

Abstract
Gold nanoparticles (Au NPs) have attracted much attention due to their potential applications in nano-medicine. While numerous studies have quantified biomolecular adsorption to Au NPs in terms of equilibrium binding constants, far less is known about biomolecular orientation on nanoparticle surfaces. In this study, the binding of the protein α-synuclein to citrate and (16-mercaptohexadecyl) trimethylammonium bromide (MTAB) coated 12 nm Au NPs is examined by heteronuclear single quantum coherence NMR spectroscopy to provide site-specific measurements of protein-nanoparticle binding. Molecular dynamics simulations support the orientation assignments, which show N-terminus binding to the Au NP for citrate-capped NPs, and C-terminus binding for the MTAB-capped NPs.
Keywords: Gold nanoparticles, α-synuclein, protein orientation, surface chemistry, protein adsorption
TOC image
Introduction
Gold nanoparticles (Au NPs) have optical and electronic properties conducive toward advanced biomedical applications such as cellular imaging1, drug delivery2, and photothermal therapy.3–8 The surface chemistry of Au NPs can be modified to meet specific needs and requirements, such as managing cellular uptake, cytotoxicity, and providing targeting capabilities.9–14 Previous work has shown that protein orientation on gold nanoparticle surfaces can modulate protein activity, and can be used as a conformation-specific antigen to raise antibodies to cytotoxic oligomeric intermediates implicated in neurodegenerative diseases.15,16 Understanding the fundamental aspects of protein adsorption to nanoscale curved surfaces is an integral part of predicting the impact of NPs on biological systems.
α-Synuclein (α-syn) is a protein that is highly localized in the presynaptic termini of human brains.17 It belongs to the synuclein family of proteins, along with β and γ synuclein, and consists of 140 amino acid residues.18 α-Syn is an intrinsically disordered protein in aqueous solutions with an atypical charge distribution; its N terminus is cationic and its C terminus is anionic at physiological pH.19 a-Synuclein’s conformation depends upon chemical environment.20 For example, it has been shown that α-syn binds to anionic lipid micelles and vesicles, via its N-terminus and seven imperfect Lys-Thr-Lys-Glu-Gly-Val motifs, adopting an α-helical secondary structure.21–27 In the presence of polyamines such as putrescine and spermidine, α-syn shows β-sheet character, which is highly indicative of the aggregation states of amyloid proteins.28 α-Syn has also been shown to have affinity for cationic metal ions such as copper(II) to its C-terminal domain, owing to the high density of acidic amino acids.29–31 The aggregation of α-synuclein is a hallmark feature of Parkinson’s disease, dementia with Lewy bodies, and other neuronal afflictions.32–34 Thus, there is great interest in measuring the conformation and aggregation state of α-syn as a function of environment and developing a detailed atomic-level understanding of these binding events.35,36
In previous work from our lab, the binding of micromolar concentrations of α-syn to ~0.1 – 1 nM anionic citrate-coated 20 nm Au NPs was extensively studied.37 Protein adsorption isotherms were measured via optical spectroscopic titrations, and a laborious trypsin digestion/mass spectrometry method was developed in order to gain information about bound protein orientation.37 Observed peptide fragments indicated that the C-terminus was more available for digestion, suggesting that α-syn bound to citrate-capped Au NPs via its N-terminus. Analogous experiments were undertaken to examine the interaction of α-syn with cationic poly(allylamine) hydrochloride (PAH) coated 20 nm Au NPs.38 In contrast to the citrate-coated Au NPs, PAH Au NPs experienced severe agglomeration upon interaction with α-syn, forming webs of entangled nanoparticles and proteins that seemed to indicate seeded beta sheet formation in solution. The effects of agglomeration were reflected in the UV-Vis spectra as massive red-shifts of the plasmon peak, preventing a proper fit to adsorption isotherms. This observation is consistent with the result of the coupling of plasmon resonances when noble metal nanoparticles are close in proximity.39 These previous results suggested that cationic nanoparticles, in general, might promote α-syn aggregation.
Aggregation effects often present an enormous obstacle to studying the interactions of ligands to nanomaterials. Especially in the case of agglomerated plasmonic nanoparticles, not only are optical techniques invalid due to inter-particle effects as described earlier, physical changes to the sample itself (such as settling) impede the ability to properly perform analytical measurements (Scheme 1). However, electrostatic forces are not the only factor in mediating protein-nanomaterial interactions; previous studies have shown that NPs with similarly charged coatings elicit different cellular responses, further emphasizing the importance of surface chemistry.40 We find that a recently-synthesized cationic ligand, (16-mercaptohexadecyl) trimethylammonium bromide (MTAB, Scheme 1), provides for a monolayer-protected surface that does not promote α-syn aggregation and therefore allows for clean interpretation of protein orientation on different Au NPs.
Scheme 1. The effect of surface chemistries of 20 nm Au Nps upon interaction with α-syn.
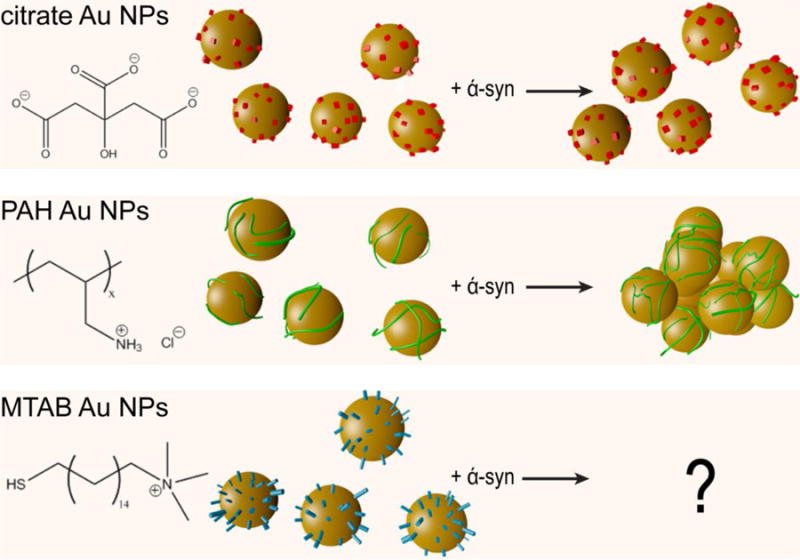
Au NPs NPs are shown as gold spheres, citrate as red cubes, PAH polymer as green lines, and (16-mercaptohexadecyl) trimethylammonium bromide (MTAB) as blue cylinders. While citrate NPs remain in a well-dispersed state upon binding to α-syn, PAH-coated NPs experienced agglomeration as a result of exposure to α-syn.38 The use of MTAB as a ligand is the subject of this work.
In this study, we employed NMR heteronuclear correlation spectroscopy, which allows for the analysis of complex chemical processes at a molecular level.41 For example, the heteronuclear single quantum coherence pulse sequence (HSQC) is an NMR experiment that provides correlations between a proton and a directly attached spin-half nucleus, typically 13C or 15N atom. A 1H-15N HSQC gives a fingerprint of the protein backbone structure. This experiment is commonly used as a starting point for protein resonance assignment and structure determination, and specifically has been used in previous studies of a-syn.22,23,26–28 Following this approach, here we use HSQC NMR to give information on α-syn’s adsorption and interaction with citrate-coated and MTAB-coated Au NPs. As far as we know, this is the first study of this kind, which combines NMR methods with nanoparticle surface chemistry to infer protein display on NP surfaces.
Materials and Methods
Synthesis of citrate-coated gold nanoparticles
All reagents were purchased from Sigma-Aldrich and used as received without further purification. 12 nm citrate nanoparticles were synthesized as previously described.42 Briefly, 2.5 mL of 0.01 M HAuCl4·3H2O was added to 97.5 mL of ultrapure water, and the solution was heated to a rolling boil. Then, 3 mL of 1% w/w sodium citrate was added, and the solution was kept at a boil for 40 more minutes. After the heat was turned off, the solution was allowed to cool naturally, and an additional 0.5 mL of 1% w/w sodium citrate was added to ensure stability of the sample. To purify, the Au NPs were centrifuged at 8,000 rcf for 20 min. The supernatant was discarded, and the remaining sample was re-dispersed in ultrapure water.
MTAB coating of gold nanoparticles
(16-mercaptohexadecyl) trimethylamoonium bromide (MTAB) was prepared according to the procedure published by Zubarev, et al.43 Starting reagents were purchased from Sigma-Aldrich and used without purification. To coat citrate-capped Au NPs with MTAB, 300 μL of 600 nM 12 nm citrate-capped Au NPs was added to a 1.5 mg/mL solution of MTAB, which had previously been heated at 60° C for 30 min. The solution was allowed to incubate at room temperature for 3 hours before excess MTAB was removed. To remove excess MTAB, the Au NPs were centrifuged twice at 8,000 rcf for 20 min each, and the supernatant was discarded. The resultant pellet was re-dispersed in ultrapure water.
Production of α-synuclein
α-Syn was prepared using E. coli BL21(DE3)/pET28a-AS grown at 37 °C as described previously using an enhanced LB medium (containing 2 mM MgCl2 and 0.2x Studier trace elements) with 30 μg/ml kanamycin.44,45 For the 15N-labeled sample, the cells were grown at 37 °C in Studier Medium M containing 3 g/L glucose and 2 g/L 15N ammonium chloride, 10 ml/L 15N BioExpress, 0.1x BME vitamins, and 30 μg/ml kanamycin.45 At a cell density of A600 = 0.8, the temperature was lowered to 25°C. When the cell density reached A600= 1.3, protein expression was induced using 0.5 mM isopropyl β-D-1-thiogalactopyranoside. Cells were harvested 14 h post-induction. 15N ammonium chloride and 15N BioExpress were obtained from Cambridge Isotope Laboratories, Tewksbury, MA. BME vitamins (no. B6891) were from Sigma-Aldrich, St. Louis, MO.
Purification of α-syn began by re-suspending a cell pellet from 1 L of culture medium in 100 mM Tris, pH 8 containing 1200 units TurboNuclease (Accelagen, San Diego, CA). The cells were lysed either by sonication or French Press. The lysate was boiled for 10 minutes, cooled, and the precipitant and cell debris removed by ultracentrifugation (1h at 100,000 rcf). The supernatant was loaded on a 60-mL anion-exhange column (Q Sepharose FF) and eluted using a linear gradient of 0–0.8 M NaCl. α-Syn eluted at ~0.3 M NaCl. Pooled fractions containing α-syn were concentrated using a stirred cell and loaded on a 26/60 Sephacryl S-200 HR gel filtration column in 10 mM Tris, pH 8, 1 mM EDTA, and 150 mM NaCl. Sodium azide (0.01%) was added to the pooled S-200 fractions containing α-syn monomer. The protein was stored at 4 °C. The yield of purified α-syn was 50–60 mg/L growth medium, for both expression conditions. For titrations involving natural abundance α-syn, the sample was dialyzed into 10 mM Na phosphate, pH 7. For NMR, 15N and 13C/15Nα-syn was dialyzed into 20 mM HEPES, pH 7. The Q Sepharose resin and the S-200 column were obtained from GE Healthcare, Piscataway, NJ.
NMR spectroscopy
All NMR spectra of α-syn were collected at 4 °C on an Agilent VNMRS 750 MHz or UnityInova 600 MHz spectrometer with a 5-mm indirect detection triple resonance 1H{13C/15N} triaxial PFG probe. For intrinsically unstructured proteins, HSQC spectra are most often collected at 4 °C, to minimize exchange of amide protons with water. The structure of α-syn remains unchanged between 4 °C and room temperature. The Biopack pulse sequences (VNMRJ3.2 or VNMRJ2.1B) were used for HNCACB, CACA(CO)NH, 1H-15N SOFAST-HMQC, and 1H-15N HSQC experiments.
Resonance assignments
15N, 13Cα and 13Cβ chemical shifts were assigned from 2D 1H-15N SOFAST HMQC, 3D 1H-13C-15N HNCACB and CBCA(CO)NH spectra. The 3D experiments were collected with a spectral width of 10 ppm in the 1H dimension (1532 points), 80 ppm in the 13C dimension (64 points), and 30 ppm in the 15N dimension (32 points). The 15N SOFAST-HMQC experiment had a spectral width of 10 ppm in the 1H dimension (1532 points), and 40 ppm in the 15N dimension (400 points). The 1H-15N HSQC spectra were recorded with a spectral width of 14 ppm and 36 ppm in the 1H and 15N dimensions, respectively. An acquisition time of 0.061 seconds was used in the direct 1H dimension, with 200 points in indirect 15N dimension (t1), and a pulse delay of 1 s. Between 168 and 200 scans per t1 increment were collected (except samples with 20 μM α-syn, which were collected with 352 scans per increment).
All spectra were processed using NMRPipe.46 In the 3D experiments, FIDs were zero-filled to 2048, 512, and 256 points in the 1H, 13C, and 15N dimensions, respectively. In the 1H-15N HSQC and 15N SOFAST-HMQC experiments, FIDs were zero-filled to 2048 points in the 1H dimension, and 512 points in the 15N dimension. The 90 degree shifted sine-bell square window functions were used in processing these spectra, as well as linear prediction to the next power of 2. The processed spectra were analyzed with SPARKY.47 Peak assignments on the spectra of bound α-syn were made by copying the entire set of assignments from the HSQC spectrum of free 30 μM α-syn, then pasting onto the HSQC of bound α-syn. Due to small changes in the chemical shifts of the backbone peaks of α-syn upon binding to the NPs, minor adjustments were made to ensure that all labels were centered.
Simulation of α-synuclein’s interactions with gold nanoparticles
Parameters for the MTAB and citrate ligands were assigned using the CHARMM general forcefield (ver. 2b8)48,49 through use of ParamChem (ver. 0.9.7.1),50,51 while α-synuclein was characterized using the CHARMM protein forcefield (ver. c32b1).52,53 The initial structure for α-synuclein was taken from the RCSB Protein Database.23 The MTAB and citrate ligand coverages used were 3.7 and 2.65 molecules/nm2, respectively. The surface density for MTAB was estimated using previously published experimental data quantifying the density of MTAB molecules on a gold nanorod at 3.7 molecules/nm2;43 recent x-ray photoelectron spectroscopy data for citrate-capped Au NPs suggests that both 1.7 citrate molecules/nm2 and 1.7 oxidized citrate molecules/nm2 exist on the surface.54 All systems were solvated in TIP3 water with 20 mM sodium chloride added to neutralize the system and to fulfill the role of the HEPES buffer. Each system was simulated using the NAMD molecular dynamics package.55 All systems were visualized and analyzed using VMD.56 The simulations used periodic boundary conditions, a particle mesh Ewald (PME) method, and Langevin dynamics with a damping constant of γLang = 1.0 ps−1. In both systems the Au NP core was kept fixed during the simulations, while the rest of the system was equilibrated. In the first 10 ns, α-synuclein was fixed in space roughly 1 nm away from the NP surface. Then the α-synuclein was released, and the citrate-NP system was simulated for 45 ns, while the MTAB-NP system was simulated for 51 ns (reasonable time for selective adsorption but not folding).
In addition, the systems were then simulated for 5 ns with bulk van der Waals (vdW) coupling added between gold NP and the rest of the system. The vdW cutoff was extended from 10 Å to 16 Å. To describe the bulk vdW coupling to all the molecules in the system, we modified the vdW coupling of the surface shell atoms of our NP to characterize the high strength of the bulk gold vdW coupling.57 Polarization and image charging of gold was not considered.
Results and Discussion
For Au NPs and other noble metal nanoparticles that exhibit strong plasmon bands, typical biomolecule-NP titration experiments consist of monitoring either plasmon shifts by UV-visible spectroscopy, or hydrodynamic size in solution by dynamic light scattering. One difference between NMR methods and these typical NP methods is the far higher NP concentrations required (300x) for NMR, as well as the need for isotopically labelled protein. However, protein NMR techniques afford single-residue resolution in situ observation of structural and dynamical changes in proteins as they are exposed to different chemical environment. Related studies at the nano-bio interface has allowed for understanding of protein interactions with various types of nanoparticles58,59; recent pioneering work using NMR has provided deeper insight into protein behavior following adsorption to nano-scale surfaces.60–63 The study of protein adsorption onto NPs using NMR benefits from the clear change in relaxation parameters of protein bound to the NP. Due to the slow tumbling rate of protein-nanoparticle complexes, the bound and rigid portions of the protein upon binding will have increased relaxation rates and broad linewidths. Protein molecules, or portions thereof, that are not bound will exhibit linewidths consistent with rapid (<10 ns) tumbling.64 In measurements described here, the NP acts as an anchor to slow down the portions of protein that are bound to it, resulting in a severe increase in linewidth and concomitant reduction of peak height, which report the portions of the protein bound to the NP.
Adsorption of α-syn to anionic citrate Au NPs
Previously, HSQC spectra of α-syn monomer in sodium phosphate buffer (pH 7.4) solution have been assigned and published by multiple research groups.22,65 However, the conditions under which experiments in this study were performed are slightly altered, due mostly to considerations of NP aggregation in high salt content. It has been previously demonstrated that α-syn and citrate/PAH coated Au NPs retain stability in 20 mM HEPES buffer (pH 7), so this was chosen as the buffer for this study.
As amino acid backbone chemical shifts are sensitive to conditions such as buffer and pH, the peaks of α-syn in an 1H-15N HSQC spectra collected in 20 mM HEPES buffer are expected to shift slightly when compared to 1H-15N HSQC spectra collected under different conditions. Being a disordered protein, the HSQC spectrum of α-syn will have regions of overlap in the 2D spectra. Thus, to perform resonance assignments of α-syn in 20 mM HEPES buffer, we utilized 3D HNCACB66 and CBCA(CO)NH67 data sets in addition to the 2D 1H-15N SOFAST-HMQC68. Using the HNCACB sequence, we observed strong correlations from NH groups to Cα and Cβ atoms within an amino acid residue, and weak correlations to Cα and Cβ atoms in the preceding residue. On the other hand, the CBCA(CO)NH experiment only shows correlations from NH groups to Cα and Cβ atoms in the preceding residue. Using a combination of this data along with the SOFAST-HMQC experiment, which shows H-N correlations, we were able to assign the chemical shifts of 15N, 13Cα, and 13Cβ atoms to 88% of amino acids in monomeric α-syn in 20 mM HEPES buffer (pH 7). A table of chemical shifts can be found in the Supporting Information (Table S1) and has been deposited to the BioMagRes Databank as accession number 26557.
In previous work, an extensive amino acid digestion was performed to quantify the number of α-syn proteins for monolayer coverage on citrate-capped Au NPs of different sizes.37 Using this data, we extrapolated that a protein concentration of 30 μM α-syn would provide monolayer coverage for 300 nM of 12 nm Au NPs. Comparison of 1H-15N HSQC spectra of 30 μM α-syn bound to 300 nM 12 nm citrate Au NPs and 1H-15N HSQC spectra of free α-syn reveals that in addition to small changes in chemical shifts (Figures 1 and S2), the major effect of NP incubation is the change in intensity of a subset of protein peaks. Analysis of the HSQC spectrum shows that a large portion of signals corresponding to amino acids located near the N-terminus experience a large reduction in signal strength compared to the spectrum of free α-syn, indicating that effective correlation time has been increased upon binding to the Au NPs, while other portions of the protein exhibit stronger signal intensities. This observation is consistent with reported mass spectrometry on partially digested α-syn adsorbed to citrate Au NPs.37 Due to the overlap of peaks in the HSQC spectrum of α-syn (Figure S2), signal intensities from certain residues such as Lys-6, Lys-10, and Ala-19 appear greater than the general trend. Although some broadening and loss of signal intensity is observed for all sites in the protein, it is notable that signals from sites near the C-terminus—such as Ala-140, Glu-139, Gly-132, Glu-130, Ser-129 etc.—have higher, and similar, normalized intensities and that a gradual decrease in intensity is observed in the signals progressively closer to the putative Au NP binding region. In particular, Asn-103, Gly-101, Leu-100, Thr-92, Ala-89 Gly-84 and Thr-81 show decreased intensities relative to the C-terminal residues (residue labels and corresponding location in bar graphs shown in green in Figure 1). We attribute this intensity pattern to the restricted diffusion of the C-terminus when tethered via the N-terminus to the Au NP. Sites closer to the bound region have the most restricted motion, and therefore longer effective correlation times and weaker signals.
Figure 1.
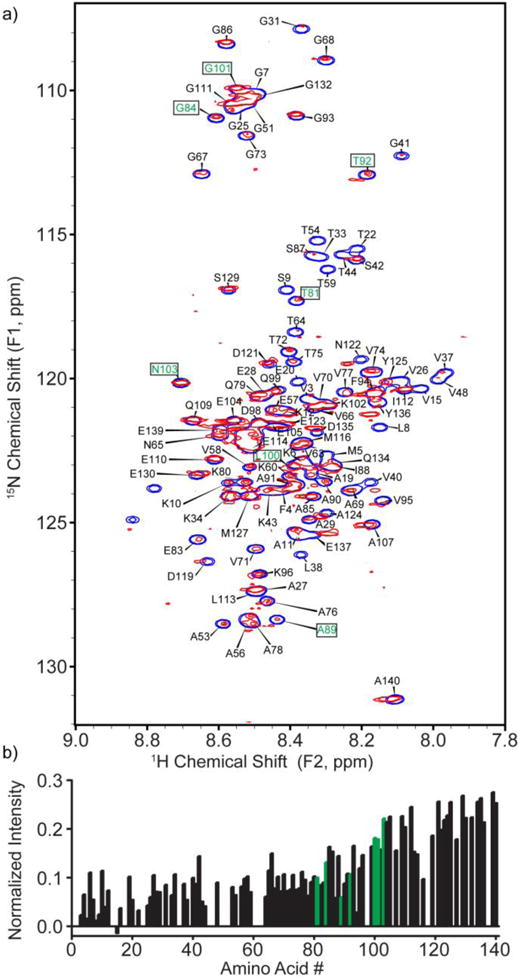
HSQC spectrum of 30 μM α-syn bound to 300 nM 12 nm citrate Au NPs (red) in 20 mM HEPES (pH 7), overlaid on an HSQC spectrum of free α-syn (blue). The concentration of protein was selected by extrapolating from previous work to provide monolayer coverage.37 b) Intensities of signals corresponding to the peaks of α-syn bound to citrate Au NPs, normalized to the spectrum of free α-syn (Figure S3). Residue labels (a) and bars (b) shown in green refer to particular amino acids mentioned in the text. Negative data heights arise from random noise.
The NP-dependent changes in the α-syn spectrum increase in proportion to the NP:α-syn stoichiometric ratio, reminiscent of the effects previously observed by Bax and co-workers upon titration of phospholipid vesicles into solutions of α-syn.26,27 For example, when we decreased the concentration of α-syn to 20 μM while maintaining the Au NP concentration at 300 nM (Figure S4), the differences in intensities of peaks arising from free and bound portions of the protein increased to where the bound residues were no longer detected (and to a greater extent than attributable to the dilution of protein). Additionally we observe that at this stoichiometry, more residues near the N-terminus vanish below the detection limit of the NMR. The assigned peaks in this HSQC spectrum show an amplified trend: signals arising from amino acids located near the N-terminus, notably residues Val-3, Met-5, Lys-6, Gly-7, and Leu-8 (residue labels shown in green in Figure S4) experience a large reduction in intensity, confirming that that binding of α-syn to 12 nm citrate Au NPs occurs via the N-terminus. Although there are a myriad of possible arrangements that α-syn could adopt, the observation of signals from amino acids near the C-terminus consistent with the known random coil chemical shifts of α-syn demonstrates that the C-terminus interacts minimally with the NP surface.
Adsorption of α-syn to cationic MTAB Au NPs
We next investigated the effect of NP surface charge on the binding of α-syn to Au NPs by NMR. Due to agglomeration effects of α-syn on PAH-coated Au NPs, we adapted previous work by Zubarev et al to synthesize MTAB as a cationic ligand for Au NPs.43 When MTAB is added to citrate-coated Au NPs, it allegedly displaces the citrate, and forms a monolayer on the surface (although recent work suggests that citrate still lingers on the surface in the form of trimers even with thiol adsorption).54 Evidence of ligand exchange can be shown through ζ potential measurements, as citrate and MTAB coated NPs will have opposite surface charges. The ζ potential of Au NPs was -13.44 ± 1.77 mV before the MTAB exchange, and +27.99 ± 1.45 mV after. Binding of α-syn to MTAB NPs was confirmed through light scattering measurements, which show an increase in the hydrodynamic diameter from 24.9 ± 1.3 nm to 36.9 ± 1.9 nm upon addition of 300 nM α-syn to 2.8 nM 12 nm MTAB Au NP, suggesting monolayer adsorption of the 5.3-nm protein.37 To study the binding of α-syn to MTAB-coated particles, spectroscopic titrations were performed to monitor the plasmon shift of the Au NP as a function of protein concentration (Figure 2). Using the same method of analysis as before,37 an equilibrium association constant (Ka) of 7.9 ± 1.1 × 106 M−1 was calculated, somewhat less than the binding constant for α-syn to citrate-capped Au NPs (6.61 ± 1.3 × 107 M−1, Figure S5).
Figure 2.
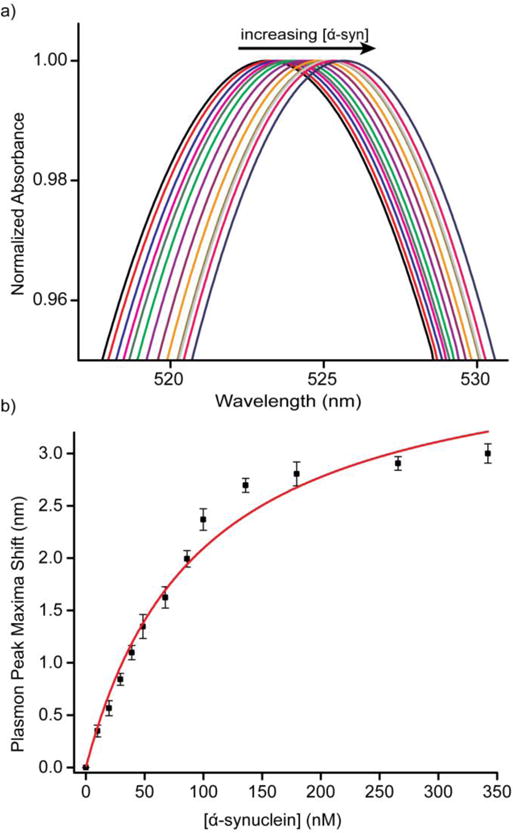
(a) UV-Vis traces of 1.4 nM MTAB Au NPs titrated with 0–350 nM α-syn (black: 0 nM, red: 9.8 nM, blue: 19.6 nM, magenta: 29.4 nM, olive: 39.1 nM, green: 48.6 nM, violet 67.6 nM, purple: 86.2 nM, orange: 99.9 nM, dark yellow: 135.9 nM, light gray: 179.4 nM, pink: 265.7 nM, navy: 342.1 nM). (b) Shifts of plasmon peak (Δλ) plotted against α-syn concentration. The red trace is a fit of the data using a Langmuir adsorption isotherm.
The HSQC spectrum for 30 μM α-syn bound to 300 nM cationic MTAB 12 nm Au NPs is both noticeably different from citrate NPs (Figure 1) and shows no indication of aggregation, even at the relatively high NP concentrations required for NMR (Figure 3). Upon peak assignment and analysis of the HSQC spectrum, we observe that signals from amino acids located near the C-terminus undergo a decrease in intensity, nearly the “mirror image” of the citrate NP data (as mentioned previously, peak overlap in the HSQC spectrum will cause amino acid such as Tyr-136 Glu-139 to have peak intensities that are higher than expected). Therefore, our data strongly support the interpretation that α-syn binds to positively charged 12 nm MTAB Au NPs via its C-terminus. The reversal of binding is consistent with the amino acid composition of α-syn, with a much greater density of acidic amino acids towards the C-terminus of α-syn, allowing for favorable electrostatic interactions with positively charged surfaces (Figure 3).38 The affinity of the C-terminus of α-syn for cationic objects has been documented in previous studies, which agrees with our observation of the reversal of binding orientation upon surface charge modification.29–31 However, the surprising result is that this flipping of the solvent-facing domains of α-syn is so cleanly resolved by switching the NP surface ligand from anionic and polar citrate to cationic yet also hydrophobic MTAB. Again, as in the case of α-syn interacting with citrate NPs, little to no changes in chemical shifts of the amino acid peaks in the HSQC spectra of α-syn and MTAB NPs support the interpretation that the protein remains disordered and does not adopt any defined secondary structure upon adsorption with the MTAB NP. The lack of defined protein structure is supported by circular dichroism measurements of α-syn with citrate and MTAB NPs, (Figure S9).
Figure 3.
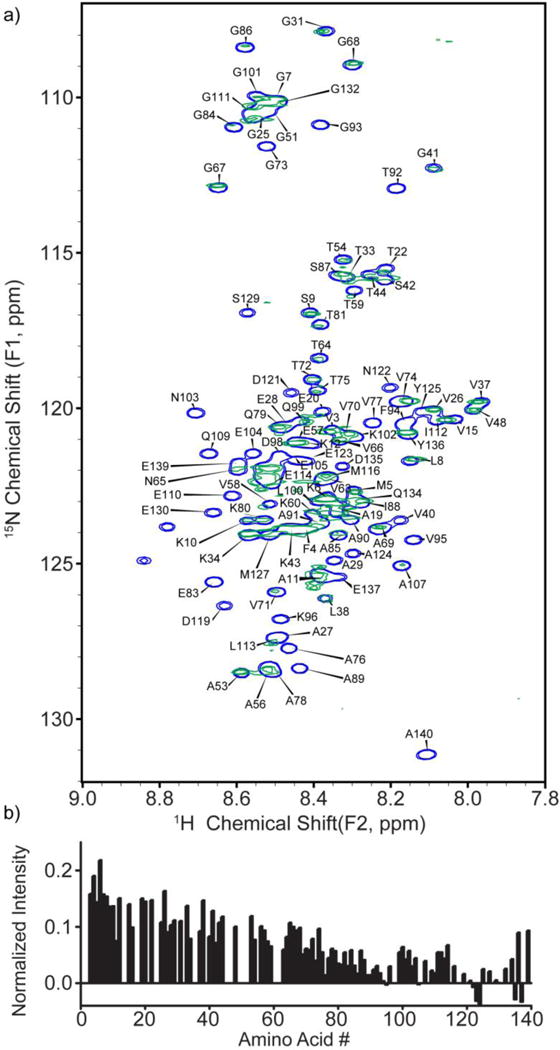
a) HSQC spectrum of 30 μM α-syn bound to 300 nM MTAB Au NPs (green) in 20 mM HEPES (pH 7), overlaid on an HSQC spectrum of free α-syn (blue). b) Intensities of signals corresponding to the peaks of α-syn bound to MTAB Au NP, normalized to the spectrum of free α-syn (Figure S3). Negative data heights arise from random noise.
Simultion of α-syn’s adsorption to Au NPs
That protein orientation on nanoscale surfaces is controlled by surface chemistry is further supported by atomistic molecular dynamics simulations. From the molecular dynamics simulations shown in Figure 4, the N-terminus region is attracted to the negatively charged citrate NP, but does not interact with the cationic MTAB Au NP. Conversely, the C-terminus region of α-syn is strongly attracted to the MTAB NP, and strongly repelled from the citrate NP due to Coulombic coupling. To quantify these effects, we calculated the distance from each amino acid (center of mass) to the closest charged or neutral ligand atom over the last 3 ns of each simulation, searching a range up to 100 Å. In order to account for the effects of local pH (crowded charged ligands), only ten percent of bound ligands were charged, while the remaining ninety percent were left neutral; calculation of distances to the closest charged or neutral ligands represent the most accurate situation of α-syn’s interaction with the actual NP. From 48 ns to 50 ns in the simulation of α-syn’s interaction with citrate-capped Au NPs, the average distance of residues Met-1 through Glu-20 to the citrate ligand shell was 14.9 Å; in contrast, the average distance of residues Pro-120 to Ala-140 to the citrate ligand was 54.1 Å. In the case of α-syn’s interaction with MTAB nanoparticles, between 54 ns and 56 ns, the average distance of residues Pro-120 through Ala-140 to the nearest ligand atom is 5.4 Å; in contrast, the average distance of residues Met-1 through Glu-20 to the nearest ligand atom is 32.3 Å. In both cases, there is a drastic difference in the average distance of either terminus from the corresponding ligand shells, which suggests preferential binding via the termini of α-syn.
Figure 4.
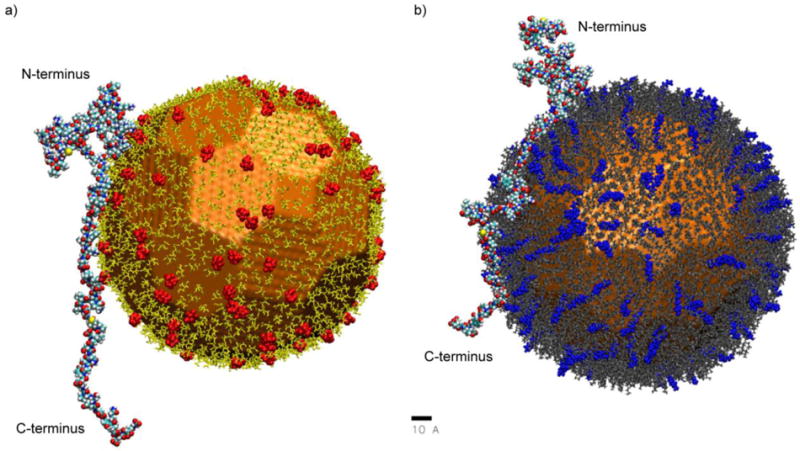
VMD rendering of α-syn’s simulated interaction with individual citrate-capped (a) and MTAB-capped (b) Au NPs, where 10 % of citrate and MTAB ligands are charged (highlighted in red and blue, respectively). Scale bar is 10 angstroms. Distances from each amino acid to the nearest ligand are shown in Figure S10.
Additionally, our simulations suggest that the center region of α-syn is strongly attracted not only to the anionic citrate NP, but also to the cationic MTAB NP. Indeed, distance calculations reveal that the average separation of residues Thr-75 through Leu-100 to the nearest citrate ligand atom is 6.5 Å (from 48–50 ns), and the average distance between residues Thr-75 and Leu-100 to the nearest MTAB ligand is 3.8 Å (from 54–56 ns) in each respective system (Figure S10). This central region of α-syn, also known as the NAC domain (non-Aβ component of Alzheimer disease amyloid), is composed of largely hydrophobic amino acid residues, and is thought to be heavily involved in the aggregation of α-syn due to intramolecular hydrophobic forces.69,70 It is possible that unfavorable hydrophilic-hydrophobic interactions are driving the NAC domain towards the surface of the Au NP in order to minimize contact between hydrophobic residues and the surrounding solvent. However, fine structural details of the simulated systems may slightly differ from the actual protein conformations on the NP surfaces, since we do not simulate the crowding effects of neighboring proteins and other potential features such as gold surface image charging. Nevertheless, the simulations seem to fully corroborate our NMR results that suggest a reversal of protein binding orientation upon the changing of surface charge of the Au NP.
The data presented here suggest that while surface charge plays a large role in dictating the binding orientation of α-syn to Au NPs, there remain some variations in the adsorption of α-syn to these NPs which depends on the chemistries of ligands with similar charges. While both PAH and MTAB act as cationic surface ligands for Au NPs, the interaction of α-syn with PAH-coated and MTAB-coated NPs is very different. Comparison of the structures of PAH and MTAB reveal chemical differences that can influence intermolecular forces, potentially resulting in an effect on protein behavior. For example, the ammonium headgroup of MTAB is unable to participate in hydrogen bonding due to the lack of protons or lone pairs, making it a poor candidate as either a hydrogen bond donor or acceptor. Furthermore, the three methyl groups present a barrier for any potential interactions with the headgroup. On the other hand, the ammonium ions of PAH allow for hydrogen bonding interactions as well as stronger electrostatic attractions due to the steric accessibility of the cation (Scheme 1).
Conclusion
Our experiments demonstrate the impacts of modifying Au NP surface chemistry on protein adsorption as observed by NMR. We were able to manipulate the surface charge of a 12 nm Au NP with citrate and MTAB to control the binding orientation of α-syn, as observed by NMR. Furthermore, this observation of the reversal of binding orientation was corroborated by molecular dynamics simulations of α-syn interacting with both anionic citrate and cationic MTAB Au NPs. The ability to precisely control the surface chemistry of Au NPs opens possibilities to tailor nanoparticles to accurately mimic nanoscale systems of interest, to study protein adsorption onto surfaces of different chemistries and curvatures, and have molecular control of protein display on organelle-sized engineered nano-objects. Knowledge of protein behavior in the presence of different surface chemistries grants predictive power in the future design and implementation of biologically functional nanomaterials.
Supplementary Material
Acknowledgments
We thank the National Science Foundation (CHE-1306596) and the National Institutes of Health (R01-GM073770) for funding this research. M.D.T. is a member of the NIH Molecular Biophysics Training Grant at the University of Illinois at Urbana-Champaign (PHS 5 T32 GM008276). The work of P.K. was supported by the NSF DMR grant No. 1309765, and by the ACS PRF grant No. 53062-ND6. We also thank Joshua Hinman for his assistance with analyzing NMR spectra, Wei Wang for his help in synthesizing MTAB, and Joseph Courtney and Alexander Barclay for the 13C/15N α-synuclein.
Footnotes
ASSOCIATED CONTENT
The 1H-15N HSQC spectra of free α-syn in 20 mM HEPES (pH 7) and 20 μM α-syn bound to 300 nM 12 nm citrate NPs in 20 mM HEPES, enlarged insets showing NMR peak signal intensities, UV-Vis titrations of α-syn to 20 nm citrate Au NPs, and plots of distances from amino acid residues to the Au NP surface as calculated from MD simulations are available in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interests.
References
- 1.Murphy CJ, Gole AM, Stone JW, Sisco PN, Alkilany AM, Goldsmith EC, Baxter SC. Gold Nanoparticles in Biology: Beyond Toxicity to Cellular Imaging. Acc Chem Res. 2008;41(12):1721–1730. doi: 10.1021/ar800035u. [DOI] [PubMed] [Google Scholar]
- 2.Ghosh P, Han G, De M, Kim CK, Rotello VM. Gold Nanoparticles in Delivery Applications. Adv Drug Deliv Rev. 2008;60(11):1307–1315. doi: 10.1016/j.addr.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 3.Alkilany AM, Thompson LB, Boulos SP, Sisco PN, Murphy CJ. Gold Nanorods: Their Potential for Photothermal Therapeutics and Drug Delivery, Tempered by the Complexity of Their Biological Interactions. Adv Drug Deliv Rev. 2012;64(2):190–199. doi: 10.1016/j.addr.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 4.Dreaden EC, Alkilany AM, Huang X, Murphy CJ, El-Sayed MA. The Golden Age: Gold Nanoparticles for Biomedicine. Chem Soc Rev. 2012;41(7):2740. doi: 10.1039/c1cs15237h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimizu T, Teranishi T, Hasegawa S, Miyake M. Size Evolution of Alkanethiol-Protected Gold Nanoparticles by Heat Treatment in the Solid State. J Phys Chem B. 2003;107(12):2719–2724. [Google Scholar]
- 6.Carbó-Argibay E, Rodríguez-González B, Pacifico J, Pastoriza-Santos I, Pérez-Juste J, Liz-Marzán LM. Chemical Sharpening of Gold Nanorods: The Rod-to-Octahedron Transition. Angew Chem Int Ed. 2007;46(47):8983–8987. doi: 10.1002/anie.200703259. [DOI] [PubMed] [Google Scholar]
- 7.Kim F, Connor S, Song H, Kuykendall T, Yang P. Platonic Gold Nanocrystals. Angew Chem Int Ed. 2004;43(28):3673–3677. doi: 10.1002/anie.200454216. [DOI] [PubMed] [Google Scholar]
- 8.Personick ML, Langille MR, Zhang J, Harris N, Schatz GC, Mirkin CA. Synthesis and Isolation of {110}-Faceted Gold Bipyramids and Rhombic Dodecahedra. J Am Chem Soc. 2011;133(16):6170–6173. doi: 10.1021/ja201826r. [DOI] [PubMed] [Google Scholar]
- 9.Shukla R, Bansal V, Chaudhary M, Basu A, Bhonde RR, Sastry M. Biocompatibility of Gold Nanoparticles and Their Endocytotic Fate Inside the Cellular Compartment: A Microscopic Overview. Langmuir. 2005;21(23):10644–10654. doi: 10.1021/la0513712. [DOI] [PubMed] [Google Scholar]
- 10.Connor EE, Mwamuka J, Gole A, Murphy CJ, Wyatt MD. Gold Nanoparticles Are Taken Up by Human Cells but Do Not Cause Acute Cytotoxicity. Small. 2005;1(3):325–327. doi: 10.1002/smll.200400093. [DOI] [PubMed] [Google Scholar]
- 11.Chithrani BD, Ghazani AA, Chan WCW. Determining the Size and Shape Dependence of Gold Nanoparticle Uptake into Mammalian Cells. Nano Lett. 2006;6(4):662–668. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 12.Brannon-Peppas L, Blanchette JO. Nanoparticle and Targeted Systems for Cancer Therapy. Adv Drug Deliv Rev. 2012;64(Supplement):206–212. doi: 10.1016/j.addr.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J, Yuan Z-F, Wang Y, Chen W-H, Luo G-F, Cheng S-X, Zhuo R-X, Zhang X-Z. Multifunctional Envelope-Type Mesoporous Silica Nanoparticles for Tumor-Triggered Targeting Drug Delivery. J Am Chem Soc. 2013;135(13):5068–5073. doi: 10.1021/ja312004m. [DOI] [PubMed] [Google Scholar]
- 14.Meyers JD, Cheng Y, Broome A-M, Agnes RS, Schluchter MD, Margevicius S, Wang X, Kenney ME, Burda C, Basilion JP. Peptide-Targeted Gold Nanoparticles for Photodynamic Therapy of Brain Cancer. Part Part Syst Charact. 2015;32(4):448–457. doi: 10.1002/ppsc.201400119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science. 2003;300(5618):486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 16.Liu F, Wang L, Wang H, Yuan L, Li J, Brash JL, Chen H. Modulating the Activity of Protein Conjugated to Gold Nanoparticles by Site-Directed Orientation and Surface Density of Bound Protein. ACS Appl Mater Interfaces. 2015;7(6):3717–3724. doi: 10.1021/am5084545. [DOI] [PubMed] [Google Scholar]
- 17.Maroteaux L, Campanelli JT, Scheller RH. Synuclein: A Neuron-Specific Protein Localized to the Nucleus and Presynaptic Nerve Terminal. J Neurosci. 1988;8(8):2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.George JM. The Synucleins. Genome Biol. 2001;3(1):3002. doi: 10.1186/gb-2001-3-1-reviews3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Croke RL, Patil SM, Quevreaux J, Kendall DA, Alexandrescu AT. NMR Determination of pKa Values in α-Synuclein. Protein Sci. 2011;20(2):256–269. doi: 10.1002/pro.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ullman O, Fisher CK, Stultz CM. Explaining the Structural Plasticity of α-Synuclein. J Am Chem Soc. 2011;133(48):19536–19546. doi: 10.1021/ja208657z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perrin RJ, Woods WS, Clayton DF, George JM. Interaction of Human α-Synuclein and Parkinson’s Disease Variants with Phospholipids STRUCTURAL ANALYSIS USING SITE-DIRECTED MUTAGENESIS. J Biol Chem. 2000;275(44):34393–34398. doi: 10.1074/jbc.M004851200. [DOI] [PubMed] [Google Scholar]
- 22.Eliezer D, Kutluay E, Bussell R, Jr, Browne G. Conformational Properties of α-Synuclein in Its Free and Lipid-Associated States. J Mol Biol. 2001;307(4):1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 23.Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and Dynamics of Micelle-Bound Human α-Synuclein. J Biol Chem. 2005;280(10):9595–9603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- 24.Jao CC, Hegde BG, Chen J, Haworth IS, Langen R. Structure of Membrane-Bound α-Synuclein from Site-Directed Spin Labeling and Computational Refinement. Proc Natl Acad Sci. 2008;105(50):19666–19671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Georgieva ER, Ramlall TF, Borbat PP, Freed JH, Eliezer D. Membrane-Bound α-Synuclein Forms an Extended Helix: Long-Distance Pulsed ESR Measurements Using Vesicles, Bicelles, and Rodlike Micelles. J Am Chem Soc. 2008;130(39):12856–12857. doi: 10.1021/ja804517m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bodner CR, Dobson CM, Bax A. Multiple Tight Phospholipid-Binding Modes of α-Synuclein Revealed by Solution NMR Spectroscopy. J Mol Biol. 2009;390(4):775–790. doi: 10.1016/j.jmb.2009.05.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bodner CR, Maltsev AS, Dobson CM, Bax A. Differential Phospholipid Binding of α-Synuclein Variants Implicated in Parkinson’s Disease Revealed by Solution NMR Spectroscopy. Biochemistry (Mosc) 2010;49(5):862–871. doi: 10.1021/bi901723p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fernández CO, Hoyer W, Zweckstetter M, Jares‐Erijman EA, Subramaniam V, Griesinger C, Jovin TM. NMR of α‐synuclein–polyamine Complexes Elucidates the Mechanism and Kinetics of Induced Aggregation. EMBO J. 2004;23(10):2039–2046. doi: 10.1038/sj.emboj.7600211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uversky VN, Li J, Fink AL. Metal-Triggered Structural Transformations, Aggregation, and Fibrillation of Human α-Synuclein A POSSIBLE MOLECULAR LINK BETWEEN PARKINSON′S DISEASE AND HEAVY METAL EXPOSURE. J Biol Chem. 2001;276(47):44284–44296. doi: 10.1074/jbc.M105343200. [DOI] [PubMed] [Google Scholar]
- 30.Rasia RM, Bertoncini CW, Marsh D, Hoyer W, Cherny D, Zweckstetter M, Griesinger C, Jovin TM, Fernández CO. Structural Characterization of copper(II) Binding to α-Synuclein: Insights into the Bioinorganic Chemistry of Parkinson’s Disease. Proc Natl Acad Sci U S A. 2005;102(12):4294–4299. doi: 10.1073/pnas.0407881102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sung Y, Rospigliosi C, Eliezer D. NMR Mapping of Copper Binding Sites in Alpha-Synuclein. Biochim Biophys Acta BBA – Proteins Proteomics. 2006;1764(1):5–12. doi: 10.1016/j.bbapap.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 32.Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM-Y. Pathological α-Synuclein Transmission Initiates Parkinson-like Neurodegeneration in Nontransgenic Mice. Science. 2012;338(6109):949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, et al. Distinct α-Synuclein Strains Differentially Promote Tau Inclusions in Neurons. Cell. 2013;154(1):103–117. doi: 10.1016/j.cell.2013.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 Years of Lewy Pathology. Nat Rev Neurol. 2013;9(1):13–24. doi: 10.1038/nrneurol.2012.242. [DOI] [PubMed] [Google Scholar]
- 35.Breydo L, Wu JW, Uversky VN. α-Synuclein Misfolding and Parkinson’s Disease. Biochim Biophys Acta BBA – Mol Basis Dis. 2012;1822(2):261–285. doi: 10.1016/j.bbadis.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 36.Galvagnion C, Buell AK, Meisl G, Michaels TCT, Vendruscolo M, Knowles TPJ, Dobson CM. Lipid Vesicles Trigger α-Synuclein Aggregation by Stimulating Primary Nucleation. Nat Chem Biol. 2015;11(3):229–234. doi: 10.1038/nchembio.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang JA, Johnson BJ, Wu S, Woods WS, George JM, Murphy CJ. Study of Wild-Type α-Synuclein Binding and Orientation on Gold Nanoparticles. Langmuir. 2013;29(14):4603–4615. doi: 10.1021/la400266u. [DOI] [PubMed] [Google Scholar]
- 38.Yang JA, Lin W, Woods WS, George JM, Murphy CJ. α-Synuclein’s Adsorption, Conformation, and Orientation on Cationic Gold Nanoparticle Surfaces Seeds Global Conformation Change. J Phys Chem B. 2014;118(13):3559–3571. doi: 10.1021/jp501114h. [DOI] [PubMed] [Google Scholar]
- 39.Sönnichsen C, Reinhard BM, Liphardt J, Alivisatos AP. A Molecular Ruler Based on Plasmon Coupling of Single Gold and Silver Nanoparticles. Nat Biotechnol. 2005;23(6):741–745. doi: 10.1038/nbt1100. [DOI] [PubMed] [Google Scholar]
- 40.Qiu Y, Liu Y, Wang L, Xu L, Bai R, Ji Y, Wu X, Zhao Y, Li Y, Chen C. Surface Chemistry and Aspect Ratio Mediated Cellular Uptake of Au Nanorods. Biomaterials. 2010;31(30):7606–7619. doi: 10.1016/j.biomaterials.2010.06.051. [DOI] [PubMed] [Google Scholar]
- 41.Bodenhausen G, Ruben DJ. Natural Abundance Nitrogen-15 NMR by Enhanced Heteronuclear Spectroscopy. Chem Phys Lett. 1980;69(1):185–189. [Google Scholar]
- 42.Turkevich J, Stevenson PC, Hillier J. A Study of the Nucleation and Growth Processes in the Synthesis of Colloidal Gold. Discuss Faraday Soc. 1951;11(0):55–75. [Google Scholar]
- 43.Vigderman L, Manna P, Zubarev ER. Quantitative Replacement of Cetyl Trimethylammonium Bromide by Cationic Thiol Ligands on the Surface of Gold Nanorods and Their Extremely Large Uptake by Cancer Cells. Angew Chem. 2012;124(3):660–665. doi: 10.1002/anie.201107304. [DOI] [PubMed] [Google Scholar]
- 44.Kloepper KD, Woods WS, Winter KA, George JM, Rienstra CM. Preparation of α-Synuclein Fibrils for Solid-State NMR: Expression, Purification, and Incubation of Wild-Type and Mutant Forms. Protein Expr Purif. 2006;48(1):112–117. doi: 10.1016/j.pep.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 45.Studier FW. Protein Production by Auto-Induction in High-Density Shaking Cultures. Protein Expr Purif. 2005;41(1):207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 46.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A Multidimensional Spectral Processing System Based on UNIX Pipes. J Biomol NMR. 1995;6(3):277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 47.Lee W, Westler WM, Bahrami A, Eghbalnia HR, Markley JL. PINE-SPARKY: Graphical Interface for Evaluating Automated Probabilistic Peak Assignments in Protein NMR Spectroscopy. Bioinformatics. 2009;25(16):2085–2087. doi: 10.1093/bioinformatics/btp345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, et al. CHARMM General Force Field (CGenFF): A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J Comput Chem. 2010;31(4):671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu W, He X, Vanommeslaeghe K, MacKerell AD. Extension of the CHARMM General Force Field to Sulfonyl-Containing Compounds and Its Utility in Biomolecular Simulations. J Comput Chem. 2012;33(31):2451–2468. doi: 10.1002/jcc.23067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vanommeslaeghe K, MacKerell AD. Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J Chem Inf Model. 2012;52(12):3144–3154. doi: 10.1021/ci300363c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vanommeslaeghe K, Raman EP, MacKerell AD. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J Chem Inf Model. 2012;52(12):3155–3168. doi: 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mackerell AD, Feig M, Brooks CL. Extending the Treatment of Backbone Energetics in Protein Force Fields: Limitations of Gas-Phase Quantum Mechanics in Reproducing Protein Conformational Distributions in Molecular Dynamics Simulations. J Comput Chem. 2004;25(11):1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 53.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J Phys Chem B. 1998;102(18):3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 54.Park J-W, Shumaker-Parry JS. Strong Resistance of Citrate Anions on Metal Nanoparticles to Desorption under Thiol Functionalization. ACS Nano. 2015;9(2):1665–1682. doi: 10.1021/nn506379m. [DOI] [PubMed] [Google Scholar]
- 55.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable Molecular Dynamics with NAMD. J Comput Chem. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Humphrey W, Dalke A, Schulten K. VMD: Visual Molecular Dynamics. J Mol Graph. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 57.Wright LB, Rodger PM, Corni S, Walsh TR. GolP-CHARMM: First-Principles Based Force Fields for the Interaction of Proteins with Au(111) and Au(100) J Chem Theory Comput. 2013;9(3):1616–1630. doi: 10.1021/ct301018m. [DOI] [PubMed] [Google Scholar]
- 58.Bayraktar H, You C-C, Rotello VM, Knapp MJ. Facial Control of Nanoparticle Binding to Cytochrome c. J Am Chem Soc. 2007;129(10):2732–2733. doi: 10.1021/ja067497i. [DOI] [PubMed] [Google Scholar]
- 59.Aubin-Tam ME, Hwang W, Hamad-Schifferli K. Site-Directed Nanoparticle Labeling of Cytochrome c. Proc Natl Acad Sci. 2009 doi: 10.1073/pnas.0807299106. pnas.0807299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Engel MFM, Visser AJWG, Mierlo CPMV. Conformation and Orientation of a Protein Folding Intermediate Trapped by Adsorption. Proc Natl Acad Sci U S A. 2004;101(31):11316–11321. doi: 10.1073/pnas.0401603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Calzolai L, Franchini F, Gilliland D, Rossi F. Protein–Nanoparticle Interaction: Identification of the Ubiquitin–Gold Nanoparticle Interaction Site. Nano Lett. 2010;10(8):3101–3105. doi: 10.1021/nl101746v. [DOI] [PubMed] [Google Scholar]
- 62.Siriwardana K, Wang A, Vangala K, Fitzkee N, Zhang D. Probing the Effects of Cysteine Residues on Protein Adsorption onto Gold Nanoparticles Using Wild-Type and Mutated GB3 Proteins. Langmuir. 2013;29(35):10990–10996. doi: 10.1021/la402239h. [DOI] [PubMed] [Google Scholar]
- 63.Shrivastava S, McCallum SA, Nuffer JH, Qian X, Siegel RW, Dordick JS. Identifying Specific Protein Residues That Guide Surface Interactions and Orientation on Silica Nanoparticles. Langmuir. 2013;29(34):10841–10849. doi: 10.1021/la401985d. [DOI] [PubMed] [Google Scholar]
- 64.Bloembergen N, Purcell EM, Pound RV. Relaxation Effects in Nuclear Magnetic Resonance Absorption. Phys Rev. 1948;73(7):679–712. [Google Scholar]
- 65.Woods WS, Boettcher JM, Zhou DH, Kloepper KD, Hartman KL, Ladror DT, Qi Z, Rienstra CM, George JM. Conformation-Specific Binding of α-Synuclein to Novel Protein Partners Detected by Phage Display and NMR Spectroscopy. J Biol Chem. 2007;282(47):34555–34567. doi: 10.1074/jbc.M705283200. [DOI] [PubMed] [Google Scholar]
- 66.Grzesiek S, Bax A. An Efficient Experiment for Sequential Backbone Assignment of Medium-Sized Isotopically Enriched Proteins. J Magn Reson 1969. 1992;99(1):201–207. [Google Scholar]
- 67.Grzesiek S, Bax A. Correlating Backbone Amide and Side Chain Resonances in Larger Proteins by Multiple Relayed Triple Resonance NMR. J Am Chem Soc. 1992;114(16):6291–6293. [Google Scholar]
- 68.Schanda P, Brutscher B. Very Fast Two-Dimensional NMR Spectroscopy for Real-Time Investigation of Dynamic Events in Proteins on the Time Scale of Seconds. J Am Chem Soc. 2005;127(22):8014–8015. doi: 10.1021/ja051306e. [DOI] [PubMed] [Google Scholar]
- 69.Uéda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T. Molecular Cloning of cDNA Encoding an Unrecognized Component of Amyloid in Alzheimer Disease. Proc Natl Acad Sci. 1993;90(23):11282–11286. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deleersnijder A, Gerard M, Debyser Z, Baekelandt V. The Remarkable Conformational Plasticity of Alpha-Synuclein: Blessing or Curse? Trends Mol Med. 2013;19(6):368–377. doi: 10.1016/j.molmed.2013.04.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.