

Abstract
Atypical hemolytic uremic syndrome (aHUS) is a disease characterized by the triad of microangiopathic hemolytic anemia, thrombocytopenia and acute kidney injury. The histopathologic lesions of aHUS include thrombotic microangiopathy involving the glomerular capillaries and thrombosis involving arterioles or interlobar arteries. Extra-renal manifestations occur in up to 20% of patients. The majority of aHUS is caused by complement system defects impairing ordinary regulatory mechanisms. Activating events therefore lead to unbridled, ongoing complement activity producing widespread endothelial injury. Pathologic mutations include those resulting in loss-of-function in a complement regulatory gene (CFH, CFI, CD46 or THBD) or gain-of-function in an effector gene (CFB or C3). Treatment with the late complement inhibitor, eculizumab – a monoclonal antibody directed against C5 – is effective.
Key words: Atypical hemolytic uremic syndrome, genetics
Competing interest statement
Conflict of interest: the authors declare no potential conflict of interest.
Introduction
Hemolytic uremic syndrome (HUS) is defined by the simultaneous occurrence of microangiopathic hemolytic anemia (MAHA), thrombocytopenia, and acute kidney injury.1 The most common cause of HUS is Shiga toxin-producing Escherichia coli (STEC). STEC HUS (i.e., that associated with infections by E.coli O157:H7) is also referred to as typical HUS whereas complement-mediated thrombotic microangiopathy (TMA) has now become equated with atypical HUS, although other etiologies of non-STEC HUS also fall under this rubric.2
Atypical HUS, for the remainder of this review referring to complement-mediated TMA, is a rare disorder with an annual incidence of one to two cases per million.3 The renal lesions of atypical HUS include TMA within the glomerular capillaries as well as thrombosis involving arterioles or interlobar arterioles; a renal limited form – C3 glomerulopathy, has also been described.4 The clinical TMA picture results from unchecked activation of the alternative pathway leading to excessive deposition and deleterious action of complement on the endothelium, often with a predilection for the renal vasculature.5
Ongoing complement activity in atypical HUS results from a failure of complement regulation (Figure 1). Complement regulatory proteins may be impaired in their action by loss-of-function mutations (CFH, CFI, CD46, and THBD) or acquired antibodies (specifically to complement factor H). Conversely, potentiation may be augmented by gain-of-function mutations in CFB or C3.6 Together, these account for 60% to 70% of atypical HUS cases.7 It is noteworthy that antibodies against complement factor H (CFH) are not strictly acquired and there is a well known-association of the homozygous CFHR1-3 deletion with occurrence of CFH-auto-antibodies.8,9 The penetrance of the disease is low, as less than half of family members carrying the same mutation as the patient with atypical HUS will be affected with the disease.1
Figure 1.
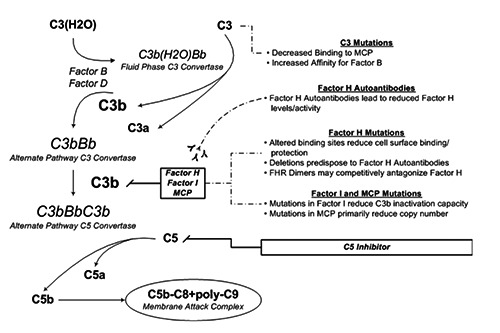
The alternative pathway of complement and pathophysiologic interactions with complement regulatory proteins.
Clinical presentation
The onset of atypical HUS is either idiopathic or secondary to potential triggers such as upper respiratory tract infections, fevers, pregnancy,10 drugs such as quinine and non E coli diarrheal illnesses.11 Similar to typical STEC-HUS, atypical HUS also manifests microangiopathic hemolytic anemia, thrombocytopenia, and renal failure. Extra-renal manifestations occur in 20% and most commonly involve derangements in the central nervous (altered conscious, seizures, or focal neurologic deficits in 8% of adults, 16% of children)12 and gastrointestinal (especially prodromic diarrhea in up to 28%)13 systems. Nonspecific findings – such as hypertension and malaise – also occur, often related to underlying renal involvement. In addition, aHUS triggering factors such as infection may contribute to the symptom complex at presentation. The presence of diarrhea, therefore, does not exclude the diagnosis of atypical HUS. The genetic contributions, clinical presentations and risk of discontinuation of eculizumab (see below) are shown in Table 1.6,14-28
Table 1.
Atypical hemolytic uremic syndrome: genetic contributions, clinical presentations and risk of discontinuation of eculizumab.
| Genes | Chr. locus | Proteins | Proportion to atypical HUS | Clinical presentations | Risk of disconti of eculizumab14 |
|---|---|---|---|---|---|
| C3 | 19p13.3 | Complement C3 | 2-8%15 | Typically present in childhood, 60% develop ESRD. 57% response to plasma exchange6 | High16 |
| CD46 | 1q32.2 | Membrane cofactor protein | 5-9%15,17 | Typically present in childhood, milder acute episode. 80% complete remission, 60-70% of individuals remain dialysis free even after several recurrences6 | Low |
| CFB | 6p21.33 | Complement factor B | Rare15,18 | Presenting both in childhood and adulthood. Higher variability. 70% eventually ESRD19 | |
| CFH | 1q31.3 | Complement factor H | 21-22%15 | High risk of relapse, 60-80% ESRD or death. Liver-kidney transplantation20 | High21 |
| CFHR1-5 | 1q31.3 | Complement factor H-related protein 1 | CFHR3/CFHR1 deletion: 28%22 CFH/CFHR1 hybrid: 3-5% CFRH1/CFHR4 deletion: rare | Poor clinical prognosis and high risk of post-transplant recurrence23-25 | Low for homozygous CFHR3/CFHR1 deletion |
| CFI | 4q25 | Complement factor I, C3b inactivator | 4-8%15 | Variable. 58% ESRD6 | Low |
| DGKE | 17q22 | Diacylglycerol kinase epsilon | ~27% of those present at age <1 year | Onset before 1-year old in homozygote patient.26 Early chronic kidney disease | |
| THBD | 20p11.21 | Thrombomodulin | ~5% | 90% present in childhood. 50% ESRD. Plasma treatment induced disease remission in about 80% of acute episodes6 | |
| CFH auto-antibody | Highly relapsing disease course. Significant gastrointestinal symptoms and/or diarrhea, thus, resembling eHUS. May have infection trigger.27 Plasma exchange and immunosuppression therapy28 |
Although earlier studies suggested that atypical HUS due to genetic mutations of the complement proteins was primarily a pediatric disorder,17 a French cohort of 214 patients reported that 58% of the cohort presented as adults.12
Atypical HUS has a poor prognosis with progression to end-stage renal disease in half the patients.29
Diagnostics
The clinical findings of atypical HUS requires consideration of other diseasemimics. Such disorders include idiopathic TTP (which is confirmed by the finding of completely suppressed ADAMTS-13, is less likely to present with profound renal impairment,30,31 and is typically more responsive to plasma exchange), STEC-HUS, Drug Induced TMA (which includes cases occurring in the post-transplant setting and often calcineurin-inhibitor related), HIV-related,32 disseminated intravascular coagulation (which can produce a MAHA pattern but as opposed to classic TMA, also engenders coagulopathy), hypertensive emergencies, malignancy-related TMA,33,34 cobalamin deficiency,35-38 pregnancy related conditions such as hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome, and others.39
Atypical HUS involves the following features:40 i) MAHA with hemoglobin levels below 10 g/dL, elevation of serum lactate dehydrogenase (LDH) level, notable decrease of serum haptoglobin level, and the presence of schistocytes on a peripheral blood smear (although degree of schistocytosis may vary and renal-limited HUS cases have been described), ii) thrombocytopenia with platelet counts less than 150 K/mcL and, iii) acute kidney injury (AKI). In pediatric patients, AKI is defined as serum creatinine levels at least 1.5 times the upper limit of the age- and sex-specific pediatric reference range. For adult patients, the diagnosis of AKI should be made according to well-established diagnostic guidelines.41
Patients with clinical features suggestive for HUS therefore deserve careful evaluation of the peripheral smear. The degree of schistocytosis and LDH elevation may vary, but suppressed haptoglobin level and hypocomplementemia are concerning. A specimen should immediately be drawn and sent for ADAMTS-13 activity testing but initiation of plasma exchange should not await results. If the ADAMTS 13 activity level is completely suppressed (i.e, <5-10%) many laboratories will reflexively perform an inhibitor study to assess for presence of an antibody directed toward ADAMTS-13. If present, a titer is performed.
To establish dysregulation of the complement system, tests based on plasma protein and screening of gene mutations are commercially available as shown in Table 2. As with ADAMTS-13 testing, treatment should not be forestalled pending results. Once available, aHUS may still be present even if a specific mutation is not determined as up to 40% of patients may lack them. Also, it is not universally the case that a bona-fide aHUS presentation will incite hypocomplementemia as deficiencies of C3 and C4 may only be present in 27-44% and 0-7% of aHUS cases, respectively.6,15
Table 2.
Commercially available diagnostic atypical hemolytic uremic syndrome test panels.
| Name | Gene panel | Plasma or serum proteins | Method | Turn-around time | Location |
|---|---|---|---|---|---|
| Machaon Diagnostics | CFH, MCP (CD46), CFI, C3, CFB, CFHR1, CFHR3, CFHR4, CFHR5, Thrombomodulin (THBD), Plasminogen (PLG) and DGKE | Next generation sequencing | <4 weeks | Oakland, CA | |
| Cincinnati Children’s Hospital Medical Center | C3, CFB, CFH, CFHR1, CFHR3, CFHR5, CFI, DGKE, MCP/CD46, and THBD | Next generation sequencing >40 fold coverage | 42 days | Cincinnati, OH | |
| Connective Tissue Gene Tests (CTGT) | ADAMTS13, C3, CD46, CFB, CFH, CFHR1, CFHR2, CFHR3, CFHR5, CFI, DGKE, MMACHC, and THBD | Next generation sequencing | Allentown, PA | ||
| Genetic Testing Registry (GTR) | ADAMTS13, C3, CD46, CFB, CFH, CFHR1, CFHR2, CFHR3, CFHR4, CFHR5, CFI, CLU and THBD | Applied Biosystem Genetic Analyzer 3130 | Weisswasser, Germany | ||
| The Blood Center of Wisconsin | CFH, CFI, MCP/CD46, CFHR1, CFHR3, CFHR4, THBD, C4BP, CFB and C3 | Complement protein studies and genetic analysis | 28 days | Milwaukee, WI | |
| Mayo Clinic Medical laboratories (Pacific Diagnostic Laboratories) | NT51, AH50, SC5B9, CBB, IC4D, COM, C3, C4, FBCA and FHCA | Automated method using liposomes as the target for the serum complement system | Varies | Rochester, MN |
Therapeutics
General approach
In patients with evidence for thrombotic microangiopathy, plasma exchange may immediately be initiated given the overlap in presenting laboratory and clinical findings among the various thrombotic microangiopathies.42-44 The clinical and biochemical response to plasma exchange, typically assessed after 5-7 daily treatments, serves as a guide to further treatment.43 While continuation of plasma exchange is pursued in responders, some patients may manifest incomplete responses or frank refractoriness. The results of initial tests now come into focus. Particularly when the ADAMTS-13 activity level is detectable and especially if accompanied by continued MAHA or absence of renal recovery, treatment with a late complement inhibitor may be indicated.
Complement inhibition
The first-line treatment of patients with atypical HUS is eculizumab (Soliris, Alexion Pharmaceuticals, Cheshire, CT, USA). Eculizumab is a humanized monoclonal antibody that blocks complement C5 activation. C5 activation is the crucial step toward assembly of the membrane attack complex (MAC) which involves C5b-9 formation.45 Endothelial damage caused by excessive complement activation underlies the rationale for therapeutic complement inhibition. Eculizumab has proven effective against atypical HUS,46 where it has been shown to interdict the hemolytic process and rescue native kidney function hence dramatically changing the prognosis of this potentially fatal syndrome.47 In Legendre et al.,46 eculizumab (dosed intravenously at 900 mg/week weeks 1-4, 1200 mg at week 5, then 1200 mg fortnightly thereafter) normalized hemolytic markers and platelet counts in nearly 90% of patients with aHUS refractory to plasma exchange. Furthermore, patients experienced reliable improvements in renal function: in trial 1 (low platelets and renal damage, n=17) 4/5 (80%) patients who were dialysis dependent at time of eculizumab initiation achieved dialysis independence and group total mean-time-dependent increase in GFR (mL/min/1.73 m2) was 32 (95%CI, 14 to 49 P=0.001) by week 26. In trial 2 (renal damage but no decline in platelet >25% for 8 weeks during low intensity plasma exchange/infusion, n=20), group total mean-time-dependent improvement in GFR (in mL/min/1.73 m2) was 6 (95%CI, 3 to 9; P<0.001) by week 26.46 Eculizumab also appears to be beneficial in patients with complement-mediated HUS due to autoantibodies to complement factor H (CFH).48,49
Since 2011, eculizumab has been licensed for the treatment of pediatric and adult patients with aHUS in the United States, Europe, and Canada.11 Maintenance treatment with eculizumab is expensive, with an estimated annual drug cost of $350,000 to $645,000.50 Furthermore, inhibition of late complement predisposes to meningococcal infections, and invasive meningococcal infections despite vaccination have been reported during eculizumab treatment.51,52 Eculizumab carries a black box warning and Risk Evaluation and Mitigation Strategy (REMS) [http://www.solirisrems.com/] advising vaccination against Neisseria meningitidis two weeks prior to initiation of eculizumab as well as counseling regarding early recognition and medical attention if signs of infection develop.53 The Advisory Committee on Immunization Practices (ACIP) recommends simultaneous vaccination with meningococcal quadrivalent conjugate vaccine (protective against serogroups A, C, W, Y – Menactra, Menveo) as well as vaccination against serogroup B (Bexsero, Trumenba).54 During the interval until protective titers are achieved, penicillin V potassium at 250 mg q12 hours or ciprofloxacin 500 mg daily have been recommended.55 In non-immune individuals, consideration could also be given to vaccination against Haemophilus influenzae type b and Pneumococcus pneumoniae.56
It is difficult to know when the medication can be discontinued and relapse after discontinuation has been described. The presence of certain genetic mutations, such as those affecting CFH or C3, may increase the risk of recurrence,16 whereas this risk may be relatively low for patients with MCP mutations, homozygous CFHR3/R1 deletions, anti-CFH antibodies, CFI mutations, and no identifiable mutations.14 The location of the mutations might also be critical since patients with mutations in short consensus repeat (SCR) 19 and 20 of the CFH are more prone to relapse,57 while mutations in the carboxy-terminal portion of CFH carry relatively low risk for eculizumab discontinuation.58
Ardissino and colleagues report a clinical strategy following eculizumab discontinuation wherein patients utilize home urine dipstick monitoring on a regular basis and when feeling unwell. The appearance of hemoglobinuria triggers immediate clinic visit for in-depth assessment for aHUS recurrence.59 A total of 16 patients with aHUS with eculizumab-induced remission participated. Eculizumab was stopped after a median of 4.3 months and home monitoring ensued. Five patients experienced relapse within 6 months of discontinuation and remission was successfully reinduced with immediate return to eculizumab therapy.60 This strategy was not recommended in patients with CFH mutations or those with poor renal function.60 Use of eculizumab trough measurements and complement activity measurements aimed at individualizing dosing frequency, and thereby reducing costs, have also been studied.61,62
Early detection of TMA recurrence and prompt retreatment with eculizumab seem to be efficient in controlling of TMA and restoration of kidney functions. Further prospective studies are needed to identify biomarkers predictive of relapse and determine the best strategy of retreatment in relapsing patients.63
CCX168, an oral administration C5aR antagonist, is currently in a phase 2 clinical trial to evaluate the effect of thrombus formation and disease activity in patients with diagnosis of Atypical HUS with or without genetic abnormalities in the complement system or thrombomodulin, on stable chronic extracorporeal or peritoneal dialysis therapy since at least 6 months.64 A slew of additional agents interfering at various stages of the complement pathway are under development.65
Plasmapheresis
Since the 1980s, plasma exchange therapy has been the mainstay method for management of aHUS. This therapy aims to eliminate abnormal complement regulatory proteins and anti-CFH antibodies, while supplementing normal complement regulatory proteins. With the current understanding of the pathological mechanism and extensive use of eculizumab, plasma exchange becomes somewhat limited.
The effectiveness of plasma based therapeutics is associated with genetic background.66 In anti-CFH antibody-positive patients, plasma exchange combined with immunosuppressants or steroids, as compared to plasma exchange alone, yielded better outcomes with reduced antibody titers.12 Responses to plasma-based therapeutics (either plasma infusion or plasma exchange) in atypical HUS appears highest among those with MCP mutations (87-97%) and lowest among those with CFI mutations (25%) with 3 year outcomes of ESRD or death as high as 77% among those with CFH and lowest among those with MCP mutations (6%).6,13 Eculizumab may be considered for treating atypical HUS accompanied by extra-renal organ injury.67
Before a firm diagnosis can be made, it is still considered as standard care to initiate total plasma exchange when TTP is suspected. The American Society for Apheresis (ASFA) publishes evidence-based guidelines for therapeutic apheresis. Indications are graded as Category I for those in which the evidence supports a 1st line indication for apheresis, Category II for those in which apheresis represents 2nd line or adjunctive therapy, Category III for those conditions in which the role of apheresis has yet been clearly established, and Category IV when the evidence suggests that apheresis either ineffective or harmful.68 Atypical HUS due to Factor H autoantibodies represents the sole Category I indication for apheresis whereas aHUS due to other disease-causing mutations represent Category III indications.68
Conclusions
Atypical hemolytic uremic syndrome is a rare, life-threatening disease characterized by the triad of microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury. Initial treatment with a course of therapeutic plasma exchange is reasonable while awaiting ADAMTS-13 activity level results in a new patient. Response to plasma exchange is a useful guide toward further treatment requirements as return of a detectable ADAMTS-13 activity level in the face of continuing hematologic, renal or other clinical manifestations indicates a potential role for eculizumab. While initiation of eculizumab has become a first-line treatment for patients with atypical HUS, there remains considerable work to be done in determining the duration of treatment. A host of emerging complement-directed therapeutics are under study and may offer additional treatment options in the future.
Acknowledgements
This study is under the support of Department of Pathology, University of California Irving Medical Center.
References
- 1.Noris M, Remuzzi G. Hemolytic uremic syndrome. J Am Soc Nephrol 2005;16:1035-50. [DOI] [PubMed] [Google Scholar]
- 2.Loirat C, Fremeaux-Bacchi V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis. 2011;6:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Constantinescu AR, Bitzan M, Weiss LS, et al. Non-enteropathic hemolytic uremic syndrome: causes and short-term course. Am J Kidney Dis 2004;43:976-82. [DOI] [PubMed] [Google Scholar]
- 4.Noris M, Remuzzi G. Glomerular diseases dependent on complement activation, including atypical hemolytic uremic syndrome, membranoproliferative glomerulonephritis, and C3 glomerulopathy: core curriculum 2015. Am J Kidney Dis 2015;66:359-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davin JC, van de Kar NC. Advances and challenges in the management of complement-mediated thrombotic microangiopathies. Ther Adv Hematol 2015;6:171-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 2010;5:1844-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kavanagh D, Richards A, Fremeaux-Bacchi V, et al. Screening for complement system abnormalities in patients with atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol 2007;2:591-6. [DOI] [PubMed] [Google Scholar]
- 8.Jozsi M, Licht C, Strobel S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood 2008;111:1512-4. [DOI] [PubMed] [Google Scholar]
- 9.Dragon-Durey MA, Blanc C, Marliot F, et al. The high frequency of complement factor H related CFHR1 gene deletion is restricted to specific subgroups of patients with atypical haemolytic uraemic syndrome. J Med Genet 2009;46:447-50. [DOI] [PubMed] [Google Scholar]
- 10.Fakhouri F, Roumenina L, Provot F, et al. Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J Am Soc Nephrol 2010;21:859-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson S, Stojanovic J, Ariceta G, et al. An audit analysis of a guideline for the investigation and initial therapy of diarrhea negative (atypical) hemolytic uremic syndrome. Pediatr Nephrol 2014;29:1967-78. [DOI] [PubMed] [Google Scholar]
- 12.Fremeaux-Bacchi V, Fakhouri F, Garnier A, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol 2013;8:554-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, et al. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol 2007;18:2392-400. [DOI] [PubMed] [Google Scholar]
- 14.Sahutoglu T, Basturk T, Sakaci T, et al. Can eculizumab be discontinued in aHUS?: case report and review of the literature. Medicine (Baltimore) 2016;95:e4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bresin E, Rurali E, Caprioli J, et al. Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol 2013;24:475-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toyoda H, Wada H, Miyata T, et al. Disease recurrence after early discontinuation of eculizumab in a patient with atypical hemolytic uremic syndrome with complement C3 I1157T mutation. J Pediatr Hematol Oncol 2016;38:e137-9. [DOI] [PubMed] [Google Scholar]
- 17.Caprioli J, Noris M, Brioschi S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood 2006;108:1267-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davin JC, Strain L, Goodship TH. Plasma therapy in atypical haemolytic uremic syndrome: lessons from a family with a factor H mutation. Pediatr Nephrol 2008;23:1517-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Funato M, Uemura O, Ushijima K, et al. A complement factor B mutation in a large kindred with atypical hemolytic uremic syndrome. J Clin Immunol 2014;34:691-5. [DOI] [PubMed] [Google Scholar]
- 20.Gonzales E, Ulinski T, Habes D, et al. Long-term successful liver-kidney transplantation in a child with atypical hemolytic uremic syndrome caused by homozygous factor H deficiency. Pediatr Nephrol 2016;31:2375-8. [DOI] [PubMed] [Google Scholar]
- 21.Quiroga B, de Lorenzo A, Vega C, de Alvaro F. A case report and literature review of eculizumab withdrawal in atypical hemolytic-uremic syndrome. Am J Case Rep 2016;17:950-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zipfel PF, Edey M, Heinen S, et al. Deletion of complement factor H-related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. PLoS Genet 2007;3:e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abarrategui-Garrido C, Martinez-Barricarte R, Lopez-Trascasa M, et al. Characterization of complement factor H-related (CFHR) proteins in plasma reveals novel genetic variations of CFHR1 associated with atypical hemolytic uremic syndrome. Blood 2009;114:4261-71. [DOI] [PubMed] [Google Scholar]
- 24.Valoti E, Alberti M, Tortajada A, et al. A novel atypical hemolytic uremic syndrome-associated hybrid CFHR1/CFH gene encoding a fusion protein that antagonizes factor H-dependent complement regulation. J Am Soc Nephrol 2015;26:209-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moore I, Strain L, Pappworth I, et al. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood 2010;115:379-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lemaire M, Fremeaux-Bacchi V, Schaefer F, et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet 2013;45:531-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hofer J, Giner T, Jozsi M. Complement factor H-antibody-associated hemolytic uremic syndrome: pathogenesis, clinical presentation, and treatment. Semin Thromb Hemost 2014;40:431-43. [DOI] [PubMed] [Google Scholar]
- 28.Sinha A, Gulati A, Saini S, et al. Prompt plasma exchanges and immunosuppressive treatment improves the outcomes of anti-factor H autoantibody-associated hemolytic uremic syndrome in children. Kidney Int 2014;85:1151-60. [DOI] [PubMed] [Google Scholar]
- 29.Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med 2009;361:1676-87. [DOI] [PubMed] [Google Scholar]
- 30.Coppo P, Schwarzinger M, Buffet M, et al. Predictive features of severe acquired ADAMTS13 deficiency in idiopathic thrombotic microangiopathies: the French TMA reference center experience. PLoS One 2010;5:e10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bendapudi PK, Li A, Hamdan A, et al. Impact of severe ADAMTS13 deficiency on clinical presentation and outcomes in patients with thrombotic microangiopathies: the experience of the Harvard TMA Research Collaborative. Br J Haematol 2015;171:836-44. [DOI] [PubMed] [Google Scholar]
- 32.George JN, Terrell DR, Vesely SK, et al. Thrombotic microangiopathic syndromes associated with drugs, HIV infection, hematopoietic stem cell transplantation and cancer. Presse Med 2012;41:e177-88. [DOI] [PubMed] [Google Scholar]
- 33.George JN. Systemic malignancies as a cause of unexpected microangiopathic hemolytic anemia and thrombocytopenia. Oncology (Williston Park) 2011;25:908-14. [PubMed] [Google Scholar]
- 34.Francis KK, Kalyanam N, Terrell DR, et al. Disseminated malignancy misdiagnosed as thrombotic thrombocytopenic purpura: a report of 10 patients and a systematic review of published cases. Oncologist 2007;12:11-9. [DOI] [PubMed] [Google Scholar]
- 35.Garderet L, Maury E, Lagrange M, et al. Schizocytosis in pernicious anemia mimicking thrombotic thrombocytopenic purpura. Am J Med 2003;114:423-5. [DOI] [PubMed] [Google Scholar]
- 36.Dalsania CJ, Khemka V, Shum M, et al. A sheep in wolf’s clothing. Am J Med 2008;121:107-9. [DOI] [PubMed] [Google Scholar]
- 37.Tadakamalla AK, Talluri SK, Besur S. Pseudo-thrombotic thrombocytopenic purpura: a rare presentation of pernicious anemia. N Am J Med Sci 2011;3:472-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noel N, Maigne G, Tertian G, et al. Hemolysis and schistocytosis in the emergency department: consider pseudothrombotic microangiopathy related to vitamin B12 deficiency. QJM 2013;106:1017-22. [DOI] [PubMed] [Google Scholar]
- 39.George JN, Charania RS. Evaluation of patients with microangiopathic hemolytic anemia and thrombocytopenia. Semin Thromb Hemost 2013;39:153-60. [DOI] [PubMed] [Google Scholar]
- 40.Campistol JM, Arias M, Ariceta G, et al. An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document. Nefrologia 2013;33:27-45. [DOI] [PubMed] [Google Scholar]
- 41.Kidney Disease: Improving Global Outcomes (KDIGO), Acute Kidney Injury Work Group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int Suppl 2012:1-138. [Google Scholar]
- 42.Raval JS, Mazepa MA, Brecher ME, Park YA. How we approach an acquired thrombotic thrombocytopenic purpura patient. Transfusion 2014;54:2375-82. [DOI] [PubMed] [Google Scholar]
- 43.Cataland SR, Wu HM. Diagnosis and management of complement mediated thrombotic microangiopathies. Blood Rev 2014;28:67-74. [DOI] [PubMed] [Google Scholar]
- 44.George JN, Al-Nouri ZL. Diagnostic and therapeutic challenges in the thrombotic thrombocytopenic purpura and hemolytic uremic syndromes. Am Soc Hematol Educ Program 2012;2012:604-9. [DOI] [PubMed] [Google Scholar]
- 45.Cofiell R, Kukreja A, Bedard K, et al. Eculizumab reduces complement activation, inflammation, endothelial damage, thrombosis, and renal injury markers in aHUS. Blood 2015;125:3253-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 2013;368:2169-81. [DOI] [PubMed] [Google Scholar]
- 47.Zuber J, Le Quintrec M, Krid S, et al. Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. Am J Transplant 2012;12:3337-54. [DOI] [PubMed] [Google Scholar]
- 48.Noone D, Waters A, Pluthero FG, et al. Successful treatment of DEAP-HUS with eculizumab. Pediatr Nephrol 2014;29:841-51. [DOI] [PubMed] [Google Scholar]
- 49.Diamante Chiodini B, Davin JC, Corazza F, et al. Eculizumab in anti-factor h antibodies associated with atypical hemolytic uremic syndrome. Pediatrics 2014;133:e1764-8. [DOI] [PubMed] [Google Scholar]
- 50.Conway EM. HUS and the case for complement. Blood 2015;126:2085-90. [DOI] [PubMed] [Google Scholar]
- 51.Struijk GH, Bouts AH, Rijkers GT, et al. Meningococcal sepsis complicating eculizumab treatment despite prior vaccination. Am J Transplant 2013;13:819-20. [DOI] [PubMed] [Google Scholar]
- 52.Applegate AO, Fong VC, Tardivel K, et al. Notes from the field: meningococcal disease in an international traveler on eculizumab therapy - United States, 2015. MMWR Morb Mortal Wkly Rep 2016;65:696-7. [DOI] [PubMed] [Google Scholar]
- 53.Soliris. Warning: serious meningococcal infections. 2016. Available form: http://www.soliris.net/download.php?file=/resources/pdf/soliris_pi_mg.pdf. Accessed: January 2017.
- 54.CDC. Meningococcal: who needs to be vaccinated? 2016. Available from: https://wwwcdcgov/vaccines/vpd/mening/hcp/who-vaccinate-hcphtml.
- 55.Benamu E, Montoya JG. Infections associated with the use of eculizumab: recommendations for prevention and prophylaxis. Curr Opin Infect Dis 2016;29:319-29. [DOI] [PubMed] [Google Scholar]
- 56.Kuchar E, Miskiewicz K, Karlikowska M. A review of guidance on immunization in persons with defective or deficient splenic function. Br J Haematol 2015;171:683-94. [DOI] [PubMed] [Google Scholar]
- 57.Wetzels JF, van de Kar NC. Discontinuation of eculizumab maintenance treatment for atypical hemolytic uremic syndrome. Am J Kidney Dis 2015;65:342. [DOI] [PubMed] [Google Scholar]
- 58.Habbig S, Bergmann C, Weber LT. Discontinuation of eculizumab in a patient with atypical hemolytic uremic syndrome due to a mutation in CFH. Am J Kidney Dis 2016;67:532-3. [DOI] [PubMed] [Google Scholar]
- 59.Ardissino G, Testa S, Possenti I, et al. Discontinuation of eculizumab maintenance treatment for atypical hemolytic uremic syndrome: a report of 10 cases. Am J Kidney Dis 2014;64:633-7. [DOI] [PubMed] [Google Scholar]
- 60.Ardissino G, Possenti I, Tel F, et al. Discontinuation of eculizumab treatment in atypical hemolytic uremic syndrome: an update. Am J Kidney Dis 2015;66:172-3. [DOI] [PubMed] [Google Scholar]
- 61.Gatault P, Brachet G, Ternant D, et al. Therapeutic drug monitoring of eculizumab: Rationale for an individualized dosing schedule. MAbs 2015;7:1205-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cugno M, Gualtierotti R, Possenti I, et al. Complement functional tests for monitoring eculizumab treatment in patients with atypical hemolytic uremic syndrome. J Thromb Haemost 2014;12:1440-8. [DOI] [PubMed] [Google Scholar]
- 63.Nantes University Hospital. Study assessing an algorithm-based strategy of eculizumab discontinuation in children and adults with aHUS (STOPECU). ClinicalTrials.gov Identifier:NCT02574403 2016.
- 64.Mario Negri Institute for Pharmacological Research. Complement Inhibition in aHUS Dialysis Patients (ACCESS). ClinicalTrials.gov Identifier: NCT02464891 2016.
- 65.Risitano AM, Marotta S. Therapeutic complement inhibition in complement-mediated hemolytic anemias: Past, present and future. Semin Immunol 2016;28:223-40. [DOI] [PubMed] [Google Scholar]
- 66.Raufi AG, Scott S, Darwish O, et al. Atypical hemolytic uremic syndrome secondary to lupus nephritis, responsive to eculizumab. Hematol Rep 2016;8:6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Loirat C, Fakhouri F, Ariceta G, et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol 2016;31:15-39. [DOI] [PubMed] [Google Scholar]
- 68.Schwartz J, Padmanabhan A, Aqui N, et al. Guidelines on the use of therapeutic apheresis in clinical practice-evidence-based approach from the writing committee of the american society for apheresis: the seventh special issue. J Clin Apher 2016;31:149-62. [DOI] [PubMed] [Google Scholar]