

Abstract
An approach to identify β-secretase 1 (BACE1) fragment binders that do not interact with the catalytic aspartate dyad is presented. A ThermoFluor (thermal shift) and a fluorescence resonance energy transfer enzymatic screen on the soluble domain of BACE1, together with a surface plasmon resonance (SPR) screen on the soluble domain of BACE1 and a mutant of one catalytic Asp (D32N), were run in parallel. Fragments that were active in at least two of these assays were further confirmed using one-dimensional NMR (WaterLOGSY) and SPR binding competition studies with peptidic inhibitor OM99-2. Protein-observed NMR (two-dimensional 15N heteronuclear single-quantum coherence spectroscopy) and crystallographic studies with the soluble domain of BACE1 identified a unique and novel binding mode for compound 12, a fragment that still occupies the active site while not making any interactions with catalytic Asps. This novel approach of combining orthogonal fragment screening techniques, for both wild-type and mutant enzymes, as well as binding competition studies could be generalized to other targets to overcome undesired interaction motifs and as a hit-generation approach in highly constrained intellectual property space.
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder and a massive societal burden.1,2 Available treatments only provide a modest delay of the cognitive decline.3 Considerable research efforts aim to intervene in disease progression.4−6 Among these, inhibition of β-secretase 1 (BACE1) is the most studied since its discovery in 1999.7−10 The approach prevents the cleavage of the amyloid precursor protein (APP) into neurotoxic Aβ40–42 peptide products, which aggregate to form the extracellular amyloid plaques found in the AD brains.11 Genetic evidence also supports BACE1 as a target for AD.12
BACE1 is a membrane-anchored aspartic protease with three domains: an N-terminal ectodomain, a single transmembrane domain, and a cytosolic C-terminus. The catalytic ectodomain has an aspartic protease fold, with the substrate-binding cleft located between the N- and C-terminal lobes (Figure 1). The crucial catalytic aspartate (Asp) dyad, D32 and D228, is located at the interface of the two lobes.7 A hairpin loop “flap” in the N-terminal lobe partially covers the cleft in a perpendicular orientation. The conformational changes in the flap control the substrate access to the active site, and open to closed conformations have been observed in crystal structures of BACE1.13,14 Loops C, D, and F in the C-lobe of the ectodomain are the epitopes for binding of a known antibody.15
Figure 1.
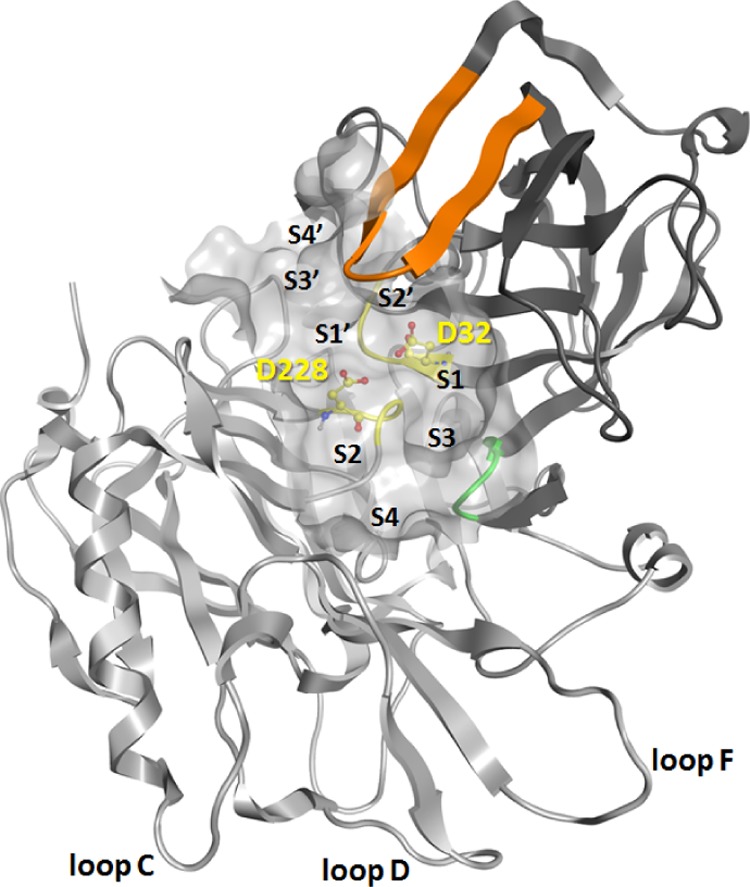
BACE1 (PDB 1XN3) in ribbon representation with the N-terminal lobe in dark gray, C-terminal lobe in light gray, active site with the catalytic Asp dyad in yellow, flap in orange, and 10S loop in green. The substrate-binding cleft is shown as a surface together with the location of subpockets S1, S2, S3, S4, S1′, S2′, S3′, and S4′.
The first BACE1 inhibitors were substrate analogues that mimicked the APP-cleavage sequence with a noncleavable peptide bond. They displayed high in vitro potency but typically had poor oral bioavailability and low brain penetration.16−18 The discovery of amidine moieties that form optimal interactions with the Asp dyad revolutionized the field of BACE1 inhibitors, as improved drug likeness became possible.19 These Asp-binding amidine and guanidine motifs have been widely explored (Chart 1A),19 including studies conducted in our labs (compounds 1, 3, and 4).20−25 Compound 1 was reported to bend back, allowing the distal N-cyclohexyl group to occupy the S1 pocket, whereas the central cyclohexyl occupied the lipophilic S1′ pocket. Compounds 2 and 3 on the other hand contain a quaternary sp3 carbon, which provides an ideal vector into the S1–S3 and S2′ pockets of the catalytic site (Figure 1).26 Although a step forward, the basicity of the amidine/guanidine function provides a formal positive charge, impacting the optimization of physicochemical parameters. In contrast, there are a few known ligands that bind to the catalytic cleft without interacting with the Asp dyad. Pyrimidine 6 was reported by Merck to bind to the S1 and S3 pockets27 (Chart 1B), and dihydroisoquinolines 7 and 8 were reported by Elan Pharmaceuticals28−30 to bind to the S2 pocket. Hence, the drawbacks of amidine inhibitors and the limited intellectual property (IP) space spurred our interest to seek alternatives to binding at the Asp dyad.
Chart 1. (A) Examples of Amidine-Based BACE1 Inhibitors Reported by AstraZeneca, Janssen, and Schering/Merck; (B) Examples of Orthosteric Non-Asp BACE1 Binders.

Fragment-based drug discovery has now become an accepted strategy for hit finding in drug discovery.31,32 The principle is that structurally smaller “fragment-like” hits (generally MW below 250 D or ∼21 heavy atoms) can be evolved into more optimal binders with superior properties than those of traditional high-throughput screen (HTS) hits. Because of their few but optimal interactions, fragment hits typically have low affinity in the high micromolar or low millimolar range. Hence, fragment screens are often performed at similar concentration ranges using sensitive biophysical techniques, such as surface plasmon resonance (SPR), ThermoFluor (TF), ligand-observed NMR, and sometimes X-ray screening. Although normally the throughput is lower than that of biochemical HTS assays, the fragment space is exponentially smaller, requiring only thousands of fragments to cover a similar chemistry space, than that in HTS decks of hundreds of thousands of compounds.33 Fragments are more likely progressed to leads when the activity is confirmed by orthogonal screening techniques, and also knowing the binding mode via crystallography can be crucial to improve potency. Numerous BACE1 leads have evolved from fragment screening,34 most binding to the catalytic Asp dyad, for instance those from Edwards et al. (Chart 1, compound 2).24 Hence, fragment screening can be successful, but careful consideration is needed to overcome the contemporary challenges of BACE1 inhibition. Here, we applied orthogonal screening approaches, leading to the identification of a non-Asp-binding fragment, for which a crystal structure was obtained showing a distinct and previously undescribed binding mode.
Results and Discussion
Overview of the Approach
We defined a strategy to identify the fragments that do not interact with the catalytic Asps, Figure 2. An SPR screen of the Janssen Fragment Library (JFL) was conducted at two pH values using the Biacore 4000 system with the mature BACE1 ectodomain and neutravidin (reference protein) immobilized. In parallel, TF and enzymatic FRET assays were run at high compound concentration. These allowed additional triage and permitted us to identify false positives from the SPR screen. The hits were further characterized in a new SPR screen by assessing their binding in the presence of the potent peptidomimetic inhibitor, OM99-2 (FRET IC50, 4.38 nM; SPR Kd, 12.7 nM),35 known to occupy most subpockets of wild-type (WT) BACE1 and the active-site mutant D32N. This approach provided additional information on the binding site and discriminated only the most promising fragments. Hits with an attractive structure and desired binding profile were then confirmed by WaterLOGSY,36 and the actives were submitted to protein-observed 1H–15N TROSY NMR and/or X-ray crystallography.37
Figure 2.
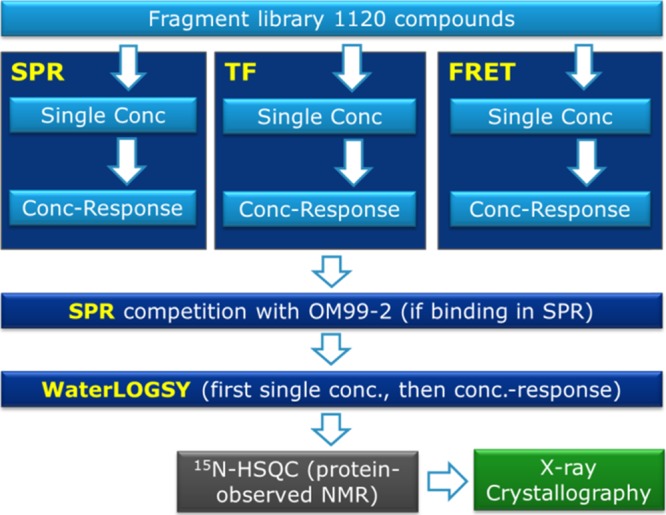
Hit identification and characterization flow chart used to identify non-Asp-binding fragments to BACE1.
Fragment Library
High-concentration screens often render assays sensitive to false positives resulting from minor impurities in the screening samples.38 Janssen R&D has therefore invested in a fragment collection in which all compounds have been analyzed by LC/MS with UV or CLND detection and their dimethyl sulfoxide (DMSO) solutions have been analyzed for structure plausibility by NMR using the Bruker CMC-Q software. The purity threshold in LC/MS and NMR was set to 95% and above. Fragments with 8–21 heavy atoms were selected from both our screening deck and internally synthesized intermediates or final compounds. These were stored in 100, 50, or 20 mM DMSO solutions depending on the solubility. The library was constructed on the basis of criteria such as 2D shape, 2D fingerprint, and scaffold diversity. Similarities to both the known drugs and the number of sp3 centers were also promoted in the selection process. The size of the JFL used in this study was 1120 compounds.
SPR Screen
For SPR-based fragment screening, the mature BACE1 ectodomain was immobilized on a CM7 sensor chip together with neutravidin (reference). First, all fragments were tested for promiscuous binding, that is, showing nonspecific interaction with the target and/or reference protein, resulting in high baseline increase or decrease, or showing irregular sensorgram shapes. Nine fragments were removed from the JFL due to promiscuous binding. Next, the JFL was screened in duplicate using a compound concentration of 0.25 mM at both pHs 7.4 and 4.5 because Dominguez et al. demonstrated that BACE1 inhibitors binding well at acidic and neutral pH values could be optimal cellular inhibitors as they maintain interaction with BACE1 throughout the transport from the neutral extracellular environment through to the acidic endosome.39 Concentration-reponse (CR) curves were subsequently generated for hits at either pH value or both pH values.
TF Screen
Thermal shift analysis of the mature BACE1 ectodomain was used to estimate binding affinities by measuring the effect of a ligand on protein stability using thermal denaturation.40,41 The JFL was tested in duplicate at a single concentration (2.1, 0.8, or 0.2 mM based on the respective haystack DMSO concentration of 100, 50, or 20 mM). Fragments showing a significant ΔTm shift compared to the average protein-only control value were confirmed by CR curves.
Enzymatic Screen
BACE1 enzymatic activity was assessed by a FRET assay using a mature BACE1 ectodomain. An APP-derived 13 amino acid substrate containing the “Swedish” Lys-Met/Asn-Leu mutation at the cleavage site was employed. 7-Methoxycoumarin-4-yl acetic acid (MCA ) was the fluorescent donor, and 2,4-dinitrophenyl (DNP) was the acceptor. The 100, 50, and 20 mM haystack samples of the JFL were first screened at 33.3-fold dilution, hence at 3.0, 1.5, and 0.6 mM, respectively. Hits with ≥49% inhibition were subsequently tested twice by CR curves. Autofluorescent compounds (sample at T0 minus mean control at T0 > 3 × σ) were removed.
SPR Competition and Mutant BACE1 Experiments
To identify fragment binding in the vicinity of the Asp’s, a competition between OM99-2 and the mature BACE1 ectodomain was performed with six overlapping hits from the prior assays. In addition, SPR binding experiments versus the D32N mutant allowed us to identify the catalytic Asp binding fragments. Fragment 10 was not tested because of its nonspecific binding behavior at pH 4.5 (very slow on/off rate constants).
Hit Analysis
The outcome from the three orthogonal primary assays, SPR, TF, and enzymatic FRET, is presented in Figure 3 and the corresponding data in Table 1. The criteria for hit selection were as follows: SPR hits showed a specific and concentration-dependent binding at one or both pHs; TF hits gave a ΔTm shift >0.5 °C at the highest concentration with no sign of autofluorescence; and FRET hits showed a robust CR curve and no autofluorescence. Of the three approaches, FRET gave 87 hits in total (7.8%), but a majority (81, 92%) did not confirm in other assays. Because of its high sensitivity, SPR gave the highest hit rate (159 hits: 77 at pH 7.4 and 106 at pH 4.5, with an overlap of 24 hits), with only 6 confirmed in other assays. Finally, TF gave the lowest amount of hits (2, 0.18%), but both confirmed in SPR and FRET. The structures of hits confirmed in at least two assays are shown in Figure 3.
Figure 3.
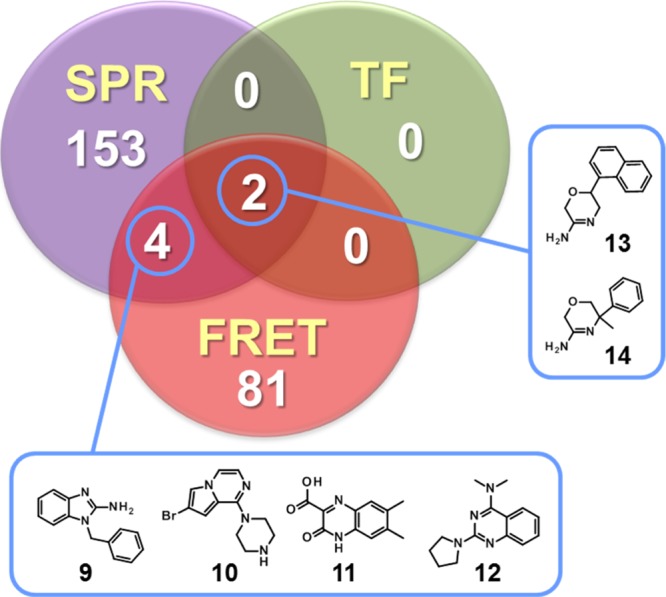
Hits identified across different fragment screens.
Table 1. FRET, SPR, TF, and WL Data for Compounds 9–14a,b.
| FRET | SPR |
TF |
WaterLOGSY |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| compd | IC50 (mM) | binding at pH 7.5 | binding at pH 4.5 | OM99-2 competition | D32N binding | ΔTm (°C) | conc (mM) | KD (mM) | signal at pH 7.5 | signal at pH 4.5 |
| 9 | 1.6 | yes | yes | minor | yes | –1.12 | 2.1 | intense positive | NT | |
| 10 | 0.6 | yes | SBd | no | yes | –0.27 | 2.1 | positive | NT | |
| 11 | 0.4 | yes | yes | minor | yes | 0.15 | 0.8 | negative | negative | |
| 12 | 0.5 | yes | yes | minor | yes | NMc | 2.1 | positive | NT | |
| 13 | 1.1 | yes | yes | minor | yes | 0.63 | 2.1 | 0.83 | NT | NT |
| 14 | 0.2 | yes | yes | yes | no | 2.78 | 2.1 | 0.12 | negative | intense positive |
Average of at least two independent runs, except WaterLOGSY, which was performed once.
NT, not tested.
NM, not measurable due to (auto)fluorescence interference.
SB, superstoechiometric binder.
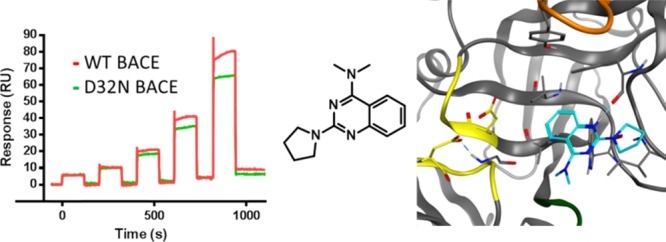
Four compounds (9–12) showed SPR and enzymatic activity but no thermal shift (Figure 3 and Table 1). Of these, 9 bound to both WT and mutant BACE1 but also showed reduced binding in the presence of OM99-2, suggesting interaction with the catalytic Asp’s. Fragment 9 contains a known guanidine motif42 and hence did not fit our criteria of novelty. Why 9 binds to the BACE1 D32N mutant despite its guanidine substructure remains elusive, but it may be indicative of nonspecific binding despite our efforts to filter out such false positives. The other fragments with overlapping SPR and FRET activity, 10–12, were not previously reported to bind to BACE1. Fragment 10 bound to the BACE1 D32N mutant alone and in the presence of OM99-2, suggesting binding outside the catalytic site. Interestingly, fragments 11 and 12 do not display any known Asp-binding motif, bound to the BACE1 D32N mutant, and showed reduced binding in the presence of OM99-2. This strongly suggested that they bind to the active site without interaction with the catalytic Asps. The sensorgrams of 12 show binding in the absence (red) and presence (green) of OM99-2 and to the BACE1 D32N mutant (Figure 4). Finally, only two hits were confirmed in all three assays: 13 and 14. However, 14 is an intermediate from a BACE1 medicinal chemistry exploration,23 whereas 13 is a structurally related but much weaker binding compound synthesized for an unrelated project. For fragments 13 and 14, KD’s of respectively 0.83 and 0.12 mM could be calculated from the TF CR data. In SPR, 14 bound strongly to the mature BACE1 ectodomain but, in contrast to 13, lost binding to the BACE1 D32N mutant (Figure 4C). Binding of 14 was also drastically reduced in the presence of OM99-2 (Figure 4A).
Figure 4.

SPR sensorgrams showing the interaction of 14 (A) and 12 (B) to WT BACE1 before (red curve) and after (green curve) OM99-2 injection. (C) Sensorgrams of the binding of 12 (green curve) and 14 (red curve) to the BACE1 D32N mutant. The compounds were tested in CR series (0.03, 0.06, 0.13, 0.25, 0.50 mM).
WaterLOGSY
In the preparation of eventual heteronuclear single-quantum coherence spectroscopy (HSQC) experiments, the binding of selected hits (9–12 and 14) was confirmed using WaterLOGSY, a ligand-observed NMR binding assessment technique (Table 1). A 30-fold excess of ligand was used at physiological pH. Experiments were repeated at pH 4.5.43 Fragment 9 showed a very intense positive peak at pH 7.5, and also clear positive signals were observed for 10 and 12. Signals for 11 and 14 were negative under these conditions. Nevertheless, when the experiments were repeated at acidic pH, a strong positive signal was obtained for 14, whereas 11 remained negative.
Protein-Observed NMR
The partial assignment of amino acids in the mature BACE1 ectodomain via 15N-HSQC has been reported.44 This technique was used to further characterize the binding of fragments 10 and 12 to BACE1 and as a selection criterion for X-ray crystallography. Being a known Asp binder, 14 was selected as a reference. Compound 9 and 13 were discarded given their similarity to previously reported hits, and 11 was excluded on the basis of WaterLOGSY experiments. BACE1 was labeled with 15N as necessary for the 15N-HSQC 2D experiment. The TROSY approach was employed as it produced better resolution, which is beneficial given the signal broadening and overlap in the spectra typically seen for large proteins. Most of the reported assignment could be annotated at the corresponding NH signals. A 5-fold excess of ligand was added to a 0.1 mM protein sample. To visualize shift changes upon interaction between the ligand and protein, the combined shift difference was calculated from the chemical shift differences in the nitrogen and the proton dimension, using the following formula.
The 2D-HSQC results of 12 and 14 are shown in Figure 5. Amino acids from mature 15N-labeled BACE1 that experience changes upon interaction are displayed on the X axis, and the combined shift difference in parts per million (ppm) is represented on the Y axis. As expected, amino acids Asp32 and Asp228 were affected by the binding of 14, as well as neighboring amino acids Gly34 and Gly120. Interestingly, 12 affected different amino acids compared to 14. The shifts are visualized on the surface of the BACE1 protein extracted from the co-crystal with lead compound 4 (PDB 5CLM; Figure 6). Whereas 14 affected the two catalytic Asp’s (shown in pink, adjacent to the amidine substructure, Figure 6A), 12 did not (Figure 6B). Instead, shifts were observed for multiple amino acids in the protein, with some close to the binding site such as Leu121, Arg128, Thr329, and Gly334. Unfortunately, a large number of amino acids in the active site were not assigned; therefore, the exact position of 12 could not be determined by NMR. In the case of 10, the observed chemical shift changes were much lower than those for compound 12 and no significant differences with 14 could be detected (data not shown).
Figure 5.

Chemical shift changes in BACE1 upon interaction with fragments 12 and 14. The three plots show the amino acids experiencing alterations in their 2D-HSQC 15N shifts calculated according to the combined shift difference equation provided in the text.
Figure 6.
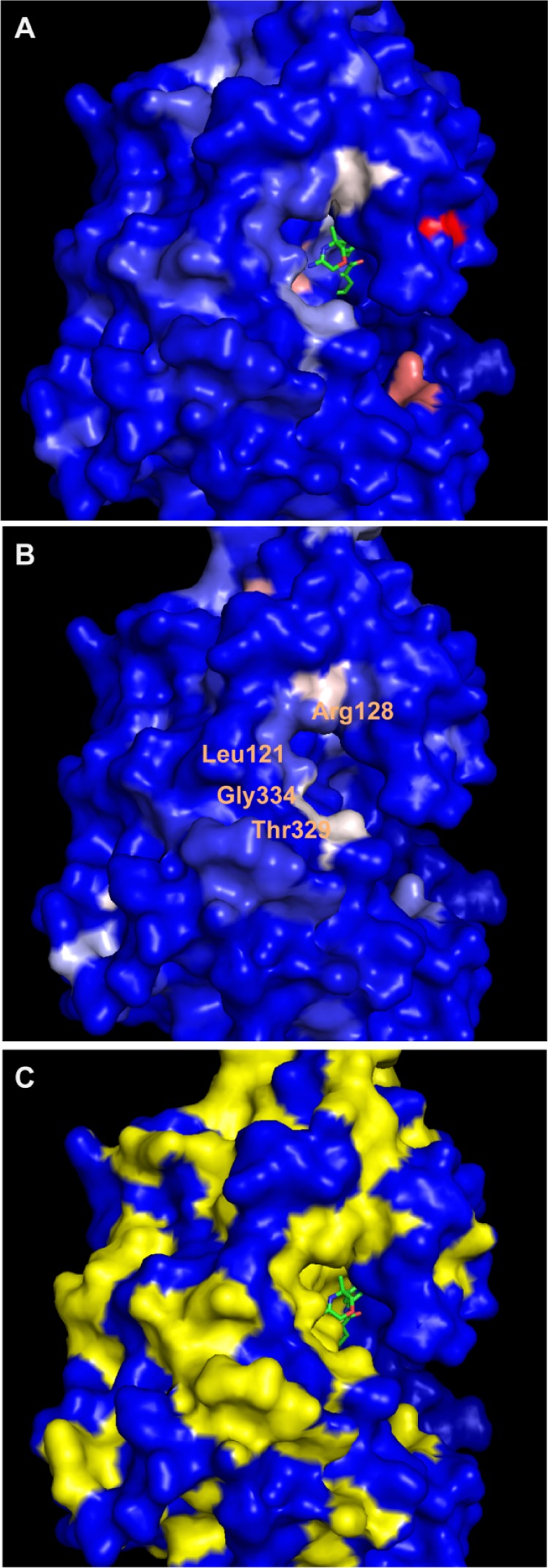
Visualization of 15N-HSQC shift changes with 14 (A) and 12 (B) using the co-structure of 4 with BACE1 (PDB 5CLM): blue, no change; light blue, 0–0.04 ppm; white, 0.05–0.08 ppm; pink, 0.05–0.11 ppm; and red, 0.11–0.15 ppm. (C) The assigned amino acids are shown in yellow. The structure of 4 is shown in green in (A) and (C).
Crystallography
Following the various primary assays and follow-up work, 12 emerged as the strongest candidate for attempting crystallography. Pleasingly, the screening, detailed analysis, and prioritization paid dividends as the crystal structure of 12 with BACE1 was readily solved at 2.5 Å resolution (PDB code 5MXD, Figure 7A). There are three copies of the BACE1–inhibitor complex in the crystallographic asymmetric unit, with each showing a similar binding mode of 12. The flexible active-site flap containing Tyr71 adopts different orientations in the three monomers.
Figure 7.

Interactions of 12 (A), interaction map of 12 (B), a comparison between the binding modes of 12 and Asp binder 4 (C), and a comparison of the binding modes of non-Asp binders 6 (magenta), 8 (green), and 12 (cyan) (D).
In contrast to that in most known BACE1 inhibitors, such as 4, no interaction between 12 and the catalytic Asp dyad (Asp32 and Asp228) either directly or via a water-mediated network was seen (Figure 7C).23 Instead, the quinazoline occupies the S1 pocket and the dimethylamine is oriented toward S3. Fragment 12 forms an H-bond between the protonated quinazoline and the backbone carbonyl of Phe108 in two of the three copies of BACE1 and forms π-stacking and hydrophobic interactions with residues Leu30, Tyr71, Phe108, Ile110, Trp115, and Ile118 (Figure 7B). The pyrolidine ring goes into a new, more-solvent-exposed subpocket. The distance between the nitrogen of the dimethylamine and the carbonyl of Gly230 is too long to form a strong hydrogen bond (3.4–3.9 Å in the three copies).
Figure 7D compares the structures of reported non-Asp binders 6 and 8 to BACE1 with that of 12. Inhibitor 6 occupies a similar region but has some differences.27 It also forms a H-bond with the carbonyl of Phe108; however, a sulfonamide is oriented toward S3 compared with the dimethylamine in 12. The dihydroisoquinolines 7 and 8 do not interact with Phe108 but, instead, form an H-bond network in S2, compensating for the absence of interaction with the catalytic Asp dyad.28,29
Conclusions
Fragment screening with SPR, TF, and enzymatic FRET approaches delivered six hits, confirmed in at least two assays, and two in all three. The use of the BACE1 D32N mutant identified fragments that bind to BACE1 but not via Asp interactions. Further SPR competition experiments with OM99-2 helped in selecting those that bind to the catalytic cleft. After confirming binding by NMR (WaterLOGSY), protein-observed NMR using 15N-labeled BACE1 led to the prioritization of 12 for X-ray crystallography, which revealed a unique binding mode in the S2 pocket of BACE1. This orthogonal screening cascade along with mutant and competition experiments is a valid general approach for finding novel ligands for drug discovery targets, especially when the IP space is dominated by similar chemical motifs. In addition, this orthogonal screening paradigm maximizes the chance to obtain X-ray structures of such ligands with unusual binding modes.
Methods
Proteins
The mature BACE1 ectodomain protein (amino acid residues −16 to 393) with a COOH-terminal 6xHis-tag (denoted “mature BACE1 ectodomain” throughout the article) was expressed by baculovirus infection of HighFive insect cells and purified from the cell culture supernatant using Ni-NTA affinity chromatography, anion exchange chromatography, and size-exclusion chromatography essentially as described by Bruinzeel et al.45 The mature BACE1 ectodomain catalytic-site mutant (D32N) protein (amino acid residues −16 to 393) with a COOH-terminal 6xHis-tag and StrepII-tag was expressed in and purified from E. coli cells by the Protein Service Facility of the University of Ghent.46 The only difference between the WT and mutant forms of the protein (apart from the mutation) is the somewhat longer tag at the COOH-terminus of the mutant form (only 9 amino acid residues for His-tag vs 21 amino acid residues for His- + Strep-tag). The BACE1 ectodomain has been tagged at the COOH-terminus in many studies, and no apparent effect on its activity or conformation has been described. Although an experimental effect of the tag and/or the difference in length between the tags cannot be excluded, it is highly unlikely. For 15N-HSQC NMR experiments, labeled BACE1 (amino acid residues −16 to 393) was expressed in minimal medium with 15NH4Cl from E. coli cells and purified by ion-exchange and size-exclusion chromatography by the Structural Biochemistry Laboratory of the CIPF.47
FRET
Primary BACE1 enzymatic activity was assessed by a FRET assay using an APP-derived 13 amino acids peptide, that contains the “Swedish” Lys-Met/Asn-Leu mutation of the APP β-secretase cleavage site, as a substrate (MCA -Ser-Glu-Val-Asn-Leu-Asp-Ala-Glu-Phe-Arg-Lys(DNP)-Arg-Arg-NH2 TFA salt, Bachem cat No. M-2465) and a mature BACE1 ectodomain (Aurigene, custom-made). The substrate contains fluorophore MCA as a fluorescent donor with excitation wavelength at 320 nm and emission wavelength at 405 nm and proprietary quencher acceptor DNP. The distance between these two groups has been selected so that upon light excitation the donor fluorescence energy is significantly quenched by the acceptor, through resonance energy transfer. Upon cleavage by BACE1, MCA is separated from DNP, restoring the full fluorescence yield of the donor. The increase in fluorescence is linearly related to the rate of proteolysis. In a 384-well format, BACE1 is incubated for 2 h with the substrate and the inhibitor. The amount of proteolysis is directly measured by fluorescence measurement in the Fluoroskan microplate fluorometer (Thermo Scientific). For the low control, no enzyme was added to the reaction mixture. The enzymatic BACE1 FRET assay was performed in a total volume of 30 μL in a buffer containing 33.3 mM citrate, pH 5.0; 0.033% PEG; 10 μM substrate; 32.4 nM BACE1; and the compound. Compound stock solutions in 100% DMSO were diluted, 1/33.3, in the reaction mixture, resulting in final DMSO concentrations of 4% (3% from compound, 1% from substrate).
SPR
Measurements were performed using a Biacore 4000 instrument and CM7 biosensor chips (GE Healthcare). The mature BACE1 ectodomain and neutravidin both were immobilized by amine coupling using standard procedures and materials provided by GE Healthcare. A mixture of Hepes (10 mM), pH 7.4, NaCl (150 mM; HBS-N), and 0.005% Tween 20 was used as the running buffer during immobilization. Interaction experiments were performed at 25 °C at both pH 7.4 (running buffer HBS-N, pH 7.4, with 0.05% Tween 20 and 5% DMSO) and pH 4.5 (running buffer 10 mM NaOAc, pH 4.5, with 150 mM NaCl, 0.05% Tween 20, and 5% DMSO). Fragments and reference compounds were injected for 30 s over the sensor surface at a flow rate of 30 μL/min. DMSO solvent correction was performed using calibration points containing between 2.5 and 6.5% DMSO. Binding response of reference compounds was followed during the fragment screen to determine potential decrease in the apparent analyte binding capacity of BACE1, which was compensated for in the data analysis. Data analysis has been performed using the Biacore 4000 Control and Evaluation software and Excel. Fragments were screened twice (forward and reverse order) at 0.25 mM and at pH 7.4 and 4.5 against BACE1, neutravidin, and a blank reference spot to identify and discard promiscuous binders. For the primary screen, fragments have been tested at 0.25 mM in duplicate both at pH 7.4 and 4.5. Top 10% screening hits were retested by CR curves, ranging between 0.03 and 0.500 mM (8 concentrations with 1.5-fold dilution range). Competition experiments (pH 4.5) were performed by injection of fragment hits before and after injection of the OM99-2 peptide (Calbiochem, 496000). Immobilization and SPR measurements using the BACE1 D32N mutant were performed under the conditions similar to those used for WT BACE1.
ThermoFluor
The BACE1 ThermoFluor assay was performed in 384-well plates in a total volume of 4 μL in a buffer containing 50 mM NaOAc pH 5.4, 150 mM NaCl, 0.0005% Tween 20, 100 μM ANS, 1.6 μM mature BACE1 ectodomain protein and compound. Compound stock solutions in 100% DMSO were diluted 1/24 in the reaction, resulting in final compound concentrations of 2.1, 0.80, or 0.2 mM (depending on the initial compound stock concentration) and a final DMSO concentration of 4.2%. Thermal shift measurements were done in a ThermoFluor instrument from 25 to 85 °C in 1 °C increments. Fluorescence at every temperature was determined using a charge-coupled device camera and subsequent image analysis using proprietary software.
WaterLOGSY and Protein-Observed NMR
All NMR experiments were performed using a Bruker Avance II 600 MHz spectrometer equipped with a 5 mm inverse cryoprobe at 27 °C. For WaterLOGSY experiments, to 480 μL of 5 μM mature BACE1 ectodomain, 20 μL of D2O and 0.15 mM of ligand were added (from a 100 mM stock in DMSO-d6), in a protein/ligand ratio of 1:30, optimal for the WaterLOGSY experiments. For each sample, 1D 1H and WaterLOGSY experiments were conducted. A total of 32 K-points were used for a sweep width of 14 ppm, and a total of 128 scans were accumulated for the WaterLOGSY experiment. For 15N-HSQC NMR experiments, protein samples were concentrated to 0.10 mM, and 0.02 mL of D2O and 0.50 mM of ligand were added (from a 100 mM stock in DMSO-d6) to 0.48 mL of protein sample. The TROSY version of 15N-HSQC with sensitivity enhancement was acquired with 200 scans, spectral widths of 14 and 40 ppm and offsets of 4.7 and 117 ppm in the 1H and the 15N dimension, respectively. Spectra were acquired and processed with Topspin 3.2 (Bruker Biospin), and 2D experiments were integrated with Sparky (T. D. Goddard and D. G. Kneller; SPARKY 3, University of California, San Francisco).
Crystal Structure Experimental Data
A structure of 12 in BACE1 (1–454) was obtained by Proteros Biostructures GmbH.48 X-ray crystallography data collection and refinement statistics are summarized in Table 2, and refinement statistics are summarized in Table 3. Human BACE-1 (amino acids 22–446 preceded by MetHis6; numbering as in UniProtKB entry P56817) was expressed in E. coli BL21(DE3) in inclusion bodies. Briefly, inclusion bodies were refolded from 8 M urea and further purified by ion-exchange and gel-filtration chromatography. After the removal of the propeptide by digestion with clostripain, the noncleaved material was removed by NiNTA chromatography and the protein was finally subjected to ion-exchange chromatography. For crystallization, the protein was concentrated to 23 mg/mL, flash-frozen in liquid nitrogen, and stored at −80 °C until further use.
Table 2. Data Collection and Processing Statistics for 12.
| ligand | 12 |
|---|---|
| PDB ID | 5MXD |
| X-ray source | PXI/X06SA (SLS)a |
| wavelength [Å] | 0.99999 |
| detector | PILATUS 6M |
| temperature [K] | 100 |
| space group | C2 |
| cell: a; b; c; [Å] | 232.91; 100.74; 59.05 |
| cell: α; β; γ; [deg] | 90.0; 102.0; 90.0 |
| resolution [Å] | 2.52 (2.77 – 2.52)b |
| unique reflections | 43 635 (10 835)b |
| multiplicity | 2.9 (2.9)b |
| completeness [%] | 96.5 (97.5)b |
| Rsym [%] | 6.6 (50.6)b |
| Rmeas [%] | 8.1 (61.7)b |
| mean(I)/sd | 13.64 (2.44)b |
Swiss Light Source (SLS, Villigen, Switzerland).
Values in parentheses refer to the resolution bin with Rsym = 50.6%.
Table 3. Refinement Statistics.
| resolution [Å] | 113.90–2.52 |
| number of reflections (working/test) | 42 644/991 |
| Rcryst [%] | 22.7 |
| Rfree [%] | 28.1 |
| total number of atoms | |
| protein | 8960 |
| water | |
| ligand | 54 |
| deviation from ideal geometry | |
| bond lengths [Å] | 0.007 |
| bond angles [deg] | 1.23 |
| bonded B’s [Å2] | 3.5 |
| Ramachandran plot | |
| most favored regions [%] | 87.5 |
| additional allowed regions [%] | 11.9 |
| generously allowed regions [%] | 0.3 |
| disallowed regions [%] | 0.3 |
For crystallization, the protein at 10 mg/mL was incubated for 1 h on ice with 2 mM of the ligand (diluted from 100 mM DMSO stock in the protein solution). The protein was crystallized from 12% (m/v) PEG4000, 100 mM MES/NaOH, pH 5.5, by hanging-drop vapor diffusion at 20 °C (0.75 μL protein + 0.75 μL reservoir solution). For cryoprotection, crystals were briefly soaked in reservoir solution mixed with 25% (v/v) glycerol and frozen in liquid nitrogen.
X-ray diffraction data were collected at the SLS (beamline PXI/X06SA) using a PILATUS 6M detector. Data were integrated, scaled, and merged using XDS.49 The structure was refined with REFMAC5.50 Manual model completion was carried out using Coot.51 The quality of the final model was verified by PROCHECK52 and the validation tools available through Coot.51
Chemistry
Compound 9 was obtained commercially from Specs and Biospecs (CAS# 43182-10-1) and has been described before.53 Compound 11 was obtained from the internal compound collection and has been previously reported.54
Analytical Methods
All final compounds were characterized by 1H NMR and LC/MS and were >95% pure by 1H NMR. 1H NMR spectra were recorded on Bruker spectrometers: DPX-360 MHz, AVI-500 MHz, and AVI-600 MHz. For 1H NMR spectra, all chemical shifts are reported in ppm (δ) units and are relative to the residual signal at 7.26 and 2.50 ppm for CHCl3 and DMSO, respectively. Different LC/MS methods were used to characterize the products. LC/MS method A. Instrument: Waters Alliance HT 2790 - DAD-MS; column: Xterra MS C18 (3.5 μm, 4.6 × 100 mm2); eluent: (A) 95% NH4OAc 25 mM + 5% ACN, (B) ACN, (C) MeOH; gradient from 100% A to 50% B and 50% C in 6.5 min, to 100% B in 1 min, 100% B for 1 min and re-equilibrate with 100% A for 1.5 min; flow 1.6 mL/min; column T: 40 °C; run time: 10 min. LC/MS Method B. Instrument: Waters Acquity UPLC - DAD and SQD; column: waters: BEH C18 (1.7 μm, 2.1 × 50 mm2); eluent: (A) 95% NH4OAc 6.5 mM + 5% ACN, (B) ACN; gradient from 95% A to 40% A in 3.8 min to 5% A in 0.8 min held for 0.4 min; flow: 1 mL/min; column T: 50 °C; run time: 5 min. LC/MS Method C. Instrument Waters Acquity UPLC - DAD and SQD; column: BEH C18 (1.7 μm, 2.1 × 50 mm2; Waters Acquity); eluent: (A) 95% NH4OAc 25 mM + 5% ACN, (B) ACN; gradient from 95% A and 5% B to 5% A and 95% B in 1.3 min and hold for 0.3 min; flow: 0.8 mL/min; column T: 55 °C; run time: 2 min. LC/MS Method D. Instrument Waters Acquity UPLC - DAD and SQD; column: Waters HSS T3 (1.8 μm, 2.1 × 100 mm2); eluent A: 10 mM NH4OAc in 95% H2O + 5% ACN, (B) ACN; gradient from 100% A to 5% A in 2.10 min, to 0% A in 0.90 min, to 5% A in 0.5 min; flow: 0.7 mL/min; column T: 55 °C; run time 3.5 min.
Experimental Procedures
Scheme 1 outlines the routes used to prepare compounds 10, 12–14.
Scheme 1. Synthesis of Compounds 10, 12–14.

Reagents and conditions: (a) 1-Boc-piperazine, K2CO3, ACN, rt, 1 h. (b) 6 N HCl/2-propanol, rt, 1 h. (c) NHMe2 (2 M in THF), THF/water, 17 h, rt. (d) pyrrolidine, ACN, microwave 110 °C, 3 h. (e) TMSCN, ZnI2, 0 °C to rt, 2 days. (f) LiAlH4, Et2O, rt, 2 days. (g) ClCH2COCl, NEt3, DCM, 0 °C to rt, 30 min. (h) NaH, 2-propanol, rt, overnight. (i) Epichlorohydrin, BF3OEt2, Et2O, rt, overnight. (j) 7 N NH3/MeOH, rt, overnight. (k) ClCH2COCl, DIPEA, THF, −10 to −5 °C. (l) t-BuOK, 5–10 °C, 60 min. (m) P2S5, THF, 50 °C, 30 min. (n) NH4OH, 40 °C, 11 h.
7-Bromo-1-piperazin-1-yl-pyrrolo[1,2-a]pyrazine (10). 7-Bromo-1-chloro-pyrrolo[1,2-a]pyrazine (0.4 g, 1.7 mmol, CAS 1597913-29-5), 1-Boc-piperazine (0.633 g, 3.4 mmol, CAS 143238-38-4) and K2CO3 (0.47 g, 3.4 mmol) in ACN (7.5 mL) were heated in a microwave oven for 15 min at 170 °C. Then, additional 1-Boc-piperazine (0.633 g, 3.4 mmol, CAS 143238-38-4) was added and the reaction was heated at 180 °C for 45 min. After cooling to rt, DCM was added, the solid was filtered off, and the filtrate solvent was evaporated affording 1.0 g of intermediate 10-1 (100%).
Intermediate 10-1 (1.0 g, 1.7 mmol) in 6 N HCl/2-propanol (10 mL) was stirred for 1 h at room temperature (rt). Then, DCM and aqueous saturated Na2CO3 were added. The organic layer was separated, dried (Na2SO4), filtered, and the solvent was evaporated. The residue was purified by column chromatography (eluent: DCM/(7 N NH3 in MeOH), 96:4), affording 430 mg of compound 10 (90%). 1H NMR (600 MHz, DMSO-d6): δ ppm 2.78–2.85 (m, 4H), 3.47–3.50 (m, 4H), 6.84 (dd, J = 1.5, 0.9 Hz, 1H), 7.12 (d, J = 4.6 Hz, 1H), 7.70 (dd, J = 4.7, 0.9 Hz, 1H), 7.74 (d, J = 1.5 Hz, 1H); LC/MS (method D); Rt 1.22 min, MH+ 281.
N,N-Dimethyl-2-pyrrolidin-1-yl-quinazolin-4-amine (12). Dimethylamine (2 M solution in THF, 22.6 mL, 45.22 mmol, CAS 607-68-1) was added dropwise to a slurry of 2,4-dichloroquinazoline (3000 mg, 15.07 mmol) in THF (10 mL)/water (15 mL). The resulting mixture was stirred for 17 h at rt and then poured into water and extracted with EtOAc (3 × 75 mL). The combined extracts were dried (MgSO4), filtered, and concentrated in vacuo, yielding intermediate 12-1 as a bright white powder (3.16 g, 100%). LC/MS (method C); rt 0.84 min, MH+ 208.
A MW-vessel was charged with intermediate 12-1 (0.25 g, 1.20 mmol), pyrrolidine (301 μL, 3.61 mmol), and ACN (5 mL). This mixture was stirred and heated under MW irradiation at 110 °C for 30 min. Next, the reaction mixture was concentrated in vacuo and purified using RP-HPLC (RP Shandon Hyperprep C18 BDS – 10 μm, 250 g, 5 cm, mobile phase (0.25% NH4HCO3 solution in water, MeOH + ACN)). The desired fractions were concentrated in vacuo and co-evaporated twice with MeOH at 60 °C. An off-white solid was obtained, which was dissolved in 10 mL of DCM. This solution was then treated with NaOH (1 mL, 1 M), dried over an Isolute HM-N tube, and rinsed with 10 mL of DCM. The collected solution was evaporated at 50 °C under a stream of nitrogen. The residue was dissolved in 15 mL of DCM, and 1.5 equiv of HCl (6 N in 2-propanol) was added dropwise. The obtained solution was then stirred for 1 h. The white crystals were collected on a filter, rinsed with 10 mL of DCM, and dried in a vacuum oven at 55 °C for 24 h, yielding compound 12 (242 mg, 83%) as a white powder. 1H NMR (600 MHz, DMSO-d6): δ ppm 1.95 (br. s, 2H), 2.04 (br. s, 2H), 3.47 (br. s, 6H), 3.67 (t, J = 6.8 Hz, 4H), 7.37 (t, J = 7.7 Hz, 1H), 7.78 (t, J = 7.7 Hz, 1H), 7.94 (d, J = 8.3 Hz, 1H), 8.17 (d, J = 8.3 Hz, 1H), 12.05 (s, 1H); LC/MS (method A); Rt 4.24 min, MH+ 243.
2-(1-Naphthyl)-3,6-dihydro-2H-1,4-oxazin-5-amine (13). Zinc(II) iodide (0.025 g, 0.078 mmol) was added to 1-naphthaldehyde (36.4 g, 0.233 mol, CAS 66-77-3), and the resulting mixture was cooled with ice. Next, TMSCN (25 g, 0.252 mol) was added dropwise. When the addition was complete, the mixture was brought to 25 °C. After the reaction was judged complete (thin-layer chromatography (TLC) monitoring), the mixture was diluted with DCM and washed with an aqueous saturated NaHCO3 solution. The organic layer was dried (Na2SO4), filtered, and concentrated to provide intermediate 13-1 as a red-orange oil that solidified upon standing (59.97 g, 100%).
In a three-necked flask fitted with a mechanical stirrer and addition funnel, LiAlH4 (9.90 g, 0.261 mol) was suspended in dry diethyl ether (205 mL) under nitrogen flow. Next, intermediate 13-1 (59.50 g, 0.233 mol) dissolved in 75 mL dry diethyl ether was added dropwise. When the addition was complete, the reaction was stirred at rt until judged complete by TLC monitoring (about 2 days). Next, 10 mL water was added dropwise, followed by 10 mL of a 15% NaOH solution. Then, 29.7 mL of water was again added carefully. The mixture was further stirred until a precipitate was formed, which was filtered over dicalite. The filtrate was concentrated, providing intermediate 13-2 as a bright orange-red oil (13.9 g, 32%).
Intermediate 13-2 (13.8 g, 0.737 mol) was dissolved in 500 mL dry DCM. Triethylamine (14.9 g, 0.147 mol) was added, and the resulting solution was cooled with ice. Next, chloroacetyl chloride (9.99 g, 0.0884 mol) was added dropwise, and after addition, the reaction mixture was allowed to warm to rt. After the reaction was judged complete (TLC monitoring), the mixture was poured into 1 N HCl (500 mL). The organic layer was separated, and the aqueous layer was extracted with DCM (3 × 250 mL). The combined organic layers were dried (Na2SO4), filtered, and evaporated to dryness. The residue was purified over silica gel using CHCl3 as eluent. The product fractions were evaporated, yielding intermediate 13-3 as a beige foam (6.10 g, 31%).
Sodium hydride (60% dispersion in mineral oil, 1.85 g, 0.463 mmol) was washed with dry hexanes, and then, 76 mL of dry 2-propanol was added. The resulting mixture was stirred for 2.5 h at 25 °C. Next, intermediate 13-3 (6.10 g, 0.0232 mol) in 78 mL of 2-propanol was added. The reaction mixture was stirred overnight at 25 °C. Then, the reaction was quenched by pouring the mixture into 110 mL of ice-water containing HOAc (1.30 mL, 0.0232 mol). The mixture was extracted with CHCl3 (2 × 250 mL). The combined organic layers were dried (Na2SO4), filtered, and evaporated to dryness. The residue was purified over silica gel using CHCl3/MeOH (99:1) as eluent, yielding intermediate 13-4 (2.88 g, 55%).
Boron trifluoride diethyl etherate (1.34 g, 0,00944 mol) was dissolved in 5.6 mL of dry diethyl ether. Epichlorohydrin (0.68 g, 0.00726 mol) was added, and the resulting mixture was stirred for 2 h at 25 °C. Next, the ether was decanted and the residue was washed with ether. DCM (11 mL) was added to the residue, and intermediate 13-4 (1.65 g, 0.00726 mol) in 11 mL DCM was subsequently added. The resulting mixture was stirred overnight at 25 °C, after which all volatiles were evaporated, providing intermediate 13-5 as a light yellow foam, which was carried as such to the next reaction step.
Intermediate 13-5 (crude from previous step, approx. 0.00726 mol) was dissolved in 15.6 mL of NH3 in MeOH (7 N, 0.109 mol), and the resulting mixture was stirred overnight at rt. Next, 1 N NaOH (50 mL) was added, and the mixture was extracted with CHCl3 (3 × 50 mL). The combined organic layers were dried (Na2SO4), filtered, and evaporated. Diethyl ether was added to the residue, followed by concentrated HCl (0.61 mL, 0.00726 mol). The resulting precipitate was filtered, and recrystallized from MeOH/Et2O, yielding compound 13 as a white solid (0.66 g, 46% two steps). 1H NMR (600 MHz, DMSO-d6): δ ppm 3.57 (dd, J = 13.2, 10.4 Hz, 1H), 3.75 (dd, J = 13.2, 3.3 Hz, 1H), 4.81 (d, J = 17.3 Hz, 1H), 4.87 (d, J = 17.3 Hz, 1H), 5.69 (dd, J = 10.4, 3.3 Hz, 1H), 7.53–7.65 (m, 4H), 7.92–8.01 (m, 2H), 8.19 (d, J = 8.4 Hz, 1H), 9.01 (br. s, 2H), 10.20 (br. s, 1H); LC/MS (method D); Rt 1.31 min, MH+ 227. Anal. Calcd for C14H14N2O. HCl: C, 64.00; H, 5.75; N, 10.66; Cl, 13.49. Found: C, 63.01; H, 5.66; N, 10.17; Cl, 13.03. Mp 213–215 °C.
5-Methyl-5-phenyl-2,6-dihydro-1,4-oxazin-3-amine (14). To 2-amino-2-phenyl-propan-1-ol (1000 mg, 6.613 mmol, CAS 90642-81-2) and DIPEA (1.37 mL, 7.94 mmol) in THF (40 mL) at −10 °C was added chloroacetyl chloride (0.527 mL, 6.61 mmol) dropwise. Then, the mixture was left stirring while slowly warming up to −5 °C for 20 min. Then, t-BuOK (1.86 g, 16.53 mmol) was added portionwise, and the mixture was left warming up slowly to 5–10 °C for 60 min. Then, the reaction was quenched with 10% aq. NH4Cl and extracted with DCM. The organic layers were dried (Na2SO4), filtered, evaporated, and purified by automated flash silica column (eluent: DCM/EtOAc, gradient 1:0 to 7:3), giving intermediate 14-1 as a transparent oil (870 mg, 69%). 1H NMR (500 MHz, chloroform-d): δ ppm 1.70 (s, 3H), 3.74 (d, J = 11.6 Hz, 1H), 3.80 (d, J = 11.6 Hz, 1H), 4.21 (d, J = 16.8 Hz, 1H), 4.26 (d, J = 16.8 Hz, 1H), 6.41 (br. s, 1H), 7.29–7.36 (m, 1H), 7.37–7.44 (m, 4H); LC/MS (method D); rt 1.27 min, MH+ 192.
Intermediate 14-1 (400 mg, 2.092 mmol) and P2S5 (511 mg, 2.301 mmol) were dissolved in THF (15 mL), and the mixture was stirred at 50 °C for 30 min. Then, the mixture was cooled to rt and filtered over cotton and evaporated. The residue was dissolved in DCM and directly injected into a flash silica column, eluting with DCM. Evaporation of the product fractions provided intermediate 14-2 as a yellow oil (360 mg, 83%). 1H NMR (500 MHz, chloroform-d): δ ppm 1.75 (s, 3H), 3.79 (d, J = 11.6 Hz, 1H), 3.84 (d, J = 11.6 Hz, 1H), 4.58 (d, J = 18.5 Hz, 1H), 4.65 (d, J = 18.5 Hz, 1H), 7.32–7.38 (m, 3H), 7.39–7.46 (m, 2H), 8.35 (br. s, 1H); LC/MS (method B); Rt 1.22 min, MH+ 208.
Intermediate 14-2 (340 mg, 1.64 mmol) was dissolved in NH4OH (15 mL), and the mixture was stirred at rt overnight and then for 11 h at 40 °C. Then, water and DCM were added, the organic layer was separated, and the aqueous layer was further extracted with DCM (3×). The combined organic layers were dried (Na2SO4), filtered, and evaporated. Then, DCM (15 mL) and TFA (0.25 mL) were added, mixed well, and evaporated. Et2O (20 mL) was added to the residue and sonicated. This formed a white precipitate, which was filtered and washed with Et2O and dried. Then, this product was further purified by automated flash silica column (eluent (1% TFA in DCM)/MeOH; gradient 100:0 to 95:5). The fractions containing the product were collected and evaporated, and the residue was precipitated in Et2O, filtered, and dried in vacuo. This gave intermediate 14 as a white solid (310 mg, 62%). 1H NMR (500 MHz, DMSO-d6): δ ppm 1.63 (s, 3H), 3.87 (d, J = 11.8 Hz, 1H), 3.93 (d, J = 12.1 Hz, 1H), 4.53–4.61 (m, 2H), 7.32–7.38 (m, 1H), 7.39–7.45 (m, 4H), 8.74 (br. s, 1H), 9.03 (br. s, 1H), 10.69 (s, 1H); LC/MS (method B); Rt 0.49 min, MH+ 191.
Acknowledgments
We thank Proteros for BACE1 crystal growth, soaking, and structure determination of 12.48
Glossary
Abbreviations
- BACE1
β-secretase 1
- DNP
2,4-dinitrophenyl
- FRET
fluorescence resonance energy transfer
- HTS
high-throughput screening
- MCA
(7-methoxycoumarin-4-yl) acetic acid
- DNP
2,4-dinitrophenyl
- SPR
surface plasmon resonance
- TF
ThermoFluor
- TROSY
transverse relaxation optimized spectroscopy
- JFL
Janssen Fragment Library
- CR
concentration-response
Author Present Address
∇ This author is presently also affiliated with the Instituto de Investigación Sanitaria La Fe, Drug Discovery Unit, Avda. Fernando Abril Martorell 106, 46026 Valencia, Spain (A.P.-L.).
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- Hebert L. E.; Weuve J.; Scherr P. A.; Evans D. A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. 10.1212/WNL.0b013e31828726f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dementia; Fact Sheet April, 2016. World Health Organization Website. http://www.who.int/mediacentre/factsheets/fs362/en/ (accessed Nov 8, 2016).
- Yiannopoulou K. G.; Papageorgiou S. G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. 10.1177/1756285612461679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüll M.; Berger M.; Heneka M. Disease-modifying therapies in Alzheimer’s disease: how far have we come?. Drugs 2006, 66, 2075–2093. 10.2165/00003495-200666160-00004. [DOI] [PubMed] [Google Scholar]
- Citron M. Alzheimer’s disease: strategies for disease modification. Nat. Rev. Drug Discovery 2010, 9, 387–398. 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- Leger G. C.; Massoud F. Novel disease-modifying therapeutics for the treatment of Alzheimer’s disease. Exp. Rev. Clin. Pharmacol. 2013, 6, 423–442. 10.1586/17512433.2013.811237. [DOI] [PubMed] [Google Scholar]
- Hussain I.; Powell D.; Howlett D. R.; Tew D. G.; Meek T. D.; Chapman C.; Gloger I. S.; Murphy K. E.; Southan C. D.; Ryan D. M.; Smith T. S.; Simmons D. L.; Walsh F. S.; Dingwall C.; Christie G. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol. Cell. Neurosci. 1999, 14, 419–427. 10.1006/mcne.1999.0811. [DOI] [PubMed] [Google Scholar]
- Vassar R.; Bennett B. D.; Babu-Khan S.; Kahn S.; Mendiaz E. A.; Denis P.; Teplow D. B.; Ross S.; Amarante P.; Loeloff R.; Luo Y.; Fisher S.; Fuller J.; Edenson S.; Lile J.; Jarosinski M. A.; Biere A. L.; Curran E.; Burgess T.; Louis J. C.; Collins F.; Treanor J.; Rogers G.; Citron M. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Yan R. Q.; Bienkowski M. J.; Shuck M. E.; Miao H. Y.; Tory M. C.; Pauley A. M.; Brashler J. R.; Stratman N. C.; Mathews W. R.; Buhl A. E.; Carter D. B.; Tomasselli A. G.; Parodi L. A.; Heinrikson R. L.; Gurney M. E. Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature 1999, 402, 533–537. 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- Sinha S.; Anderson J. P.; Barbour R.; Basi G. S.; Caccavello R.; Davis D.; Doan M.; Dovey H. F.; Frigon N.; Hong J.; Jacobson-Croak K.; Jewett N.; Keim P.; Knops J.; Lieberburg I.; Power M.; Tan H.; Tatsuno G.; Tung J.; Schenk D.; Seubert P.; Suomensaari S. M.; Wang S. W.; Walker D.; Zhao J.; McConlogue L.; John V. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 1999, 402, 537–540. 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- Hardy J. A.; Higgins G. A. Alzheimer’s disease: the amyloid cascade hypothesis. Science 1992, 256, 184–185. 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Karran E.; Mercken M.; De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discovery 2011, 10, 698–712. 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- Shimizu H.; Tosaki A.; Kaneko K.; Hisano T.; Sakurai T.; Nukina N. Crystal structure of an active form of BACE1, an enzyme responsible for amyloid beta protein production. Mol. Cell. Biol. 2008, 28, 3663–3671. 10.1128/MCB.02185-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y. C.; Li M. J.; Greenblatt H.; Chen W. Y.; Paz A.; Dym O.; Peleg Y.; Chen T. T.; Shen X.; He J. H.; Jiang H. L.; Silman I.; Sussman J. L. Flexibility of the flap in the active site of BACE1 as revealed by crystal structures and molecular dynamics simulations. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 13–25. 10.1107/S0907444911047251. [DOI] [PubMed] [Google Scholar]
- Wang W. R.; Liu Y. C.; Lazarus R. A. Allosteric inhibition of BACE1 by an exosite-binding antibody. Curr. Opin. Struct. Biol. 2013, 23, 797–805. 10.1016/j.sbi.2013.08.001. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Brindisi M.; Tang J. Developing beta-secretase inhibitors for treatment of Alzheimer’s disease. J. Neurochem. 2012, 120, 71–83. 10.1111/j.1471-4159.2011.07476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Kumaragurubaran N.; Tang J. Recent developments of structure based beta-secretase inhibitors for Alzheimer’s disease. Curr. Top. Med. Chem. 2005, 5, 1609–1622. 10.2174/156802605775009711. [DOI] [PubMed] [Google Scholar]
- Hong L.; Koelsch G.; Lin X.; Wu S.; Terzyan S.; Ghosh A. K.; Zhang X. C.; Tang J. Structure of the protease domain of memapsin 2 (beta-secretase) complexed with inhibitor. Science 2000, 290, 150–153. 10.1126/science.290.5489.150. [DOI] [PubMed] [Google Scholar]
- Oehlrich D.; Prokopcova H.; Gijsen H. J. The evolution of amidine-based brain penetrant BACE1 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 2033–2045. 10.1016/j.bmcl.2014.03.025. [DOI] [PubMed] [Google Scholar]
- Baxter E. W.; Conway K. A.; Kennis L.; Bischoff F.; Mercken M. H.; De Winter H. L.; Reynolds C. H.; Tounge B. A.; Luo C.; Scott M. K.; Huang Y.; Braeken M.; Pieters S. M. A.; Berthelot D. J. C.; Masure S.; Bruinzeel W. D.; Jordan A. D.; Parker M. H.; Boyd R. E.; Qu J.; Alexander R. S.; Brenneman D. E.; Reitz A. B. 2-Amino-3,4-dihydroquinazolines as inhibitors of BACE-1 (beta-site APP cleaving enzyme): Use of structure based design to convert a micromolar hit into a nanomolar lead. J. Med. Chem. 2007, 50, 4261–4264. 10.1021/jm0705408. [DOI] [PubMed] [Google Scholar]
- Tresadern G.; Delgado F.; Delgado O.; Gijsen H.; Macdonald G. J.; Moechars D.; Rombouts F.; Alexander R.; Spurlino J.; Van Gool M.; Vega J. A.; Trabanco A. A. Rational design and synthesis of aminopiperazinones as beta-secretase (BACE) inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 7255–7260. 10.1016/j.bmcl.2011.10.050. [DOI] [PubMed] [Google Scholar]
- Mateu N.; Ciordia M.; Delgado O.; Sanchez-Rosello M.; Trabanco A. A.; Van Gool M.; Tresadern G.; Perez-Benito L.; Fustero S. A versatile approach to CF3-containing 2-pyrrolidones by tandem michael addition-cyclization: exemplification in the synthesis of amidine class BACE1 inhibitors. Chemistry 2015, 21, 11719–11726. 10.1002/chem.201501662. [DOI] [PubMed] [Google Scholar]
- Rombouts F. J. R.; Tresadern G.; Delgado O.; Martínez-Lamenca C.; Van Gool M.; García-Molina A.; Alonso de Diego S. A.; Oehlrich D.; Prokopcova H.; Alonso J. M.; Austin N.; Borghys H.; Van Brandt S.; Surkyn M.; De Cleyn M.; Vos A.; Alexander R.; Macdonald G.; Moechars D.; Gijsen H.; Trabanco A. A. 1,4-Oxazine β-secretase 1 (BACE1) inhibitors: from hit generation to orally bioavailable brain penetrant leads. J. Med. Chem. 2015, 58, 8216–8235. 10.1021/acs.jmedchem.5b01101. [DOI] [PubMed] [Google Scholar]
- Edwards P. D.; Albert J. S.; Sylvester M.; Aharony D.; Andisik D.; Callaghan O.; Campbell J. B.; Carr R. A.; Chessari G.; Congreve M.; Frederickson M.; Folmer R. H.; Geschwindner S.; Koether G.; Kolmodin K.; Krumrine J.; Mauger R. C.; Murray C. W.; Olsson L. L.; Patel S.; Spear N.; Tian G. Application of fragment-based lead generation to the discovery of novel, cyclic amidine beta-secretase inhibitors with nanomolar potency, cellular activity, and high ligand efficiency. J. Med. Chem. 2007, 50, 5912–25. 10.1021/jm070829p. [DOI] [PubMed] [Google Scholar]
- Forman M.; Palcza J.; Tseng J.; Leempoels J.; Ramael S.; Jhee S.; Ereshefsky L.; Tanen M.; Laterza O.; Dockendorf M.; Krishna G.; Ma L.; Wagner J.; Troyer M. The novel BACE inhibitor MK-8931 dramatically lowers cerebrospinal fluid Aβ peptides in healthy subjects following single- and multiple-dose administration. Alzheimer’s Dement. 2012, 8, P704. 10.1016/j.jalz.2012.05.1900. [DOI] [Google Scholar]
- Malamas M. S.; Erdei J.; Gunawan I.; Turner J.; Hu Y.; Wagner E.; Fan K.; Chopra R.; Olland A.; Bard J.; Jacobsen S.; Magolda R. L.; Pangalos M.; Robichaud A. J. Design and synthesis of 5,5′-disubstituted aminohydantoins as potent and selective human beta-secretase (BACE1) inhibitors. J. Med. Chem. 2010, 53, 1146–1158. 10.1021/jm901414e. [DOI] [PubMed] [Google Scholar]
- Steele T. G.; Hills I. D.; Nomland A. A.; de León P.; Allison T.; McGaughey G.; Colussi D.; Tugusheva K.; Haugabook S. J.; Espeseth A. S.; Zuck P.; Graham S. L.; Stachel S. J. Identification of a small molecule β-secretase inhibitor that binds without catalytic aspartate engagement. Bioorg. Med. Chem. Lett. 2009, 19, 17–20. 10.1016/j.bmcl.2008.11.027. [DOI] [PubMed] [Google Scholar]
- Bowers S.; Xu Y. Z.; Yuan S.; Probst G. D.; Hom R. K.; Chan W.; Konradi A. W.; Sham H. L.; Zhu Y. L.; Beroza P.; Pan H.; Brecht E.; Yao N.; Lougheed J.; Tam D.; Ren Z.; Ruslim L.; Bova M. P.; Artis D. R. Structure-based design of novel dihydroisoquinoline BACE-1 inhibitors that do not engage the catalytic aspartates. Bioorg. Med. Chem. Lett. 2013, 23, 2181–2186. 10.1016/j.bmcl.2013.01.103. [DOI] [PubMed] [Google Scholar]
- Xu Y. Z.; Yuan S.; Bowers S.; Hom R. K.; Chan W.; Sham H. L.; Zhu Y. L.; Beroza P.; Pan H.; Brecht E.; Yao N.; Lougheed J.; Yan J.; Tam D.; Ren Z.; Ruslim L.; Bova M. P.; Artis D. R. Design and synthesis of thiophene dihydroisoquinolines as novel BACE1 inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 3075–3080. 10.1016/j.bmcl.2013.03.009. [DOI] [PubMed] [Google Scholar]
- Ren Z.; Tam D.; Xu Y. Z.; Wone D.; Yuan S. D.; Sham H. L.; Cheung H.; Regnstrom K.; Chen X. H.; Rudolph D.; Jobling M. F.; Artis D. R.; Bova M. P. Development of a novel beta-secretase binding assay using the AlphaScreen platform. J. Biomol. Screening 2013, 18, 695–704. 10.1177/1087057113482138. [DOI] [PubMed] [Google Scholar]
- Zartler E.; Shapiro M.. Fragment-Based Drug Discovery: a Practical Approach; Wiley: Hoboken, 2008. [Google Scholar]
- Erlanson D. A. Introduction to fragment-based drug discovery. Top. Curr. Chem. 2012, 317, 1–32. 10.1007/128_2011_180. [DOI] [PubMed] [Google Scholar]
- Leach A. R.; Hann M. M. Molecular complexity and fragment-based drug discovery: ten years on. Curr. Opin. Chem. Biol. 2011, 15, 489–496. 10.1016/j.cbpa.2011.05.008. [DOI] [PubMed] [Google Scholar]
- Stamford A.; Strickland C. Inhibitors of BACE for treating Alzheimer’s disease: a fragment-based drug discovery story. Curr. Opin. Chem. Biol. 2013, 17, 320–328. 10.1016/j.cbpa.2013.04.016. [DOI] [PubMed] [Google Scholar]
- a De Simone A.; Mancini F.; Real Fernàndez F.; Rovero P.; Bertucci C.; Andrisano V. Surface plasmon resonance, fluorescence, and circular dichroism studies for the characterization of the binding of BACE-1 inhibitors. Anal. Bioanal. Chem. 2013, 405, 827–835. 10.1007/s00216-012-6312-0. [DOI] [PubMed] [Google Scholar]; b In house generated Kd (SPR) and IC50 (FRET) values were in line with those reported by De Simone et al.
- Dalvit C.; Fogliatto G.; Stewart A.; Veronesi M.; Stockman B. WaterLOGSY as a method for primary NMR screening: Practical aspects and range of applicability. J. Biomol. NMR 2001, 21, 349–359. 10.1023/A:1013302231549. [DOI] [PubMed] [Google Scholar]
- Pervushin K. The use of TROSY for detection and suppression of conformational exchange NMR line broadening in biological macromolecules. J. Biomol. NMR 2001, 20, 275–285. 10.1023/A:1011208109853. [DOI] [PubMed] [Google Scholar]
- Keserű G. M.; Erlanson D. A.; Ferenczy G. G.; Hann M. M.; Murray C. W.; Pickett S. D. Design Principles for Fragment Libraries: Maximizing the Value of Learnings from Pharma Fragment-Based Drug Discovery (FBDD) Programs for Use in Academia. J. Med. Chem. 2016, 59, 8189–8206. 10.1021/acs.jmedchem.6b00197. [DOI] [PubMed] [Google Scholar]
- Domínguez J. L.; Christopeit T.; Villaverde M. C.; Gossas T.; Otero J. M.; Nystrom S.; Baraznenok V.; Lindstrom E.; Danielson U. H.; Sussman F. Effect of the protonation state of the titratable residues on the inhibitor affinity to BACE-1. Biochemistry 2010, 49, 7255–7263. 10.1021/bi100637n. [DOI] [PubMed] [Google Scholar]
- Pantoliano M. W.; Petrella E. C.; Kwasnoski J. D.; Lobanov V. S.; Myslik J.; Graf E.; Carver T.; Asel E.; Springer B. A.; Lane P.; Salemme F. R. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screening 2001, 6, 429–440. 10.1177/108705710100600609. [DOI] [PubMed] [Google Scholar]
- Matulis D.; Kranz J. K.; Salemme F. R.; Todd M. J. Thermodynamic stability of carbonic anhydrase: Measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry 2005, 44, 5258–5266. 10.1021/bi048135v. [DOI] [PubMed] [Google Scholar]
- Fuchs K.; Dorner-Ciossek C.; Handschuh S.; Heine N.; Hoerer S.; Klinder K.. Preparation of Substituted Amino-Benzimidazoles for the Treatment of Alzheimer’s Disease. WO2009092566, 30 Jul, 2009.
- Vassar R.; Kovacs D. M.; Yan R.; Wong P. C. The beta-secretase enzyme BACE in health and Alzheimer’s disease: regulation, cell biology, function, and therapeutic potential. J. Neurosci. 2009, 29, 12787–11794. 10.1523/JNEUROSCI.3657-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. S.; Beyer B. M.; Senior M. M.; Wyss D. F. Characterization of autocatalytic conversion of precursor BACE1 by heteronuclear NMR spectroscopy. Biochemistry 2005, 44, 16594–16601. 10.1021/bi051040o. [DOI] [PubMed] [Google Scholar]
- Bruinzeel W.; Yon J.; Giovannelli S.; Masure S. Recombinant insect cell expression and purification of human beta-secretase (BACE-1) for X-ray crystallography. Protein Expression Purif. 2002, 26, 139–148. 10.1016/S1046-5928(02)00516-8. [DOI] [PubMed] [Google Scholar]
- VIB Protein Service Facility , UGent (PSF), UGent-VIB Research Building FSVM, Technologiepark 927, 9052 Zwijnaarde, Belgium. http://www.dmbr.ugent.be/index.php?id=proteinservicehome (accessed Nov 8, 2016).
- Centro de investigation Principe Felipe (CIPF) , C/ Eduardo Primo Yúfera 3, Valencia 46012, Spain. http://www.cipf.es/ (accessed Nov 8, 2016).
- Proteros GMBH, Bunsenstr . 7a, 82152 Martinsried, Germany. http://www.proteros.com/ (accessed Nov 8, 2016).
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A. A.; Steiner R. S.; Lebedev A. A.; Potterton L.; McNicholas S.; Long F.; Murshudov G. N. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2184–2295. 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Moss D. S.; Thornton J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. 10.1107/S0021889892009944. [DOI] [Google Scholar]
- Beesu M.; Malladi S. S.; Fox L. M.; Jones C. D.; Dixit A.; David S. A. Human toll-like receptor 8-selective agonistic activities in 1-alkyl-1H-benzimidazol-2-amines. J. Med. Chem. 2014, 57, 7325–7341. 10.1021/jm500701q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley D. W.; Stewart J. M. Synthesis of oncolytic analogs of 1,2-dimethyl-4,5-diaminobenzene. J. Med. Chem. 1963, 6, 599–601. 10.1021/jm00341a030. [DOI] [PubMed] [Google Scholar]