

Abstract
Chronic non-healing venous leg ulcers (VLUs) are widespread and debilitating, with high morbidity and associated costs; approximately $15 billion is spent annually on the care of VLUs in the US. Despite this, there is a paucity of treatments for VLUs, due to the lack of pathophysiologic insight into ulcer development as well as the lack of knowledge regarding biologic actions of existing VLU-targeted therapies. The bioengineered bilayered living cellular construct (BLCC) skin substitute is an FDA-approved biologic treatment for healing VLUs. To elucidate the mechanisms through which the BLCC promotes healing of chronic VLUs, we conducted a clinical trial (NCT01327937) in which patients with non-healing VLUs were treated with either standard care (compression therapy) or the BLCC together with standard care. Tissue was collected from the VLU edge before and 1 week after treatment, and samples underwent comprehensive microarray, mRNA, and protein analyses. Ulcers treated with the BLCC skin substitute displayed three distinct transcriptomic patterns, suggesting that BLCC induced a shift from a non-healing to a healing tissue response involving modulation of inflammatory and growth factor signaling, keratinocyte activation, and attenuation of Wnt/β-catenin signaling. In these ways, BLCC application orchestrated a shift from the chronic non-healing ulcer microenvironment to a distinctive healing milieu resembling that of an acute, healing wound. Our findings provide in vivo evidence in patient VLU biopsies of pathways that can be targeted in the design of new therapies to promote healing of chronic VLUs.
Introduction
Chronic non-healing venous leg ulcers (VLUs) continue to be a cause of substantial morbidity, straining healthcare budgets and negatively impacting quality of life. Over 70% of VLUs fail to heal with standard care compression therapy and have high recurrence rates, posing additional burden to wound care professionals. The chronicity, frequent relapses and associated complications of non-healing VLUs heavily impact patients’ quality of life and increase healthcare expenditures for millions of people worldwide.
Deciphering the network of de-regulated wound healing processes present in chronic VLUs is challenging, and many therapies showing promise in the laboratory and in initial clinical trials have failed to improve clinical outcomes. The histologic hallmark of chronic VLUs is a hyperproliferative wound edge, which is characterized by non-migratory keratinocytes, decreased angiogenesis, an increase in proteases, increased bacterial colonization and/or infection, and inflammatory infiltrates (1, 2). We have shown that the non-healing VLU edge displays loss of genes controlling the fate of local stem cells and their niche, as well as aberrant activation of ß-catenin and c-Myc (2, 3). Moreover, genomic profiling of VLUs has revealed de-regulation of epidermal activation and differentiation, including attenuation of EGF and TGF-beta receptor signaling (4). However, the molecular pathophysiology of VLUs has not yet been fully elucidated, which has slowed development and validation of targeted therapies (5). There is an urgent need for therapeutic approaches which target multiple aberrantly regulated cellular processes simultaneously, successfully converting the non-healing VLU to a healing wound phenotype. Furthermore, enhanced understanding of the molecular pathophysiology of chronic VLUs is critical in identifying relevant clinical trial endpoints that can be used to evaluate new treatments, paving the way for delivery of maximally efficacious therapies to VLU patients.
An FDA-approved bioengineered bilayered living cellular construct (BLCC) has demonstrated efficacy in promoting healing of chronic ulcers (6, 7). The BLCC skin substitute consists of human foreskin-derived neonatal fibroblasts in a bovine type I collagen matrix below a layer of human foreskin-derived neonatal epidermal keratinocytes. The BLCC has been suggested to interact with the surrounding environment to promote wound healing. In vitro, the BLCC produces growth factors and cytokines that are indispensable for a successful wound healing process (8–10), but the precise in vivo mechanism of action is unknown. To this end, we designed a randomized controlled post-marketing clinical trial to investigate the effects of a commercially available BLCC (Apligraf, Organogenesis, Inc.) on gene expression in chronic VLUs. We analyzed human wound edge biopsies obtained from non-healing VLUs at baseline and one week after BLCC treatment. We hypothesized that treatment with the BLCC might activate responsiveness to cellular signals similar to those that facilitate successful healing of acute wounds, thus changing a non-healing to a healing phenotype.
Results
Randomized controlled trial of the non-healing VLU edge response to BLCC treatment
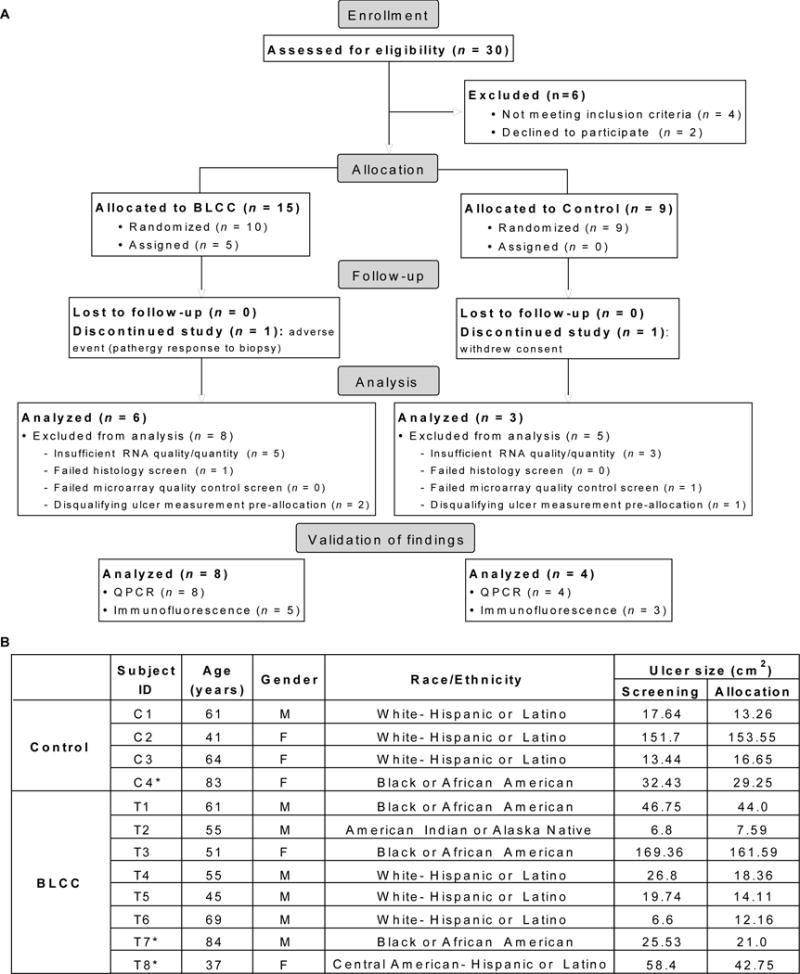
The study design and participant information for clinical trial no. NCT01327937 is summarized in Figure 1 and described further in the supplementary materials and methods. Briefly, potential study participants with VLUs (n = 30) were treated with the standard of care compression therapy for 4 weeks. Patients with non-healing VLUs, defined as those that did not have 40% reduction in ulcer size with compression therapy over this time period, were enrolled and randomly assigned to receive either ongoing standard of care treatment with compression dressings (control group, n = 9) or up to five weekly BLCC applications in addition to the standard of care (treatment group, n = 15). Biopsies from the wound edge were obtained at baseline (week 0) and week 1 for all ulcers, capturing the interval of the first week after study allocation, and ulcer size was monitored over time. At this time point, insufficient RNA quality or quantity was one of the exclusion criteria. Of the patients from whom tissue specimens were obtained, approximately one third yielded sufficient high quality RNA from paired week 0/week 1 biopsies to enable microarray analysis (Fig. 1; tables S1–S2).
Fig. 1. Participant characteristics.
(A) Flow diagram of subjects that were assessed and analyzed for the primary outcome of ClinicalTrials.gov NCT01327937. (B). Subject demographics; ulcer size at screening was determined at time of enrollment (study week –4), whereas size at allocation was determined at week 0, when patients were randomly assigned to BLCC or control treatment groups. Asterisk (*) indicates the sample biomaterial was of insufficient quality for sensitive microarray analysis but was available for validation of study findings.
BLCC skin substitute provides immune signals and growth stimuli to chronic VLUs
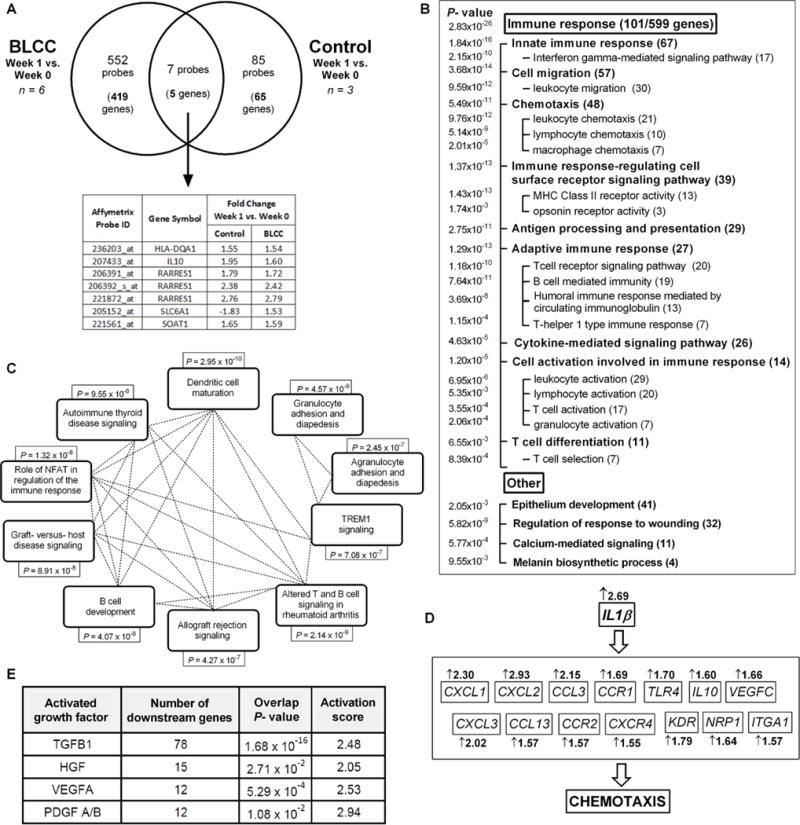
The primary outcome measure of our clinical trial was to evaluate changes in gene expression in wound biopsies from subjects with non-healing VLUs one week after BLCC application compared to gene expression in wound biopsies from subjects with non-healing VLUs one week after standard care. To this end, we examined paired microarray profiles of biopsies from the patients with non-healing VLUs before and after a single application of the BLCC (week 0 to week 1) and compared those to biopsies from the VLU controls receiving standard care compression therapy (Fig. 1). The peak of the BLCC-mediated effect was expected in the first week of treatment, based on dosing in previous clinical trials (7, 8). Importantly, to exclude the possibility that BLCC as opposed to wound tissue was evaluated, DNA genotypes from week-1 biopsies were examined and compared to cellular genotypes of the BLCC used, to ensure that no cells from the BLCC were detectable in the biopsy specimens at the 1-week time point. We found 559 microarray probes (corresponding to 424 unique genes) significantly differentially expressed between week 0 and week 1 after BLCC treatment, compared to differential expression in control biopsies, which showed a change in only 92 probes (70 genes) (paired t-test, P <0.05; fold change >1.5). There were 5 genes that overlapped between BLCC treated and control biopsies (Fig. 2A, table S3).
Fig. 2. Transcriptional response of chronic VLUs to BLCC treatment or compression therapy.
(A) Venn diagram of significantly regulated probes and corresponding genes in BLCC and Control-treated VLUs (fold change >1.5; paired t-test P<0.05). (B) Gene ontology analysis of enriched biological processes among the BLCC-modulated genes. Using Broad Institute GSEA Algorithm, significantly overrepresented functions are grouped by category and listed with enrichment P values (left column) as well as number of corresponding BLCC-influenced genes (parentheses). (C) Ingenuity Pathway Analysis (IPA) of BLCC-modulated genes. The top 10 pathways are shown with corresponding Benjamini-Hochberg-corrected enrichment P values; dotted lines represent genes that are common to the linked pathways. See also table S4. (D) IPA-predicted mechanistic network for BLCC stimulation of immune cell chemotaxis via IL1β; fold changes in IL1β target gene expression after BLCC treatment are shown. (E) Growth factors with IPA-predicted active downstream signaling in response to BLCC treatment, as indicated by overlap of their known targets with BLCC-modulated genes (Fisher’s exact test, P<0.05) as well as by activation (Z) score > 2 reflecting consistent gene expression changes in response to BLCC treatment (11). TGFB1= transforming growth factor beta 1; HGF=hepatocyte growth factor; VEGFA= vascular endothelial growth factor; PDGF=platelet-derived growth factor. See also fig. S2 and table S5.
Although some variability in gene expression among individual study subjects was evident, a clear BLCC-specific consensus transcriptional signature emerged (fig. S1). Gene ontology analysis of differentially expressed entities post-BLCC application highlighted a strong enrichment in immune system biological processes, with 18.1% (101/559) of the modulated genes related to the immune response (P=2.83 × 10−26) (Fig. 2B). The highly enriched biological processes also included “regulation of response to wounding”, as well as “epithelium development,” consistent with the demonstrated clinical role for the BLCC in accelerating wound healing.
Ingenuity pathway analysis (IPA) of the differentially expressed genes post-BLCC treatment identified multiple enriched pathways (Fig. 2C, table S4), all of which related to various aspects of the innate and adaptive immune response. In contrast, in the standard of care VLUs, no significantly enriched pathways were identified using this approach. Using published algorithms (11), IPA Core Analysis generated predicted networks connecting upstream regulators to downstream biological processes that were significantly enriched among the BLCC-stimulated genes. One such network is shown in Figure 2D, in which the upstream regulator interleukin-1 beta (IL1β) induced a set of chemokines, which were significantly upregulated after BLCC treatment, to increase chemotaxis, an enriched and activated biological process (Fig. 2B). These pathway and network data provide initial support for the hypothesis that BLCC application introduces an alternate inflammatory response distinct from chronic inflammatory infiltrates typically found in non-healing VLUs (12).
IPA analysis also predicted multiple upstream regulators whose targets were enriched among BLCC-modulated genes, including such key wound healing factors as transforming growth factor beta (TGFβ), hepatocyte growth factor (HGF), vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) (Fig. 2E). Moreover, the directionality of downstream gene expression (i.e., induction vs. inhibition) supported active signaling states of these growth factors (table S5 and fig. S2).
Generation of acute and chronic wound reference gene expression profiles
We hypothesized that the inflammation triggered by BLCC application was similar to that invoked during the acute wound healing response. To explore this possibility, we needed to differentiate the inflammation during acute wound healing, which leads to successful wound closure, from the chronic inflammation present in the background of non-healing VLUs. To do so, we generated three reference gene expression profiles comparing intact skin, acute wounds at day 3 post-wounding, and chronic VLUs (Fig. 3A, table S7). We used these reference gene expression profiles to explore possible mechanisms through which application of BLCC might uniquely shift chronic, non-healing VLUs to an acute wound healing-like phenotype.
Fig. 3. Models for BLCC-triggered acute wound healing response.
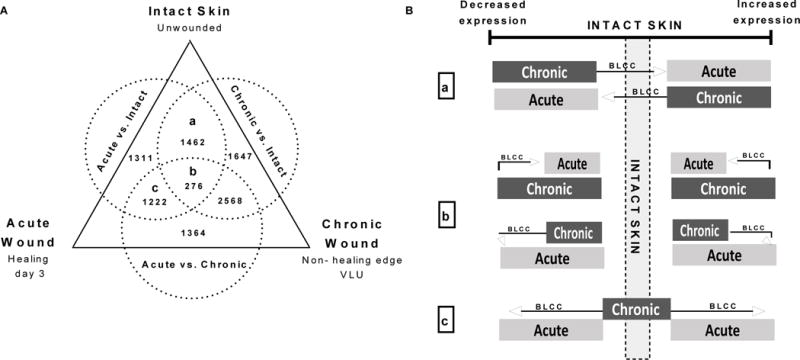
(A) Venn Diagram of three reference profiles generated using microarray data from intact unwounded skin (n=6), healing acute wounds on post-injury day 3 (n=6), and the non-healing edge of chronic venous leg ulcers (n=3) as described in tables S6–S7. Letters a–c indicate aggregates from which genes in Fig. 2B were obtained. (B) Proposed mechanisms by which BLCC might accelerate healing of chronic VLUs: (a) reversing expression of genes with divergent expression in acutely healing wounds vs. chronic VLUs; (b) shifting genes that are pathologically hyper-regulated in chronic VLUs back to acute healing wound levels; or (c) inducing expression of genes modulated in acute healing wounds that are quiescent in chronic VLUs.
To obtain an acute wound healing profile that could be coordinately analyzed head-to-head with our chronic VLU profiles, we used the raw in vivo full-thickness acute wound microarray data previously generated from human skin graft donor site wounds ((13), GEO accession number GSE28914). We downloaded paired data from 6 patients, comparing intact vs. healing skin at day 3 post-wounding (13), to capture the early response and inflammatory phase of acute wound healing (acute vs. intact, Fig. 3A and table S6–S7). A reference expression profile of the prototypic non-healing chronic VLUs was obtained from the pre-treatment (week 0) biopsies of 3 patients with VLUs that displayed poorest healing trajectories during the 4-week screening period prior to randomization (fig. S3, patients C2, C3, and T6), as compared with biopsies of healthy (intact, unwounded) skin (chronic vs. intact, Fig. 3A and table S6–S7). Finally, we compared the acute wound profiles to transcriptional profiles of the baseline chronic VLUs to enable discrimination of the acute response from the background of chronic VLU inflammation (acute vs. chronic, Fig. 3A and table S6). The three reference profiles contained distinct as well as overlapping genes (Fig. 3A).
We used these reference profiles to propose the following mechanisms for BLCC effects. For genes modulated in both acute and chronic wounds, changes may occur in expression but in opposite directions when compared to intact skin, but BLCC treatment might revert the expression in VLUs towards the acute wound phenotype (Fig. 3B, “a”). For genes with consistently altered expression in both acute wounds and chronic VLUs, but pathologically over- or under-expressed in VLUs, the BLCC may normalize this perturbed gene expression back to expression levels observed in acute healing wounds (Fig. 3B, “b”). For genes modulated during acute wound healing, but not in chronic VLUs, the BLCC may regulate the response in the direction consistent with acute wound levels, which are associated with a beneficial healing outcome (Fig. 3B, “c”).
BLCC reverses gene dysregulation in non-healing chronic VLUs towards an acute wound healing phenotype
We proceeded to examine the BLCC and control gene expression profiles (week 1 vs. week 0, table S3) for evidence of the proposed mechanisms in Figure 3B. We identified caspase 14 (CASP14) as a BLCC-modulated gene representing model “a” in Figure 3B. CASP14 is a non-apoptotic caspase with a critical role in the terminal differentiation of keratinocytes (14). As expected, its expression was lower in acutely healing wounds and, consistent with previous reports (4), was higher in non-healing chronic VLUs (Fig. 4A). CASP14 transcript levels were down-regulated in VLUs after BLCC treatment, adopting the direction of acutely healing wounds (Fig. 4A).
Fig. 4. Model validation for BLCC-induced gene expression changes in chronic VLUs.
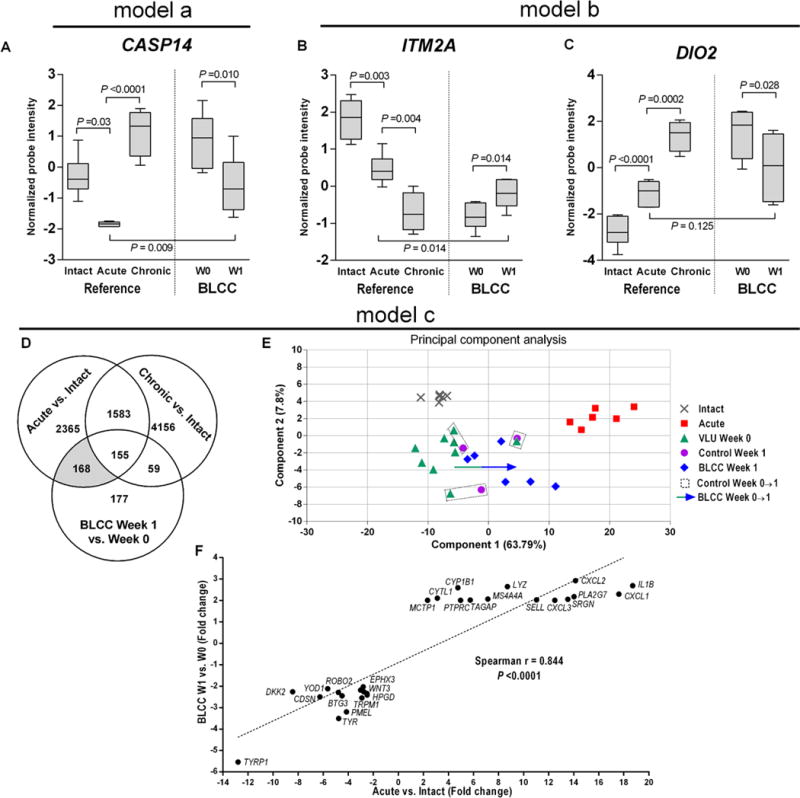
Expressions of gene aggregates identified in Fig. 2A were explored in the context of the models proposed in Fig. 2B by comparing reference profiles of intact unwounded skin, healing acute wounds on post-injury day 3, and the non-healing edge of chronic venous leg ulcers to pairs of non-healing VLUs before and after BLCC treatment. Data are represented as box and whisker plots of microarray probe expression intensities in the unwounded skin (“Intact”, n=6), healing acute wounds (“Acute”, n=6), non-healing chronic VLUs (“Chronic”, n=3), as well as 6 pairs of BLCC-treated VLUs (“W0” and “W1”). (A) Model “a”: Caspase 14 (CASP14) expression in intact, acute, and chronic reference samples compared to BLCC-treated VLUs at W0 and W1. (B- C). Model “b”: Integral membrane protein 2A (ITM2A) and thyroid hormone deiodinase (DIO2) expression in intact, acute, and chronic reference samples compared to BLCC-treated VLUs at W0 and W1. P values were determined by 2-tailed paired t-test (Acute vs. Intact, BLCC W1 vs W0) or 2-tailed moderated t-test (Acute vs. Chronic, BLCC W1 vs. Acute). (D–E) Model “c”: 4-component Principal Component Analysis (PCA) of 168 microarray probes common to healing acute wounds and chronic VLUs post-BLCC (shaded in grey); plot of components 1 and 2 which comprise over 70% of the variation. Arrow represents the trend towards acute wound expression in 1+ PCA components for pairs of BLCC-treated VLUs (BLCC Week 0 to 1) which is absent in Control-treated pairs (dotted rectangles). (F) Plot of changes in gene expression for BLCC-treated VLUs (fold change, W0 to W1) and changes in gene expression between acute wounds and intact skin as determined by Spearman’s nonparametric rank order correlation (2-tailed P<0.0001).
Model “b” in Figure 3B was represented by the expression of integral membrane protein 2A (ITM2A) and type 2 thyroid deiodinase (DIO2) (Fig. 4B–C). ITM2A is an integral transmembrane protein that is variably expressed in T-cell lineage hematopoietic cells, with high expression in select subsets of activated T cells and lower expression in stimulated regulatory T cells (15, 16). BLCC application induced expression of ITM2A in the chronic VLUs to an expression level consistent with an acutely healing wound. DIO2 was induced during acute wounding and even further overexpressed in chronic non-healing VLUs (Fig. 4C). DIO2 catalyzes the conversion of thyroid hormone to its active form (T3) and functions as the key regulator of thyroid hormone action on target tissues, including the skin (17). Thyroid hormone broadly influences epidermal development and function and has a demonstrated role in wound healing (18). DIO2 expression in BLCC-treated VLUs was similar to that in healing acute wounds (Fig. 4C). ITM2A and DIO2 expressions were unchanged in the VLUs from control patients receiving the standard of care; in fact, none of the genes expressed in the group receiving compression therapy alone showed the expression pattern in model “b” of Figure 3B (table S3).
We identified multiple up-regulated and down-regulated genes (Fig. 4D) corresponding to model “c” of Figure 3B. Principal Component Analysis (PCA) demonstrated a shift in BLCC-treated VLUs but not control VLUs towards a gene expression profile consistent with acute wounds during the inflammatory wound healing phase (day 3) (Fig. 4E). Moreover, when plotted against each other, changes in gene expression in acute wounds correlated significantly with changes in gene expression in BLCC-treated VLUs (Fig. 4F).
BLCC recapitulates biological processes of acute wound healing
Pathway analysis also supported the global hypothesis that BLCC treatment, in contrast to standard-of-care compression therapy alone, reverted the chronic non-healing VLUs to an acute wound healing phenotype. We performed IPA on the reference gene profiles established in Figure 3A and compared them with biological pathways enriched in BLCC-treated and control VLUs (week 1 vs. week 0) (see tables S3 and S7). Although a small number of genes were modulated in both BLCC- and control-treated VLUs, acute wound healing biologic pathways including innate and adaptive immunity and their interaction were enriched exclusively in acute and BLCC- treated VLUs (Fig. 5A, table S8). Biological processes of B cell proliferation, antibody production, calcium mobilization, and T cell differentiation, activation, migration, and signaling were present in BLCC-treated wounds but absent in chronic VLUs at baseline or after compression treatment (Fig. 5B, table S9).
Fig. 5. Biologic pathways and processes common to acutely healing wounds and BLCC-treated VLUs. (A).
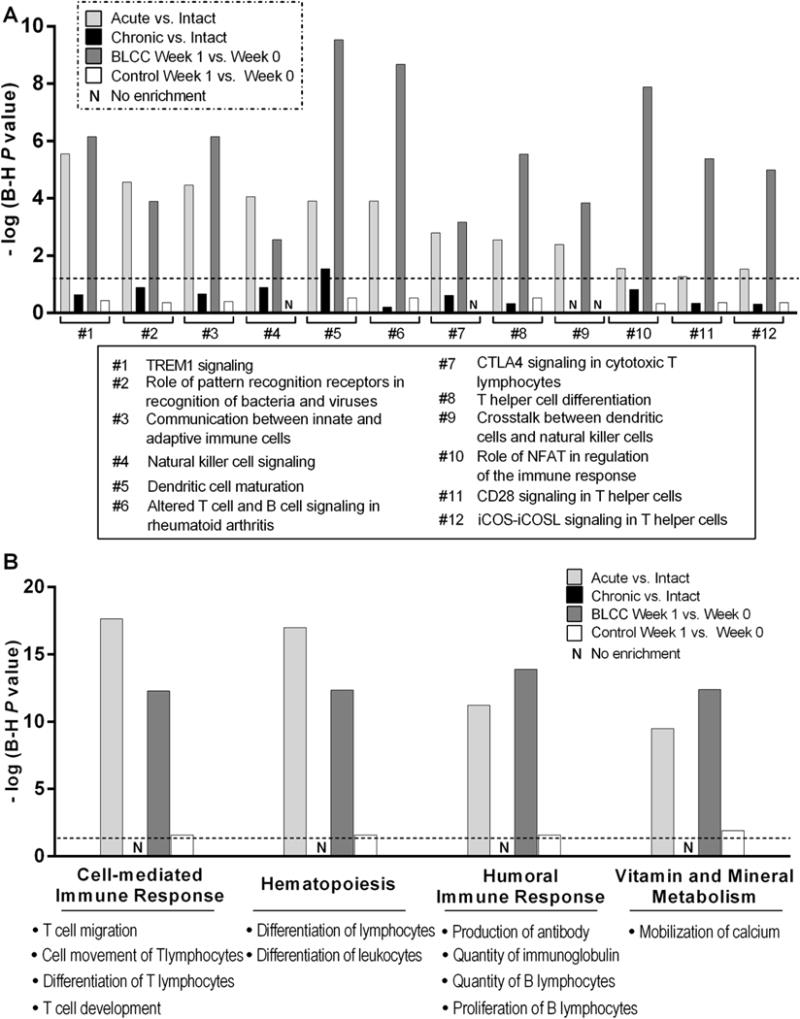
IPA identified pathways and (B) biologic processes significantly enriched in acutely healing wounds (Acute vs. Intact, n=6 pairs) and in chronic VLUs after 1 week of BLCC application (BLCC Week 1 vs. Week 0, n=6 pairs), but not enriched in VLUs treated with standard of care compression alone (Control Week 1 vs. Week 0, n=3 pairs) nor in non-healing VLUs at baseline (n=3 chronic VLU vs n=8 intact (unwounded) skin). Histograms reflect P values of enrichment for each pathway or process assessed by Fisher’s exact test after Benjamini-Hochberg (B–H) correction for multiple testing. Dotted line denotes thresholds for significance at B–H P=0.05. See tables S8–S9 for corresponding genes.
BLCC treatment induces acute inflammatory wound healing in chronic VLUs
To validate our microarray findings, we used Ingenuity knowledge base to identify a literature-supported network of genes and biological processes enriched in acute healing and BLCC-treated wounds but not chronic VLUs at baseline or after standard-care treatment (Fig. 6A). An expanded pool of biopsies from BLCC-treated (n=8) and control treated (n=4) VLUs were used to validate the expression of IL1β, CXCL1, CXCL2, and CXCL3 using qPCR (Fig. 6B). We used a well-characterized ex vivo model of acute wound healing (19) using healthy human skin samples (n=2) to confirm the induction of all four genes during the acute wounding response (Fig. 6C; fig. S4). Qualitative immunofluorescence analysis of protein tyrosine phosphatase receptor type C (PTPRC), which encodes the CD45 receptor expressed on the surfaces of leukocytes, identified an increased dermal infiltrate of CD45+ cells in BLCC-treated tissue samples (Fig. 6D). We confirmed that BLCC treatment induced toll-like receptor 4 (TLR4) expression in the upper spinous and granular layers of the epidermis (Fig. 6E). TLR4 functions in both innate and adaptive immunity and is an important player in the inflammatory phase of early wound healing (20).
Fig. 6. Recapitulation of immune features of the acute wound healing phenotype in chronic VLUs following BLCC treatment.
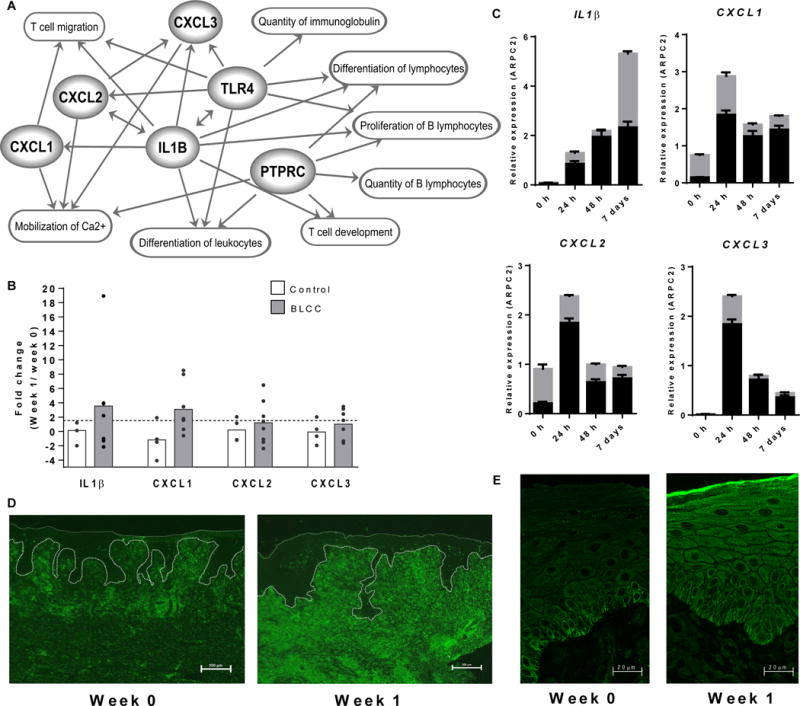
(A) Interplay of genes and biological functions jointly enriched in acute wounds (n=6) and in BLCC-treated VLUs (n=6). Links (arrows) are based on IPA-categorized literature findings. (B) qPCR expression of IL1B, CXCL1, CXCL2 and CXCL3 in non-healing VLUs one week after standard of care compression therapy (control, n=4, unshaded bars) or compression therapy plus BLCC (n=8, shaded bars). Dots represent fold expression change in individual paired samples one week post treatment, with bar height at the group mean. See also fig. S4. (C) IL1B, CXCL1, CXCL2, and CXCL3 expression in an ex vivo model of acute wound healing in skin from n=2 healthy donors, as determined by qPCR at 0, 1, 2, and 7 days post-wounding. Gray and black bars represent expression in individual donors (mean ± SEM of n=3 technical replicates). (D) PTPRC-encoded CD45 receptor immunofluorescence staining of wound edge sections before (week 0) and after (week 1) BLCC treatment. Scale bar, 200 um. (E) Immunofluorescence staining of wound edge sections for epidermal TLR4 expression before (week 0) and after (week 1) BLCC treatment. Scale bar, 20 um. Images in D–E are representative of n=5 study subjects before and after BLCC application (Week 0 vs. Week 1).
Healing BLCC-treated VLUs are distinguished by attenuated Wnt/β-catenin signaling
The BLCC skin substitute has demonstrated efficacy in healing chronic ulcers in conjunction with standard therapy (6, 7), but VLUs that do not respond to initial applications of BLCC are unlikely to derive additional clinical benefit from repeated BLCC treatments (21). To determine whether gene expression could be retrospectively correlated with a successful 4-week healing trajectory, we performed regression analysis on the healing curves of the control and BLCC-treated VLUs from week 0 through week 4 (Fig. 7). We identified changes in gene expression from week 0 to week 1 in the BLCC-treated VLUs that correlated with the trajectory slope of the “healers,” but not with the slope of the “non-healers” or controls receiving compression therapy alone (table S10). One of these genes was CSNK2A2, a casein kinase with an essential role in Wnt/β-catenin signaling (22). BLCC treatment decreased CSNK2A2 expression in VLUs and the magnitude of down regulation correlated with healing extent in the “healers” (R2=0.9980, correlation P=0.02) but not in “non-healers” and/or control VLUs (Fig. 7A). Interestingly, reduced expression of CSNK2A2 is a shared feature of BLCC-healing VLUs and acute wound healing, as CSNK2A2 expression decreased in vivo by approximately 3-fold at 72 hours post-wounding (Fig. 7B; table S7, “Acute vs. Intact”). Furthermore, WNT3, which regulates CSNK2A2 activity (22), decreased in BLCC-treated chronic VLUs and in vivo and ex vivo models of acute wound healing (Fig. 7C). Finally, while immunofluorescence of phosphorylated β-catenin in non-healing VLUs confirmed previously described intense nuclear staining in wound edge keratinocytes (2), nuclear β-catenin was absent in portions of BLCC-treated “healer” VLUs (Fig. 7D).
Fig. 7. WNT/β-catenin signaling pathway in BLCC-treated VLUs and correlation with healing trajectory.
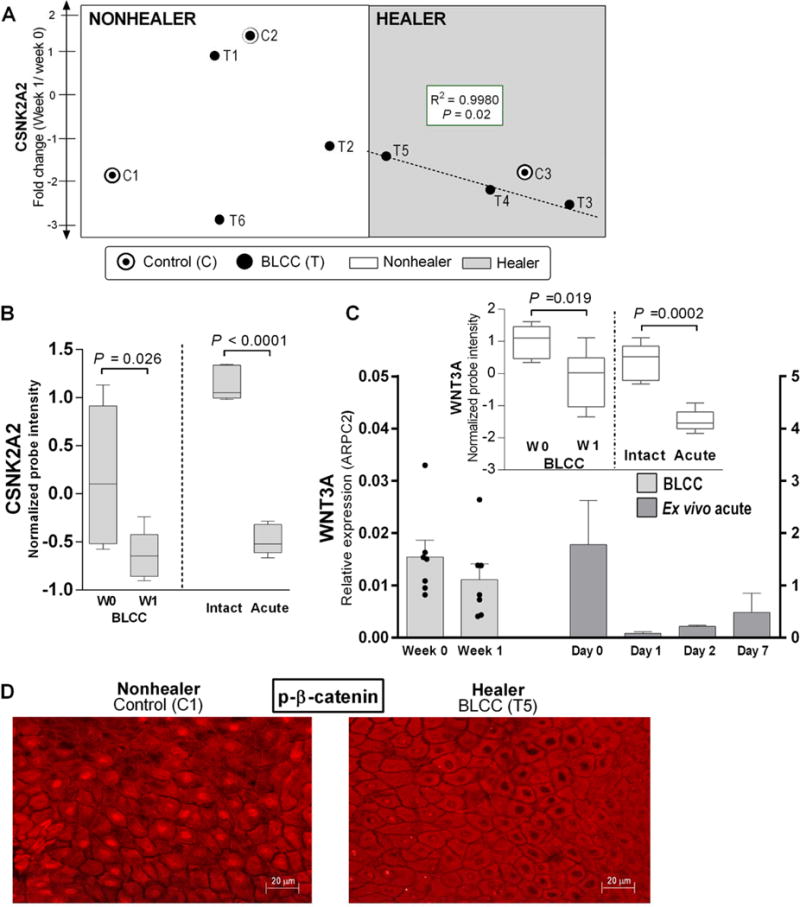
(A) CSNK2A2 expression changes and correlation with wound closure trajectory slope in 3 BLCC-responsive “Healers” (shaded area, dashed line) compared to 3 “Nonhealers” and 3 controls. See also fig. S5. (B) CSNK2A2 expression in healing acute wounds (n=6) and in chronic VLUs treated with BLCC (n=6), as represented by box-and-whisker plots of microarray probe intensity; paired t-test. (C) Relative WNT3 expression in chronic VLUs (n=7) before and after BLCC treatment and in acute wound healing (n=2 healthy donor skin, ex vivo assay). Bars represent mean and SEM of 3 technical replicates after normalization to ARPC2 internal control. Inset: WNT3 microarray probe expression in paired biopsies of chronic VLUs pre- vs. post-BLCC treatment (n=6) as well as acute wounds pre- vs. 3 days post-wounding (n=6); paired t-test P<0.05. (D) Immunofluorescence of phospho-β catenin cellular localization in keratinocytes at the wound edge of a “Nonhealer” VLU treated with standard of care (control), versus a BLCC-treated “Healer” VLU. Images are representative of 3 controls and 3 BLCC-treated VLU “Healers”. Scale bar 20μm.
Discussion
Our clinical trial was designed to understand the mechanisms of action of an FDA-approved skin substitute that has already demonstrated clinical efficacy in healing chronic ulcers (6). We found that BLCC application induces genes and processes of acute inflammatory healing, activates keratinocytes, and diminishes WNT/β-catenin signaling, thus recapitulating features of the acute wound phenotype. Our data therefore suggest that therapeutic approaches which can successfully activate an acute wound healing response have the greatest likelihood of being efficacious in the clinical setting. As such, our findings provide representative testable genomic and molecular endpoints that can be incorporated into the design of the future clinical trials testing the efficacy of VLU therapies.
Our data demonstrate that BLCC application triggers an inflammatory response that is distinct from the chronic inflammation present in the non-healing VLUs at baseline. It is unlikely that we are capturing acute rejection of the allogeneic cells of the BLCC, i.e., graft versus host response, for several reasons. First, well-documented clinical signs of rejection such as pain, erythema, and necrosis are absent from both acute and chronic wounds that have been treated with BLCC (6, 23). Second, the BLCC does not contain resident skin antigen-producing cells such as Langerhans cells or melanocytes, and keratinocytes and fibroblasts do not express HLA Class II antigens or other immune co-stimulatory molecules (6). Third, antibodies signifying a BLCC-specific immune rejection (e.g., anti-HLA type I BLCC alloantigens) were not detectable in VLU patients treated with BLCC (6).
Rather than reflecting a rejection response, comparison of the BLCC gene expression profile to in vivo acute wound profiles illustrated that this BLCC-invoked inflammation recapitulated features of the inflammatory phase of acute wound healing which is an essential stage of successful wound closure. We demonstrated this at the level of individual genes as well as at the level of coordinately enriched pathways and biologic processes in acute wounds and BLCC-treated chronic ulcers. Their expression in healing vs. non-healing wounds might thus reflect fine-tuning of the adaptive immune response. Moreover, our data may indicate that prolonged inflammation present in non-healing VLUs does not facilitate progression of healing found in acute wounds. Further, our data support the hypothesis that BLCC application reverts this sub-optimal inflammation to activate acute inflammatory signaling similar to that seen in an acute wound healing environment.
The gene expression changes in paired VLU biopsies before and after treatment illustrated that BLCC triggered therapeutic reprogramming, reversing the non-healing phenotype and activating pro-healing pathways. Importantly, these changes were not present in the control VLU group treated with standard-care compression therapy alone, indicating BLCC-specific effects. The only exception was RARRES1 (retinoic acid receptor responder 1), an acute wound healing gene that was upregulated in both BLCC and control groups one week after treatment, perhaps reflecting continued optimal standard-care compression therapy, which both groups received, rather than a BLCC-unique mechanism. There are three possible mechanisms by which BLCC-initiated activation of healing may occur: 1) as demonstrated in vitro (8–10), the BLCC secretes growth factors and cytokines common to the pro-healing pathways; 2) the BLCC stimulates patients’ VLU cells to activate pro-healing signaling pathways; or 3) the BLCC both secretes growth factors and activates pro-healing signaling pathways within the VLUs. Although it is not possible to test which of these three options occurred in our study, our gene expression data confirm that these important wound healing signaling pathways are activated and successfully executed in VLUs only upon BLCC and not standard compression treatment. Similarly, our findings do not pinpoint the exact cellular source(s) of the acute wound healing signals captured by our post-BLCC microarrays, as there are several cell types which could contribute to the gene expression changes detected in the VLU edge post-BLCC. However, because no BLCC DNA material was present in the VLUs after 1 week of treatment, our data indicate that the resident chronic VLU cells altered their transcriptomes in response to BLCC application.
Our study design was limited by the lack of repeated sampling and profiling of BLCC-treated VLUs at later time points. However, consistent with previous reports (24), we note again that no detectable BLCC cellular DNA remained in any of the VLU edge biopsies after one week of treatment. This observation implies that any direct BLCC-triggered effects are more likely to be detected within the initial week of treatment, which was the interval captured by our study, than at later time points. Interestingly, given that original clinical trials of the BLCC’s efficacy in VLUs only demonstrated a healing response months (rather than weeks) after treatment (6), the BLCC’s early in vivo disappearance suggests that its initial therapeutic activity must be of sufficient intensity to effect sustained changes in the resident cells of the chronic wound, which then go on to gradually orchestrate successful wound closure.
The drawbacks of our study highlight a question that is of great importance to the field of chronic wound healing: should one perform in vivo “mechanism of action” studies in humans knowing that direct demonstration of causality cannot be attributed to anything more precise than the tissue response to presence or absence of a particular therapeutic intervention? More to the point, the field of chronic wound healing is challenged by lack of validated pre-clinical models, a factor that severely impedes translation of therapies to clinical practice. Given this reality, we believe that there are two choices: to continue to use products without attempting to elucidate their mechanisms of action in this complex multi-factorial disease and thus impede development of second-generation products for chronic wounds; or, to accept the limitations of more descriptive, less mechanistic approaches to deciphering how patients’ tissues respond to therapies in order to gain potential valuable clinical insights.
We argue that our clinical trial findings have direct clinical relevance. We were able to provide initial validation for specific biologic processes through which placement of the BLCC converts chronic non-healing VLUs into an acute wound healing phenotype. These biologic processes may serve as specific targets and testable endpoints in the design and testing of future VLU therapies, which are sorely needed in the clinical setting. Moreover, we identified early changes in gene expression that, upon further validation in larger patient groups, may be predictive of healing outcomes after BLCC application. These early indicator genes could help identify subsets of patients with chronic wounds who are most likely to derive clinical benefit from BLCC therapy. For instance, Wnt/β-catenin signaling is highly dysregulated in chronic VLUs (2, 3), and we found that expression of Wnt family member CSNK2A2 correlated with healing in BLCC-treated ulcers. CSNK2A2 might thus serve as a molecular marker that indicates commencement of healing with an initial application of the BLCC, warranting repeated treatments.
Taken together, fifteen years after initial pivotal clinical trials demonstrated efficacy of BLCC in healing chronic VLUs, data from our study provide new insights into how and why this occurs. We believe that similar studies can be integrated into clinical trials, providing new foundations upon which existing and novel therapeutic and diagnostic approaches for chronic wound healing can be examined. This has the potential to positively impact the lives of millions of patients that suffer from non-healing VLUs.
Materials and Methods
Study design
Study participants (ClinicalTrial.gov NCT01327937) were recruited from patients presenting to the wound clinic at the University of Miami (Miami, FL) with VLUs. Written informed consent was obtained from all subjects enrolled in the clinical trial and the study protocol and informed consent were approved by the Institutional Review Board of the University of Miami. All subjects (n=30), from screening (day −28) through day 0 visit, received the standard-of-care including a dressing regimen of a foam dressing and a 4 layered compression bandage system. Participants with non-infected target ulcers of >5 cm2 that had not reduced in area by >40% during the 4-week screening period were randomized to either the control group receiving standard of care compression therapy (foam dressing plus four layered compression bandage system, changed weekly by the investigator) (n=9), or to the treatment group receiving weekly BLCC applications (Apligraf, Organogenesis, Inc.) along with standard of care compression therapy (n=15). Prior to all applications, the BLCC was fenestrated in a standardized manner using a #11 blade with 6 fenestrations per 44 cm2. Skin biopsy specimens were obtained from the non-healing edges of VLUs at the time of randomization (week 0) as well as one week later (week 1). Specimens were clinically designated by a physician as the most proximal skin edge to the ulcer bed. All patients were debrided and local lidocaine injection was used for anesthesia. After week 5, all patients were monitored in the wound clinic for 12 weeks or until wound closure was achieved. In addition, to confirm the acute wound profile, ex vivo human skin experiments were performed on discarded human skin tissue obtained from voluntary surgeries (n=2 donors) at the University of Miami Hospital and were found to be exempt under 45 CFR46.101.2 by the IRB at the University Of Miami Miller School Of Medicine. See supplementary materials and methods for full clinical study protocol and ex vivo experiment details.
Sample processing
Skin biopsies were processed as follows: (a) samples were embedded in OCT compound (Fisher Scientific) and/or (b) stored in formalin for paraffin embedding and/or (c) stored in RNAlater or homogenized in Trizol (Ambion/Applied Biosystems) or snap frozen for subsequent RNA/protein isolation. Tissue morphology was evaluated using hematoxylin and eosin staining. Genotyping of a portion of each week 1 biopsy in the BLCC treatment group was performed by an outside laboratory (Esoterix Clinical Trials Services).
VLU morphology assessment
Biopsies obtained from VLU wound edges were formalin fixed and paraffin embedded. 5μm thick sections were stained using hematoxylin & eosin following standard protocol and assessed for the presence of epidermis and dermis thus confirming characteristic VLUs morphology as previously described (2).
Gene expression microarrays
Microarray experiments were performed at the University of Miami’s Genomic Facility Core. RNA was amplified, fragmented and hybridized to arrays using GeneChip 3′ IVT Express kits by following the manufacturer’s protocol (Affymetrix). 100 ng of total RNA were used as input for the Ambion WT Expression Kit (Ambion, Austin, TX) to produce labeled single-stranded cDNA according to the manufacturer’s instructions. Labeled products were hybridized to Affymetrix Human Genome U133 Plus 2.0 Arrays (Affymetrix, USA). The staining, washing and scanning of the arrays was carried out using a Fluidics 450 station, GeneChip Operating Software and GeneChip Scanner 3000 7G (Affymetrix, USA). For quality control, Bioanalyzer analysis after the generation of cRNA, cDNA, and fragmentation was carried out using the Nano 6000 kit (Agilent). Microarray data were analyzed as described in the supplementary methods.
Pathway analysis
Gene set enrichment analysis was performed using the Gene Ontology tool contained within the Genespring 13.0 Suite. Further pathway analysis and downstream target/functional predictions were performed using Ingenuity Pathway Analysis (IPA; Qiagen; www.ingenuity.com). Microarray probes were mapped to corresponding genes using Ingenuity software; if multiple probes mapped to the same gene, the gene was used only once in enrichment calculations. Counts and lists of genes used as inputs for pathway analysis are found in tables S3 and S6. Statistical tools within the IPA software package used Fisher’s exact test to detect the reported significantly enriched pathways, biologic processes, and upstream regulators; in all cases, enrichment P-values were Benjamini-Hochberg-corrected for multiple testing.
Ex vivo acute wound model
Skin from two independent healthy donors was wounded and processed as described (19). RNA and formalin-fixed/paraffin-embedded sections were used for qPCR analyses. Please refer to the supplementary methods for full details of experimental design and donor demographics.
Statistical Analysis
Statistics for microarray data were performed as described in the supplementary methods. For qPCR validation studies, technical triplicates were included and groups were compared using 2-sided paired t-test. Correlation of nonparametric data was assessed using Spearman’s test. Gene ontology enrichment P-values were calculated within the Genespring 13.0 software package which utilizes Broad Institute’s Gene Set Enrichment Analysis algorithms. Pathway enrichment statistics were calculated within the Ingenuity software package using Fisher’s exact test with Benjamini-Hochberg correction for multiple testing. Growth factor upstream regulator overlap P-values were similarly calculated within IPA using Fisher’s exact test. Two-sided testing was performed with alpha <0.05 for all reported analyses, as specified in corresponding figure legends.
Supplementary Material
Fig. S1. Expression heatmaps of top BLCC-regulated genes in clinical trial subjects.
Fig. S2. Networks of growth factors and their known effects on gene targets that are also regulated in chronic VLUs post-BLCC application.
Fig. S3. Clinical trajectories of VLU study subjects.
Fig. S4. Subject-specific expression of cytokines.
Fig. S5. Post-treatment healing trajectories.
Table S1. Subject disposition.
Table S2. Exclusions from analysis of primary outcome.
Table S3. Significantly regulated entities in the BLCC and control treatment groups.
Table S4. Genes corresponding to top 10 enriched pathways post-BLCC treatment.
Table S5. Evidence for TGFB1 activation post-BLCC treatment.
Table S6. Establishment of reference gene expression profiles for acute and chronic VLU wound healing.
Table S7. Significantly regulated entities in the acute and chronic wound reference profiles.
Table S8. Regulated genes corresponding to enriched pathways shared by acute wounds and BLCC-treated VLUs.
Table S9. Regulated genes corresponding to enriched biological processes shared by acute wounds and BLCC-treated VLUs.
Table S10. Genes correlating with healing trajectory in VLUs after BLCC treatment.
One Sentence Summary.
A bioengineered bilayered living cellular construct promotes ulcer healing by modulating inflammation, stimulating wound edge keratinocytes, and attenuating Wnt/β-catenin signaling to activate an acute wound response.
Editor’s Summary: Activating healing in chronic wounds.
Effective therapies for chronic venous leg ulcers (VLUs) remain elusive, in part due to incomplete understanding of the pathophysiology of non-healing wounds. Stone et al. conducted a post-market clinical trial using transcriptomics to understand the mechanisms of action of an FDA-approved bilayered living cell construct (BLCC) in non-healing VLUs. After one week of BLCC treatment in addition to standard of care compression therapy, non-healing VLUs showed changes in inflammation and gene expression characteristic of acutely healing wounds. This study provides mechanistic insight into how the acute healing process can be activated by a cell therapy in chronic, non-healing wounds.
Acknowledgments
We are very grateful to many of our colleagues: H. Brem (Winthrop Medical Center), R. S. Kirsner (University of Miami Miller School of Medicine), C. Attinger (Georgetown University), J.Steinberg (Georgetown University), and the Organogenesis representatives: P. Golden, M. Sabolinski T. Bollenbach D. Bates, N. Parsons and K. Giovino for their helpful suggestions and contributions to clinical design and execution of this trial. We are very grateful to the members of the Wound Healing Clinical Research Team of the University of Miami-Miller School of Medicine especially to A. Espinosa, C. Kittles, and A.Vivas for their work on all aspects of the clinical protocol. We are also very grateful to the members of the Dermatopathology Unit including C. Perez for their dedicated service and support in tissue specimen processing. Finally, we thank E. Capobianco of the Center for Computational Science at the University of Miami for his expert review of the experimental design and statistical analyses reported in this work.
Funding: This study was funded in part by a research grant from Organogenesis Inc., 09-MOA-002-AG (to EB and MTC), the NIH (NR015649, DK098055, NR013881 to MTC), and the UMSDRC Department of Dermatology and Cutaneous Surgery of the University of Miami Miller School of Medicine, University of Miami SAC-2013-19 award (to MTC) and SAC-2016-9R1 award (to RCS).
Footnotes
Overline: Wound healing
Author contributions: Clinical trial design: EB, MTC, OS. Execution of clinical trial protocol: EB. Specimen collection: EB, OS. Experimental design: RCS, OS, MTC. Execution of experiments: RCS, OS, AMR, HR, MTC. Analysis of microarray data: RCS, HR, MB, MTC. Pathway analysis: RCS. Writing of manuscript: RCS, OS, EB, MB, AMR, HR, MTC.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: Microarray data files are Minimum Information About a Microarray Experiment (MIAME) compliant and have been deposited in NCBI’s Gene Expression Omnibus under accession number GSE84571, accessible at http://www.ncbi.nlm.nih.gov/geo.
References and Notes
- 1.Brem H, Stojadinovic O, Diegelmann RF, Entero H, Lee B, Pastar I, Golinko M, Rosenberg H, Tomic-Canic M. Molecular markers in patients with chronic wounds to guide surgical debridement. Molecular medicine. 2007;13:30–39. doi: 10.2119/2006-00054.Brem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stojadinovic O, Brem H, Vouthounis C, Lee B, Fallon J, Stallcup M, Merchant A, Galiano RD, Tomic-Canic M. Molecular pathogenesis of chronic wounds: the role of beta-catenin and c-myc in the inhibition of epithelialization and wound healing. The American journal of pathology. 2005;167:59–69. doi: 10.1016/s0002-9440(10)62953-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stojadinovic O, Pastar I, Nusbaum AG, Vukelic S, Krzyzanowska A, Tomic-Canic M. Deregulation of epidermal stem cell niche contributes to pathogenesis of nonhealing venous ulcers. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society. 2014;22:220–227. doi: 10.1111/wrr.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stojadinovic O, Pastar I, Vukelic S, Mahoney MG, Brennan D, Krzyzanowska A, Golinko M, Brem H, Tomic-Canic M. Deregulation of keratinocyte differentiation and activation: a hallmark of venous ulcers. Journal of cellular and molecular medicine. 2008;12:2675–2690. doi: 10.1111/j.1582-4934.2008.00321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eming SA, Martin P, Tomic-Canic M. Wound repair and regeneration: mechanisms, signaling, and translation. Science translational medicine. 2014;6:265sr266. doi: 10.1126/scitranslmed.3009337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Falanga V, Margolis D, Alvarez O, Auletta M, Maggiacomo F, Altman M, Jensen J, Sabolinski M, Hardin-Young J. Rapid healing of venous ulcers and lack of clinical rejection with an allogeneic cultured human skin equivalent. Human Skin Equivalent Investigators Group. Archives of dermatology. 1998;134:293–300. doi: 10.1001/archderm.134.3.293. [DOI] [PubMed] [Google Scholar]
- 7.Falanga V, Sabolinski M. A bilayered living skin construct (APLIGRAF) accelerates complete closure of hard-to-heal venous ulcers. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society. 1999;7:201–207. doi: 10.1046/j.1524-475x.1999.00201.x. [DOI] [PubMed] [Google Scholar]
- 8.Brem H, Young J, Tomic-Canic M, Isaacs C, Ehrlich HP. Clinical efficacy and mechanism of bilayered living human skin equivalent (HSE) in treatment of diabetic foot ulcers. Surgical technology international. 2003;11:23–31. [PubMed] [Google Scholar]
- 9.Spiekstra SW, Breetveld M, Rustemeyer T, Scheper RJ, Gibbs S. Wound-healing factors secreted by epidermal keratinocytes and dermal fibroblasts in skin substitutes. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society. 2007;15:708–717. doi: 10.1111/j.1524-475X.2007.00280.x. [DOI] [PubMed] [Google Scholar]
- 10.Falanga V, Isaacs C, Paquette D, Downing G, Kouttab N, Butmarc J, Badiavas E, Hardin-Young J. Wounding of bioengineered skin: cellular and molecular aspects after injury. The Journal of investigative dermatology. 2002;119:653–660. doi: 10.1046/j.1523-1747.2002.01865.x. [DOI] [PubMed] [Google Scholar]
- 11.Kramer A, Green J, Pollard J, Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosner K, Ross C, Karlsmark T, Petersen AA, Gottrup F, Vejlsgaard GL. Immunohistochemical characterization of the cutaneous cellular infiltrate in different areas of chronic leg ulcers. APMIS : acta pathologica, microbiologica, et immunologica Scandinavica. 1995;103:293–299. doi: 10.1111/j.1699-0463.1995.tb01109.x. [DOI] [PubMed] [Google Scholar]
- 13.Nuutila K, Siltanen A, Peura M, Bizik J, Kaartinen I, Kuokkanen H, Nieminen T, Harjula A, Aarnio P, Vuola J, Kankuri E. Human skin transcriptome during superficial cutaneous wound healing. Wound repair and regeneration: official publication of the Wound Healing Society [and] the European Tissue Repair Society. 2012;20:830–839. doi: 10.1111/j.1524-475X.2012.00831.x. [DOI] [PubMed] [Google Scholar]
- 14.Eckhart L, Ban J, Fischer H, Tschachler E. Caspase-14: analysis of gene structure and mRNA expression during keratinocyte differentiation. Biochemical and biophysical research communications. 2000;277:655–659. doi: 10.1006/bbrc.2000.3698. [DOI] [PubMed] [Google Scholar]
- 15.Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, MacIsaac KD, Levine SS, Fraenkel E, von Boehmer H, Young RA. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. 2007;445:931–935. doi: 10.1038/nature05478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kirchner J, Bevan MJ. ITM2A is induced during thymocyte selection and T cell activation and causes downregulation of CD8 when overexpressed in CD4(+)CD8(+) double positive thymocytes. The Journal of experimental medicine. 1999;190:217–228. doi: 10.1084/jem.190.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams GR, Bassett JH. Deiodinases: the balance of thyroid hormone: local control of thyroid hormone action: role of type 2 deiodinase. The Journal of endocrinology. 2011;209:261–272. doi: 10.1530/JOE-10-0448. [DOI] [PubMed] [Google Scholar]
- 18.Safer JD. Thyroid hormone and wound healing. Journal of thyroid research. 2013;2013:124538. doi: 10.1155/2013/124538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stojadinovic O, Tomic-Canic M. Human ex vivo wound healing model. Methods in molecular biology. 2013;1037:255–264. doi: 10.1007/978-1-62703-505-7_14. [DOI] [PubMed] [Google Scholar]
- 20.Chen L, Guo S, Ranzer MJ, DiPietro LA. Toll-like receptor 4 has an essential role in early skin wound healing. The Journal of investigative dermatology. 2013;133:258–267. doi: 10.1038/jid.2012.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cavorsi J, Vicari F, Wirthlin DJ, Ennis W, Kirsner R, O’Connell SM, Steinberg J, Falanga V. Best-practice algorithms for the use of a bilayered living cell therapy (Apligraf) in the treatment of lower-extremity ulcers. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society. 2006;14:102–109. doi: 10.1111/j.1743-6109.2006.00098.x. [DOI] [PubMed] [Google Scholar]
- 22.Gao Y, Wang HY. Casein kinase 2 Is activated and essential for Wnt/beta-catenin signaling. The Journal of biological chemistry. 2006;281:18394–18400. doi: 10.1074/jbc.M601112200. [DOI] [PubMed] [Google Scholar]
- 23.Donohue KG, Carson P, Iriondo M, Zhou L, Saap L, Gibson K, Falanga V. Safety and efficacy of a bilayered skin construct in full-thickness surgical wounds. The Journal of dermatology. 2005;32:626–631. doi: 10.1111/j.1346-8138.2005.tb00811.x. [DOI] [PubMed] [Google Scholar]
- 24.Hu S, Kirsner RS, Falanga V, Phillips T, Eaglstein WH. Evaluation of Apligraf persistence and basement membrane restoration in donor site wounds: a pilot study. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society. 2006;14:427–433. doi: 10.1111/j.1743-6109.2006.00148.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Expression heatmaps of top BLCC-regulated genes in clinical trial subjects.
Fig. S2. Networks of growth factors and their known effects on gene targets that are also regulated in chronic VLUs post-BLCC application.
Fig. S3. Clinical trajectories of VLU study subjects.
Fig. S4. Subject-specific expression of cytokines.
Fig. S5. Post-treatment healing trajectories.
Table S1. Subject disposition.
Table S2. Exclusions from analysis of primary outcome.
Table S3. Significantly regulated entities in the BLCC and control treatment groups.
Table S4. Genes corresponding to top 10 enriched pathways post-BLCC treatment.
Table S5. Evidence for TGFB1 activation post-BLCC treatment.
Table S6. Establishment of reference gene expression profiles for acute and chronic VLU wound healing.
Table S7. Significantly regulated entities in the acute and chronic wound reference profiles.
Table S8. Regulated genes corresponding to enriched pathways shared by acute wounds and BLCC-treated VLUs.
Table S9. Regulated genes corresponding to enriched biological processes shared by acute wounds and BLCC-treated VLUs.
Table S10. Genes correlating with healing trajectory in VLUs after BLCC treatment.