PPA.
(From Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011)
|
I. Clinical diagnosis of nonfluent/agrammatic variant PPA: At least one of the following core features must be present:
|
At least 2 of 3 of the following other features must be present:
|
|
II. Imaging-supported nonfluent/agrammatic variant diagnosis Both of the following criteria must be present:
|
|
III. Nonfluent/agrammatic variant PPA with definite pathology Clinical diagnosis (criterion A below) and either criterion B or C must be present:
|
Figure 5. nfvPPA MRI.
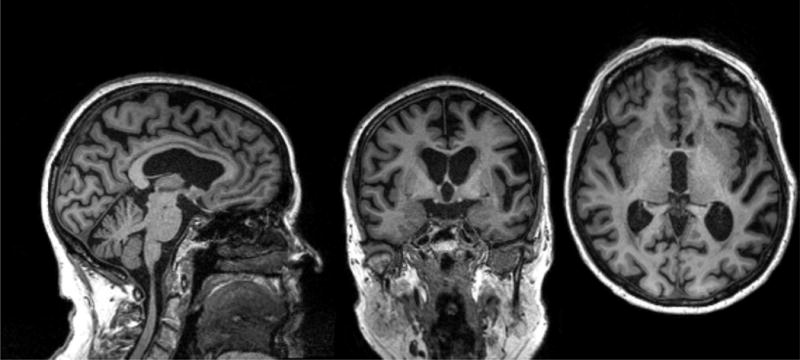
MRI showing predominant left posterior fronto-insular atrophy.
The patient is an 84 year old woman, whose first symptom was word finding difficulties 7 years prior. Over time she developed impaired grammar and a speech apraxia as her speech output diminished.
Related FTD syndromes
The core FTD disorders are bvFTD, svPPA (right and left temporal variants), and nfvPPA, but there are other disorders within the FTD spectrum that include FTD-MND, PSP-S and CBS.
Frontotemporal Dementia with Motor Neuron Disease
The relationship of dementia, motor neuron disease (MND) and parkinsonism was first noted in the Guamanian Chamorros, after World War II, in the ALS-parkinsonism dementia complex of Guam (Lomen-Hoerth, Anderson, & Miller, 2002; McGeer, Schwab, McGeer, Haddock, & Steele, 1997). Up to 15% of FTD patients and up to 30% of patients with motor neuron disease experience overlap between the two syndromes (Lomen-Hoerth, 2011). The coexistence of two disorders may be under recognized, as patients tend to present to either a neuromuscular disease clinic or a dementia clinic (Lomen-Hoerth, 2011). To meet criteria for MND, an FTD patient needs upper and lower motor neuron signs on physical examination or electromyogram (EMG) (Geevasinga et al., 2016). The El Escorial criteria are one of the most widely accepted criteria for the diagnosis of ALS but the Awaji criteria are also used which deemphasizes the use of EMG to make an earlier diagnosis (Brooks, 1994; Carvalho & Swash, 2009; Geevasinga et al., 2016). If an FTD patient presents with fasciculations, muscle weakness, trouble swallowing, spastic tone, hyperreflexia, or pathological laughing or crying, this should raise concern for MND and prompt a neuromuscular work up. If patients with ALS presents with symptoms concerning for bvFTD, this should prompt a cognitive work up. There is a shorter survival in patients with both FTD and MND since patients with FTD symptoms are less compliant with standard ALS treatments (Olney et al., 2005).
The overlap story between FTD and MND became much richer in 2005, when Mackenzie used ubiquitin immunohistochemistry to show that MND, FTD-MND and FTD were related pathologically and fit on a spectrum (Mackenzie & Feldman, 2005). Then in 2006, TDP-43 was shown to be the major disease protein in the neuropathology of tau-negative FTD and MND (Arai et al., 2006; Mackenzie, Neumann, et al., 2011; Neumann et al., 2006). In 2009, human genetic studies and post mortem analysis linked mutations in the Fused in sarcoma (FUS) gene to a familial form of MND (Kwiatkowski et al., 2009; Neumann et al., 2011; Vance et al., 2009). This finding prompted researchers to look for FUS pathology in FTD patients. FUS inclusions were found in patients with FTD that did not have a mutation in the FUS gene (Mackenzie, Rademakers, & Neumann, 2010; Neumann, Rademakers, et al., 2009; J. S. Snowden et al., 2011; Urwin et al., 2010). A genetic linkage between FTD -MND and chromosome 9q21-q22 had been known since 2000 (Hosler et al., 2000) but a hexanucleotide repeat, GGGGCC, in the non-coding region of chromosome 9 open reading frame 72 (C9ORF72) was not discovered until 2011 (DeJesus-Hernandez et al., 2011; Renton et al., 2011).
Corticobasal Degeneration
Corticobasal Degeneration (CBD) was first described by Rebeiz in 1968, in three patients with progressive asymmetric symptoms of movement and posturing that were found to have “corticodentatonigral degeneration with neuronal achromasia” on autopsy (Rebeiz, Kolodny, & Richardson, 1968). Despite numerous revisions in diagnostic criteria, predicting CBD neuropathology has proven to be a diagnostic challenge for clinicians with the correct CBD pathology predicted before death in only 56% of cases (Armstrong et al., 2013). Currently, the term corticobasal syndrome (CBS) is used to describe the “canonical” clinical entity associated with CBD, understanding that many cases of CBS are not associated with CBD neuropathology and vice versa. Many cases of CBD pathology have early clinical presentations different than CBS (Boeve et al., 1999). The clinical syndrome of CBS has been associated with Alzheimer’s disease, PSP, CBD, Pick’s disease, TDP type A, Lewy Body and rarely prion pathology (Armstrong et al., 2013; Boeve et al., 1999; Josephs et al., 2006; S. E. Lee et al., 2011; Ling et al., 2010). The term CBD is today only used to refer to the 4R tau specific neuropathologic entity.
To meet the most recent clinical criteria for probable CBS, a patient must have an asymmetric presentation with two of the following motor symptoms: limb rigidity or akinesia, limb dystonia, or limb myoclonus, as well as two of the following higher cortical symptoms: orobuccal or limb apraxia, cortical sensory deficit, or alien limb phenomena (Armstrong et al., 2013). Although asymmetry occurs with CBD, data have shown that CBD is not more likely to be asymmetric than any other neurodegenerative condition (Hassan et al., 2010; S. E. Lee et al., 2011). CBD tends to affect a dorsal frontal network involved with drive, movement, language and behavior. In a paper by Lee and colleagues CBD was predicted by clinical syndromes bvFTD, nfvPPA and an executive motor disorder (S. E. Lee et al., 2011). Typically, patients first present with a behavioral, language or executive dysfunction, leading to suspicion of bvFTD or nfvPPA. Eventually they exhibit motor dysfunction, often parkinsonism with axial rigidity (Kertesz, Davidson, McCabe, Takagi, & Munoz, 2003; Kertesz, McMonagle, Blair, Davidson, & Munoz, 2005). The absence of early motor symptoms should not exclude CBD (S. E. Lee et al., 2011). There is significant overlap with PSP clinically and in some cases series, over 50% of patients diagnosed with CBS in life had PSP pathology (Wadia & Lang, 2007). Patients with an H1H1 haplotype of the MAPT gene are associated with an increased risk for CBD and PSP pathology (Myers et al., 2007). In general, if the disease begins in the parietal region or there is a parietally-predominant syndrome the diagnosis is more likely to be Alzheimer’s disease than CBD.
There are no specific biomarkers that allow the prediction of CBD pathology. In patients who meet CBS criteria, current PET scan technology and CSF biomarkers can identify patients with Alzheimer’s pathology and those considered amyloid negative can be considered more likely to have 4R CBD neuropathology (Rabinovici et al., 2011). Neuroimaging with MRI is associated with more posterior atrophy in CBS associated with Alzheimer’s pathology and there is more frontal atrophy associated with CBD tau pathology (S. E. Lee et al., 2011; Whitwell et al., 2010). Dorsal atrophy can help predict underlying CBD 4R tau pathology especially in cases presenting as bvFTD (Boxer et al., 2006; Rankin et al., 2011).
Progressive Supranuclear Palsy
Progressive supranuclear palsy syndrome (PSP-S), previously known as Steele-Richardson-Olszewski syndrome was first described in 1964 (Steele, Richardson, & Olszewski, 1964). The initial description of nine patients displayed vertical greater than horizontal supranuclear gaze palsy, axial rigidity, dysarthria, frequent falls, pseudobulbar affect, lack of tremor, and mild dementia with vague changes in personality and psychiatric symptoms (Steele et al., 1964). Neuropathologically, nerve cells loss and neurofibrillary tangles in the basal ganglia, brainstem and cerebellum were reported decades before the identification of PSP as a 4R tau pathology (Chambers, Lee, Troncoso, Reich, & Muma, 1999). PSP-S can be a difficult diagnosis to make in the early stages since it can overlap with many syndromes but by the final exam, clinicians have had accuracy as high as over 80% in predicting the correct PSP pathology (Osaki 2004, Hughes 2002). At UCSF nearly all cases (>92%) with a clinical diagnosis of PSP-S showed PSP pathology. Most of the other PSP-S cases had CBD.
The 1996 consensus clinical research criteria for possible PSP defines the following core features: onset after age 40, gradual progression, either vertical supranuclear gaze palsy or slow vertical saccades, and postural instability with falls in the first year (Litvan, Agid, et al., 1996). Patients must also have no exclusion criteria. Probable PSP is inclusive of both a vertical supranuclear gaze palsy and postural instability with falls (Litvan, Agid, et al., 1996). Severe impairment of postural reflexes and bradykinesia make PSP falls extremely dangerous since patients are often unable to protect themselves from objects during the fall. The falls in PSP can be multifactorial from either the impaired eye movements, postural instability, impulsivity or all three symptoms.
Patients can present with predominant executive dysfunction, personality changes, reduced mental speed and attention deficits giving a more frontal behavior syndrome that is not emphasized in the current PSP criteria (Donker Kaat et al., 2007). Patients presenting with frontal behavior symptoms and without falls or supranuclear gaze palsy in their first year of presentation are likely to be among the 20% of PSP patients that are clinically misdiagnosed (Donker Kaat et al., 2007; Williams & Lees, 2009). Clinical presentations of bvFTD or nfvPPA can progress to PSP or alternatively, an initial presentation of PSP can progress into bvFTD or nfvPPA (Donker Kaat et al., 2007; Kertesz & McMonagle, 2010; Kertesz et al., 2005). Williams has proposed a system to further differentiate PSP. He classifies Richardson’s syndrome as the classic PSP-S described above. PSP-Parkinsonism (PSP-P), is the most common non Steele-Richardson PSP subtype, which is similar to Richardson’s syndrome but has tremor and mild responsiveness to Levodopa. PSP-pure akinesia with gait freezing (PSP-PAGF) is a PSP-S subtype which can have a slower disease course in spite of severe atrophy in the globus pallidus, substantia nigra and subthalamic nucleus. There are also PSP-Corticobasal Syndrome (PSP-CBS) as well as PSP-progressive nonfluent aphasia (PSP-PNFA) (Williams & Lees, 2009).
There are no definitive biomarkers for PSP. Recently, levels of neurofilament light chain in serum have been correlated with PSP disease severity but this marker is not specific for PSP and has been associated with other neurodegenerative disorders (Lu et al., 2015; Meeter et al., 2016; Rohrer et al., 2016; Rojas et al., 2016). Midbrain atrophy and the hummingbird sign on neuroimaging have been associated with PSP. The utility of the hummingbird sign in predicting midbrain atrophy has been debated but there is evidence showing it can help differentiate PSP from idiopathic Parkinson’s disease and multisystem atrophy (Kim, Kang, Ma, Ju, & Kim, 2015). Boxer et al showed that with neuroimaging, one can differentiate CBD from PSP with 93% accuracy using only the differences between atrophy of the midbrain and left frontal eye fields (Boxer et al., 2006).
Neuropathology of FTLD
“Frontotemporal lobar degeneration” (FTLD) is defined as the neurodegenerative process causing selective neuronal loss and gliosis of the frontal and temporal lobes of the brain (Mackenzie et al., 2009). The term is also used, less strictly, to encompass the diverse group of neuropathologic substrates of disease associated with the clinical phenotypes of FTD, although some of these pathological processes display a wider range of neurodegeneration than the one limited to the frontal and temporal lobes of the brain. The current classification of “FTLD” subtypes is based on the immunohistochemical staining for specific intracellular protein accumulations associated with distinct molecular defects (Mackenzie, Neumann, et al., 2011; Mackenzie, Neumann, et al., 2010). We will discuss the basic groupings of FTLD-tau, FTLD-TDP, FTLD-FET and FTLD-UPS. We will also discuss clinicopathological correlations that are known for these neuropathologic entities (Figure 6: Clinical and pathological correlations in FTD spectrum disorders).
Figure 6. Clinical and pathological correlations in FTD spectrum disorders.
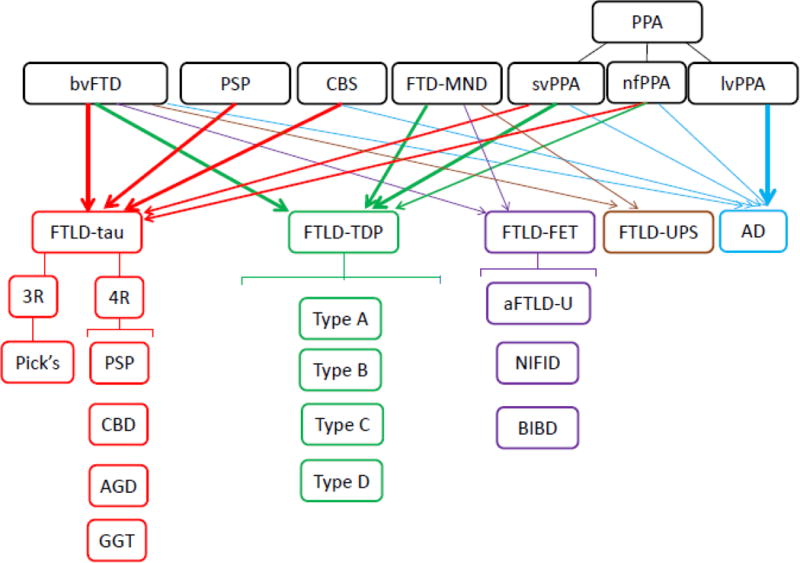
This figure summarizes the overlap of FTD spectrum disorders (bvFTD, PSP, CBS, FTD-MND, svPPA, nfPPA) and their neuropathology (FTLD-tau, FTLD-TDP, FTLD-FET, FTLD-UPS) with a small portion of clinical syndromes being caused by AD pathology. The clinical syndrome of lvPPA is highly correlated with AD pathology.
FTLD-Tau
The protein tau was first discovered in 1975 as an essential protein in microtubule assembly (Weingarten, Lockwood, Hwo, & Kirschner, 1975). It was later found to assist in cellular transport and stabilization of the structure of the cell (Mandelkow & Mandelkow, 2012). More recently it has been established that tau is also involved in cellular signaling pathways (Jenkins & Johnson, 1998; Leugers & Lee, 2010; Morris, Maeda, Vossel, & Mucke, 2011). The tau gene, MAPT, was found to be on chromosome 17q21 and over fifty mutations have been linked to FTLD-tau pathology (Clark et al., 1998; Hutton, Lendon, Rizzu, Baker, Froelich, Houlden, Pickering-Brown, Chakraverty, Isaacs, Grover, Hackett, Adamson, Lincoln, Dickson, Davies, Petersen, Stevens, de Graaff, Wauters, van Baren, Hillebrand, Joosse, Kwon, Nowotny, Che, et al., 1998; Mandelkow & Mandelkow, 2012; Poorkaj et al., 1998; Spillantini et al., 1998; K. C. Wilhelmsen, T. Lynch, E. Pavlou, M. Higgins, & T. G. Nygaard, 1994). Alternative splicing of MAPT generates six different isoforms of tau. Alternative inclusion of exon 10 generates tau isoform with either four (4R tau) (3R tau) microtubule binding domain repeats (Mandelkow & Mandelkow, 2012). Neurodegenerative diseases characterized by predominant accumulation of hyperphosphorylated tau inclusions are also called tauopathies. Neurofibrillary tangles in Alzheimer’s disease are composed of both hyperphosphorylated 4R tau and 3R tau inclusions. In FTLD-tau, Pick’s disease is characterized by predominant deposition of 3R tau aggregates, while CBD, PSP, globular glial tauopathy (GGT) and argyrophilic grain disease (AGD) are 4R tauopathies. Distinct tauopathies have unique patterns of immunohistochemistry and western blot profiles (Ghetti et al., 2015).
Pick’s Disease (3R Tau)
The neuropathology of Pick’s disease displays ballooned neurons, known as Pick’s cells, as well as large spherical argyrophilic neuronal cytoplasmic inclusions, known as Pick bodies (Figure 7: Pick’s 3R tau). Pick’s bodies are predominantly composed of 3R tau, although 4R can be present (Zhukareva et al., 2002). Pick’s disease is associated with severe atrophy of the ventral regions of the frontal and temporal lobes as well as anterior cingulate gyrus and insula. Pick’s disease occurs predominantly sporadically, although it has been rarely reported in association with specific MAPT mutations: L315R, S320F, Q336H and G389R (Ghetti et al., 2015; Tacik et al., 2015). Pick’s presents more commonly as bvFTD, though nfvPPA and svPPA phenotypes are not infrequent (Graff-Radford et al., 1990; Irwin, 2016; Irwin et al., 2016). In autopsy confirmed cases within the UCSF cohort, 23% of patients with a clinical diagnosis of nfvPPA and 9% of svPPA had Pick’s 3R tau.
Figure 7. 3R tau in Pick’s Disease.
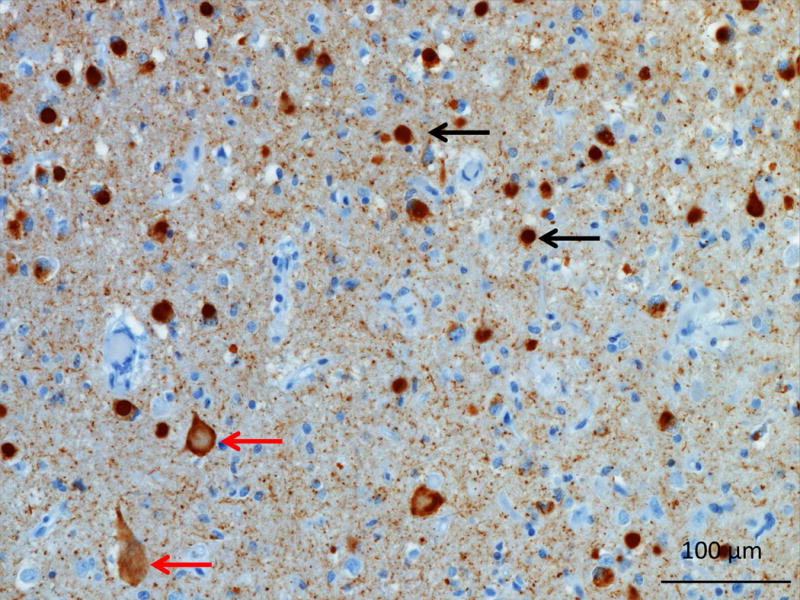
Photomicrograph of the middle frontal gyrus from a patient with Pick’s disease. Tau immunohistochemistry demonstrates numerous Pick’s bodies (black arrow) and ballooned neurons or Pick’s cells (red arrows). Image courtesy of Salvatore Spina MD, PhD UCSF
PSP (4R Tau)
The neuropathology of PSP is characterized by the presence of tufted astrocytes, thorny astrocytes and spherical ‘globose’ neurofibrillary tangles in subcortical nuclei (Figure 8: PSP 4R tau) (D. W. Dickson et al., 2002; Josephs et al., 2011; Josephs et al., 2006; Litvan, Hauw, et al., 1996). Outside the brainstem, flame shaped neurofibrillary tangles are seen, along with diffuse granular ‘pre-tangles (D. Dickson & ebrary Inc., 2011). Oligodendroglial cytoplasmic inclusions known as coiled bodies are seen in the cortex and more abundantly in the subcortical white matter (D. Dickson & ebrary Inc., 2011). The tau inclusions are predominantly composed of 4R tau and are present in the brainstem more than the cortex (V. M. Lee, Goedert, & Trojanowski, 2001). Cortical involvement can be variable, with the primary motor cortex and the frontal eye fields being more often involved while worse cognitive performance is associated with wider cortical involvement (Bigio, Brown, & White, 1999).
Figure 8. 4R tau in PSP.
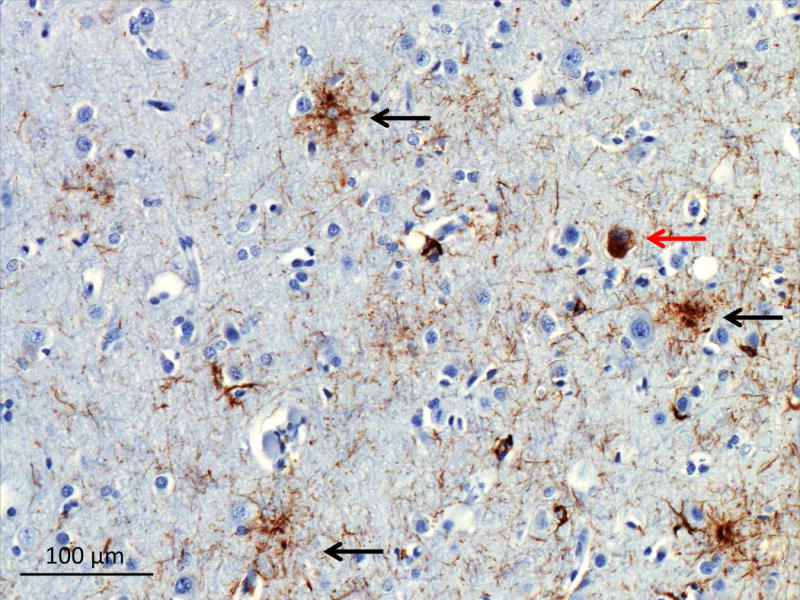
Tau immunohistochemistry of the superior frontal sulcus showing tufted astrocytes (black arrows), and a globose tangle (red arrow).
Image courtesy of Salvatore Spina MD, PhD UCSF
CBD (4R Tau)
Astrocytic plaques (Figure 9: CBD 4R tau) are the most characteristic neuropathological hallmark of CBD (Feany & Dickson, 1996). Just like in tufted astrocytes of PSP, astrocytic plaques are made of hyperphosphorylated 4R tau deposits in astrocytic processes (V. M. Lee et al., 2001). In astrocytic plaques these deposits localize in the most distal portion of the process as opposed to the localization of deposits (proximal to the cell body) in tufted astrocytes (D. Dickson & ebrary Inc., 2011). Neuronal inclusions in the form of granular cytoplasmic accumulation of tau or neurofibrillary tangles are seen in the cortex (D. W. Dickson et al., 2002). Ballooned neurons are a common feature of CBD, although they are also seen in Pick’s disease (D. W. Dickson et al., 2002) A distinctive feature of CBD is the extensive involvement of the subcortical white matter with tau deposits in neurites and oligodendroglial coiled bodies (D. Dickson & ebrary Inc., 2011). CBD pathology affects a wider range of cortical involvement than PSP pathology involving more dorsal regions and tends to be found in the pre and post central gyri (D. W. Dickson et al., 2002).
Figure 9. 4R tau in CBD.
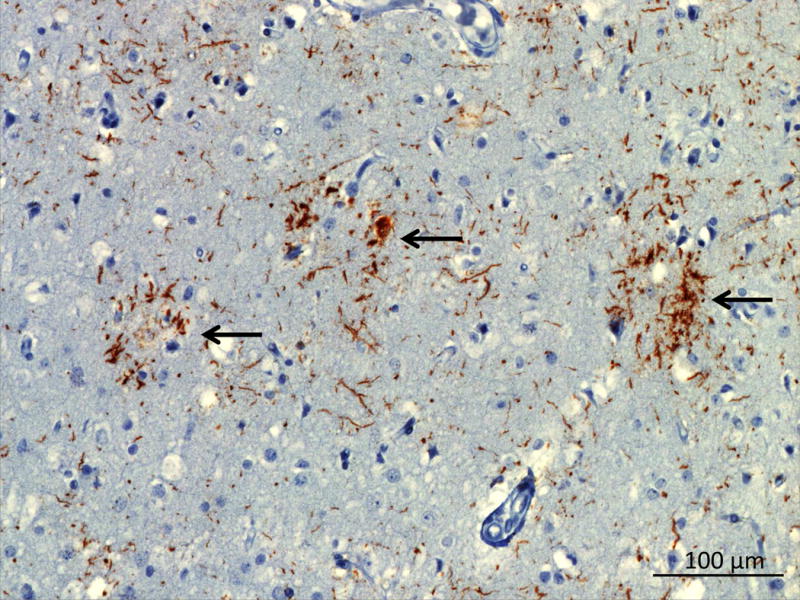
Tau immunohistochemistry in the paracentral gyrus showing astrocytic plaques (black arrows).
Image courtesy of Salvatore Spina MD, PhD UCSF
Globular Glial Tauopathy (GGT)
Globular Glial Tauopathy (GGT) is a rare 4R tauopathy, whose pathological classification has been recently revised. Proposed criteria describe three different pathological subtypes that are variably associated with the clinical phenotypes of bvFTD and/or MND, with or without extrapyramidal signs (Ahmed et al., 2013). Microscopically, the disease is characterized by the presence of large 4R tau inclusions with globular morphology in oligodendrocytes and astrocytes, particularly abundant within the white matter (Ahmed et al., 2013).
Argyrophilic Grain Disease (AGD)
Argyrophilic gain disease (AGD) is a 4R tauopathy mainly characterized by the accumulation of small dot like, comma-like argyrophilic 4R tau positive inclusions (“grains”), predominantly seen in the temporal lobes and limbic structures. AGD can occur alone or contextually to other neurodegenerative diseases, more often Alzheimer’s disease pathology (Fujino et al., 2005; Thal et al., 2005). The clinical presentation ranges from mild cognitive impairment to AD-like dementia and a slowly progressive bvFTD (Grinberg et al., 2013; Ishihara et al., 2005; Tsuchiya et al., 2001). Tau deposits in AGD lack acetylation, a post-translational modification step thought to be essential for the acquisition of pathogenic features by tau species in AD, hence the hypothesis of a potential protective role of AGD in AD that may explain the more benign clinical phenotype (slow disease course) often associated with this underlying neuropathology (Grinberg et al., 2013).
FTLD-TDP
In 2006, TDP-43 was shown to be the major disease protein in the neuropathology of both FTLD-U (tau negative, ubiquitin positive FTLD) and amyotrophic lateral sclerosis (ALS) (Arai et al., 2006; Mackenzie, Neumann, et al., 2011; Neumann et al., 2006). TAR DNA-binding protein 43 (TDP-43) belongs to the heterogeneous nuclear ribonucleoproteins family, and has been shown to be involved with micro RNA processing, mRNA stabilization, transport and translation (Baralle, Buratti, & Baralle, 2013). The exact role of TDP-43 is unknown but it has been associated with thousands of mRNA targets and thought to be vital for cell function (Polymenidou et al., 2011). Pathological TDP-43 in neurodegenerative diseases is found as aggregates in the cytoplasm leading to phosphorylation, ubiquitination and it’s degradation (Buratti & Baralle, 2009). In 2011, a harmonized classification of FTLD-TDP pathology was proposed to categorize TDP-43 neuropathology into 4 groups (A, B, C, D) which has since been widely accepted (Mackenzie, Neumann, et al., 2011).
TDP-43 type A neuropathology is characterized by abundance of neuronal rounded or crescent-like cytoplasmic inclusions, and short dystrophic neurites and rare lentiform neuronal intranuclear inclusions (Figure 10: TDP-43 Type A) which are more commonly found in cortical layer II (Mackenzie, Neumann, et al., 2011). TDP-43 type A neuropathology can present with the clinical phenotypes of bvFTD, nfvPPA, and CBS while MND is less common (Mackenzie, Neumann, et al., 2011). Pathogenic mutations in GRN on chromosome 17 are associated with TDP-43 type A pathology (Cairns, Neumann, et al., 2007).
Figure 10. TDP-43 Type A.
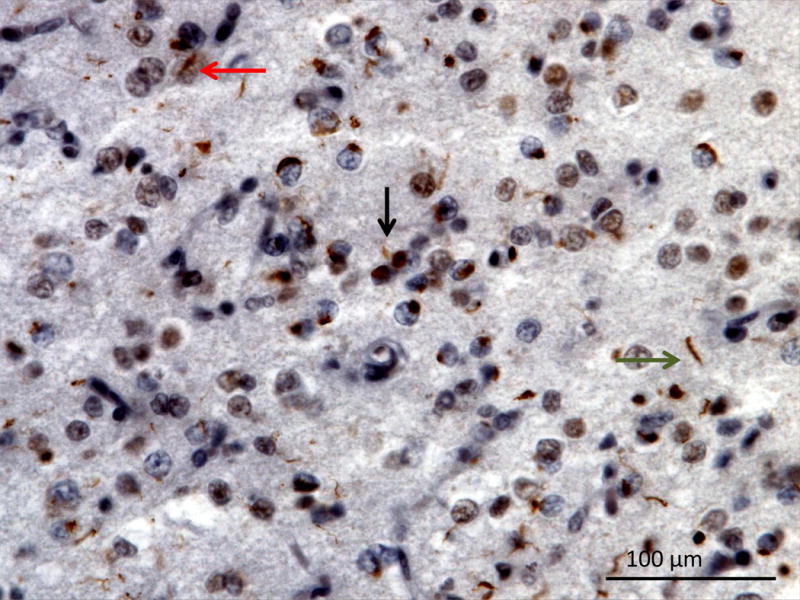
Orbitofrontal gyrus showing many short dystrophic neurites (black arrow), neuronal cytoplasmic inclusions (green arrow) and a lentiform neuronal intranuclear inclusions (red arrow). TDP immunohistochemistry. Image courtesy of Lea Grinberg MD, PhD UCSF
TDP-43 type B neuropathology is characterized by much less frequent neuronal cytoplasmic inclusions and dystrophic neurites found in both superficial and deep cortical layers (Figure 11: TDP-43 Type B) (Mackenzie, Neumann, et al., 2011). TDP-43 type B pathology is the most common subtype associated with presence of neuronal cytoplasmic inclusions in lower motor neurons, with or without clinical signs of MND (Mackenzie, Neumann, et al., 2011). Clinical phenotypes of FTD, MND and FTD-MND can have TDP-43 type B neuropathology (Mackenzie, 2007b; Mackenzie, Neumann, et al., 2011). Patients with C9ORF72 repeat expansions most often have TDP-43 type B although TDP-43 type A is seen as well (J. S. Snowden et al., 2015).
Figure 11. TDP-43 Type B.
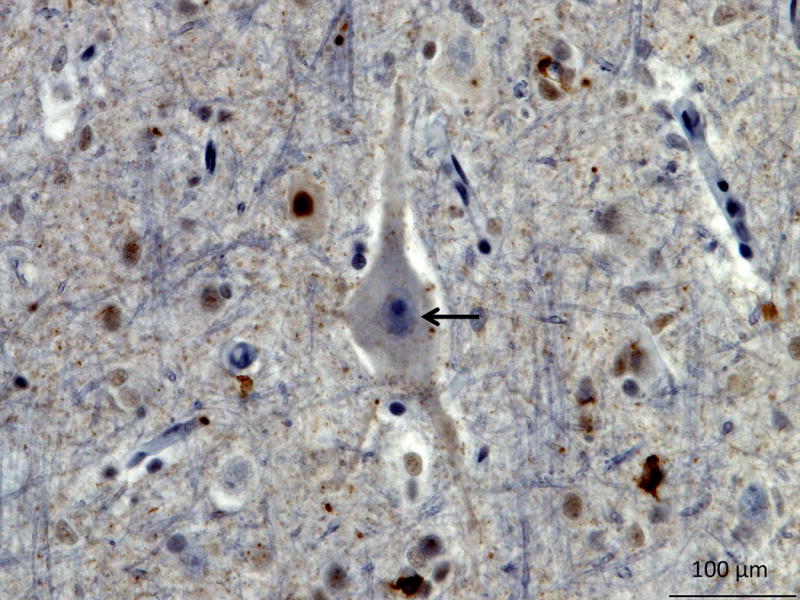
TDP immunohistochemistry of the precentral gyrus showing moderate neuronal cytoplasmic inclusions (black arrow). TDP immunohistochemistry.
Image courtesy of Lea Grinberg MD, PhD UCSF
TDP-43 type C neuropathology is characterized by long tortuous dystrophic neurites, and few neuronal intracytoplasmic inclusions (Figure 12: TDP-43-C) (Mackenzie, Neumann, et al., 2011), while neuronal intranuclear inclusions are uncommon. TDP-43 type C is the most common neuropathology substrate of svPPA and in the UCSF autopsy cohort it is pathology confirmed in 90% of the cases.
Figure 12. TDP-43 Type C.
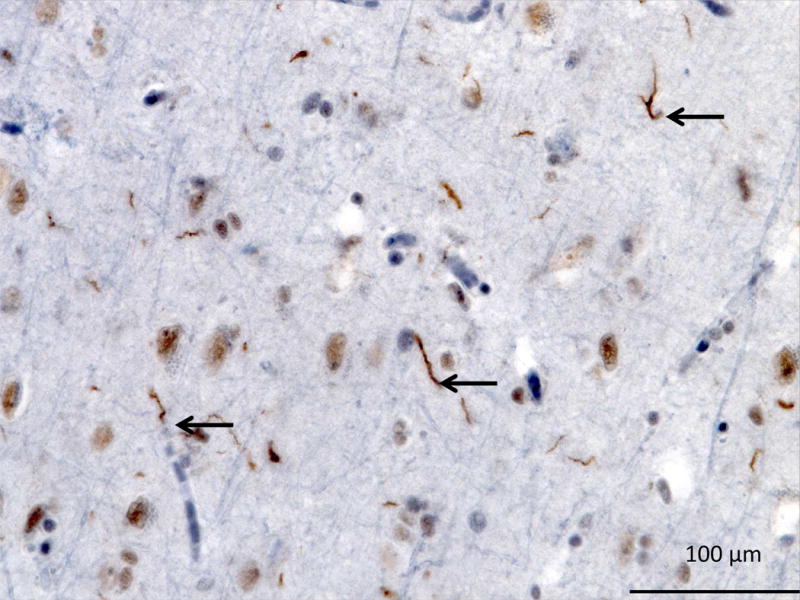
TDP immunohistochemistry of the insula showing long tortuous dystrophic neurites (black arrows), and few neuronal intranuclear inclusions.
Image courtesy of Lea Grinberg MD, PhD UCSF
TDP-43 type D is characterized by the presence of frequent lentiform neuronal intranuclear inclusions as well as short dystrophic neurites and rare neuronal cytoplasmic inclusions in all layers (Mackenzie, Neumann, et al., 2011), though these findings are more common in superficial layers. This subtype of FTLD-TDP is uniquely associated with mutations in the VCP gene and the clinical phenotype of Inclusion Body Myopathy with Paget’s disease and FTD (Cairns, Bigio, et al., 2007; Mackenzie, Neumann, et al., 2011).
FTLD-FET
In 2009, human genetic studies found mutations in the Fused in sarcoma (FUS) gene were linked to familial MND and FUS cytoplasmic inclusions were found in post mortem analyses (Kwiatkowski et al., 2009; Neumann et al., 2011; Vance et al., 2009). The role of FUS is not fully understood but it has DNA/RNA binding properties and is thought to be similar to TDP-43 (Baloh, 2012; Mackenzie & Neumann, 2012; Vance et al., 2009). Given the known overlap of MND and FTD, involvement of FUS in FTD was explored and FUS cytoplasmic inclusions were found in patients with FTD (Mackenzie, Rademakers, et al., 2010; Neumann, Rademakers, et al., 2009; Urwin et al., 2010). MND were correlated with mutations in the FUS gene, but FUS pathology in FTD was shown to be sporadic and not affiliated with a mutation in FUS (J. S. Snowden et al., 2011). FUS is a member of the FET protein family, which also consists of other RNA/DNA binding proteins Ewing’s sarcoma (EWS) and TATA-binding protein-associated factor 15 (TAF15) (Mackenzie & Neumann, 2012). In 2011, immunohistochemical analysis of FUS inclusions in FTLD showed that the whole FET family (FUS, EWS and TAF15) were found in FTLD pathology but not MND FUS inclusions (Neumann et al., 2011). This suggests different mechanism for FTLD and MND with FUS pathology (Mackenzie & Neumann, 2012). FET proteins were also found in neuronal intermediate filament inclusion disease (NIFID) and basophilic inclusion body disease (BIBD) (Munoz et al., 2009) (Neumann et al., 2011; Neumann, Roeber, et al., 2009). Previously aFTLD-U, NIFID, BIBD were considered FTLD-FUS subtypes but since they are all positive for FET-positive inclusions, they can now grouped together under FTLD-FET (Mackenzie, Munoz, et al., 2011; Mackenzie & Neumann, 2012).
Atypical FTLD-U (aFTLD-U) neuropathology is characterized by neuronal cytoplasmic inclusions containing FET proteins and most distinctively by the presence of rare and unique neuronal intranuclear inclusions with a peculiar vermiform shape (Mackenzie, Munoz, et al., 2011) (Figure 13: aFTLD-U binding FUS).
Figure 13. aFTLD-U binding FUS.
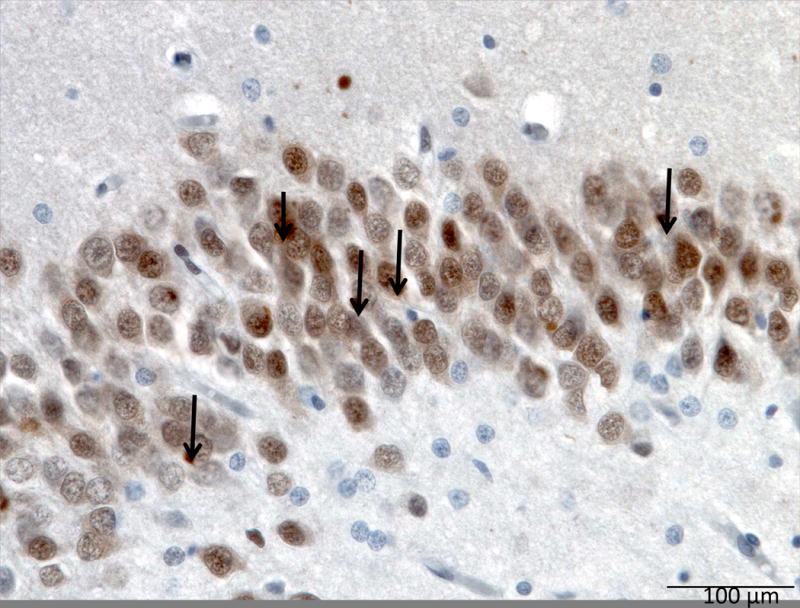
FUS immunohistochemistry of the dentate fascia (hippocampus) showing many FUS positive neuronal cytoplasmic inclusions (black arrows).
Image courtesy of Lea Grinberg MD, PhD UCSF
Neuronal intermediate filament inclusion disease (NIFID) is a rare neurodegenerative disorder with distinct neuropathology due to immunoreactive neurofilament inclusions that were later found to bind all class IV intermediate filaments (Neumann, Roeber, et al., 2009). In 2009, NIFID was found to have intracellular FUS accumulation (Neumann, Roeber, et al., 2009) and in 2011 it was found to have widespread FET proteins accumulation (Neumann et al., 2011).
Basophilic inclusion body disease (BIBD), displays pathognomonic basophilic round inclusions, when stained with hematoxylin and eosin. Basophilic inclusions are more common in subcortical regions such as the basal ganglia and brainstem tegmentum. There is severe frontotemporal atrophy in FTD cases and spinal cord involvement in MND cases (E. B. Lee et al., 2013).
FTLD-UPS
A rare form of FTD has been linked to chromosome 3 in a large Danish family (Holm, Isaacs, & Mackenzie, 2009), and was later associated with a mutation in the CHMP2B (charged multivesicular body protein 2B) gene (Skibinski et al., 2005). CHMP2B mutations have been associated with familial FTD and ALS. The neuropathology is characterized by the presence of inclusions that are immunoreactive for ubiquitin and are negative for tau, TDP-43 and FET proteins, though the exact nature of an eventual pathogenic protein remains unknown (Holm et al., 2009).
Genetics
Frontotemporal dementia frequently has a strong genetic component contributing to its pathogenesis. Over half of FTD cases are sporadic, but up to 40% of cases have a family history of dementia, psychiatric disease or motor symptoms, with at least 10% of cases having an autosomal dominant pattern (Chow, Miller, Hayashi, & Geschwind, 1999; Goldman et al., 2005). Of the clinical syndromes, FTD-MND is the most heritable and svPPA is the least heritable (Goldman et al., 2005). The three most common genes associated with FTD are C9ORF72, MAPT and GRN. Other less common genes associated with FTD include: VCP, CHMP2B, TARDBP, FUS, EXT2, TBK1 and SQSTM1.
Microtubule Associated Protein Tau (MAPT)
In the mid 1900’s, FTD was observed by Malamud, Waggoner and Sjogren, to have a genetic contribution and some families with an autosomal dominant pattern of inheritance (Bruce L. Miller, 2014). In 1994, Wilhelmsen linked a family that had an autosomal dominant pattern of FTD, which he called disinhibition-dementia-parkinsonism-amyotrophy-complex (DDPAC), to a region on chromosome 17 (K.C. Wilhelmsen, T. Lynch, E. Pavlou, M. Higgins, & T. G. Nygaard, 1994). In 1998, as more FTD families were linked to chromosome 17, missense mutations in tau exons 9–13 and a splice site mutation in intron 10 was linked to increased 4R tau production as a possible mechanism for the tau pathology (Grover et al., 1999; Hutton, Lendon, Rizzu, Baker, Froelich, Houlden, Pickering-Brown, Chakraverty, Isaacs, Grover, Hackett, Adamson, Lincoln, Dickson, Davies, Petersen, Stevens, de Graaff, Wauters, van Baren, Hillebrand, Joosse, Kwon, Nowotny, Heutink, et al., 1998).
Clinical presentations of patients with MAPT mutations vary with bvFTD, nfPPA, PSP-S, and CBS all described (Rohrer & Warren, 2011). Symptoms of motor neuron disease are rarely associated with MAPT mutations (J. S. Snowden et al., 2015). The phenotype can vary between family members affected with the same mutation (Yasuda et al., 2005). The neuroimaging of patients with MAPT mutations tends to show a more symmetric pattern of atrophy than other genetic causes of FTD (Rohrer & Warren, 2011; Whitwell et al., 2009).
An H1H1 haplotype of the MAPT gene increases a patient’s risk for PSP and CBD (Conrad et al., 1997; Myers et al., 2007). The rare sequence variant in MAPT, p. A152T was found initially in patients with PSP, but in a larger study was shown to increase the risk of developing AD and FTD spectrum disorders, when compared to controls (Coppola et al., 2012). This is the first time a tau polymorphism has been linked to both FTD and AD.
Progranulin (GRN)
In 2006, mutations in GRN, also on chromosome 17, were linked to FTLD (Baker et al., 2006; Cruts et al., 2006). Further research of GRN, revealed that the haploinsufficiency was the mechanism for the neurodegeneration (Petkau & Leavitt, 2014; Ward & Miller, 2011). Patients with mutations in GRN, have 50% less GRN mRNA progranulin levels in both CSF and plasma supporting the haploinsufficiency theory (Coppola et al., 2008; Van Damme et al., 2008). The exact role of progranulin is not fully understood, but it has been linked to neuronal growth, lysosomal function plus inflammation and stress response (Petkau & Leavitt, 2014; Ward & Miller, 2011). The neuropathology associated with GRN is predominantly TDP-43 type A (Cairns, Neumann, et al., 2007; Mackenzie, 2007a).
Patients with GRN mutations most often present with bvFTD or nfvPPA, but CBS is also common and MND is rare (Le Ber et al., 2008; Mackenzie, 2007a). On average patients tend to be older with age of onset, mean age 60, and apathy is often a predominant symptom (J. S. Snowden et al., 2015). MRI imaging can show asymmetry with atrophy in the fronto-temporo-parietal atrophy, suggesting a GRN mutation (Rohrer & Warren, 2011). Only 70–90% of families with a GRN mutation have a family history of neurodegenerative disease suggesting it has lower penetrance than other FTD genes (van Swieten & Heutink, 2008). Homozygote GRN carriers develop a syndrome suggestive of neuronal ceroid lipofuscinosis with early onset retinal degeneration, seizures and cerebellar ataxia (Smith et al., 2012).
Chromosome 9 Open Reading Frame 72 (C9ORF72)
A genetic linkage between FTD -MND and chromosome 9q21-q22 had been known since 2000 (Hosler et al., 2000) but a mutation was not discovered until 2011 (DeJesus-Hernandez et al., 2011; Renton et al., 2011). This mutation is a hexanucleotide repeat, GGGGCC, in a non-coding region of chromosome 9 open reading frame 72 (C9ORF72) (DeJesus-Hernandez et al., 2011; Renton et al., 2011). The pathologic C9ORF72 hexanucleotide repeat expansion is the most frequent genetic cause of familial FTD (11.7%) and familial ALS (23.5%) (DeJesus-Hernandez et al., 2011). The maximum size of hexanucleotide repeats in controls was 2–23. Expansion sizes from 700 to 1600 have been associated with pathogenesis causing FTD, MND or FTD-MND (DeJesus-Hernandez et al., 2011). The exact role of C9ORF72 is still being studied, but the repeat expansions of C9ORF72 are thought to cause loss of function in transcription as well as possible gain of function due to toxic RNA foci (DeJesus-Hernandez et al., 2011). Dipeptides are produced by the abnormal expansions that also likely contribute to the neurodegeneration (Mann, 2015).
The most common neuropathology associated with the pathologic C9ORF72 hexanucleotide repeat expansions (C9+) is TDP-43 type B, (Mackenzie et al., 2013; J. S. Snowden et al., 2015). There is current debate over the significance of inclusions with dipeptide repeat (DPR) proteins that are specifically found in all C9+ phenotypes (Mackenzie et al., 2013; Mackenzie & Neumann, 2016; Mann, 2015; Vatsavayai et al., 2016). The DPR proteins have not been correlated specifically with neurodegeneration and cases of C9+ patients that died prematurely had DPR pathology, no TDP-43 pathology and no symptoms of FTD or MND suggesting DPR accumulates early (Baborie et al., 2015; Mackenzie & Neumann, 2016; Proudfoot et al., 2014). C9+ patients show RNA aggregates, and these RNA foci have sparked theories about mechanism of neurotoxicity (DeJesus-Hernandez et al., 2011; Mackenzie & Neumann, 2016).
The most common clinical phenotype with C9+ patients is bvFTD, although MND and FTD-MND are also common (Boeve et al., 2012; J. S. Snowden et al., 2015). When compared to other gene FTD gene carriers, C9+ patients showed a higher likelihood of exhibiting psychotic symptoms, and delusions (Sha et al., 2012; J. S. Snowden et al., 2015). In one study C9+ patients were less likely to exhibit dietary changes but were more socially appropriate and warm (J. S. Snowden et al., 2015). C9+ patients can have a long disease course (Khan et al., 2012; Sha et al., 2012).
MRI imaging of C9+ patients have shown typical FTD atrophy patterns when compared to controls but when compared to other FTD genes the atrophy pattern is atypical (Sha et al., 2012; Whitwell et al., 2012). Patients with FTD-MND have shown more atrophy in dorsal frontal, parietal, the thalamus and cerebellum (Sha et al., 2012; Whitwell et al., 2012). Functional connectivity studies associated diminished salience network connectivity with atrophy of the left medial pulvinar thalamic nucleus in C9+ patients (S. E. Lee et al., 2014).
Rare genetic causes of FTD
A rare autosomal dominant disorder with inclusion body myositis, Paget’s disease of the bone and FTD has been linked to a mutation in VCP on chromosome 9 (Neumann et al., 2007; Watts et al., 2007). Exome studies have linked VCP mutations to MND (Johnson et al., 2010). Mutations in VCP are linked to a unique form of neuropathology, TDP-43 type D (Neumann et al., 2007).
Mutations in FUS are rare and predominantly associated with familial ALS. Neuropathologically, they present as FTLD-FUS diseases with inclusions made of FUS protein in the absence of other FET proteins (Kwiatkowski et al., 2009; Neumann et al., 2011).
There are numerous other genes that have been associated with FTLD which are uncommon. These include mutations in: TARDBP (Borghero et al., 2011; Kovacs et al., 2009; Mosca et al., 2012), CHMP2B (Skibinski et al., 2005), TBK1 (Freischmidt et al., 2015; Pottier et al., 2015), OPTN (Pottier et al., 2015; Pottier, Ravenscroft, Sanchez-Contreras, & Rademakers, 2016), SQMSTM1 (Fecto et al., 2011; Pottier et al., 2016), UBQLN2 (Deng et al., 2011), and EXT-2 (Narvid et al., 2009)
Treatments
There are currently no FDA approved treatments for FTD, but off-label pharmacological and behavioral modification techniques can be used to manage symptoms in FTD.
The FDA approved treatments for Alzheimer’s disease have not shown benefit in FTD. There is evidence that acetylcholinesterase inhibitors actually make symptoms in FTD worse (Kimura & Takamatsu, 2013; Mendez, Shapira, McMurtray, & Licht, 2007). Memantine was tolerated in patients with FTD but in a double-blind, placebo-controlled trial there was no benefit to behavior or cognition (Boxer, Knopman, et al., 2013; Vercelletto et al., 2011).
It is accepted that FTD symptoms and behavior can improve with selective serotonin uptake inhibitors (SSRIs) (Herrmann et al., 2012; Ikeda et al., 2004; Mendez, Shapira, & Miller, 2005; Swartz, Miller, Lesser, & Darby, 1997). A small randomized, placebo controlled double blinded trial with Trazadone showed improvement in NPI scores of FTD patients but no change in MMSE (Lebert, Stekke, Hasenbroekx, & Pasquier, 2004).
Atypical antipsychotics should be used with caution in patients with FTLD given that they may have increase vulnerability to the extrapyramidal side effects and there is a black box warning for use in the elderly (Pijnenburg, Sampson, Harvey, Fox, & Rossor, 2003). There is little evidence for benefit from mood stabilizers in FTLD and evidence is limited (Chow & Mendez, 2002; Cruz, Marinho, Fontenelle, Engelhardt, & Laks, 2008). Oxytocin has been proposed as a potential therapy targeting the emotional changes in FTD and a small study showed mild improvement on the Neuropsychiatric Inventory after its use (Finger, 2011; Jesso et al., 2011).
There are also important non-pharmacological therapies to treat FTD symptoms. FTD symptoms can improve with caregiver education about behavioral, environmental and physical techniques to minimize or redirect unwanted behaviors (Merrilees, 2007). Benefits from physical exercise have been shown to delay cognitive decline and should be recommended to all FTD patients that can safely tolerate it (Cheng et al., 2014). Patients with nfvPPA, svPPA, and language deficits, may benefit from speech therapy (Kortte & Rogalski, 2013).
Although there are no FDA-approved treatments for FTD, this is a hopeful time for FTD treatments to come to fruition. There are currently active clinical trials targeting specific FTD mechanisms and pathology. There are trials targeted at tau pathology with therapeutics aimed at preventing tau aggregation, tau microtubule stabilization and removal of tau with tau-targeted antibodies (Karakaya, Fusser, Prvulovic, & Hampel, 2012; Schneider & Mandelkow, 2008; Tsai & Boxer, 2016). There are trials currently targeting the haploinsufficiency in GRN gene expression by using different methods of increasing progranulin (Boxer, Gold, Huey, Gao, et al., 2013; Boxer, Gold, Huey, Hu, et al., 2013; Lagier-Tourenne et al., 2013). Antisense oligonucleotide (ASO) therapies are being developed to target the toxin gain of function with C9ORF72 genetic mutation (Lagier-Tourenne et al., 2013). Molecular based treatments for FTD are closer than they have ever been.
Key Points.
-
-
The core FTD spectrum disorders include: behavioral variant FTD (bvFTD), nonfluent/agrammatic variant primary progressive aphasia (nfvPPA), and semantic variant PPA (svPPA).
-
-
Related FTD disorders include frontotemporal dementia with motor neuron disease (FTD-MND), progressive supranuclear palsy (PSP) and corticobasal syndrome (CBS).
-
-
The most common neuropathologic substrates of frontotemporal lobar degeneration (FTLD) are: FTLD-tau, FTLD-TDP, and FTLD-FET.
-
-
The three genes most commonly associated with FTD are C9ORF72, MAPT and GRN.
-
-
There are currently no FDA approved treatments for FTD
Acknowledgments
Dr. Olney has received grant support from the National Institute of Health/National Institute of Aging T32AG023481-11S1 grant. Dr. Spina receives research support from the National Institute of Health/National Institute of Aging grant K08AG052648. Dr. Miller has served as an Advisor/Director to The Tau Consortium, The John Douglas French Foundation, The Larry L. Hillblom Foundation, Medical Advisory Board, National Institute for Health Research, Cambridge Biomedical Research Centre and its subunit, the Biomedical Research Unit in Dementia (UK); he has served as an External Advisor to University of Washington ADRC, Stanford University ADRC, and University of Pittsburgh ADRC; he has received grant/research support from National Institute of Health/National Institute of Aging grants P50AG023501, P01AG019724, and P50 AG1657303, Centers for Medicare & Medicaid Services (CMS) Dementia Care Ecosystem 1C1CMS3313460100, and a UCSF/Quest Diagnostics Dementia Pathway Collaboration Research Grant; he receives royalties from Cambridge University Press, Guilford Publications, Inc., and Neurocase.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The Authors have nothing to disclose.
Contributor Information
Nicholas T. Olney, Clinical Fellow in the Department of Neurology, UCSF School of Medicine, where he holds the A.W. & Mary Margaret Clausen Distinguished Chair, and directs the UCSF Memory and Aging Center.
Salvatore Spina, An Assistant Adjunct Professor of Neurology at the UCSF School of Medicine, where he holds the A.W. & Mary Margaret Clausen Distinguished Chair, and directs the UCSF Memory and Aging Center.
Bruce L. Miller, Professor of Neurology at the UCSF School of Medicine, where he holds the A.W. & Mary Margaret Clausen Distinguished Chair, and directs the UCSF Memory and Aging Center.
References
- Ahmed Z, Bigio EH, Budka H, Dickson DW, Ferrer I, Ghetti B, Kovacs GG. Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol. 2013;126(4):537–544. doi: 10.1007/s00401-013-1171-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer A. Uber eigenartige Krankheitsfalle des spateren Alters. Z Gesamte Neruol Psychiatr. 1911;4:356–385. [Google Scholar]
- Ames D, Cummings JL, Wirshing WC, Quinn B, Mahler M. Repetitive and compulsive behavior in frontal lobe degenerations. Journal of Neuropsychiatry and Clinical Neurosciences. 1994;6(2):100–113. doi: 10.1176/jnp.6.2.100. [DOI] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, Weiner WJ. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013;80(5):496–503. doi: 10.1212/WNL.0b013e31827f0fd1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baborie A, Griffiths TD, Jaros E, Perry R, McKeith IG, Burn DJ, Mann DM. Accumulation of dipeptide repeat proteins predates that of TDP-43 in frontotemporal lobar degeneration associated with hexanucleotide repeat expansions in C9ORF72 gene. Neuropathol Appl Neurobiol. 2015;41(5):601–612. doi: 10.1111/nan.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- Baloh RH. How do the RNA-binding proteins TDP-43 and FUS relate to amyotrophic lateral sclerosis and frontotemporal degeneration, and to each other? Curr Opin Neurol. 2012;25(6):701–707. doi: 10.1097/WCO.0b013e32835a269b. [DOI] [PubMed] [Google Scholar]
- Baralle M, Buratti E, Baralle FE. The role of TDP-43 in the pathogenesis of ALS and FTLD. Biochem Soc Trans. 2013;41(6):1536–1540. doi: 10.1042/BST20130186. [DOI] [PubMed] [Google Scholar]
- Bigio EH, Brown DF, White CL. Progressive supranuclear palsy with dementia: cortical pathology. J Neuropathol Exp Neurol. 1999;58(4):359–364. doi: 10.1097/00005072-199904000-00006. [DOI] [PubMed] [Google Scholar]
- Boeve BF, Boylan KB, Graff-Radford NR, DeJesus-Hernandez M, Knopman DS, Pedraza O, Rademakers R. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain. 2012;135(Pt 3):765–783. doi: 10.1093/brain/aws004. doi:aws004 [pii] 10.1093/brain/aws004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeve BF, Maraganore DM, Parisi JE, Ahlskog JE, Graff-Radford N, Caselli RJ, Petersen RC. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology. 1999;53(4):795–800. doi: 10.1212/wnl.53.4.795. [DOI] [PubMed] [Google Scholar]
- Borghero G, Floris G, Cannas A, Marrosu MG, Murru MR, Costantino E, Chio A. A patient carrying a homozygous p.A382T TARDBP missense mutation shows a syndrome including ALS, extrapyramidal symptoms, and FTD. Neurobiol Aging. 2011;32(12):2327 e2321–2325. doi: 10.1016/j.neurobiolaging.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer AL, Geschwind MD, Belfor N, Gorno-Tempini ML, Schauer GF, Miller BL, Rosen HJ. Patterns of brain atrophy that differentiate corticobasal degeneration syndrome from progressive supranuclear palsy. Arch Neurol. 2006;63(1):81–86. doi: 10.1001/archneur.63.1.81. [DOI] [PubMed] [Google Scholar]
- Boxer AL, Gold M, Huey E, Gao FB, Burton EA, Chow T, Cummings J. Frontotemporal degeneration, the next therapeutic frontier: molecules and animal models for frontotemporal degeneration drug development. Alzheimers Dement. 2013;9(2):176–188. doi: 10.1016/j.jalz.2012.03.002. S1552-5260(12)01750-5 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer AL, Gold M, Huey E, Hu WT, Rosen H, Kramer J, Cummings JL. The advantages of frontotemporal degeneration drug development (part 2 of frontotemporal degeneration: the next therapeutic frontier) Alzheimers Dement. 2013;9(2):189–198. doi: 10.1016/j.jalz.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer AL, Knopman DS, Kaufer DI, Grossman M, Onyike C, Graf-Radford N, Miller BL. Memantine in patients with frontotemporal lobar degeneration: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet neurology. 2013;12(2):149–156. doi: 10.1016/S1474-4422(12)70320-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J Neurol Sci. 1994;124(Suppl):96–107. doi: 10.1016/0022-510x(94)90191-0. [DOI] [PubMed] [Google Scholar]
- Brun A. Frontal lobe degeneration of non-Alzheimer type revisited. Dementia. 1993;4:126–131. doi: 10.1159/000107311. [DOI] [PubMed] [Google Scholar]
- Buratti E, Baralle FE. The molecular links between TDP-43 dysfunction and neurodegeneration. Adv Genet. 2009;66:1–34. doi: 10.1016/S0065-2660(09)66001-6. [DOI] [PubMed] [Google Scholar]
- Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, Mann DM. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol (Berl) 2007;114(1):5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, Mackenzie IR. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol. 2007;171(1):227–240. doi: 10.2353/ajpath.2007.070182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho MD, Swash M. Awaji diagnostic algorithm increases sensitivity of El Escorial criteria for ALS diagnosis. Amyotroph Lateral Scler. 2009;10(1):53–57. doi: 10.1080/17482960802521126. [DOI] [PubMed] [Google Scholar]
- Chambers CB, Lee JM, Troncoso JC, Reich S, Muma NA. Overexpression of four-repeat tau mRNA isoforms in progressive supranuclear palsy but not in Alzheimer’s disease. Ann Neurol. 1999;46(3):325–332. doi: 10.1002/1531-8249(199909)46:3<325::aid-ana8>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Cheng ST, Chow PK, Song YQ, Yu EC, Chan AC, Lee TM, Lam JH. Mental and physical activities delay cognitive decline in older persons with dementia. Am J Geriatr Psychiatry. 2014;22(1):63–74. doi: 10.1016/j.jagp.2013.01.060. [DOI] [PubMed] [Google Scholar]
- Chow TW, Binns MA, Cummings JL, Lam I, Black SE, Miller BL, van Reekum R. Apathy symptom profile and behavioral associations in frontotemporal dementia vs dementia of Alzheimer type. Arch Neurol. 2009;66(7):888–893. doi: 10.1001/archneurol.2009.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow TW, Mendez MF. Goals in symptomatic pharmacologic management of frontotemporal lobar degeneration. Am J Alzheimers Dis Other Demen. 2002;17(5):267–272. doi: 10.1177/153331750201700504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow TW, Miller BL, Hayashi VN, Geschwind DH. Inheritance of frontotemporal dementia. Arch Neurol. 1999;56(7):817–822. doi: 10.1001/archneur.56.7.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark LN, Poorkaj P, Wszolek Z, Geschwind DH, Nasreddine ZS, Miller B, Wilhelmsen KC. Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(22):13103–13107. doi: 10.1073/pnas.95.22.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinical and neuropathological criteria for frontotemporal dementia. The Lund and Manchester Groups. J Neurol Neurosurg Psychiatry. 1994;57(4):416–418. doi: 10.1136/jnnp.57.4.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad C, Andreadis A, Trojanowski JQ, Dickson DW, Kang D, Chen X, Saitoh T. Genetic evidence for the involvement of tau in progressive supranuclear palsy [see comments] Ann Neurol. 1997;41(2):277–281. doi: 10.1002/ana.410410222. [DOI] [PubMed] [Google Scholar]
- Constantinidis J, Richard J, Tissot R. Pick’s disease: histological and clinical correlations. European Neurology. 1974;11:208–217. doi: 10.1159/000114320. [DOI] [PubMed] [Google Scholar]
- Coppola G, Chinnathambi S, Lee JJ, Dombroski BA, Baker MC, Soto-Ortolaza AI, Geschwind DH. Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer’s diseases. Hum Mol Genet. 2012;21(15):3500–3512. doi: 10.1093/hmg/dds161. dds161 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola G, Karydas A, Rademakers R, Wang Q, Baker M, Hutton M, Geschwind DH. Gene expression study on peripheral blood identifies progranulin mutations. Ann Neurol. 2008;64(1):92–96. doi: 10.1002/ana.21397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle-Gilchrist IT, Dick KM, Patterson K, Vazquez Rodriquez P, Wehmann E, Wilcox A, Rowe JB. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology. 2016;86(18):1736–1743. doi: 10.1212/WNL.0000000000002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Van Broeckhoven C. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442(7105):920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- Cruz M, Marinho V, Fontenelle LF, Engelhardt E, Laks J. Topiramate may modulate alcohol abuse but not other compulsive behaviors in frontotemporal dementia: case report. Cogn Behav Neurol. 2008;21(2):104–106. doi: 10.1097/WNN.0b013e31816bdf73. [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Siddique T. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477(7363):211–215. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson D, ebrary Inc. Neurodegeneration the molecular pathology of dementia and movement disorders. 2011:xvii, 477. p. chiefly col. ill.). Retrieved from http://site.ebrary.com/lib/princeton/Doc?id=10501281.
- Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, Litvan I. Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. 2002;61(11):935–946. doi: 10.1093/jnen/61.11.935. [DOI] [PubMed] [Google Scholar]
- Diehl-Schmid J, Perneczky R, Koch J, Nedopil N, Kurz A. Guilty by suspicion? Criminal behavior in frontotemporal lobar degeneration. Cogn Behav Neurol. 2013;26(2):73–77. doi: 10.1097/WNN.0b013e31829cff11. [DOI] [PubMed] [Google Scholar]
- Donker Kaat L, Boon AJ, Kamphorst W, Ravid R, Duivenvoorden HJ, van Swieten JC. Frontal presentation in progressive supranuclear palsy. Neurology. 2007;69(8):723–729. doi: 10.1212/01.wnl.0000267643.24870.26. [DOI] [PubMed] [Google Scholar]
- Edwards-Lee T, Miller BL, Benson DF, Cummings JL, Russell GL, Boone K, Mena I. The temporal variant of frontotemporal dementia. Brain. 1997;120(Pt 6):1027–1040. doi: 10.1093/brain/120.6.1027. [DOI] [PubMed] [Google Scholar]
- Feany MB, Dickson DW. Neurodegenerative Disorders with Extensive Tau Pathology: A Comparative Study and Review. Annals of Neurology. 1996;40(2):139–148. doi: 10.1002/ana.410400204. [DOI] [PubMed] [Google Scholar]
- Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, Siddique T. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68(11):1440–1446. doi: 10.1001/archneurol.2011.250. [DOI] [PubMed] [Google Scholar]
- Finger EC. New potential therapeutic approaches in frontotemporal dementia: oxytocin, vasopressin, and social cognition. J Mol Neurosci. 2011;45(3):696–701. doi: 10.1007/s12031-011-9550-2. [DOI] [PubMed] [Google Scholar]
- Flanagan EP, Baker MC, Perkerson RB, Duffy JR, Strand EA, Whitwell JL, Josephs KA. Dominant frontotemporal dementia mutations in 140 cases of primary progressive aphasia and speech apraxia. Dement Geriatr Cogn Disord. 2015;39(5–6):281–286. doi: 10.1159/000375299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Muller K, Weishaupt JH. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18(5):631–636. doi: 10.1038/nn.4000. [DOI] [PubMed] [Google Scholar]
- Fujino Y, Wang DS, Thomas N, Espinoza M, Davies P, Dickson DW. Increased frequency of argyrophilic grain disease in Alzheimer disease with 4R tau-specific immunohistochemistry. J Neuropathol Exp Neurol. 2005;64(3):209–214. doi: 10.1093/jnen/64.3.209. [DOI] [PubMed] [Google Scholar]
- Geevasinga N, Loy CT, Menon P, de Carvalho M, Swash M, Schrooten M, Vucic S. Awaji criteria improves the diagnostic sensitivity in amyotrophic lateral sclerosis: A systematic review using individual patient data. Clin Neurophysiol. 2016;127(7):2684–2691. doi: 10.1016/j.clinph.2016.04.005. [DOI] [PubMed] [Google Scholar]
- Ghetti B, Oblak AL, Boeve BF, Johnson KA, Dickerson BC, Goedert M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathology and applied neurobiology. 2015;41(1):24–46. doi: 10.1111/nan.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman JS, Farmer JM, Wood EM, Johnson JK, Boxer A, Neuhaus J, Miller BL. Comparison of family histories in FTLD subtypes and related tauopathies. Neurology. 2005;65(11):1817–1819. doi: 10.1212/01.wnl.0000187068.92184.63. [DOI] [PubMed] [Google Scholar]
- Gorno-Tempini ML, Dronkers NF, Rankin KP, Ogar JM, Phengrasamy L, Rosen HJ, Miller BL. Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol. 2004;55(3):335–346. doi: 10.1002/ana.10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, Grossman M. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006–1014. doi: 10.1212/WNL.0b013e31821103e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorno-Tempini ML, Ogar JM, Brambati SM, Wang P, Jeong JH, Rankin KP, Miller BL. Anatomical correlates of early mutism in progressive nonfluent aphasia. Neurology. 2006;67(10):1849–1851. doi: 10.1212/01.wnl.0000237038.55627.5b. [DOI] [PubMed] [Google Scholar]
- Graff-Radford NR, Damasio AR, Hyman BT, Hart MN, Tranel D, Damasio H, Rezai K. Progressive aphasia in a patient with Pick’s disease: a neuropsychological, radiologic, and anatomic study. Neurology. 1990;40:620–626. doi: 10.1212/wnl.40.4.620. [DOI] [PubMed] [Google Scholar]
- Gregory CA, Serra-Mestres J, Hodges JR. Early diagnosis of the frontal variant of frontotemporal dementia: how sensitive are standard neuroimaging and neuropsychologic tests? Neuropsychiatry, Neuropsychology, and Behavioral Neurology. 1999;12(2):128–135. [PubMed] [Google Scholar]
- Grinberg LT, Wang X, Wang C, Sohn PD, Theofilas P, Sidhu M, Seeley WW. Argyrophilic grain disease differs from other tauopathies by lacking tau acetylation. Acta neuropathologica. 2013;125(4):581–593. doi: 10.1007/s00401-013-1080-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover A, Houlden H, Baker M, Adamson J, Lewis J, Prihar G, Hutton M. 5’ splice site mutations in tau associated with the inherited dementia FTDP-17 affect a stem-loop structure that regulates alternative splicing of exon 10. J Biol Chem. 1999;274(21):15134–15143. doi: 10.1074/jbc.274.21.15134. [DOI] [PubMed] [Google Scholar]
- Hassan A, Whitwell JL, Boeve BF, Jack CR, Jr, Parisi JE, Dickson DW, Josephs KA. Symmetric corticobasal degeneration (S-CBD) Parkinsonism Relat Disord. 2010;16(3):208–214. doi: 10.1016/j.parkreldis.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann N, Black SE, Chow T, Cappell J, Tang-Wai DF, Lanctot KL. Serotonergic function and treatment of behavioral and psychological symptoms of frontotemporal dementia. Am J Geriatr Psychiatry. 2012;20(9):789–797. doi: 10.1097/JGP.0b013e31823033f3. [DOI] [PubMed] [Google Scholar]
- Hodges JR, Davies R, Xuereb J, Kril J, Halliday G. Survival in frontotemporal dementia. Neurology. 2003;61(3):349–354. doi: 10.1212/01.wnl.0000078928.20107.52. [DOI] [PubMed] [Google Scholar]
- Hodges JR, Mitchell J, Dawson K, Spillantini MG, Xuereb JH, McMonagle P, Patterson K. Semantic dementia: demography, familial factors and survival in a consecutive series of 100 cases. Brain. 2010;133(Pt 1):300–306. doi: 10.1093/brain/awp248. [DOI] [PubMed] [Google Scholar]
- Hodges JR, Patterson K, Ward R, Garrard P, Bak T, Perry R, Gregory C. The differentiation of semantic dementia and frontal lobe dementia (temporal and frontal variants of frontotemporal dementia) from early Alzheimer’s disease: a comparative neuropsychological study. Neuropsychology. 1999;13(1):31–40. doi: 10.1037//0894-4105.13.1.31. [DOI] [PubMed] [Google Scholar]
- Hogan DB, Jette N, Fiest KM, Roberts JI, Pearson D, Smith EE, Maxwell CJ. The Prevalence and Incidence of Frontotemporal Dementia: a Systematic Review. Can J Neurol Sci. 2016;43(Suppl 1):S96–S109. doi: 10.1017/cjn.2016.25. [DOI] [PubMed] [Google Scholar]
- Holm IE, Isaacs AM, Mackenzie IR. Absence of FUS-immunoreactive pathology in frontotemporal dementia linked to chromosome 3 (FTD-3) caused by mutation in the CHMP2B gene. Acta Neuropathol. 2009;118(5):719–720. doi: 10.1007/s00401-009-0593-1. [DOI] [PubMed] [Google Scholar]
- Holroyd CB, Yeung N. Motivation of extended behaviors by anterior cingulate cortex. Trends Cogn Sci. 2012;16(2):122–128. doi: 10.1016/j.tics.2011.12.008. [DOI] [PubMed] [Google Scholar]
- Hosler BA, Siddique T, Sapp PC, Sailor W, Huang MC, Hossain A, Brown RH., Jr Linkage of familial amyotrophic lateral sclerosis with frontotemporal dementia to chromosome 9q21–q22. JAMA. 2000;284(13):1664–1669. doi: 10.1001/jama.284.13.1664. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Heutink P. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Shigenobu K, Fukuhara R, Hokoishi K, Maki N, Nebu A, Tanabe H. Efficacy of fluvoxamine as a treatment for behavioral symptoms in frontotemporal lobar degeneration patients. Dement Geriatr Cogn Disord. 2004;17(3):117–121. doi: 10.1159/000076343. [DOI] [PubMed] [Google Scholar]
- Ingvar DH, Gustafson L. Regional cerebral blood flow in organic dementia with early onset. Acta Neurol Scand. 1970;46(Suppl 43):42+. doi: 10.1111/j.1600-0404.1970.tb02156.x. [DOI] [PubMed] [Google Scholar]
- Irwin DJ. Tauopathies as clinicopathological entities. Parkinsonism Relat Disord. 2016;22(Suppl 1):S29–33. doi: 10.1016/j.parkreldis.2015.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin DJ, Brettschneider J, McMillan CT, Cooper F, Olm C, Arnold SE, Trojanowski JQ. Deep clinical and neuropathological phenotyping of Pick disease. Ann Neurol. 2016;79(2):272–287. doi: 10.1002/ana.24559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara K, Araki S, Ihori N, Shiota J, Kawamura M, Yoshida M, Nakano I. Argyrophilic grain disease presenting with frontotemporal dementia: a neuropsychological and pathological study of an autopsied case with presenile onset. Neuropathology. 2005;25(2):165–170. doi: 10.1111/j.1440-1789.2005.00598.x. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. The enigma of vascular cognitive disorder and vascular dementia. Acta Neuropathol. 2007;113(4):349–388. doi: 10.1007/s00401-006-0185-2. [DOI] [PubMed] [Google Scholar]
- Jenkins SM, Johnson GV. Tau complexes with phospholipase C-gamma in situ. NeuroReport. 1998;9(1):67–71. doi: 10.1097/00001756-199801050-00014. [DOI] [PubMed] [Google Scholar]
- Jesso S, Morlog D, Ross S, Pell MD, Pasternak SH, Mitchell DG, Finger EC. The effects of oxytocin on social cognition and behaviour in frontotemporal dementia. Brain. 2011;134(Pt 9):2493–2501. doi: 10.1093/brain/awr171. [DOI] [PubMed] [Google Scholar]
- Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, Traynor BJ. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68(5):857–864. doi: 10.1016/j.neuron.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Hodges JR, Snowden JS, Mackenzie IR, Neumann M, Mann DM, Dickson DW. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. 2011;122(2):137–153. doi: 10.1007/s00401-011-0839-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Petersen RC, Knopman DS, Boeve BF, Whitwell JL, Duffy JR, Dickson DW. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology. 2006;66(1):41–48. doi: 10.1212/01.wnl.0000191307.69661.c3. [DOI] [PubMed] [Google Scholar]
- Josephs KA, Whitwell JL, Jack CR., Jr Anatomic correlates of stereotypies in frontotemporal lobar degeneration. Neurobiol Aging. 2008;29(12):1859–1863. doi: 10.1016/j.neurobiolaging.2007.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakaya T, Fusser F, Prvulovic D, Hampel H. Treatment options for tauopathies. Curr Treat Options Neurol. 2012;14(2):126–136. doi: 10.1007/s11940-012-0168-7. [DOI] [PubMed] [Google Scholar]
- Katzman R. Editorial: The prevalence and malignancy of Alzheimer disease. A major killer. Arch Neurol. 1976;33(4):217–218. doi: 10.1001/archneur.1976.00500040001001. [DOI] [PubMed] [Google Scholar]
- Kertesz A, Davidson W, McCabe P, Takagi K, Munoz D. Primary progressive aphasia: diagnosis, varieties, evolution. J Int Neuropsychol Soc. 2003;9(5):710–719. doi: 10.1017/S1355617703950041. [DOI] [PubMed] [Google Scholar]
- Kertesz A, McMonagle P. Behavior and cognition in corticobasal degeneration and progressive supranuclear palsy. J Neurol Sci. 2010;289(1–2):138–143. doi: 10.1016/j.jns.2009.08.036. [DOI] [PubMed] [Google Scholar]
- Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG. The evolution and pathology of frontotemporal dementia. Brain. 2005;128(Pt 9):1996–2005. doi: 10.1093/brain/awh598. [DOI] [PubMed] [Google Scholar]
- Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem. 2012;287(23):19440–19451. doi: 10.1074/jbc.M112.346072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan BK, Yokoyama JS, Takada LT, Sha SJ, Rutherford NJ, Fong JC, Miller BL. Atypical, slowly progressive behavioural variant frontotemporal dementia associated with C9ORF72 hexanucleotide expansion. J Neurol Neurosurg Psychiatry. 2012;83(4):358–364. doi: 10.1136/jnnp-2011-301883. doi:jnnp-2011-301883 [pii] 10.1136/jnnp-2011-301883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YE, Kang SY, Ma HI, Ju YS, Kim YJ. A Visual Rating Scale for the Hummingbird Sign with Adjustable Diagnostic Validity. J Parkinsons Dis. 2015;5(3):605–612. doi: 10.3233/JPD-150537. [DOI] [PubMed] [Google Scholar]
- Kimura T, Takamatsu J. Pilot study of pharmacological treatment for frontotemporal dementia: risk of donepezil treatment for behavioral and psychological symptoms. Geriatr Gerontol Int. 2013;13(2):506–507. doi: 10.1111/j.1447-0594.2012.00956.x. [DOI] [PubMed] [Google Scholar]
- Kipps CM, Hodges JR, Hornberger M. Nonprogressive behavioural frontotemporal dementia: recent developments and clinical implications of the ‘bvFTD phenocopy syndrome’. Curr Opin Neurol. 2010;23(6):628–632. doi: 10.1097/WCO.0b013e3283404309. [DOI] [PubMed] [Google Scholar]
- Knopman DS, Roberts RO. Estimating the number of persons with frontotemporal lobar degeneration in the US population. J Mol Neurosci. 2011;45(3):330–335. doi: 10.1007/s12031-011-9538-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortte KB, Rogalski EJ. Behavioural interventions for enhancing life participation in behavioural variant frontotemporal dementia and primary progressive aphasia. Int Rev Psychiatry. 2013;25(2):237–245. doi: 10.3109/09540261.2012.751017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, Murrell JR, Horvath S, Haraszti L, Majtenyi K, Molnar MJ, Spina S. TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Mov Disord. 2009;24(12):1843–1847. doi: 10.1002/mds.22697. [DOI] [PubMed] [Google Scholar]
- Kramer JH, Jurik J, Sha SJ, Rankin KP, Rosen HJ, Johnson JK, Miller BL. Distinctive neuropsychological patterns in frontotemporal dementia, semantic dementia, and Alzheimer disease. Cogn Behav Neurol. 2003;16(4):211–218. doi: 10.1097/00146965-200312000-00002. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Brown RH., Jr Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–1208. doi: 10.1126/science.1166066. 323/5918/1205 [pii] [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, Ravits J. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A. 2013;110(47):E4530–4539. doi: 10.1073/pnas.1318835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanata SC, Miller BL. The behavioural variant frontotemporal dementia (bvFTD) syndrome in psychiatry. J Neurol Neurosurg Psychiatry. 2016;87(5):501–511. doi: 10.1136/jnnp-2015-310697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Ber I, Camuzat A, Hannequin D, Pasquier F, Guedj E, Rovelet-Lecrux A, Brice A. Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain. 2008;131(Pt 3):732–746. doi: 10.1093/brain/awn012. awn012 [pii] [DOI] [PubMed] [Google Scholar]
- Lebert F, Stekke W, Hasenbroekx C, Pasquier F. Frontotemporal dementia: a randomised, controlled trial with trazodone. Dement Geriatr Cogn Disord. 2004;17(4):355–359. doi: 10.1159/000077171. [DOI] [PubMed] [Google Scholar]
- Lee EB, Russ J, Jung H, Elman LB, Chahine LM, Kremens D, McCluskey LF. Topography of FUS pathology distinguishes late-onset BIBD from aFTLD-U. Acta Neuropathol Commun. 2013;1(9):1–11. doi: 10.1186/2051-5960-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SE, Khazenzon AM, Trujillo AJ, Guo CC, Yokoyama JS, Sha SJ, Seeley WW. Altered network connectivity in frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. Brain. 2014;137(Pt 11):3047–3060. doi: 10.1093/brain/awu248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SE, Rabinovici GD, Mayo MC, Wilson SM, Seeley WW, DeArmond SJ, Miller BL. Clinicopathological correlations in corticobasal degeneration. Ann Neurol. 2011;70(2):327–340. doi: 10.1002/ana.22424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Leugers CJ, Lee G. Tau potentiates nerve growth factor-induced mitogen-activated protein kinase signaling and neurite initiation without a requirement for microtubule binding. J Biol Chem. 2010;285(25):19125–19134. doi: 10.1074/jbc.M110.105387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liljegren M, Naasan G, Temlett J, Perry DC, Rankin KP, Merrilees J, Miller BL. Criminal behavior in frontotemporal dementia and Alzheimer disease. JAMA Neurol. 2015;72(3):295–300. doi: 10.1001/jamaneurol.2014.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling H, O’Sullivan SS, Holton JL, Revesz T, Massey LA, Williams DR, Lees AJ. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain. 2010;133(Pt 7):2045–2057. doi: 10.1093/brain/awq123. [DOI] [PubMed] [Google Scholar]
- Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, Zee DS. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47(1):1–9. doi: 10.1212/wnl.47.1.1. [DOI] [PubMed] [Google Scholar]
- Litvan I, Hauw JJ, Bartko JJ, Lantos PL, Daniel SE, Horoupian DS, Anderson DW. Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol. 1996;55(1):97–105. doi: 10.1097/00005072-199601000-00010. [DOI] [PubMed] [Google Scholar]
- Liu A, Werner K, Roy S, Trojanowski JQ, Morgan-Kane U, Miller BL, Rankin KP. A case study of an emerging visual artist with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurocase. 2009;15(3):235–247. doi: 10.1080/13554790802633213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomen-Hoerth C. Clinical phenomenology and neuroimaging correlates in ALS-FTD. J Mol Neurosci. 2011;45(3):656–662. doi: 10.1007/s12031-011-9636-x. [DOI] [PubMed] [Google Scholar]
- Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59(7):1077–1079. doi: 10.1212/wnl.59.7.1077. [DOI] [PubMed] [Google Scholar]
- Lu CH, Macdonald-Wallis C, Gray E, Pearce N, Petzold A, Norgren N, Malaspina A. Neurofilament light chain: A prognostic biomarker in amyotrophic lateral sclerosis. Neurology. 2015;84(22):2247–2257. doi: 10.1212/WNL.0000000000001642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR. The neuropathology and clinical phenotype of FTD with progranulin mutations. Acta Neuropathol. 2007a;114(1):49–54. doi: 10.1007/s00401-007-0223-8. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR. The neuropathology of FTD associated With ALS. Alzheimer Dis Assoc Disord. 2007b;21(4):S44–49. doi: 10.1097/WAD.0b013e31815c3486. 00002093-200710000-00016 [pii] [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Arzberger T, Kremmer E, Troost D, Lorenzl S, Mori K, Neumann M. Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol. 2013;126(6):859–879. doi: 10.1007/s00401-013-1181-y. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Feldman HH. Ubiquitin immunohistochemistry suggests classic motor neuron disease, motor neuron disease with dementia, and frontotemporal dementia of the motor neuron disease type represent a clinicopathologic spectrum. J Neuropathol Exp Neurol. 2005;64(8):730–739. doi: 10.1097/01.jnen.0000174335.27708.0a. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Munoz DG, Kusaka H, Yokota O, Ishihara K, Roeber S, Neumann M. Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol. 2011;121(2):207–218. doi: 10.1007/s00401-010-0764-0. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Neumann M. FET proteins in frontotemporal dementia and amyotrophic lateral sclerosis. Brain Res. 2012;1462:40–43. doi: 10.1016/j.brainres.2011.12.010. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Neumann M. Molecular neuropathology of frontotemporal dementia: insights into disease mechanisms from postmortem studies. J Neurochem. 2016 doi: 10.1111/jnc.13588. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Lee VM. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122(1):111–113. doi: 10.1007/s00401-011-0845-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Mann DM. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 2009;117(1):15–18. doi: 10.1007/s00401-008-0460-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Mann DM. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119(1):1–4. doi: 10.1007/s00401-009-0612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9(10):995–1007. doi: 10.1016/S1474-4422(10)70195-2. S1474-4422(10)70195-2 [pii] 10.1016/S1474-4422(10)70195-2. [DOI] [PubMed] [Google Scholar]
- Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med. 2012;2(7):a006247. doi: 10.1101/cshperspect.a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann DM. Dipeptide repeat protein toxicity in frontotemporal lobar degeneration and in motor neurone disease associated with expansions in C9ORF72-a cautionary note. Neurobiol Aging. 2015;36(2):1224–1226. doi: 10.1016/j.neurobiolaging.2014.10.011. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Schwab C, McGeer EG, Haddock RL, Steele JC. Familial nature and continuing morbidity of the amyotrophic lateral sclerosis-parkinsonism dementia complex of Guam. Neurology. 1997;49(2):400–409. doi: 10.1212/wnl.49.2.400. [DOI] [PubMed] [Google Scholar]
- Meeter LH, Dopper EG, Jiskoot LC, Sanchez-Valle R, Graff C, Benussi L, van Swieten JC. Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol. 2016;3(8):623–636. doi: 10.1002/acn3.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez MF, Shapira JS. Loss of emotional insight in behavioral variant frontotemporal dementia or “frontal anosodiaphoria”. Conscious Cogn. 2011;20(4):1690–1696. doi: 10.1016/j.concog.2011.09.005. S1053-8100(11)00213-3 [pii] 10.1016/j.concog.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez MF, Shapira JS, McMurtray A, Licht E. Preliminary findings: behavioral worsening on donepezil in patients with frontotemporal dementia. Am J Geriatr Psychiatry. 2007;15(1):84–87. doi: 10.1097/01.JGP.0000231744.69631.33. [DOI] [PubMed] [Google Scholar]
- Mendez MF, Shapira JS, Miller BL. Stereotypical movements and frontotemporal dementia. Mov Disord. 2005;20(6):742–745. doi: 10.1002/mds.20465. [DOI] [PubMed] [Google Scholar]
- Merrilees J. A model for management of behavioral symptoms in frontotemporal lobar degeneration. Alzheimer Dis Assoc Disord. 2007;21(4):S64–69. doi: 10.1097/WAD.0b013e31815bf774. [DOI] [PubMed] [Google Scholar]
- Merrilees J, Hubbard E, Mastick J, Miller BL, Dowling GA. Rest-activity and behavioral disruption in a patient with frontotemporal dementia. Neurocase. 2009;15(6):515–526. doi: 10.1080/13554790903061371. 914594846 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam MM. Primary progressive aphasia: differentiation from Alzheimer’s disease. Ann Neurol. 1987;37:1448–1553. doi: 10.1002/ana.410220414. [DOI] [PubMed] [Google Scholar]
- Mesulam M, Wicklund A, Johnson N, Rogalski E, Leger GC, Rademaker A, Bigio EH. Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Ann Neurol. 2008;63(6):709–719. doi: 10.1002/ana.21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam MM. Slowly progressive aphasia without generalized dementia. Annals of Neurology. 1982;11(6):592–598. doi: 10.1002/ana.410110607. [DOI] [PubMed] [Google Scholar]
- Mesulam MM. Primary progressive aphasia. Ann Neurol. 2001;49(4):425–432. [PubMed] [Google Scholar]
- Miller BL. Frontotemporal Dementia. Oxford University Press; 2014. [Google Scholar]
- Miller BL, Boone K, Cummings JL, Read SL, Mishkin F. Functional correlates of musical and visual ability in frontotemporal dementia. Br J Psychiatry. 2000;176:458–463. doi: 10.1192/bjp.176.5.458. [DOI] [PubMed] [Google Scholar]
- Miller BL, Chang L, Mena I, Boone K, Lesser IM. Progressive right frontotemporal degeneration: clinical, neuropsychological and SPECT characteristics. Dementia. 1993;4(3–4):204–213. doi: 10.1159/000107324. [DOI] [PubMed] [Google Scholar]
- Miller BL, Cummings J, Mishkin F, Boone K, Prince F, Ponton M, Cotman C. Emergence of artistic talent in frontotemporal dementia. Neurology. 1998;51(4):978–982. doi: 10.1212/wnl.51.4.978. [DOI] [PubMed] [Google Scholar]
- Miller BL, Darby AL, Swartz JR, Yener GG, Mena I. Dietary changes, compulsions and sexual behavior in frontotemporal degeneration. Dementia. 1995;6(4):195–199. doi: 10.1159/000106946. [DOI] [PubMed] [Google Scholar]
- Miller BL, Ponton M, Benson DF, Cummings JL, Mena I. Enhanced artistic creativity with temporal lobe degeneration [letter] Lancet. 1996;348(9043):1744–1745. doi: 10.1016/s0140-6736(05)65881-3. [DOI] [PubMed] [Google Scholar]
- Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70(3):410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosca L, Lunetta C, Tarlarini C, Avemaria F, Maestri E, Melazzini M, Penco S. Wide phenotypic spectrum of the TARDBP gene: homozygosity of A382T mutation in a patient presenting with amyotrophic lateral sclerosis, Parkinson’s disease, and frontotemporal lobar degeneration, and in neurologically healthy subject. Neurobiol Aging. 2012;33(8):1846 e1841–1844. doi: 10.1016/j.neurobiolaging.2012.01.108. [DOI] [PubMed] [Google Scholar]
- Munoz DG, Neumann M, Kusaka H, Yokota O, Ishihara K, Terada S, Mackenzie IR. FUS pathology in basophilic inclusion body disease. Acta Neuropathol. 2009;118(5):617–627. doi: 10.1007/s00401-009-0598-9. [DOI] [PubMed] [Google Scholar]
- Myers AJ, Pittman AM, Zhao AS, Rohrer K, Kaleem M, Marlowe L, Hardy J. The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol Dis. 2007;25(3):561–570. doi: 10.1016/j.nbd.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Narvid J, Gorno-Tempini ML, Slavotinek A, Dearmond SJ, Cha YH, Miller BL, Rankin K. Of brain and bone: the unusual case of Dr. A. Neurocase. 2009;15(3):190–205. doi: 10.1080/13554790802632967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Benson DF. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- Neumann M, Bentmann E, Dormann D, Jawaid A, DeJesus-Hernandez M, Ansorge O, Mackenzie IR. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain. 2011;134(Pt 9):2595–2609. doi: 10.1093/brain/awr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Mackenzie IR, Cairns NJ, Boyer PJ, Markesbery WR, Smith CD, Forman MS. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol. 2007;66(2):152–157. doi: 10.1097/nen.0b013e31803020b9. [DOI] [PubMed] [Google Scholar]
- Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132(Pt 11):2922–2931. doi: 10.1093/brain/awp214. doi:awp214 [pii] 10.1093/brain/awp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Roeber S, Kretzschmar HA, Rademakers R, Baker M, Mackenzie IR. Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol. 2009;118(5):605–616. doi: 10.1007/s00401-009-0581-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Ogar J, Willock S, Baldo J, Wilkins D, Ludy C, Dronkers N. Clinical and anatomical correlates of apraxia of speech. Brain Lang. 2006;97(3):343–350. doi: 10.1016/j.bandl.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Olney RK, Murphy J, Forshew D, Garwood E, Miller BL, Langmore S, Lomen-Hoerth C. The effects of executive and behavioral dysfunction on the course of ALS. Neurology. 2005;65(11):1774–1777. doi: 10.1212/01.wnl.0000188759.87240.8b. [DOI] [PubMed] [Google Scholar]
- Onyike CU, Diehl-Schmid J. The epidemiology of frontotemporal dementia. Int Rev Psychiatry. 2013;25(2):130–137. doi: 10.3109/09540261.2013.776523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry DC, Whitwell JL, Boeve BF, Pankratz VS, Knopman DS, Petersen RC, Josephs KA. Voxel-based morphometry in patients with obsessive-compulsive behaviors in behavioral variant frontotemporal dementia. Eur J Neurol. 2012;19(6):911–917. doi: 10.1111/j.1468-1331.2011.03656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkau TL, Leavitt BR. Progranulin in neurodegenerative disease. Trends Neurosci. 2014;37(7):388–398. doi: 10.1016/j.tins.2014.04.003. [DOI] [PubMed] [Google Scholar]
- Pick A. Uber die Beziehungen der senilen Hirnatrophie zur Aphasie. Prager Medizinische Wochenschrift. 1892;17:165–167. [Google Scholar]
- Piguet O. Eating disturbance in behavioural-variant frontotemporal dementia. J Mol Neurosci. 2011;45(3):589–593. doi: 10.1007/s12031-011-9547-x. [DOI] [PubMed] [Google Scholar]
- Pijnenburg YA, Sampson EL, Harvey RJ, Fox NC, Rossor MN. Vulnerability to neuroleptic side effects in frontotemporal lobar degeneration. Int J Geriatr Psychiatry. 2003;18(1):67–72. doi: 10.1002/gps.774. [DOI] [PubMed] [Google Scholar]
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Cleveland DW. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14(4):459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Schellenberg GD. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Annals of neurology. 1998;43(6):815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- Pottier C, Bieniek KF, Finch N, van de Vorst M, Baker M, Perkersen R, Rademakers R. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 2015;130(1):77–92. doi: 10.1007/s00401-015-1436-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottier C, Ravenscroft TA, Sanchez-Contreras M, Rademakers R. Genetics of FTLD: overview and what else we can expect from genetic studies. J Neurochem. 2016;138(Suppl 1):32–53. doi: 10.1111/jnc.13622. [DOI] [PubMed] [Google Scholar]
- Proudfoot M, Gutowski NJ, Edbauer D, Hilton DA, Stephens M, Rankin J, Mackenzie IR. Early dipeptide repeat pathology in a frontotemporal dementia kindred with C9ORF72 mutation and intellectual disability. Acta Neuropathol. 2014;127(3):451–458. doi: 10.1007/s00401-014-1245-7. [DOI] [PubMed] [Google Scholar]
- Rabinovici GD, Jagust WJ, Furst AJ, Ogar JM, Racine CA, Mormino EC, Gorno-Tempini ML. Abeta amyloid and glucose metabolism in three variants of primary progressive aphasia. Ann Neurol. 2008;64(4):388–401. doi: 10.1002/ana.21451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovici GD, Rosen HJ, Alkalay A, Kornak J, Furst AJ, Agarwal N, Jagust WJ. Amyloid vs FDG-PET in the differential diagnosis of AD and FTLD. Neurology. 2011;77(23):2034–2042. doi: 10.1212/WNL.0b013e31823b9c5e. doi:WNL.0b013e31823b9c5e [pii] 10.1212/WNL.0b013e31823b9c5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranasinghe KG, Rankin KP, Lobach IV, Kramer JH, Sturm VE, Bettcher BM, Miller BL. Cognition and neuropsychiatry in behavioral variant frontotemporal dementia by disease stage. Neurology. 2016;86(7):600–610. doi: 10.1212/WNL.0000000000002373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin KP, Baldwin E, Pace-Savitsky C, Kramer JH, Miller BL. Self awareness and personality change in dementia. J Neurol Neurosurg Psychiatry. 2005;76(5):632–639. doi: 10.1136/jnnp.2004.042879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin KP, Gorno-Tempini ML, Allison SC, Stanley CM, Glenn S, Weiner MW, Miller BL. Structural anatomy of empathy in neurodegenerative disease. Brain. 2006;129(Pt 11):2945–2956. doi: 10.1093/brain/awl254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin KP, Mayo MC, Seeley WW, Lee S, Rabinovici G, Gorno-Tempini ML, Miller BL. Behavioral variant frontotemporal dementia with corticobasal degeneration pathology: phenotypic comparison to bvFTD with Pick’s disease. J Mol Neurosci. 2011;45(3):594–608. doi: 10.1007/s12031-011-9615-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, Miller BL. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(Pt 9):2456–2477. doi: 10.1093/brain/awr179. doi:awr179 [pii] 10.1093/brain/awr179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebeiz JJ, Kolodny EH, Richardson EP., Jr Corticodentatonigral degeneration with neuronal achromasia. Arch Neurol. 1968;18(1):20–33. doi: 10.1001/archneur.1968.00470310034003. [DOI] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Traynor BJ. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268. doi: 10.1016/j.neuron.2011.09.010. doi:S0896-6273(11)00797-5 [pii] 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer JD, Rosen HJ. Neuroimaging in frontotemporal dementia. Int Rev Psychiatry. 2013;25(2):221–229. doi: 10.3109/09540261.2013.778822. [DOI] [PubMed] [Google Scholar]
- Rohrer JD, Warren JD. Phenotypic signatures of genetic frontotemporal dementia. Curr Opin Neurol. 2011;24(6):542–549. doi: 10.1097/WCO.0b013e32834cd442. [DOI] [PubMed] [Google Scholar]
- Rohrer JD, Woollacott IO, Dick KM, Brotherhood E, Gordon E, Fellows A, Zetterberg H. Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology. 2016 doi: 10.1212/WNL.0000000000003154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas JC, Karydas A, Bang J, Tsai RM, Blennow K, Liman V, Boxer AL. Plasma neurofilament light chain predicts progression in progressive supranuclear palsy. Ann Clin Transl Neurol. 2016;3(3):216–225. doi: 10.1002/acn3.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman G. Vascular dementia: a historical background. Int Psychogeriatr. 2003;15(Suppl 1):11–13. doi: 10.1017/S1041610203008901. [DOI] [PubMed] [Google Scholar]
- Rosen HJ, Allison SC, Schauer GF, Gorno-Tempini ML, Weiner MW, Miller BL. Neuroanatomical correlates of behavioural disorders in dementia. Brain. 2005;128(Pt 11):2612–2625. doi: 10.1093/brain/awh628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen HJ, Gorno-Tempini ML, Goldman WP, Perry RJ, Schuff N, Weiner M, Miller BL. Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology. 2002;58(2):198–208. doi: 10.1212/wnl.58.2.198. [DOI] [PubMed] [Google Scholar]
- Rosen HJ, Lengenfelder J, Miller B. Frontotemporal dementia. Neurol Clin. 2000;18(4):979–992. doi: 10.1016/s0733-8619(05)70235-8. [DOI] [PubMed] [Google Scholar]
- Rosen HJ, Perry RJ, Murphy J, Kramer JH, Mychack P, Schuff N, Miller BL. Emotion comprehension in the temporal variant of frontotemporal dementia. Brain. 2002;125(Pt 10):2286–2295. doi: 10.1093/brain/awf225. [DOI] [PubMed] [Google Scholar]
- Rosso SM, Roks G, Stevens M, de Koning I, Tanghe HLJ, Kamphorst W, van Swieten JC. Complex compulsive behaviour in the temporal variant of frontotemporal dementia. J Neurol. 2001;248(11):965–970. doi: 10.1007/s004150170049. [DOI] [PubMed] [Google Scholar]
- Ryan NS, Rossor MN, Fox NC. Alzheimer’s disease in the 100 years since Alzheimer’s death. Brain. 2015;138(Pt 12):3816–3821. doi: 10.1093/brain/awv316. [DOI] [PubMed] [Google Scholar]
- Schneider A, Mandelkow E. Tau-based treatment strategies in neurodegenerative diseases. Neurotherapeutics. 2008;5(3):443–457. doi: 10.1016/j.nurt.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeter ML, Raczka K, Neumann J, von Cramon DY. Neural networks in frontotemporal dementia–a meta-analysis. Neurobiol Aging. 2008;29(3):418–426. doi: 10.1016/j.neurobiolaging.2006.10.023. [DOI] [PubMed] [Google Scholar]
- Seeley WW. Selective functional, regional, and neuronal vulnerability in frontotemporal dementia. Curr Opin Neurol. 2008;21(6):701–707. doi: 10.1097/WCO.0b013e3283168e2d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley WW, Bauer AM, Miller BL, Gorno-Tempini ML, Kramer JH, Weiner M, Rosen HJ. The natural history of temporal variant frontotemporal dementia. Neurology. 2005;64(8):1384–1390. doi: 10.1212/01.WNL.0000158425.46019.5C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley WW, Carlin DA, Allman JM, Macedo MN, Bush C, Miller BL, Dearmond SJ. Early frontotemporal dementia targets neurons unique to apes and humans. Ann Neurol. 2006;60(6):660–667. doi: 10.1002/ana.21055. [DOI] [PubMed] [Google Scholar]
- Seeley WW, Crawford R, Rascovsky K, Kramer JH, Weiner M, Miller BL, Gorno-Tempini ML. Frontal paralimbic network atrophy in very mild behavioral variant frontotemporal dementia. Arch Neurol. 2008;65(2):249–255. doi: 10.1001/archneurol.2007.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley WW, Menon V, Schatzberg AF, Keller J, Glover GH, Kenna H, Greicius MD. Dissociable intrinsic connectivity networks for salience processing and executive control. J Neurosci. 2007;27(9):2349–2356. doi: 10.1523/JNEUROSCI.5587-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley WW, Zhou J, Kim EJ. Frontotemporal Dementia: What Can the Behavioral Variant Teach Us about Human Brain Organization? Neuroscientist. 2012;18(4):373–385. doi: 10.1177/1073858411410354. doi:1073858411410354 [pii] 10.1177/1073858411410354. [DOI] [PubMed] [Google Scholar]
- Sha SJ, Takada LT, Rankin KP, Yokoyama JS, Rutherford NJ, Fong JC, Boxer AL. Frontotemporal dementia due to C9ORF72 mutations: Clinical and imaging features. Neurology. 2012;79(10):1002–1011. doi: 10.1212/WNL.0b013e318268452e. doi:WNL.0b013e318268452e [pii] 10.1212/WNL.0b013e318268452e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, Collinge J. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37(8):806–808. doi: 10.1038/ng1609. [DOI] [PubMed] [Google Scholar]
- Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, Berkovic SF. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet. 2012;90(6):1102–1107. doi: 10.1016/j.ajhg.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snowden JS, Adams J, Harris J, Thompson JC, Rollinson S, Richardson A, Pickering-Brown S. Distinct clinical and pathological phenotypes in frontotemporal dementia associated with MAPT, PGRN and C9orf72 mutations. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(7–8):497–505. doi: 10.3109/21678421.2015.1074700. [DOI] [PubMed] [Google Scholar]
- Snowden JS, Goulding PJ, Neary D. Semantic dementia: a form of circumscribed cerebral atrophy. Behavioural Neurology. 1989;2:167–182. [Google Scholar]
- Snowden JS, Hu Q, Rollinson S, Halliwell N, Robinson A, Davidson YS, Mann DM. The most common type of FTLD-FUS (aFTLD-U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS gene. Acta Neuropathol. 2011;122(1):99–110. doi: 10.1007/s00401-011-0816-0. [DOI] [PubMed] [Google Scholar]
- Snowden JS, Neary D, Mann DM. Frontotemporal dementia. Br J Psychiatry. 2002;180:140–143. doi: 10.1192/bjp.180.2.140. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(13):7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele JC, Richardson JC, Olszewski J. Progressive Supranuclear Palsy. Arch Neurol. 1964 Apr;10:333–360. doi: 10.1001/archneur.1964.00460160003001. [DOI] [PubMed] [Google Scholar]
- Suarez J, Tartaglia MC, Vitali P, Erbetta A, Neuhaus J, Laluz V, Miller BL. Characterizing radiology reports in patients with frontotemporal dementia. Neurology. 2009;73(13):1073–1074. doi: 10.1212/WNL.0b013e3181b9c8a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz JR, Miller BL, Lesser IM, Darby AL. Frontotemporal dementia: treatment response to serotonin selective reuptake inhibitors. J Clin Psychiatry. 1997;58(5):212–216. [PubMed] [Google Scholar]
- Tacik P, DeTure M, Hinkle KM, Lin WL, Sanchez-Contreras M, Carlomagno Y, Dickson DW. A Novel Tau Mutation in Exon 12, p.Q336H, Causes Hereditary Pick Disease. Journal of neuropathology and experimental neurology. 2015;74(11):1042–1052. doi: 10.1097/NEN.0000000000000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekin S, Cummings JL. Frontal-subcortical neuronal circuits and clinical neuropsychiatry: an update. J Psychosom Res. 2002;53(2):647–654. doi: 10.1016/s0022-3999(02)00428-2. [DOI] [PubMed] [Google Scholar]
- Thal DR, Schultz C, Botez G, Del Tredici K, Mrak RE, Griffin WS, Ghebremedhin E. The impact of argyrophilic grain disease on the development of dementia and its relationship to concurrent Alzheimer’s disease-related pathology. Neuropathol Appl Neurobiol. 2005;31(3):270–279. doi: 10.1111/j.1365-2990.2005.00635.x. [DOI] [PubMed] [Google Scholar]
- Thibodeau MP, Miller BL. ‘Limits and current knowledge of Pick’s disease: its differential diagnosis’. A translation of the 1957 Delay, Brion, Escourolle article. Neurocase. 2013;19(5):417–422. doi: 10.1080/13554794.2012.667133. [DOI] [PubMed] [Google Scholar]
- Tranel D, Bechara A, Denburg NL. Asymmetric functional roles of right and left ventromedial prefrontal cortices in social conduct, decision-making, and emotional processing. Cortex. 2002;38(4):589–612. doi: 10.1016/s0010-9452(08)70024-8. [DOI] [PubMed] [Google Scholar]
- Tsai RM, Boxer AL. Therapy and clinical trials in frontotemporal dementia: past, present, and future. J Neurochem. 2016;138(Suppl 1):211–221. doi: 10.1111/jnc.13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya K, Mitani K, Arai T, Yamada S, Komiya T, Esaki Y, Ikeda K. Argyrophilic grain disease mimicking temporal Pick’s disease: a clinical, radiological, and pathological study of an autopsy case with a clinical course of 15 years. Acta Neuropathol. 2001;102(2):195–199. doi: 10.1007/s004010100365. [DOI] [PubMed] [Google Scholar]
- Urwin H, Josephs KA, Rohrer JD, Mackenzie IR, Neumann M, Authier A, Isaacs AM. FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol. 2010;120(1):33–41. doi: 10.1007/s00401-010-0698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Van Hoecke A, Lambrechts D, Vanacker P, Bogaert E, van Swieten J, Robberecht W. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol. 2008;181(1):37–41. doi: 10.1083/jcb.200712039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Swieten JC, Heutink P. Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia. Lancet Neurol. 2008;7(10):965–974. doi: 10.1016/S1474-4422(08)70194-7. [DOI] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Shaw CE. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatsavayai SC, Yoon SJ, Gardner RC, Gendron TF, Vargas JN, Trujillo A, Seeley WW. Timing and significance of pathological features in C9orf72 expansion-associated frontotemporal dementia. Brain. 2016 doi: 10.1093/brain/aww250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercelletto M, Boutoleau-Bretonniere C, Volteau C, Puel M, Auriacombe S, Sarazin M, French research network on Frontotemporal, d. Memantine in behavioral variant frontotemporal dementia: negative results. J Alzheimers Dis. 2011;23(4):749–759. doi: 10.3233/JAD-2010-101632. [DOI] [PubMed] [Google Scholar]
- Wadia PM, Lang AE. The many faces of corticobasal degeneration. Parkinsonism Relat Disord. 2007;13(Suppl 3):S336–340. doi: 10.1016/S1353-8020(08)70027-0. [DOI] [PubMed] [Google Scholar]
- Walker LC, Jucker M. Neurodegenerative diseases: expanding the prion concept. Annu Rev Neurosci. 2015;38:87–103. doi: 10.1146/annurev-neuro-071714-033828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward ME, Miller BL. Potential mechanisms of progranulin-deficient FTLD. J Mol Neurosci. 2011;45(3):574–582. doi: 10.1007/s12031-011-9622-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts GD, Thomasova D, Ramdeen SK, Fulchiero EC, Mehta SG, Drachman DA, Kimonis VE. Novel VCP mutations in inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia. Clin Genet. 2007;72(5):420–426. doi: 10.1111/j.1399-0004.2007.00887.x. [DOI] [PubMed] [Google Scholar]
- Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72(5):1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Jack CR, Jr, Boeve BF, Parisi JE, Ahlskog JE, Drubach DA, Josephs KA. Imaging correlates of pathology in corticobasal syndrome. Neurology. 2010;75(21):1879–1887. doi: 10.1212/WNL.0b013e3181feb2e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Jack CR, Jr, Boeve BF, Senjem ML, Baker M, Rademakers R, Josephs KA. Voxel-based morphometry patterns of atrophy in FTLD with mutations in MAPT or PGRN. Neurology. 2009;72(9):813–820. doi: 10.1212/01.wnl.0000343851.46573.67. 72/9/813 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Sampson EL, Loy CT, Warren JE, Rossor MN, Fox NC, Warren JD. VBM signatures of abnormal eating behaviours in frontotemporal lobar degeneration. Neuroimage. 2007;35(1):207–213. doi: 10.1016/j.neuroimage.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Whitwell JL, Weigand SD, Boeve BF, Senjem ML, Gunter JL, DeJesus-Hernandez M, Josephs KA. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain. 2012;135(Pt 3):794–806. doi: 10.1093/brain/aws001. doi:aws001 [pii] 10.1093/brain/aws001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsen KC, Lynch T, Pavlou E, Higgins M, Nygaard TG. Localization of disinhibition-dementia-parkinsonism-amyotrophy complex to 17q21-22. American journal of human genetics. 1994;55(6):1159–1165. [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsen KC, Lynch T, Pavlou E, Higgins M, Nygaard TG. Localization of Disinhibition-Dementia-Parkinsonism-Amyotrophy Complex to 17q21-22. American Journal of Human Genetics. 1994;55:1159–1165. [PMC free article] [PubMed] [Google Scholar]
- Williams DR, Lees AJ. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol. 2009;8(3):270–279. doi: 10.1016/S1474-4422(09)70042-0. S1474-4422(09)70042-0 [pii] [DOI] [PubMed] [Google Scholar]
- Woolley JD, Gorno-Tempini ML, Seeley WW, Rankin K, Lee SS, Matthews BR, Miller BL. Binge eating is associated with right orbitofrontal-insular-striatal atrophy in frontotemporal dementia. Neurology. 2007;69(14):1424–1433. doi: 10.1212/01.wnl.0000277461.06713.23. [DOI] [PubMed] [Google Scholar]
- Woolley JD, Khan BK, Murthy NK, Miller BL, Rankin KP. The diagnostic challenge of psychiatric symptoms in neurodegenerative disease: rates of and risk factors for prior psychiatric diagnosis in patients with early neurodegenerative disease. J Clin Psychiatry. 2011;72(2):126–133. doi: 10.4088/JCP.10m06382oli. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda M, Nakamura Y, Kawamata T, Kaneyuki H, Maeda K, Komure O. Phenotypic heterogeneity within a new family with the MAPT p301s mutation. Ann Neurol. 2005;58(6):920–928. doi: 10.1002/ana.20668. [DOI] [PubMed] [Google Scholar]
- Zhukareva V, Mann D, Pickering-Brown S, Uryu K, Shuck T, Shah K, Lee VM. Sporadic Pick’s disease: a tauopathy characterized by a spectrum of pathological tau isoforms in gray and white matter. Ann Neurol. 2002;51(6):730–739. doi: 10.1002/ana.10222. [DOI] [PubMed] [Google Scholar]