

Abstract
Introduction
The technologies used to design, create and use microphysiological systems (MPS, “tissue chips” or “organs-on-chips”) have progressed rapidly in the last 5 years, and validation studies of the functional relevance of these platforms to human physiology, and response to drugs for individual model organ systems, are well underway. These studies are paving the way for integrated multi-organ systems that can model diseases and predict drug efficacy and toxicology of multiple organs in real-time, improving the potential for diagnostics and development of novel treatments of rare diseases in the future.
Areas covered
This review will briefly summarize the current state of tissue chip research and highlight model systems where these microfabricated (or bioengineered) devices are already being used to screen therapeutics, model disease states, and provide potential treatments in addition to helping elucidate the basic molecular and cellular phenotypes of rare diseases.
Expert opinion
Microphysiological systems hold great promise and potential for modeling rare disorders, as well as for their potential use to enhance the predictive power of new drug therapeutics, plus potentially increase the statistical power of clinical trials while removing the inherent risks of these trials in rare disease populations.
Keywords: clinical trials, drug development, induced pluripotent stem cells (iPSCs), microphysiological systems/platforms, rare diseases, tissue chips, toxicity screening
1. Introduction
As many stakeholders in the pharmaceutical industry, as well as academia, patient advocacy groups and government funders are aware, the inadequacies of current methods of drug discovery and testing can be expensive and failure rates are high (up to 90% of all compounds entering phase I clinical trials in man, according to Pearson1). The high attrition rate of drug development is a recurring problem and means that money, time and effort goes into the research and development of compounds that may fail toxicity screening tests as early barriers, or worse, inflict harm as treatments enter clinical trials and unexpected consequences become manifest. One of the major causes of drug failures in early human trials is efficacy – the issue that once a drug has undergone costly and time-consuming preclinical research, it fails to show effectiveness for the indicated disorder 2-4. Cook et al4 examined the results from 250 small molecule drug projects at AstraZeneca between 2005 and 2010 and found that over 88% of project failures in phase IIb (200-400 patients) were due to lack of efficacy. The authors discuss a theoretical framework to increase project success and pipeline quality - ‘five R's – the right target, the right patient, the right tissue, the right safety, the right commercial potential (and an extra, the right culture) – all of which were deemed crucial in aiding the decision-making process. While these analyses could improve pharmaceutical companies’ success in preclinical-to-clinical drug development if applied successfully, drug failure due to lack of safety in early stages of therapeutic development, and efficacy (in later stages), remains a huge problem largely because current in vitro and in vivo models are poorly predictive of human response.
Currently, the pharmaceutical industry relies heavily on 2-dimensional cell culture models and testing in animals for preclinical studies. These cell culture models are useful for basic toxicity screens, and animal models will remain critical for gaining in vivo data, but these model systems remain limited - cell culture does not recapitulate an in vivo system and lacks the complexity of human tissues and their connectivity, as well as blood and fluid perfusion and biomechanical shear forces; and rodent or other animal studies suffer from the limitation that animal physiology differs from humans in ways that may not even be known until a compound enters clinical trials5. Particularly important is the observation that certain metabolizing enzymes in rodents and humans differ, creating radically different metabolites with differing toxicity profiles6-9.
Another issue with drug and therapeutic compound development lies in the fact that negative results are, as standard, not published. This means that large numbers of compounds developed by industry, that have failed early toxicity screening or later shown poor efficacy, may be available for testing for other syndromes or diseases, but are unknown to the community unless proactive steps are taken by pharma companies to engage with non-profit communities. This lack of available data contributes to the difficulties in therapeutic drug development.
These difficulties are particularly amplified for rare diseases. The EU deems “rare” as “not more than 5 in 10,000”, while Japan says “4 per 10,000”, and the US as affecting <200,000 patients nationwide 10. These non-standardized definitions aside, at this point less than 5% of around 7000 currently identified rare diseases have effective drug therapies 11, 12. There are a huge number of challenges in rare disease research, including the difficulties of diagnoses in populations that may be geographically dispersed, plus due to their low prevalence may have poorly understood natural histories, diverse pathologies, and little medical literature devoted to them. Additionally, a lack of information accessible either by healthcare providers or patients means sufferers can remain undiagnosed and untreated for many years, adding to the burden of a rare disorder. Some diseases are identified at birth and have genetic components, some are geographically or ethnically linked, tied to age or gender, but successful treatment or management of all ultimately depend on access to a well-informed and functional healthcare system. While the number of afflicted individuals themselves may be scarce for any given rare disease, it is not just patients who are affected – family, friends, co-workers, employers, teachers, healthcare workers and others also carry the burden, meaning the ramifications can extend far beyond immediate family and have larger economic as well as emotional impacts.
Microphysiological systems (MPS), tissue chips (TCs), or “organs-on-chips” can play a unique role in rare disease research and treatment studies, as we shall go on to discuss in this review. These systems utilize microfluidic technology to create bioengineered ‘chips’ that can be seeded with human cells to model functional units of human organs, such as the kidney glomerulus, in both healthy and diseased states (see Figure 1). For example, a liver chip may contain stellate, Kupffer and hepatic cells in a physiologically relevant architecture that mimics the microenvironment of the liver and its processing capabilities – creating a helpful tool that could be utilized at early stages of drug development, and perhaps help improve the therapeutic development pipeline, when used together with standard tools and model systems.
Figure 1. current Tissue Chip platforms.
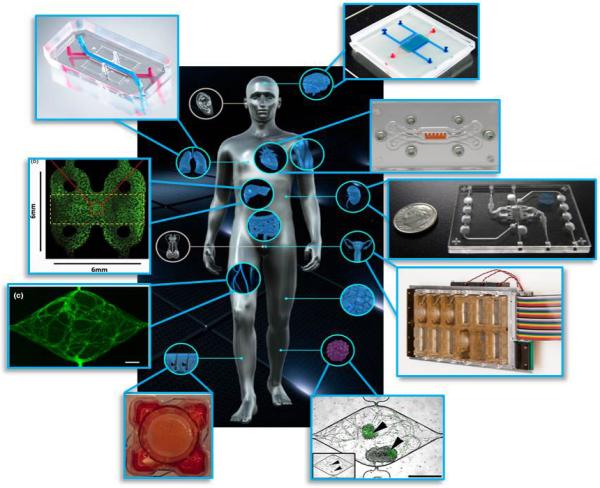
Some of the many tissue chips now developed and now in use, including (clockwise from top right) a blood-brain barrier, cardiac muscle, kidney, female reproductive tract, tumor, epidermis, vasculature, liver, and lung.
All images reproduced with permission. Acknowledgements: Blood brain barrier – Wikswo lab at Vanderbilt; Cardiac chip – Parker lab at Harvard; Kidney chip - www.nortisbio.com; female reproductive tract - Woodruff lab at Northwestern and DRAPER; tumor image - George lab at Washington University in St. Louis; epidermis - Christiano lab at Columbia; vasculature - George lab at Washington University in St. Louis; liver chip – Taylor lab at Pittsburgh; and lung chip from www.emulatebio.com.
In the last decade, the technology to develop these tools, and the range of human organs that are capable of being modelled, has progressed tremendously. Building on impressive advances in the field during the 90s and 00s, the National Institutes of Health (NIH), Defense Advanced Research Projects Agency (DARPA), Defence Threat Reduction Agency (DTRA) and Food and Drug Administration (FDA), have all been involved in funding opportunities designed to advance the field substantially over the past 5 years13, 14. This investment, along with expanding advances by a number of microfluidic device companies, is leading to more sophisticated organ modeling, and the beginnings of functional and physical coupling of platforms to move towards the goals of creating a “human-on-a-chip” 15, 16, enabling comprehensive testing of candidate drug targets in healthy and diseased tissues and perhaps even paving the way for clinical trials to be conducted on chips in the future. Importantly, government-funded programs have placed a heavy emphasis on the use of human tissues in the platforms, to ensure physiologically appropriate responses are seen when compounds are tested within the systems. This helps pave the way for translational research speeding up drug development pipelines, but could also reduce the need for animal testing and introduces the possibility of the use of these chips for precision medicine efforts i.e. individualized treatments for patients.
The potential for tissue chip technology for use in screening currently available drugs could also be very helpful in the translation of treatments from bench to bedside, as libraries of compounds that have failed previous trials, or been developed originally for other conditions, could be tested on human systems and potentially repurposed for other diseases. As the field progresses, questions of how to continue development, optimization and validation of the technology, and then commercialization and mass production for availability for the wider research community14, will become more pertinent. In the future, investment in the technology and the contribution of expertise from pharmaceutical and biotechnology companies will become necessary for effective and efficient development of cheaper, reproducible and reliable tissue chip platforms for high throughput drug screening and safety and efficacy testing - perhaps ultimately helping reduce the high attrition rate.
2. Tissue chips as an alternative to current in vitro and in vivo models
2.1 2D Cell Culture Models
Table 1 shows a brief comparison of 2D, 3D and microphysiological model system pros and cons. Classic 2-dimensional cell cultures in well plates are a straightforward, standard and reliable assay for high throughput drug toxicity screens, and have been tremendously successful in eliminating toxic compounds before they reach animal or human trials. However, these cell culture assays are limited in a number of ways. One major point is that they cannot accurately mimic a physiological environment: these cultures cannot often be engineered to provide the matrices of endothelial cells that comprise most body tissues, nor can a 2D well plate mimic the shear force and stresses that tissue experiences in vivo. Often 2D cultures are made up of a homogeneous cell type, which is unlike the heterogeneous cell composition of many tissues. Critically, the tissues sit in a static media, rather than being subject to the flow of blood and media that all tissues experience in a complete living organism. Cell culture techniques also have limited possibilities for functional coupling to other systems – i.e. a drug that metabolizes in the liver may produce metabolites needing to be screened through the kidney for toxicity. This is difficult to accomplish in a well-plate system designed for standard high-throughput assays.
Table 1.
A basic comparison of pros and cons for 2-dimensional, 3-dimensional/organoid, and microphysiological model systems.
| Model systems | Pros | Cons |
|---|---|---|
| 2-dimensional cell cultures | Straightforward, cheap, reliable | Not physiologically accurate – no endothelial support or scaffolding, shear forces, etc |
| Good for high throughput screening | Homogeneous or limited cell types | |
| Useful first line for toxicity screens | Static media | |
| Limitations for functional coupling | ||
| Short modeling timelines (<1 month) | ||
| 3-dimensional/organoids | Self-organization of iPSCs allows more physiologically relevant cellular architectures | Large heterogeneity in spontaneous self-organization leads to variability |
| Ability for disease modeling from primary cells or iPSCs | Hard to model functional units of an organ | |
| Multiple cell types represented | Internal cells lack vascularization for nutrient supply/removal of waste | |
| Possibilities for real-time cellular monitoring and repeated sampling | Short modeling timelines (<1 month) | |
| Microphysiological systems | Ability to mimic physiology via prescriptive designs (fluidics, stretch and shear forces etc) | Higher cost, lower throughput |
| Allow for vascularization, nutrient supply and waste removal | Require validation with known compounds | |
| Ability for disease modeling from primary cells or iPSCs | Short modeling timelines (<1 month) | |
| Multiple cell types represented | Technically challenging to link organ systems | |
| Possibilities for real-time cellular monitoring and repeated sampling | ||
| Possibilities for linkage of organ systems |
2.2 Organoids for 3D Modeling
In a move towards 3-dimensional organ modeling, much excellent work has been done to develop self-assembling tissue organoids (see Lancaster and Knoblich17 for a review) – where induced pluripotent stem cells (iPSCs) are cultured in an environment where cells can expand into three dimensions (often using a scaffold to induce cell polarization), and prompted to differentiate into target organoid tissue. Organoids self-organize into structures of cells of multiple different types, often around a scaffold of hydrogels or synthetic polymers, through the processes of cell sorting and spatially restricted lineage commitment in a process similar to that seen in vivo, and are a large step forward in terms of model complexity versus traditional cell culture models17. Organoids developed thus far have included those of stomach18, 19, intestine20, 21, brain22, eye23, inner ear24, liver25, 26, bone27, and kidney28, amongst many others.
One great advantage to the use of organoid models is the ability to generate disease models, as samples from patients or from primary tissues such as tumors can be induced to generate organoids specific to a disease state (e.g. 29). However, there are multiple limitations with organoids that need to be addressed in order to increase their translational research potential. Firstly, the self-organization of cells within an organoid more accurately mimics embryonic development in vivo than a 2-dimensional cell culture, but there is large heterogeneity in the self-organization process for each individual organoid, meaning reproducibility of cytoarchitecture and structural organization in organoids is low. The lack of a defined cytoarchitecture also means that the organoids do not necessarily model functional units of organs - for example, kidney organoids may contain multiple relevant cell types, but cannot mimic a functional glomerulus that filters blood at high flow rates. Related to this, organoids cannot model the natural forces that tissues in vivo are subject to, mainly because they lack vascularization for blood and nutrient supply, resulting in oxygen-starved cells in the center of the structures that do not have methods for removal of debris, metabolites and normal cell homeostasis mechanisms. One final drawback of organoid use, which is common to all technologies using induced pluripotent stem cells, is the issue of cell maturation – at this stage, it is difficult to prompt cells to mature to adult-like phenotypes, and only human intestinal organoids have shown adult phenotypes so far21. The continued research efforts of the community will help address these issues30.
2.3 Microphysiological Systems (MPS)
Microphysiological tissue chip platforms (Figure 1) differ from organoids in a number of ways, chiefly that cell cultures and chips are bioengineered to represent functional units of organs or systems rather than allowing for self-organization, by the design of chips that allow multicellular architecture and interaction between tissues and chemical microenvironments that are similar to those seen in vivo. The designs of these platforms also enable detailed and often real-time analysis of the microenvironments by imaging or sampling techniques, allowing researchers a ‘window’ into human tissue functionality in a way that other model systems cannot provide. Different types of differentiated cells may be added to the chips in separate subsections or compartments to mimic organ functionality, or cell progenitors may be placed into the chips, and then incubated to allow for the cells to differentiate and self-assemble into target organ tissues. Additionally, the microfluidic designs of some of these chips are engineered to provide fluids and nutrients to cells in ways that mimic provision by vascularization. In turn, this engineered fluid flow can remove products of cell metabolism, and exposes cells to the types of shear and compression forces that would be seen in vivo.
Building on seminal bioengineering work by pioneers in the field such as Michael Shuler31-34, and many others 35-39, the highly publicized “lung-on-a-chip” model, developed by Don Ingber and colleagues at the Wyss Institute, modelled the alveolar-capillary interface in the human lung and helped bring the field to greater public prominence. Huh et al40 designed a microphysiological system consisting of 2 closely aligned microchambers, populated with human alveolar epithelial cells in one and human pulmonary microvascular endothelial cells in the other, separated by a 10μm porous flexible membrane made of polydimethylsiloxane (PDMS). The PDMS membrane was covered in an extracellular matrix to enable cell adhesion, and cells were cultured so that after 16 days, the alveolar epithelial cell chamber contained cells in contact with endothelial cells on their basolateral side to mimic the structure of the alveolus for gas exchange, and exposed to the air on the other side, to mimic the structure of the alveolus for gas exchange. Parallel chambers running alongside the cell chambers then had a rhythmic vacuum applied, consequently stretching the PDMS layer (and attached cells) to model the stretch forces the alveolus undergoes as the lung expands and contracts.
Importantly, this “lung-on-a-chip” was able to model pulmonary inflammatory responses to the pro-inflammatory mediator tumor necrosis factor α (TNF-α), and show capture and engulfment by neutrophils of foreign E. Coli bacteria introduced into the system, recapitulating the in vivo response to bacterial infection. Furthermore, it's since been used to model drug toxicity-induced edema41. This chip development has since led to use of the chip design to model a number of other organs (including gut, a kidney proximal tubule and bone marrow), as well as the launch of a spin-off biotechnology company (Emulate Inc.) that is applying the chip for uses as diverse as agriculture, cosmetics, and food product research.
Another organ in which the pharmaceutical industry has shown particular interested is a liver chip. A number of these already exist. For example, Vernetti et al42 designed a microfluidic device layering hepatic cell types sequentially (“SQL”) and allowing self-assembly during incubation periods (“SAL”) to create an “SQL-SAL” model with many of the functions of a liver sinusoidal unit. In this model, investigators seed a single chamber commercially available device from Nortis Inc (Seattle, WA) with hepatocytes, endothelial, Kupffer-like immune cells and stellate cells, and incubate. At 7 days, endothelial and stellate cells are found in a layer above hepatocytes, as well as some being localized between hepatocytes. Validation of this chip using various drugs to monitor toxicity and metabolites has been successful, and its development continues.
The ever-expanding list of tissues represented on chips thus far developed include liver, kidney, muscle (cardiac and skeletal), blood vessels and vasculature, reproductive tissues including fallopian tube, uterus and cervix, blood-brain-barrier, lung, gut, bone marrow and skin, amongst others (see Bhatia and Ingber43 for a review), and this fast-evolving field continues to produce more tissue models every year.
3. How can tissue chips be used for drug screening and development?
Tissue Chips provide a unique opportunity for use in drug discovery and therapeutic compound development, and offer the opportunity to significantly extend upon the abilities of standard 2-dimensional toxicity screening assays.
3.1 Tissue Chips for Toxicity Screening
One obvious application, already mentioned, is toxicity screening in target tissues in early compound and preclinical development phases, whereby chips can be used to determine hepatotoxicity, nephrotoxicity, and neurotoxicity, amongst others. Developmental neurotoxicity is an important field for screening of chemicals and compounds. Currently, these chemical safety screens may include intergenerational studies in animal models to see longer term effects, in addition to in vitro cell culture methods, but their high cost, low concordance for toxicity between species, and difficulties in modeling subtle cognitive alterations such as autism give them limited power to uncover some potentially harmful effects. Hou et al44 point out that use of human fetal-derived neurospheres captures some early neural development events better than in vitro cell culture, but the neurospheres lack microglia and vascular cells – both crucial neural components – and in common with other organoids, lack the physiologically relevant architectural complexity needed to supply and remove nutrients from oxygen-demanding tissues. Utilizing advances in stem cell research and the reductions in cost in high-throughput gene sequencing, Schwartz et al45 devised a neural MPS system seeded with multiple types of neural cells and progenitors, which used machine learning algorithms to produce gene expression readouts to 60 chemicals of known toxicity. When tested on 10 blinded compounds, the model correctly predicted the toxicity of 9 of the 10, with the remaining compound later identified as a false positive. Tissue chip applications such as this hold vast potential for use in large-scale predictive toxicology screens, helping reduce costs and streamline the processes of safety testing.
3.2 Validation and Optimization of Current Therapeutics
Tissue chips may also be useful for validation and optimization of current therapeutics, for example chemotherapeutics. Tumor cells respond to their microenvironment and adjust their cellular responses accordingly, making it critical to understand the integrated environment in which they are found for successful treatment. A tissue chip modeling perfused vascular systems46 could be used to model the vasculature surrounding tumor cells and to further understand the mechanisms of metastasis, as well as for modeling and understanding chemotherapeutic cardiac and bone marrow toxicity47. Hepatic microphysiological systems are being utilized to model the toxicity of chemotherapeutics, but also for studying metastases, due to the liver being a main site of metastatic seeding48.
3.3 Real-time Feedback on Cellular Events
All compounds, chemicals and therapeutics have biological effects, and an advantage of using MPS to screen them is the ability of the chips to report back in real-time what cellular events may be occurring. To do this, many chips are fabricated from the clear plastic PDMS, which allows microscopic visualization of where cells are located, as well as, for example, the movement of fluorescent microbeads through vascularized tissue46 or even fluorescently tagged compounds of interest. Other systems incorporate ‘reporter’ cells that have been transfected with viral vectors which fluoresce when products of apoptosis or excessive hydrogen peroxide are present42, giving real-time readouts on cell health or distress. The technology in this field is fast-paced, and reporting mechanisms for these systems are improving rapidly; for example, detectors embedded in the PDMS structure of the chip may in the future be able to measure trans-epithelial electric resistance (TEER) more accurately than the currently standard, yet variable, Ag/AgCl electrodes49.
Compounds and treatments that may have fallen by the wayside for treatment of one particular disease, or shown limited efficacy in a particular cellular pathway, could also be tested in microphysiological systems to find new target pathways, investigate side-effects deemed unsafe, or screen drugs against other pathways or diseases for alternatively efficacious effects.
3.4 Multiple Organ Drug Screening
The ultimate aim in the field of tissue chip development is to move to functional and physical coupling of platforms, and the creation of a ‘human-on-a-chip’. The connection of chips is important to model the sequential metabolism of drugs as they move through the body, and the effects of compounds and metabolites on non-target tissues. A liver module that feeds fluids or metabolites to a kidney module is useful for determining whether compounds that have been metabolized in the liver create nephrotoxic metabolites that would cause kidney damage during excretion (as has been successfully demonstrated in animal models31). Currently, safety and risk assessments of nephrotoxicity that rely heavily on animal studies cause generation of dosage data that is extrapolated to humans despite the knowledge that animal and human models can differ greatly37. This can, and has, led to the detection of nephrotoxicity during late phase clinical trials and after regulatory approval, and could be a leading cause of acute kidney injury (AKI) in vulnerable patient populations50.
The field is well underway in its efforts of coupling of organ systems, although there are many technical challenges that remain to be overcome. Alternative approaches to reach the goals of physical (and therefore functional) coupling will be important, as successful approaches are integrated and publicized, and unsuccessful ones abandoned as progress continues. The DARPA-funded Wyss Institute has developed a machine, “Interrogator”, to combine ten model organ systems and support them for up to four weeks, favoring a “plug-and-play” approach where modules can be interchangeable and switched in and out where desired. Vunjak-Novakovic and colleagues at Columbia are creating a “HeLiVa” module of heart, liver and vasculature tissue into a single integrated module, with the aim of accruing real-time biological readouts (via imaging) of human vasculature, metabolizing liver lobules, and functional cardiac muscle51.
Important features of the HeLiVa platform are its design for screening of multiple compounds concurrently, with a view towards use in high-throughput drug screening and toxicity testing, and its ability for extra tissue platforms to be integrated into the design, for example skin and tumor modules.
4. Tissue Chips for modeling of rare diseases, and therapeutic development and treatments
4.1 Tissue Chips and Rare Disease Modeling
The potential of tissue chip technology for rare disease research is wide-ranging. With recent advances in stem cell technology, MPS platforms containing organ tissues from individual disease sufferers are now possible, and new doors in terms of precision medicine have begun to be opened. Public health funding agencies such as the NIH are investing in precision medicine initiatives, and in terms of rare diseases, it is now possible to populate MPS platforms with cells from a single individual suffering from a disease, leading to chips modeling genetically-identical phenotypes of those patients52. This phenotypic modeling on chips can then be used to understand the pathology of a disease in vitro in ways never before possible, and also opens new avenues for potential treatments and therapeutics for that disease, as well as predicting drug responses and efficacies in safer ways than via underpowered clinical trials in vulnerable populations.
One example of this use of tissue engineering in a microphysiological system is work done to model Barth syndrome. This rare X-chromosome-linked cardiac and skeletal mitochondrial myopathy is caused by mutation of the TAZ gene, and leads to muscle and cardiac weakness, immune deficiencies and growth delays. Wang et al53 generated cardiac iPSCs from skin biopsies of two patients and seeded them onto muscular thin film (MTF) PDMS strips, then leaving cells to culture over 5 days. MTF constructs were ‘peeled’ from their glass coverslips to allow them to take a curved form, and then stimulated with electrodes, causing cardiac muscle cell contraction. The degree of contractile forces measured by quantification of the ‘twitching’ of the strips, when compared to controls, showed that the contractile force was reduced in the Barth syndrome genotype. Importantly, the researchers then used Cas-9 gene editing techniques to restore TAZ function, and also restored contractile stress. This paper heralds the way for not only modeling of rare disease phenotypes, but the use of novel genetic editing techniques such as CRISPR-Cas to normalize tissues and contribute to preclinical work being done on these diseases. Other rare congenital disorders such as long-QT syndrome, which causes ventricular tachyarrhythmias, are being modeled using patient-derived iPSCs54 and could likely benefit from applications on MPS platforms in the future.
4.2 Tissue Chips and Rare Disease Treatments
Until recently, with the advent of the internet and electronic access to medical information, clinical practices for rare disease treatments may have been based on case studies of individual patients. These are undoubtedly important for the reporting of uncommon diseases, but mean that evidence-based medicine becomes anecdotal. The utilization of MPS platforms raises the possibility of creating chips populated with tissues from individual patients enrolled in clinical trials. This would enhance the safety of these small and disparate groups of patients, as toxicology and efficacy testing could be done on the chips to predict drug contraindications prior to in vivo administration, helping prevent potentially hazardous or fatal outcomes.
The modeling of both rare and common diseases is difficult in any ex vivo system for diseases with complex pathogeneses, such as diabetes, metabolic disorders and most neurodegenerative diseases. However, when using stem cells to create complex disease models, 3-dimensional systems provide a more physiologically relevant environment than standard wellplates, as cell-cell interactions during differentiation can be subject to mechanical forces (fluid flow, stretch and shear), which promotes greater cell maturation than standard 2-D cultures30. For example, iPSCs created from patients with amyotrophic lateral sclerosis (ALS)52 could, in the future, be used on organ-on-chip platforms to model the disease more effectively than currently possible.
Biobanking of tissues from patients could also be transformative in the process of therapeutic development for rare diseases. Currently, animal models and 2-dimensional cell culture techniques may not recapitulate the full phenotype of a rare disorder, and clinical trials can be performed only on patients currently living with a condition at any one time, leading to statistically underpowered trials and disproportionate risks to patients enrolled in trials. Biobanking of tissues or cell lines raises the possibility of recreating tissues from patients that may have died many years before (when the appropriate consent was given before death), and immediately increases the number of phenotypes for MPS screening.
Rare disease sufferers are also limited in the number of clinical trials they can take part in, and may find themselves disqualified from promising trials due to their involvement in previous trials that may ‘taint’ their outcomes. The use of MPS platforms could not only help uncover what effects previous unsuccessful trials may have had on a patient, but could also create chip ‘libraries’ of naïve populations of new patients before they receive treatments. This also opens up the possibilities of many compounds being screened for toxicity and efficacy across these chip ‘libraries’, and lead to repurposing of not only known compounds, but also orphan drugs with unknown efficacies but potential therapeutic promise.
5. Future of tissue chips and their application
Tissue chips, or MPS systems, are a promising technology for a number of applications, from toxicity screening to disease modeling to a future where clinical trials may be run on chips. As research continues to advance these technologies, a number of challenges need to be overcome to allow the application of the technology amongst the community.
5.1 Future Challenges
Firstly, even though researchers are pushing towards maintaining cell viability in their systems, the relatively short life spans of many MPS systems (mainly less than one month) limits the predictive value of the systems for delayed toxicity13. Such short timescales cannot mimic chronic illnesses such as liver disease, which is driven by stellate-cell activation and collagen remodeling and evolves over years.
Other challenges currently facing those involved in MPS development include scientific, mechanical, and even ethical issues. If the MPS technology is to be widely adopted, one of the first issues to address will be how to produce the cells needed to seed them. While the progression of stem cell science has markedly advanced in recent years, it is still technically difficult, time consuming and expensive to reliably produce large quantities of high quality stem cells and progenitors, and ensure their differentiation into tissues of interest. Once cells are available and chips are functional, the physical coupling of different platforms will need to address microfluidic issues such as how to adjust for differing flow rates between platforms and deal with the presence of bubbles, and there will also be challenges due to the materials of which platforms are made. For example, PDMS readily absorbs lipophilic compounds, meaning concentrations that are introduced into the systems currently sometimes may be as many as a hundred times higher than a clinically relevant dose55-57. Additionally, developers will be faced with decisions as to whether MPS platforms are designed to carefully and highly accurately mimic in vivo conditions, or whether designs should be modified to simplify them but make them more amenable to increased production and higher throughput. Once chips are designed and created, validation of the chips with existing drugs of known actions, toxicities and therapeutic indices will need to be performed to ensure the platforms recapitulate the in vivo state.
Functional and physical coupling efforts currently continue and are making great strides to be able to create healthy and diseased ‘humans-on-a-chip’, and will be able to explore the in-depth biochemical, molecular, cellular and genetic influences of a disease on multiple organ systems – crucially, in real time, and in a highly replicable manner. In order for decisions regarding lower throughput but more complex systems to be developed versus higher throughput but simpler systems, it will be critical for the pharmaceutical and biotechnology industries to become involved. These stakeholders have the expertise needed for mass production and commercialization of drugs and technologies, and it is in their interests to aid MPS development to streamline their own product development and profit from this promising field.
6. Conclusion
Tissue Chip platform development has advanced quickly in the last 5 years and holds promise for multiple applications in the future, including streamlining of drug development pipelines, opportunities for repurposing of orphan drugs, and uses in rare disease modeling and therapeutic research. These applications could yield important insights into disease mechanisms and treatments, but investment from the pharmaceutical and biotechnology sectors will be critical for advancing the development, validation and commercialization of MPS technology to aid its adoption by the community. At this juncture, classical 2-dimensional cell culture methods will remain critical for high throughput early screening, and 3-dimensional organoid systems are proving themselves extremely valuable for showing how cells respond differently in 2 versus 3-dimensions. Furthermore, animal in vivo models will remain crucial in the forseeable future for whole-organism therapeutic testing – tissue chips could help reduce or refine this usage, but are not likely to replace animal testing entirely within the next decade or more. Moreover, with data and resources coming from the Precision Medicine Initiative, it is conceivable that iPSC-derived tissues from many genotypically diverse individuals can be used to form the basis for clinical trials on chips. Nonetheless, the utility of tissue chip technology for advancing rare disease research shows great promise for sufferers of rare diseases, and their families and loved ones.
7. Expert Opinion
- What are the key findings and weaknesses in the research done in the field so far? What are the key lessons for the industry?
The last few years of MPS development have seen rapid progression in terms of both iPS technology, and the speed and extent of platform development and linkage. This has led to a surge in interest from potential stakeholders, including the pharmaceutical industry. For now, these platforms are still at a relatively early stage of development and yet to be commercialized and readily available to research community, but they are a step closer to modeling functioning organs than organoid technology. Work remains to be done on optimization and validation of current platforms, as well as progress made on increasing ease of use and accessibility for researchers, and decisions made on complexity versus throughput.
- What potential does this research hold? What's the ultimate goal in the field?
There is great potential use of this research for disease modeling, and toxicity and efficacy testing for therapeutic development, particularly for rare diseases. Compared to alternative 2D and 3D organoid models, MPS platforms hold promise for functional modeling of rare diseases in ways not possible before. Ultimate goals in the field include the advent of a ‘human on a chip’ for full phenotypic screening, the use of chips for personalized precision medicine, and even clinical trials on chips in the future. As far as therapeutic development goes, goals for the technology include improved drug pipelines as well as reductions in animal use, development costs, and timelines for drug deliverables.
- What research or knowledge is needed to achieve this goal, and what's the biggest challenge?
One of the biggest challenges facing the field is how to streamline and optimize the technology for iPS differentiation, making the processes cheaper and more widely available to non-experts who may wish to use the technology. Groups are continuing to address cell sourcing and differentiation protocols, and we expect to see continued progress in this area, alongside strides in the organoid development field as researchers develop vascularization and oxygenation protocols for organoid systems.
Other challenges involve functional and physical coupling efforts, as chip designs need to be modified in order to connect them. Work is continuing in this area and the field is expecting progress to continue. Collaborative research efforts are crucial for this to occur.
The field will now also require input from industry stakeholders for commercialization and development. The trade-offs between simple/high throughput versus complex/low throughput systems will need addressing by experts from many fields.
- Where is the field going in the coming years? What's going to happen?
In the short term, we can expect to see more chips being developed to represent a growing constellation of tissue types, including lymph, fat, various cancers, retina, etc etc. As discussed, advances in optimizing stem cell differentiation techniques will help with this generation of new chips, and collaborative efforts with researchers developing organoid systems, which also rely on stem cell technology, will be fruitful in this endeavor. A critical next path for the short term is validation of the existing chips with drugs of known action and toxicities, to ensure a relevant in vivo modeling of chips, diseases, and treatments. Efforts are occurring in this regard; for example, the NIH is funding a Tissue Chips Testing Center initiative to validate the NIH-funded Tissue Chip program platforms, and European institutions and worldwide organizations are engaged in similar efforts. These validation efforts will be crucial in moving the field forward and require input from multiple stakeholders.
In the mid- to longer- term, we expect to see the advent of more streamlined devices for commercialization and aim to see the adoption of the technology by the wider community, where its use will become more standard. We also expect to see pharmaceutical companies investing in the technology too, and collaborations between academia and industry proving fruitful for advancing development.
Long-term events will include representation of multiple organ phenotypes on linked chips for disease modeling and drug testing, including use in precision medicine efforts and clinical trials. We also expect to see the development and commercialization of simple platforms for use in a variety of applications – i.e. chip development for field testing of environmental toxins, for example.
- How will this research on rare diseases or orphan drugs impact the management and treatment of patients in the long term?
This research technology could provide new potential treatments for patients through the application of MPS platforms for screening of orphan drugs, repurposing of failed compounds, and efficacy testing of standard alternative therapeutics that have not yet been testing in different disease models. Furthermore, if biobanking of tissues from rare disease patients is possible, the creation of chip ‘libraries’ becomes a reality for increasing the numbers of phenotypes in a clinical trial, and the move towards clinical trials on chips for vulnerable disease populations.
Article Highlights.
Current therapeutic development has a high attrition rate due to toxicity of compounds in early stages and lack of efficacy in later stages of development
Tissue chips hold promise for toxicity screening and therefore aiding the screening of promising therapeutics
Tissue chips also hold promise for modeling of rare diseases as human tissues can be used in the platforms to elucidate disease pathologies
Individualized chips for rare disease patients could be created to test promising therapeutics in an in vivo-like environment and reduce the risks for vulnerable and scarce populations
Many challenges remain to be faced and input from multiple stakeholders will be important for continued development of this technology.
Acknowledgments
Funding
This paper was funded by National Center for Advancing Translational Services (NCATS).
Footnotes
Declaration of Interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
Reference annotations
* Of interest
** Of considerable interest
- 1.Pearson H. The bitterest pill. Nature. 2006;444(7119):532–33. doi: 10.1038/444532a. [DOI] [PubMed] [Google Scholar]
- 2.Arrowsmith J. Trial watch: Phase III and submission failures: 2007–2010. Nat Rev Drug Discov. 2011;10(2):87–87. doi: 10.1038/nrd3375. [DOI] [PubMed] [Google Scholar]
- 3.Arrowsmith J. Trial watch: Phase II failures: 2008–2010. Nat Rev Drug Discov. 2011;10(5):328–29. doi: 10.1038/nrd3439. [DOI] [PubMed] [Google Scholar]
- 4*.Cook D, Brown D, Alexander R, March R, Morgan P, Satterthwaite G, et al. Lessons learned from the fate of AstraZeneca's drug pipeline: a five-dimensional framework. Nat Rev Drug Discov. 2014;13(6):419–31. doi: 10.1038/nrd4309. [In-depth analysis of therapeutic research development from AstraZeneca] [DOI] [PubMed] [Google Scholar]
- 5*.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proceedings of the National Academy of Sciences. 2013;110(9):3507–12. doi: 10.1073/pnas.1222878110. [Highlights the lack of concurrence between mouse and human models] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsukamoto T. Animal disease models for drug screening: the elephant in the room? Drug Discovery Today. 2016;21(4):529–30. doi: 10.1016/j.drudis.2016.02.012. [DOI] [PubMed] [Google Scholar]
- 7.Fashe MM, Juvonen RO, Petsalo A, Räsänen J, Pasanen M. Species-Specific Differences in the in Vitro Metabolism of Lasiocarpine. Chemical Research in Toxicology. 2015;28(10):2034–44. doi: 10.1021/acs.chemrestox.5b00253. [DOI] [PubMed] [Google Scholar]
- 8.Sasahara K, Shimokawa Y, Hirao Y, Koyama N, Kitano K, Shibata M, et al. Pharmacokinetics and Metabolism of Delamanid, a Novel Anti-Tuberculosis Drug, in Animals and Humans: Importance of Albumin Metabolism In Vivo. Drug Metabolism and Disposition. 2015;43(8):1267–76. doi: 10.1124/dmd.115.064527. [DOI] [PubMed] [Google Scholar]
- 9.Huan J-Y, Miranda CL, Buhler DR, Cheeke PR. Species differences in the hepatic microsomal enzyme metabolism of the pyrrolizidine alkaloids. Toxicology Letters. 1998;99(2):127–37. doi: 10.1016/s0378-4274(98)00152-0. [DOI] [PubMed] [Google Scholar]
- 10.de la Paz MP, Villaverde-Hueso A, Alonso V, János S, Zurriaga Ó , Pollán M, et al. Rare Diseases Epidemiology Research. In: Posada de la Paz M, Groft CS, editors. Rare Diseases Epidemiology. Springer Netherlands; Dordrecht: 2010. pp. 17–39. [DOI] [PubMed] [Google Scholar]
- 11.Fajgenbaum DC, Ruth JR, Kelleher D, Rubenstein AH. The collaborative network approach: a new framework to accelerate Castleman's disease and other rare disease research. The Lancet Haematology. 2016;3(4):e150–e52. doi: 10.1016/S2352-3026(16)00007-7. [DOI] [PubMed] [Google Scholar]
- 12.Miyamoto BE, Kakkis ED. The potential investment impact of improved access to accelerated approval on the development of treatments for low prevalence rare diseases. Orphanet Journal of Rare Diseases. 2011;6(1):1–13. doi: 10.1186/1750-1172-6-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sutherland ML, Fabre KM, Tagle DA. The National Institutes of Health Microphysiological Systems Program focuses on a critical challenge in the drug discovery pipeline. Stem Cell Research & Therapy. 2013;4(1):1–5. doi: 10.1186/scrt361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fabre KM, Livingston C, Tagle DA. Organs-on-chips (microphysiological systems): tools to expedite efficacy and toxicity testing in human tissue. Experimental Biology and Medicine. 2014;239(9):1073–77. doi: 10.1177/1535370214538916. [DOI] [PubMed] [Google Scholar]
- 15.Maschmeyer I, Lorenz AK, Schimek K, Hasenberg T, Ramme AP, Hubner J, et al. A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab on a Chip. 2015;15(12):2688–99. doi: 10.1039/c5lc00392j. [DOI] [PubMed] [Google Scholar]
- 16.Kim J-Y, Fluri DA, Marchan R, Boonen K, Mohanty S, Singh P, et al. 3D spherical microtissues and microfluidic technology for multi-tissue experiments and analysis. Journal of Biotechnology. 2015;205:24–35. doi: 10.1016/j.jbiotec.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 17*.Lancaster MA, Knoblich JA. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science. 2014;345(6194) doi: 10.1126/science.1247125. [Helpful review of organoids] [DOI] [PubMed] [Google Scholar]
- 18.Barker N, Huch M, Kujala P, van de Wetering M, Snippert HJ, van Es JH, et al. Lgr5+ve Stem Cells Drive Self-Renewal in the Stomach and Build Long-Lived Gastric Units In Vitro. Cell Stem Cell. 2010;6(1):25–36. doi: 10.1016/j.stem.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 19.Stange Daniel E, Koo B-K, Huch M, Sibbel G, Basak O, Lyubimova A, et al. Differentiated Troy+ Chief Cells Act as Reserve Stem Cells to Generate All Lineages of the Stomach Epithelium. Cell. 2013;155(2):357–68. doi: 10.1016/j.cell.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt– villus structures in vitro without a mesenchymal niche. Nature. 2009;459(7244):262–65. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 21.Spence JR, Mayhew CN, Rankin SA, Kuhar MF, Vallance JE, Tolle K, et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature. 2011;470(7332):105–09. doi: 10.1038/nature09691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lancaster MA, Renner M, Martin C-A, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501(7467):373–79. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakano T, Ando S, Takata N, Kawada M, Muguruma K, Sekiguchi K, et al. Self-Formation of Optic Cups and Storable Stratified Neural Retina from Human ESCs. Cell Stem Cell. 2012;10(6):771–85. doi: 10.1016/j.stem.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 24.Koehler KR, Mikosz AM, Molosh AI, Patel D, Hashino E. Generation of inner ear sensory epithelia from pluripotent stem cells in 3D culture. Nature. 2013;500(7461):217–21. doi: 10.1038/nature12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takebe T, Sekine K, Enomura M, Koike H, Kimura M, Ogaeri T, et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature. 2013;499(7459):481–84. doi: 10.1038/nature12271. [DOI] [PubMed] [Google Scholar]
- 26.Huch M, Gehart H, van Boxtel R, Hamer K, Blokzijl F, Verstegen Monique MA, et al. Long-Term Culture of Genome-Stable Bipotent Stem Cells from Adult Human Liver. Cell. 2015;160(1):299–312. doi: 10.1016/j.cell.2014.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kale S, Biermann S, Edwards C, Tarnowski C, Morris M, Long MW. Three-dimensional cellular development is essential for ex vivo formation of human bone. Nat Biotech. 2000;18(9):954–58. doi: 10.1038/79439. [DOI] [PubMed] [Google Scholar]
- 28.Xia Y, Nivet E, Sancho-Martinez I, Gallegos T, Suzuki K, Okamura D, et al. Directed differentiation of human pluripotent cells to ureteric bud kidney progenitor-like cells. Nat Cell Biol. 2013;15(12):1507–15. doi: 10.1038/ncb2872. [DOI] [PubMed] [Google Scholar]
- 29.Drost J, van Jaarsveld RH, Ponsioen B, Zimberlin C, van Boxtel R, Buijs A, et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature. 2015;521(7550):43–47. doi: 10.1038/nature14415. [DOI] [PubMed] [Google Scholar]
- 30.Gjorevski N, Ranga A, Lutolf MP. Bioengineering approaches to guide stem cell-based organogenesis. Development. 2014;141(9):1794–804. doi: 10.1242/dev.101048. [DOI] [PubMed] [Google Scholar]
- 31.Sweeney LM, Shuler ML, Babish JG, Ghanem A. A cell culture analogue of rodent physiology: Application to naphthalene toxicology. Toxicology in Vitro. 1995;9(3):307–16. doi: 10.1016/0887-2333(95)00007-u. [DOI] [PubMed] [Google Scholar]
- 32.Sin A, Chin KC, Jamil MF, Kostov Y, Rao G, Shuler ML. The Design and Fabrication of Three-Chamber Microscale Cell Culture Analog Devices with Integrated Dissolved Oxygen Sensors. Biotechnology Progress. 2004;20(1):338–45. doi: 10.1021/bp034077d. [DOI] [PubMed] [Google Scholar]
- 33.Sung JH, Dhiman A, Shuler ML. A combined pharmacokinetic–pharmacodynamic (PK–PD) model for tumor growth in the rat with UFT administration. Journal of Pharmaceutical Sciences. 2009;98(5):1885–904. doi: 10.1002/jps.21536. [DOI] [PubMed] [Google Scholar]
- 34.Dance A. News Feature: Building benchtop human models. Proceedings of the National Academy of Sciences. 2015;112(22):6773–75. doi: 10.1073/pnas.1508841112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meinel L, Karageorgiou V, Fajardo R, Snyder B, Shinde-Patil V, Zichner L, et al. Bone Tissue Engineering Using Human Mesenchymal Stem Cells: Effects of Scaffold Material and Medium Flow. Annals of Biomedical Engineering. 2004;32(1):112–22. doi: 10.1023/b:abme.0000007796.48329.b4. [DOI] [PubMed] [Google Scholar]
- 36.Huh D, Fujioka H, Tung Y-C, Futai N, Paine R, Grotberg JB, et al. Acoustically detectable cellular-level lung injury induced by fluid mechanical stresses in microfluidic airway systems. Proceedings of the National Academy of Sciences. 2007;104(48):18886–91. doi: 10.1073/pnas.0610868104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hartung T. Toxicology for the twenty-first century. Nature. 2009;460(7252):208–12. doi: 10.1038/460208a. [DOI] [PubMed] [Google Scholar]
- 38.Xu T, Molnar P, Gregory C, Das M, Boland T, Hickman JJ. Electrophysiological characterization of embryonic hippocampal neurons cultured in a 3D collagen hydrogel. Biomaterials. 2009;30(26):4377–83. doi: 10.1016/j.biomaterials.2009.04.047. [DOI] [PubMed] [Google Scholar]
- 39.Domansky K, Inman W, Serdy J, Dash A, Lim MHM, Griffith LG. Perfused multiwell plate for 3D liver tissue engineering. Lab on a Chip. 2010;10(1):51–58. doi: 10.1039/b913221j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40**.Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, Ingber DE. Reconstituting Organ-Level Lung Functions on a Chip. Science. 2010;328(5986):1662–68. doi: 10.1126/science.1188302. [Seminal paper highlighting lung-on-a-chip development] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huh D, Leslie DC, Matthews BD, Fraser JP, Jurek S, Hamilton GA, et al. A Human Disease Model of Drug Toxicity–Induced Pulmonary Edema in a Lung-on-a-Chip Microdevice. Science Translational Medicine. 2012;4(159):159ra47–59ra47. doi: 10.1126/scitranslmed.3004249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vernetti LA, Senutovitch N, Boltz R, DeBiasio R, Ying Shun T, Gough A, et al. A human liver microphysiology platform for investigating physiology, drug safety, and disease models. Experimental Biology and Medicine. 2016;241(1):101–14. doi: 10.1177/1535370215592121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43*.Bhatia SN, Ingber DE. Microfluidic organs-on-chips. Nat Biotech. 2014;32(8):760–72. doi: 10.1038/nbt.2989. [Useful review on MPS technologies] [DOI] [PubMed] [Google Scholar]
- 44.Hou Z, Zhang J, Schwartz MP, Stewart R, Page CD, Murphy WL, et al. A human pluripotent stem cell platform for assessing developmental neural toxicity screening. Stem Cell Research & Therapy. 2013;4(1):1–5. doi: 10.1186/scrt373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45**.Schwartz MP, Hou Z, Propson NE, Zhang J, Engstrom CJ, Costa VS, et al. Human pluripotent stem cell-derived neural constructs for predicting neural toxicity. Proceedings of the National Academy of Sciences. 2015;112(40):12516–21. doi: 10.1073/pnas.1516645112. [MPS technology use for predictive toxicology testing] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moya ML, Hsu Y-H, Lee AP, Hughes CCW, George SC. In Vitro Perfused Human Capillary Networks. Tissue Engineering Part C, Methods. 2013;19(9):730–737. doi: 10.1089/ten.tec.2012.0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heylman C, Sobrino A, Shirure VS, Hughes CCW, George SC. A strategy for integrating essential 3D microphysiological systems of human organs for realistic anti-cancer drug screening. Experimental biology and medicine (Maywood, NJ) 2014;239(9):1240–54. doi: 10.1177/1535370214525295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clark AM, Ma B, Taylor DL, Griffith L, Wells A. Liver metastases: Microenvironments and ex-vivo models. Experimental Biology and Medicine. 2016 Jul 6;:2016. doi: 10.1177/1535370216658144. DOI:10.1177/1535370216658144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Odijk M, van der Meer AD, Levner D, Kim HJ, van der Helm MW, Segerink LI, et al. Measuring direct current trans-epithelial electrical resistance in organ-on-a-chip microsystems. Lab on a Chip. 2015;15(3):745–52. doi: 10.1039/c4lc01219d. [DOI] [PubMed] [Google Scholar]
- 50.Fuchs TC, Hewitt P. Biomarkers for Drug-Induced Renal Damage and Nephrotoxicity—An Overview for Applied Toxicology. The AAPS Journal. 2011;13(4):615–31. doi: 10.1208/s12248-011-9301-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vunjak-Novakovic G, Bhatia S, Chen C, Hirschi K. HeLiVa platform: integrated heart-liver-vascular systems for drug testing in human health and disease. Stem Cell Research & Therapy. 2013;4(1):1–6. doi: 10.1186/scrt369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Richard J-P, Maragakis NJ. Induced pluripotent stem cells from ALS patients for disease modeling. Brain Research. 2015;1607:15–25. doi: 10.1016/j.brainres.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53**.Wang G, McCain ML, Yang L, He A, Pasqualini FS, Agarwal A, et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med. 2014;20(6):616–23. doi: 10.1038/nm.3545. [Application of MPS platform for rare disease research] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spencer C I, Baba S, Nakamura K, Hua Ethan A, Sears Marie A, Fu C-c, et al. Calcium Transients Closely Reflect Prolonged Action Potentials in iPSC Models of Inherited Cardiac Arrhythmia. Stem Cell Reports. 2014;3(2):269–81. doi: 10.1016/j.stemcr.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Markov DA, Lillie EM, Garbett SP, McCawley LJ. Variation in diffusion of gases through PDMS due to plasma surface treatment and storage conditions. Biomedical microdevices. 2014;16(1):91–96. doi: 10.1007/s10544-013-9808-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang JD, Douville NJ, Takayama S, ElSayed M. Quantitative Analysis of Molecular Absorption into PDMS Microfluidic Channels. Annals of Biomedical Engineering. 2012;40(9):1862–73. doi: 10.1007/s10439-012-0562-z. [DOI] [PubMed] [Google Scholar]
- 57.Toepke MW, Beebe DJ. PDMS absorption of small molecules and consequences in microfluidic applications. Lab on a Chip. 2006;6(12):1484–86. doi: 10.1039/b612140c. [DOI] [PubMed] [Google Scholar]