

Abstract
NMDA receptor (NMDAR) is known for its ionotropic function. But recent evidence suggests that NMDAR also has a non-ionotropic property. To determine the role of non-ionotropic activity of NMDARs in clinical relevant conditions, we tested the effect of glycine, a co-agonist of NMDARs, in rat middle cerebral artery occlusion (MCAO), an animal model of cerebral ischemia-reperfusion injury after the animals were injected with the NMDAR channel blocker MK-801 and the glycine receptor antagonist strychnine. We show that glycine reduces the infarct volume in the brain of ischemic stroke animals pre-injected with MK-801 and strychnine. The effect of glycine is sensitive to the antagonist of glycine-GluN1 binding site and blocked by Akt inhibition. In the neurobehavioral tests, glycine improves the functional recovery of stroke animals pre-injected with MK-801 and strychnine. This study suggests that glycine-induced neuroprotection is mediated in part by the non-ionotropic activity of NMDARs via Akt activation in cerebral ischemia-reperfusion injury.
Introduction
The ionotropic glutamate receptors mediate the vast majority of excitatory neurotransmission in the mammalian central nervous system (CNS)1, 2. The N-methyl-D-aspartate receptor (NMDAR) is a subtype of ionotropic glutamate receptors that are ligand-gated Ca2+-permeable channels. NMDARs consist of GluN1, GluN2 (GluN2A-GluN2D) and GluN3 (GluN3A-GluN3B) subunits. In the CNS, GluN2A- and GluN2B-containing NMDARs (GluN2ARs and GluN2BRs) are the major combinations of NMDARs2, 3. Activation of GluN2ARs and GluN2BRs requires the binding of agonist glutamate to GluN2 subunits and the co-agonist glycine to GluN1 subunits, which play essential roles in synaptic plasticity, neural development and glutamate-induced neurotoxicity4, 5.
The GluN2 subunits confer distinct roles of NMDAR subtypes and link them with different intracellular signaling pathways6, 7. It has been reported that GluN2BR-mediated neurotoxicity induces neuronal death, and that enhancement of GluN2AR activity promotes neuronal survival8, 9. However, the molecular mechanisms underlying the differential effects of GluN2ARs and GluN2BRs in neuronal survival and death are not fully understood10.
Recent studies demonstrate that NMDARs have non-ionotropic activity that confers both physiological and pathophysiological effects11–16. For example, ligand binding to NMDARs is sufficient to induce long-term depression (LTD), but does not require ion flow through NMDARs12. A non-ionotropic activity is found to be mediated through GluN2BR and is required for β-amyloid–induced synaptic depression13. The non-ionotropic activity of NMDARs is shown to drive structural shrinkage at spiny synapses and couple Src family kinases to pannexin-1 in excitotoxic injury14. We recently report that glycine potentiates AMPA receptor function through non-ionotropic activation of GluN2ARs15. We also show that glycine protects against glutamate neurotoxicity-induced neuronal injury in cultured cortical neurons independent of the channel activity of GluN2ARs16. As glutamate-induced neurotoxicity plays critical role in many neurological disorders including ischemic stroke, in the present study we tested the effect of glycine in rat middle cerebral artery occlusion (MCAO)17, 18, an animal model of cerebral ischemia-reperfusion injury after the animals were injected with the NMDAR channel blocker MK-801 and the glycine receptor antagonist strychnine. We demonstrate that glycine treatment reduces the infarct volume and improves the functional recovery of stroke animals, suggesting that glycine-induced neuroprotection is mediated in part by the non-ionotropic activity of NMDARs in cerebral ischemia-reperfusion injury.
Results
Glycine protects against ischemia-reperfusion injury through non-ionotropic activity of NMDARs
We recently reported that glycine protects against glutamate neurotoxicity-induced neuronal injury through non-ionotropic activity of GluN2ARs16. To determine the functional effect of non-ionotropic activity of NMDARs, we tested whether the observed glycine effect in vitro occurred in rat MCAO model in vivo. Glycine (100 µg/100 g) was administered into the lateral ventricles at 1.5, 3, 6, 9 or 12 h following ischemia-reperfusion (Fig. 1 and S1). To suppress both the channel activity of NMDARs and the activation of glycine receptors, at 30 min before glycine injection we injected MK-801 (8.0 µg/100 g)19, 20 and strychnine (1.2 µg/100 g) into lateral ventricles21–24, which was referred to as NMDAR channel inactivation procedure (Fig. 1A). TTC staining assay revealed that treatment of glycine at 1.5, 3 or 6 h after ischemia-reperfusion significantly decreased the infarct volume at 24 h after ischemia onset compared with the groups with the injection of MK-801+ strychnine at the same time points (Fig. 1B and C). In the same experimental conditions, we also demonstrated that glycine (100 µg/100 g) reduced the infarct volume in a mouse model of MCAO at 24 h after ischemia onset (Fig. S2A and S2B).
Figure 1.
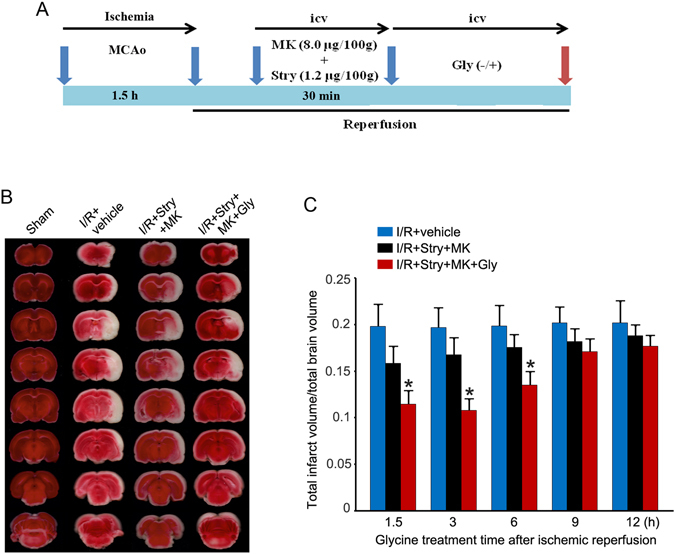
Glycine treatment reduces the infarct volume of ischemic brain independent of glycine receptor activation and the channel activity of NMDARs. (A) A schematic diagram showing rat cerebral ischemia-reperfusion injury and glycine treatment procedure. (B) Sample images of TTC strained-brain sections collected at 24 h after ischemia onset. Glycine (100 µg/100 g, icv) was administered at 3 h following ischemia-reperfusion (I/R). At 30 min prior to glycine injection, MK-801 (8.0 µg/100 g, icv) and strychnine (1.2 µg/100 g, icv) were injected. (C) Summarized quantification data indicate that glycine treatment at 1.5, 3, or 6 h following I/R reduces infarct volume after glycine receptors and NMDARs are inhibited (n = 10 for each group; ANOVA test, *P < 0.05 vs. I/R + Stry + MK). Stry: Strychnine; Gly: glycine; MK: MK-801.
To determine the neuroprotective effect of glycine in a late stage after ischemia-reperfusion, we performed Fluoro-Jade C (FJC) straining at 3 days after rat MCAO onset. Glycine (100 µg/100 g, icv) was administered at 3 h following ischemia-reperfusion. At 30 min prior to glycine injection, MK-801 (8.0 µg/100 g, icv) and strychnine (1.2 µg/100 g, icv) were injected. We showed that the number of FJC-positive degenerating neurons in the ischemic area in glycine treatment group was remarkably lower than the group without glycine injection (Fig. 2A–C). Together, these results suggest that glycine is neuroprotective in ischemia-reperfusion injury and the neuroprotection is mediated by the non-ionotropic activity of NMDARs.
Figure 2.

Glycine treatment reduces cell loss in the ischemic brain where NMDAR channel activity and glycine receptors are inhibited. (A) Sample images of FJC strained-brain sections collected at 3 d after ischemia onset. Glycine (100 µg/100 g, icv) was administered at 3 h following I/R. At 30 min prior to glycine injection, MK-801 (8.0 µg/100 g, icv) and strychnine (1.2 µg/100 g, icv) were injected. (B) The bottom panel is a higher magnification of three regions in the upper panel marked with a frame. Bar = 100 μm. (C) Quantitative analysis of FJC-positive degenerating neurons in the ischemic brain area at 3 d after ischemia onset (n = 6, ANOVA test, *P < 0.05 vs. I/R + Stry + MK). Stry: Strychnine; Gly: glycine; MK: MK-801.
Glycine-GluN1 binding mediates glycine-induced neuroprotection after glycine receptors and the channel activity of NMDARs are suppressed
To provide further evidence for how glycine induced neuroprotection following ischemia-reperfusion injury, we examined the effect of glycine-GluN1 binding site antagonist L-689560 in the rat MCAO model. L-689560 (0.1 mg/100 g, ip) was injected with MK-801 and strychnine at 30 min prior to glycine treatment as described previously25–27. Glycine (100 µg/100 g) was administered into the lateral ventricles at 3 h after ischemia-reperfusion. We showed that L-689560 significantly reduced the neuroprotective effect of glycine after glycine receptors and the channel activity of NMDARs are suppressed (Fig. 3A and B). These data indicate that glycine acts on glycine-GluN1 binding site to elicit non-ionotropic activation of NMDARs and exert neuroprotective effect in ischemic stroke animals.
Figure 3.

Glycine-GluN1 binding antagonist and Akt inhibitor attenuate the effect of glycine in reducing the infarct volume after glycine receptors and the channel activity of NMDARs are inhibited. (A) Sample images of TTC strained-brain sections collected at 24 h after ischemia onset. Glycine (100 µg/100 g, icv) was administered at 3 h following I/R. Glycine-GluN1 binding antagonist L-689560 (L68; 0.1 mg/100 g, ip) or Akt inhibitor IV (100 µM, 2 µl, icv) is injected with MK-801 and strychnine at 30 min before glycine injection. (B) Summarized quantification data indicate that glycine-GluN1 binding antagonist and Akt inhibitor treatment attenuate the effect of glycine in reducing the infarct volume after glycine receptors and NMDARs are inhibited (n = 6, ANOVA test, *P < 0.05 vs. I/R + Stry + MK; # P < 0.05 vs. I/R + Stry + MK + Gly). Stry: Strychnine; Gly: glycine; MK: MK-801.
Glycine-elicited non-ionotropic activity of NMDARs confers neuroprotection through Akt activation after ischemia-reperfusion injury
Our data thus far support the notion that glycine elicits a non-ionotropic activity of NMDARs to confer neuroprotection after ischemia-reperfusion injury. To uncover downstream signaling that mediates the effect of non-ionotropic activity of NMDARs in ischemia-reperfusion injury, we tested the role of Akt in the rat MCAO model. Akt inhibitor IV (100 μM, 2 μl, icv) was injected with MK-801 and strychnine at 30 min prior to glycine treatment as described above28. Glycine (100 µg/100 g) was administered into the lateral ventricles at 3 h after ischemia-reperfusion. As shown in Fig. 3A and B, Akt inhibitor IV reduced the neuroprotective effect of glycine after glycine receptors and the channel activity of NMDARs are suppressed. In the same experimental conditions, we showed that L-689560 and IV blocked glycine-induced increase of Akt phosphorylation in the MCAO model (Fig. 4A–C). Thus, Akt is a downstream signal to mediate the neuroprotective role of non-ionotropic activation of NMDARs by glycine in ischemic stroke animals.
Figure 4.

Glycine treatment prevents p-Akt reduction in ischemic penumbra where NMDAR channel activity and glycine receptors are inhibited. (A) Glycine (100 µg/100 g, icv) treatment at 3 h following I/R prevents p-Akt reduction in ischemic penumbra where NMDAR channel activity and glycine receptors are inhibited (n = 6 animals for each group; ANOVA test, *P < 0.05 vs. Sham; # P < 0.05 vs. I/R + vehicle; **P < 0.05 vs. I/R + Stry + MK). Samples were collected at 24 h after ischemia onset. (B) Akt inhibitor IV (100 µM, 2 µl, icv) prevents glycine (100 µg/100 g, icv)-induced increase of p-Akt in ischemic penumbra where NMDAR channel activity and glycine receptors are inhibited (n = 6, ANOVA test, *P < 0.05 vs. I/R + Stry + MK; # P < 0.05 vs. I/R + Stry + MK; **P < 0.05 vs. I/R + Stry + MK + Gly). The IV was injected with MK-801 and strychnine at 30 min before glycine injection. Samples were collected at 24 h after ischemia onset. (C) The L-689560 (0.1 mg/100 g, ip) prevents glycine (100 µg/100 g, icv)-induced increase of p-Akt in ischemic penumbra where NMDAR channel activity and glycine receptors are inhibited (n = 6, ANOVA test, *P < 0.05 vs. I/R + Stry + MK; # P < 0.05 vs. I/R + Stry + MK; **P < 0.05 vs. I/R + Stry + MK + Gly). The L-689560 was injected with MK-801 and strychnine at 30 min before glycine injection. Samples were collected at 24 h after I/R. Stry: Strychnine; Gly: glycine; MK: MK-801; L-68: L-689560.
Glycine promotes functional recovery of ischemic stroke animals through non-ionotropic activity of NMDARs
A battery of neurobehavioral tests including modified neurological severity scores (mNSS) test, beam-walking test and modified sticky-tape (MST) test were performed to further test the functional role of glycine-induced neuroprotection (Tables 1 and 2)29, 30. The neurological function of stroke animals was evaluated one day before MCAO, and 1, 3, 7 and 14 days after MCAO. Glycine (100 µg/100 g, icv) was injected at 3 h after ischemia-reperfusion. At 30 min prior to glycine injection we injected MK-801 (8.0 µg/100 g) and strychnine (1.2 µg/100 g) into the lateral ventricles. These tests were performed by the investigator who was blinded to the experimental groups. Our data showed that compared with rats treated with strychnine and MK-801 treatment (I/R + Stry + MK group), rats treated with glycine (I/R + Stry + MK + Gly group) had significantly lower scores of mNSS test at day 7 and 14 after MCAO (Fig. 5A), lower scores of beam-walking test at day 3, 7 and 14 after MCAO (Fig. 5B), and higher ratio of MST test at day 7 and 14 after MCAO (Fig. 5C). These results provide functional evidence for the role of non-ionotropic activity of NMDARs in mediating the neuroprotective effect of glycine.
Table 1.
Modified Neurological Severity Score (mNSS).
| Points | |
|---|---|
| Motor tests | |
| Raising rat by tail | 3 |
| Flexion of forelimb | 1 |
| Flexion of hindlimb | 1 |
| Head moved > 10° to vertical axis within 30 s | 1 |
| Placing rat on floor (normal = 0; maximum = 3) | 3 |
| Normal walk | 0 |
| Inability to walk straight | 1 |
| Circling toward the paretic side | 2 |
| Fall down to the paretic side | 3 |
| Sensory tests | 2 |
| Placing test (visual and tactile test) | 1 |
| Proprioceptive test (deep sensation, pushing paw against table edge to stimulate limb muscles) | 1 |
| Beam balance tests (normal = 0; maximum = 6) | 6 |
| Balances with steady posture | 0 |
| Grasps side of beam | 1 |
| Hugs beam and 1 limb falls down from beam | 2 |
| Hugs beam and 2 limbs fall down from beam, or spins on beam (å 60 s) | 3 |
| Attempts to balance on beam but falls off (å 40 s) | 4 |
| Attempts to balance on beam but falls off (å 20 s) | 5 |
| Falls off; No attempt to balance or hang on to beam (20 s) | 6 |
| Reflex absence and abnormal movements | 4 |
| Pinna reflex (head shake when auditory meatus is touched) | 1 |
| Corneal reflex (eye blink when cornea is lightly touched with cotton) | 1 |
| Startle reflex (motor response to a brief noise from snapping a clipboard paper) | 1 |
| Seizures, myoclonus, myodystony | 1 |
| Maximum points | 18 |
One point is awarded for the inability to perform the tasks or for lack of a tested reflex, 13–18, severe injury; 7–12, moderate injury; 1–6, mild injury.
Table 2.
The Beam Walk Test Scoring Criteria.
| Complex Neuromotor Function | Score | Time on Beam |
|---|---|---|
| 0 | −4 s or less. | |
| 1.0 | −5 to 7 s. | |
| 2.0 | −8 to 10 s. | |
| 3.0 | −11 to 15 s. | |
| 4.0 | -greater than 15 s. | |
| 5.0 | -not able to run | |
| Maximum Score | 5 |
Figure 5.
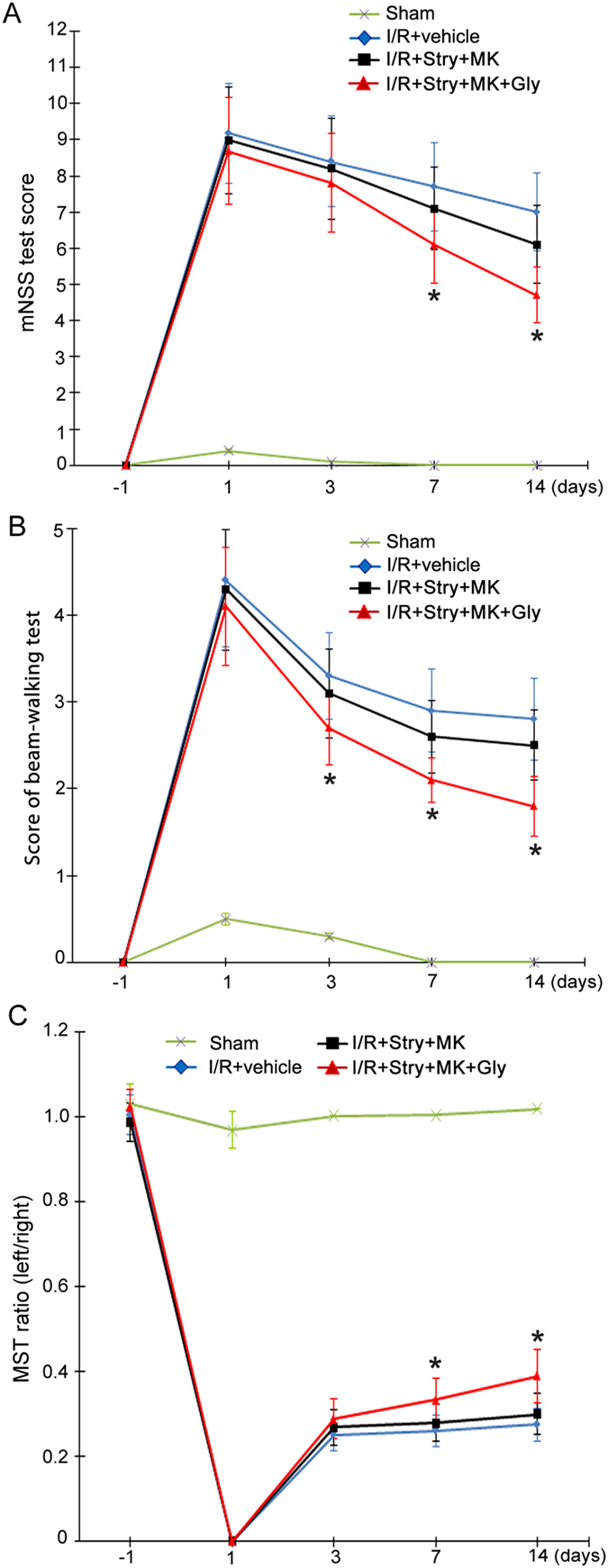
Glycine promotes functional recovery of ischemic stroke animals independent of glycine receptor activation and the channel activity of NMDARs. For all the three experiments, Glycine (100 µg/100 g, icv) was administered at 3 h following I/R. At 30 min prior to glycine injection, MK-801 (8.0 µg/100 g, icv) and strychnine (1.2 µg/100 g, icv) were injected. (A) Animals treated with glycine have lower scores of mNSS test at day 7 and 14 after I/R compared with I/R + Stry + MK group (n = 10; ANOVA test, *P < 0.05 vs. I/R + Stry + MK). (B) Animals treated with glycine has lower scores of beam-walking test at day 3, 7 and 14 after I/R compared with I/R + Stry + MK group (n = 10; ANOVA test, *P < 0.05 vs. I/R + Stry + MK). (C) Animals treated with glycine has higher ratio in MST test at day 7 and 14 after I/R compared with I/R + Stry + MK group (n = 10; ANOVA test, *P < 0.05 vs. I/R + Stry + MK). Stry: Strychnine; MK: MK-801; Gly: glycine.
Glycine-GluN1 binding mediates glycine-induced functional recovery of ischemic stroke animals after glycine receptors and the channel activity of NMDARs are suppressed
To determine whether glycine-GluN1 binding mediates glycine-induced functional recovery in the ischemic stroke animals, in the same experimental conditions, L-689560 (0.1 mg/100 g, ip) was injected with MK-801 (8.0 µg/100 g) and strychnine (1.2 µg/100 g) at 30 min prior to glycine treatment. As shown in Fig. 6, L-689560 prevented glycine-induced recovery in stroke animals after glycine receptors and NMDAR channel activities were inhibited, suggesting that glycine binds to GluN1 site to elicit non-ionotropic activity of NMDARs and promote functional recovery in ischemic stroke animals.
Figure 6.
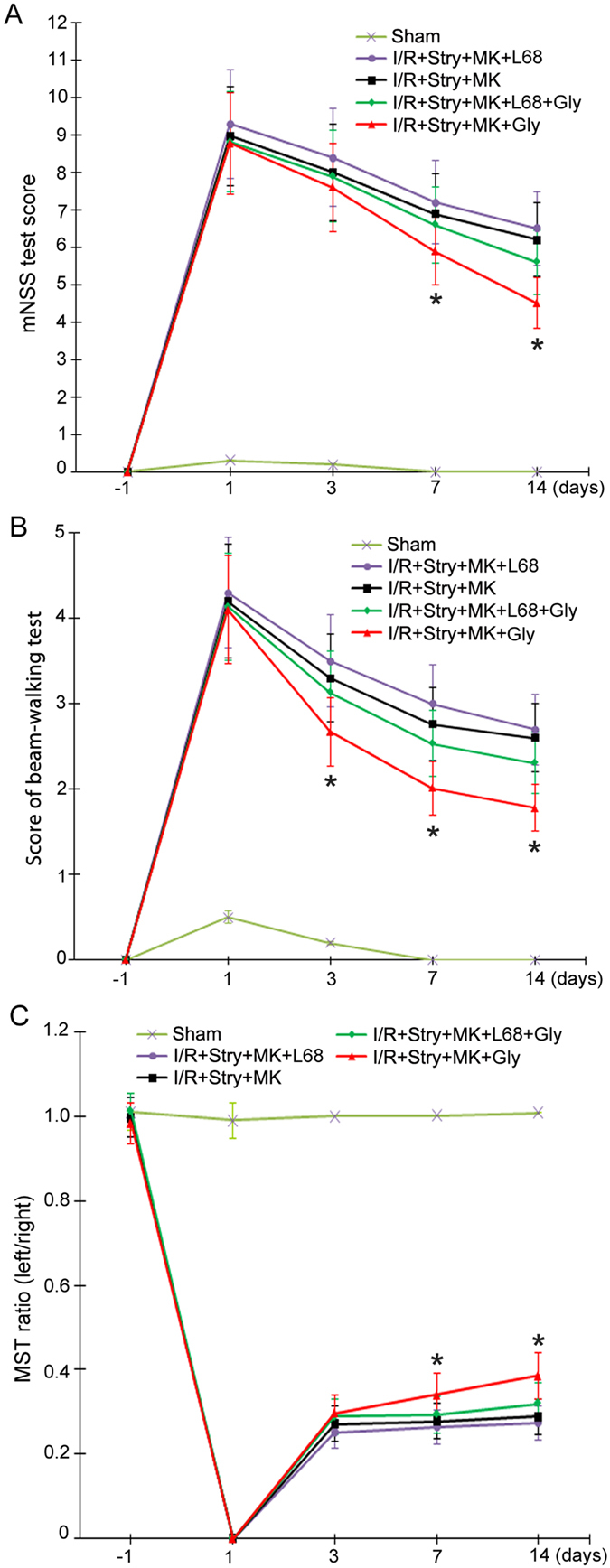
Glycine-GluN1 binding site antagonist prevents glycine-induced functional recovery of ischemic stroke animals. For all the three experiments, Glycine (100 µg/100 g, icv) was administered at 3 h following I/R. Glycine-GluN1 binding antagonist L-689560 (L68; 0.1 mg/100 g, ip) is injected with MK-801 and strychnine at 30 min before glycine injection. (A) Animals treated with glycine (100 µg/100 g, icv) has lower scores of mNSS test at day 7 and 14 compared with I/R + Stry + MK + L68 + Gly group (n = 10; ANOVA test, *P < 0.05, vs. I/R + Stry + MK + L68 + Gly). (B) Animals treated with glycine has lower scores of beam-walking test at day 3, 7 and 14 compared with I/R + Stry + MK + L68 + Gly group (n = 10; ANOVA test, *P < 0.05, vs. I/R + Stry + MK + L68 + Gly). (C) Animals treated with glycine (100 µg/100 g, icv) treatment has higher ratio in MST test at day 7 and 14 compared with I/R + Stry + MK + L68 + Gly group (n = 10; ANOVA test, *P < 0.05, vs. I/R + Stry + MK + L68 + Gly). Stry: Strychnine; Gly: glycine; MK: MK-801.
Glycine-elicited non-ionotropic activity of NMDARs confers functional recovery in ischemic stroke animals through Akt activation
To provide evidence for the role of Akt activation in mediating glycine-induced functional recovery in the ischemic stroke animals, we tested the effect of Akt inhibition in the same experimental conditions as described above31, 32. Akt inhibitor IV (100 μM, 2 μl, icv) was injected with MK-801 (8.0 µg/100 g) and strychnine (1.2 µg/100 g) at 30 min prior to glycine treatment. We found that Akt inhibitor IV blocked glycine-induced recovery of stroke animals after glycine receptors and NMDAR channel activities were inhibited (Fig. 7). These results support the conclusion that glycine triggers a non-ionotropic activation of NMDARs and induces a functional recovery in ischemic stroke animals via Akt activation.
Figure. 7.

Akt inhibitor IV prevents glycine-induced functional recovery of ischemic stroke animals. For all the three experiments, Glycine (100 µg/100 g, icv) was administered at 3 h following I/R. Akt inhibitor IV (100 µM, 2 µl, icv) is injected with MK-801 and strychnine at 30 min before glycine injection. (A) Animals treated with glycine (100 µg/100 g. icv) has lower scores of mNSS test at day 7 and 14 compared with I/R + Stry + MK + IV + Gly group (n = 10; ANOVA test, *P < 0.05, vs. I/R + Stry + MK + IV + Gly). (B) Animals treated with glycine has lower scores of beam-walking test at day 3, 7 and 14 compared with I/R + Stry + MK + IV + Gly group (n = 10; ANOVA test, *P < 0.05, vs. I/R + Stry + MK + IV + Gly). (C) Animals treated with glycine (100 µg/100 g, icv) treatment has higher ratio in MST test at day 7 and 14 compared with I/R + Stry + MK + IV + Gly group (n = 10; ANOVA test, *P < 0.05, vs. I/R + Stry + MK + IV + Gly). Stry: Strychnine; Gly: glycine; MK: MK-801.
Discussion
Glycine is a co-agonist of ionotropic NMDARs1. We have previously shown that glycine enhances the activation of cell survival-promoting kinase Akt in cultured cortical neurons in which both the channel activity of NMDARs and the glycine receptors are pre-inhibited16. The effect of glycine is reduced by shRNA-mediated knockdown of GluN2ARs, suggesting that a non-ionotropic activity of GluN2ARs mediates glycine-induced Akt activation. In support of this finding, glycine enhances Akt activation in HEK293 cells over-expressing GluN2ARs. The effect of glycine on Akt activation is sensitive to the antagonist of glycine-GluN1 binding site. As a functional consequence, glycine protects against glutamate excitotoxicity-induced neuronal death through the non-ionotropic activity of GluN2ARs and the neuroprotective effect is attenuated by Akt inhibition. Thus, we demonstrated a role of glycine in eliciting a non-ionotropic activity of GluN2ARs to confer neuroprotection via Akt activation in vitro 16.
Since glutamate-induced neurotoxicity is known to mediate neuronal death in ischemia-reperfusion injury, according to our in vitro findings we tested the effect of glycine in the rat middle cerebral artery occlusion (MCAO), an animal model of cerebral ischemia-reperfusion injury after the animals were injected with the NMDAR channel blocker MK-801 and the glycine receptor antagonist strychnine1, 8, 33. We demonstrate that glycine treatment reduces the infarct volume and improves the functional recovery of stroke animals. These data support our in vitro finding that glycine-induced neuroprotection is mediated in part by the non-ionotropic activity of NMDARs and provide a functional evidence for the neuroprotective role of non-ionotropic activity of NMDARs in cerebral ischemia-reperfusion injury. Due to the technical limitation in the in vivo stroke model, we were unable to provide direct evidence to determine whether the non-ionotropic activity of NMDARs is mediated by GluN2ARs and whether the non-ionotropic activity of GluN2ARs mediates glycine-induced neuroprotection in the animal model of cerebral ischemia-reperfusion injury. However, we showed that both glycine-GluN1 binding site antagonist and Akt inhibitor attenuated glycine-induced neuroprotection in the ischemic brain tissues where NMDAR channel activities and glycine receptors were inhibited, which is consistent with our in vitro findings. These data provide indirect evidence to support the role of non-ionotropic activity of GluN2ARs mediates glycine-induced neuroprotection in cerebral ischemia-reperfusion injury.
In the experimental conditions in which NMDAR channel activity and glycine receptors were inhibited, we found that glycine treatment prevented neuronal loss in ischemic penumbra at 7 d after ischemia-reperfusion, and that glycine promoted functional recovery of stroke animals. These results provide further evidence to support glycine as a potential neuroprotectant in stroke therapy.
How the non-ionotropic activity of NMDARs enhances Akt activation is unclear34, 35. It is possible that the C-terminal domain of NMDARs may mediate the effect of glycine on Akt activation. Indeed, recent study provides evidence that agonist binding to the NMDAR drives movement of its cytoplasmic domain independent of ion flow, and changes the interaction between the cytoplasmic domain and its downstream signaling such as protein phosphatase 1 (PP1) and calcium-calmodulin-dependent protein kinase II (CaMKII), which regulates synaptic depression36, 37.
Akt deactivation is believed to be a causal mediator of cell death38. Enhancement of Akt activity exerts pro-survival effect in neuronal injury and neurodegenerative diseases31, 39. In this study, we identify Akt as a downstream neuroprotective signal of glycine that activates non-ionotropic NMDARs. Akt is known to influence neuronal survival through activation or inhibition of substrates. For example, activated Akt promotes survival through phosphorylation of transcription factors forkhead/FOXO, NF-κ B and mdm2 or through phosphorylation of Bcl-2 family members Bad and Bim23. Further study is needed to determine which Akt-dependent signal pathway mediates the activation of non-ionotropic NMDARs by glycine.
NMDAR-mediated neurotoxicity induces neuronal death and neurodegeneration in various CNS disorders including ischemic stroke, traumatic brain injury and neurodegenerative diseases4, 9, 40–42. However, the use of NMDAR antagonists as neuroprotective agents was disappointing in clinical trials10. A simple possibility is that these antagonists, while suppressing NMDAR-mediated neurotoxicity, block the biological and/or neural survival-promoting effects of NMDARs. Thus, identification of molecular mechanisms by which specific NMDAR subtype selectively exerts its effect on neuronal survival or death would provide a critical basis for the development of potent therapy for CNS injuries and neurodegenerative diseases. Substantial evidence suggests that GluN2ARs and GluN2BRs play different role in neuronal survival or death4, 5, 9. But the underlying molecular mechanism remains unclear.
Recent studies demonstrate that NMDAR has non-ionotropic activity that plays an excitotoxic role12, 13, 15–18. A non-ionotropic activity is found to be mediated through GluN2BR and is required for β-amyloid–induced synaptic depression and synaptic loss43. The non-ionotropic activity of NMDARs is shown to drive structural shrinkage at spiny synapses and couple Src family kinases to pannexin-1 in excitotoxic injury14. These data provide new evidence for the involvement of GluN2BRs in mediating neurotoxicity. More recently, we report that glycine protects against glutamate neurotoxicity-induced neuron injury in cultured cortical neurons independent of the channel activity of GluN2ARs, indicating a non-ionotropic activation of GluN2ARs selectively by glycine16. The non-ionotropic activity of GluN2AR and GluN2BR explains in part why GluN2AR plays a different role than GluN2BR in neuronal survival. The neuroprotective effect of glycine in stroke animals observed in the present experimental conditions supports the involvement of non-ionotropic activity of GluN2ARs.
Experimental Procedures
General methods
Adult male Sprague-Dawley (SD) rats were housed in cages on a 12 h light/dark cycle in a temperature-controlled room (23 °C–25 °C) with free access to food and water. Animals were allowed at least 3 days to acclimatize before experimentation. All animal use and experimental protocols were approved and carried out in compliance with the IACUC guidelines and the Animal Care and Ethics Committee of Wuhan University School of Medicine. Randomization was used to assign samples to the experimental groups, and to collect and process data. The experiments were performed by investigators blinded to the groups for which each animal was assigned.
Focal cerebral ischemia and infarct measurement
Transient focal cerebral ischemia was induced using the suture occlusion technique. Male Sprague-Dawley rats weighing 250–300 g were anesthetized with 4% isoflurane in 70% N2O and 30% O2 using a mask. The animals were anesthetized during surgery and drug injection at different time points after MCAO. The Control and Sham animals were all subjected to the same procedures. A midline incision was made in the neck, the right external carotid artery (ECA) was carefully exposed and dissected, and a 3–0 monofilament nylon suture was inserted from the ECA into the right internal carotid artery to occlude the origin of the right middle cerebral artery (MCA) (approximately 22 mm). After 90 minutes of occlusion, the suture was removed to allow reperfusion, the ECA was ligated, and the wound was closed. Sham-operated rats underwent identical surgery and/or intracerebroventricular injections except that the suture was inserted and withdrawn immediately. Rectal temperature was maintained at 37.0 ± 0.5 °C using a heating pad and heating lamp. Rats were killed at various times after reperfusion after being anesthetized, and the brains were removed for TTC (2,3,5 -triphenyltetrazolium chloride) staining. The brain was placed in a cooled matrix and 2 mm coronal sections were cut. Individual sections were placed in 10 cm petri dishes and incubated for 30 min in a solution of 2% TTC in phosphate buffered saline at 37 °C. The slices were fixed in 4% paraformaldehyde at 4 °C for 24 h. All image collection, processing and analysis were performed in a blind manner and under controlled environmental lighting. The scanned images were analyzed using image analysis software (Image-Pro Plus Version 6.0, USA). The infarct volume was calculated to correct for edema. The normal volume of contralateral hemisphere and the normal volume of ipsilateral hemisphere were measured, and the infarct percentage was calculated as % contralateral structure to avoid mismeasurement secondary to edema44–46.
Intracerebroventricular administration
For intracerebroventricular injections, the rats were placed on ear bars of a stereotaxic instrument under anesthesia. Drug infusion to the cerebral ventricle (from the bregma: posterior, 0.8 mm; lateral, 1.5 mm; depth, 3.5 mm) was performed using a 23-gauge needle attached via polyethylene tubing to a Hamilton microsyringe at a rate of 1.0 μl/min. The injection needle was left in place for an additional 5 min to allow diffusion, it was then slowly withdrawn47, 48.
Fluoro Jade–C (FJC) Staining
Mice were anesthetized with an overdose of isoflurane, followed by intracardiac perfusion with normal saline, followed by 4% paraformaldehyde post-fixed at 4 °C for 24 h, and then transferred into 30% sucrose solution in 0.1 mol/L phosphate buffer at 4 °C for 72 h. The brains were removed by decapitation and kept overnight in 4% paraformaldehyde solution at 4 °C. Brains were cut into 20 μm coronal sections by a Leica VT1000S vibratome (Leica Micro-systems AG, Nussloch, Germany). FJC labelling was performed using a standard protocol with modification49. Slides were immersed in 1% sodium hydroxide in 80% ethanol for 5 min, followed by rinsing in 70% ethanol for 2 min, in distilled water for 2 min, and then incubated in 0.06% potassium permanganate solution for 10 min. After 2 min in distilled water, the slides were transferred into 0.0001% solution of FJC (Sigma Aldrich, USA) which was dissolved in 0.1% acetic acid. Slides were rinsed with distilled water for 1 min, three times, and then dried at 50 °C for 5 min, immersed in xylene for 1 min. The slices were mounted with DPX media (Sigma Aldrich, USA). The sections were photographed by a blinded investigator using an Olympus fluorescent microscope (IX51, Olympus, Japan). Series of microphotographs were taken from three region of the ipsilateral cerebral cortex, with a ×20 objective and FJC-positive cells were counted by ImageJ software (ImageJ, USA). The data were expressed as cells/mm2.
Western blotting analysis
Rats were killed at the indicated time points, and right ischemia penumbra were isolated, beginning 3 mm from the anterior tip of the frontal lobe, a 4 mm thick coronal slice was chosen, then made a longitudinal cut (from top to bottom) approximately 1 mm from the midline through right hemisphere, and then made a transverse diagonal cut at approximately the “2 o’clock” position to separate the core from the penumbra. The tissues were homogenized in lysis buffer, which contained a protease and phosphatase inhibitor cocktail. Protein concentration was determined using a bicinchoninic acid protein assay (Thermo Scientific). Samples were separated by a sodium dodecyl sulfate–polyacrylamide gel and transferred to a polyvinylidene difluoride membrane (Millipore). For the detection of phospho-Akt, the samples prepared in the same day were used. The polyvinylidene difluoride membrane (Millipore, Bedford, MA, USA) was incubated with primary antibody against phospho-Akt (Ser473) (Cell Signaling Technology, Beverly, MA) or Akt (Cell Signaling). Primary antibodies were labeled with horseradish peroxidase-conjugated secondary antibody, and protein bands were imaged using SuperSignal West Femto Maximum Sensitivity Substrate (Pierce, Rockford, IL, USA). The EC3 Imaging System (UVP, LLC, Upland, CA) was used to obtained blot images directly from the polyvinylidene difluoride membrane. For the detection of total Akt, the same polyvinylidene difluoride membrane was stripped and then reprobed with primary antibody against total Akt (Cell Signaling Technology). The quantification of Western blot data was performed using ImageJ software16, 50.
Neurological Severity Scores
The rats were subjected to a modified neurological severity score (mNSS) test as reported previously (Table 1). These tests are a battery of motor, sensory, reflex, and balance tests, which are similar to the contralateral neglect tests in humans. Neurological function was graded on a scale of 0 to 18 (normal score, 0; maximal deficit score, 18)51.
Beam walk test
The beam walk test measures the animals’ complex neuromotor function. The animal was timed as it walked a (90 × 4 × 1.5 cm) beam. A box for the animal to feel safe was placed at one end of the beam. A loud noise was created to stimulate the animal to walk toward and into the box. Scoring was based upon the time it took the rat to go into the box. The higher the score, the more severe is the neurological deficit (Table 2)52.
Adhesive-removal test
A modified sticky-tape (MST) test was performed to evaluate forelimb function. A sleeve was created using a 3.0 × 1.0-cm piece of yellow paper tape and was subsequently wrapped around the forepaw so that the tape attached to itself and allowed the digits to protrude slightly from the sleeve. The typical response is for the rat to vigorously attempt to remove the sleeve by either pulling at the tape with its mouth or brushing the tape with its contralateral paw. The rat was placed in its cage and observed for 30 s. Two timers were started: the first ran without interruption and the second was turned on only while the animal attempted to remove the tape sleeve. The ratio of the left (affected)/right (unaffected) forelimb performance was recorded. The contralateral and ipsilateral limbs were tested separately. The test was repeated three times per test day, and the best two scores of the day were averaged. The lower the ratio, the more severe is the neurological deficit29.
Statistics
Student’s t test or ANOVA test was used where appropriate to examine the statistical significance of the differences between groups of data. Newman–Keuls tests were used for post-hoc comparisons when appropriate. All results are presented as mean ± SE. Significance was placed at p < 0.05.
Electronic supplementary material
Acknowledgements
This work was supported by China Key Project of Basic Research (“973” Project; 2014CB541606), Natural Science Foundation of China (NSFC; 81470599) and Collaborative Innovation Center for Brain Science to Q.W.
Author Contributions
J.C., R.H., H.L., Y.Z., R.L., Z.Z. and Y.Z. performed Western blot experiments. R.H. and J.C. wrote the draft of the paper. Y.W. and P.J. designed the experiments. H.F. and Q.W. supervised the experiments and wrote the paper.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Juan Chen and Rong Hu contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-03909-0
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Juan Chen, Email: chenjuan0828@163.com.
Qi Wan, Email: qwanwh@hotmail.com.
References
- 1.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacological reviews. 1999;51:7–61. [PubMed] [Google Scholar]
- 2.Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Current opinion in neurobiology. 2001;11:327–335. doi: 10.1016/S0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- 3.Fan X, Jin WY, Wang YT. The NMDA receptor complex: a multifunctional machine at the glutamatergic synapse. Frontiers in cellular neuroscience. 2014;8:160. doi: 10.3389/fncel.2014.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anastasio NC, Xia Y, O’Connor ZR, Johnson KM. Differential role of N-methyl-D-aspartate receptor subunits 2A and 2B in mediating phencyclidine-induced perinatal neuronal apoptosis and behavioral deficits. Neuroscience. 2009;163:1181–1191. doi: 10.1016/j.neuroscience.2009.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayashi T, Thomas GM, Huganir RL. Dual palmitoylation of NR2 subunits regulates NMDA receptor trafficking. Neuron. 2009;64:213–226. doi: 10.1016/j.neuron.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foster KA, et al. Distinct roles of NR2A and NR2B cytoplasmic tails in long-term potentiation. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:2676–2685. doi: 10.1523/JNEUROSCI.4022-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ulbrich MH, Isacoff EY. Rules of engagement for NMDA receptor subunits. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:14163–14168. doi: 10.1073/pnas.0802075105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y, et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martel MA, et al. The subtype of GluN2 C-terminal domain determines the response to excitotoxic insults. Neuron. 2012;74:543–556. doi: 10.1016/j.neuron.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lipton SA. Failures and successes of NMDA receptor antagonists: molecular basis for the use of open-channel blockers like memantine in the treatment of acute and chronic neurologic insults. NeuroRx: the journal of the American Society for Experimental NeuroTherapeutics. 2004;1:101–110. doi: 10.1602/neurorx.1.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vissel B, Krupp JJ, Heinemann SF, Westbrook GL. A use-dependent tyrosine dephosphorylation of NMDA receptors is independent of ion flux. Nat Neurosci. 2001;4:587–596. doi: 10.1038/88404. [DOI] [PubMed] [Google Scholar]
- 12.Nabavi S, et al. Metabotropic NMDA receptor function is required for NMDA receptor-dependent long-term depression. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:4027–4032. doi: 10.1073/pnas.1219454110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kessels HW, Nabavi S, Malinow R. Metabotropic NMDA receptor function is required for beta-amyloid-induced synaptic depression. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:4033–4038. doi: 10.1073/pnas.1219605110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weilinger NL, et al. Metabotropic NMDA receptor signaling couples Src family kinases to pannexin-1 during excitotoxicity. Nat Neurosci. 2016;19:432–442. doi: 10.1038/nn.4236. [DOI] [PubMed] [Google Scholar]
- 15.Li LJ, et al. Glycine Potentiates AMPA Receptor Function through Metabotropic Activation of GluN2A-Containing NMDA Receptors. Frontiers in molecular neuroscience. 2016;9:102. doi: 10.3389/fnmol.2016.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu R, et al. Glycine triggers a non-ionotropic activity of GluN2A-containing NMDA receptors to confer neuroprotection. Scientific reports. 2016;6:34459. doi: 10.1038/srep34459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wexler EJ, et al. An objective procedure for ischemic area evaluation of the stroke intraluminal thread model in the mouse and rat. Journal of neuroscience methods. 2002;113:51–58. doi: 10.1016/S0165-0270(01)00476-9. [DOI] [PubMed] [Google Scholar]
- 18.Uluc, K., Miranpuri, A., Kujoth, G. C., Akture, E. & Baskaya, M. K. Focal cerebral ischemia model by endovascular suture occlusion of the middle cerebral artery in the rat. Journal of visualized experiments: JoVE, doi:10.3791/1978 (2011). [DOI] [PMC free article] [PubMed]
- 19.Deiana S, et al. MK-801-induced deficits in social recognition in rats: reversal by aripiprazole, but not olanzapine, risperidone, or cannabidiol. Behavioural pharmacology. 2015;26:748–765. doi: 10.1097/FBP.0000000000000178. [DOI] [PubMed] [Google Scholar]
- 20.Hurtubise JL, et al. MK-801-induced impairments on the trial-unique, delayed nonmatching-to-location task in rats: effects of acute sodium nitroprusside. Psychopharmacology. 2017;234:211–222. doi: 10.1007/s00213-016-4451-2. [DOI] [PubMed] [Google Scholar]
- 21.Kehne JH, Davis M. Strychnine increases acoustic startle amplitude but does not alter short-term or long-term habituation. Behavioral neuroscience. 1984;98:955–968. doi: 10.1037/0735-7044.98.6.955. [DOI] [PubMed] [Google Scholar]
- 22.Muth-Selbach U, et al. Antinociceptive effects of systemic lidocaine: involvement of the spinal glycinergic system. European journal of pharmacology. 2009;613:68–73. doi: 10.1016/j.ejphar.2009.04.043. [DOI] [PubMed] [Google Scholar]
- 23.Cronin JN, Bradbury EJ, Lidierth M. Laminar distribution of GABAA- and glycine-receptor mediated tonic inhibition in the dorsal horn of the rat lumbar spinal cord: effects of picrotoxin and strychnine on expression of Fos-like immunoreactivity. Pain. 2004;112:156–163. doi: 10.1016/j.pain.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 24.de Paula HM, Hoshino K. Potentiation of panic-like behaviors of the rat by subconvulsive doses of strychnine. Physiology & behavior. 2004;80:459–464. doi: 10.1016/j.physbeh.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 25.Vignes M, Collingridge GL. The synaptic activation of kainate receptors. Nature. 1997;388:179–182. doi: 10.1038/40639. [DOI] [PubMed] [Google Scholar]
- 26.Baptista V, Varanda WA. Glycine binding site of the synaptic NMDA receptor in subpostremal NTS neurons. Journal of neurophysiology. 2005;94:147–152. doi: 10.1152/jn.00927.2004. [DOI] [PubMed] [Google Scholar]
- 27.Javitt DC, Sershen H, Hashim A, Lajtha A. Inhibition of striatal dopamine release by glycine and glycyldodecylamide. Brain Res Bull. 2000;52:213–216. doi: 10.1016/S0361-9230(00)00258-6. [DOI] [PubMed] [Google Scholar]
- 28.Song YS, et al. The role of Akt signaling in oxidative stress mediates NF-kappaB activation in mild transient focal cerebral ischemia. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2008;28:1917–1926. doi: 10.1038/jcbfm.2008.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sughrue ME, et al. An improved test of neurological dysfunction following transient focal cerebral ischemia in rats. Journal of neuroscience methods. 2006;151:83–89. doi: 10.1016/j.jneumeth.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 30.Lim SH, et al. The functional effect of epigallocatechin gallate on ischemic stroke in rats. Acta neurobiologiae experimentalis. 2010;70:40–46. doi: 10.55782/ane-2010-1772. [DOI] [PubMed] [Google Scholar]
- 31.Rowe DD, et al. Human umbilical cord blood cells protect oligodendrocytes from brain ischemia through Akt signal transduction. The Journal of biological chemistry. 2012;287:4177–4187. doi: 10.1074/jbc.M111.296434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rowe DD, et al. Leukemia inhibitor factor promotes functional recovery and oligodendrocyte survival in rat models of focal ischemia. The European journal of neuroscience. 2014;40:3111–3119. doi: 10.1111/ejn.12675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsukawa N, et al. Therapeutic targets and limits of minocycline neuroprotection in experimental ischemic stroke. BMC neuroscience. 2009;10:126. doi: 10.1186/1471-2202-10-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tu W, et al. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell. 2010;140:222–234. doi: 10.1016/j.cell.2009.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dore K, Aow J, Malinow R. Agonist binding to the NMDA receptor drives movement of its cytoplasmic domain without ion flow. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:14705–14710. doi: 10.1073/pnas.1520023112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aow J, Dore K, Malinow R. Conformational signaling required for synaptic plasticity by the NMDA receptor complex. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:14711–14716. doi: 10.1073/pnas.1520029112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luo HR, et al. Akt as a mediator of cell death. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:11712–11717. doi: 10.1073/pnas.1634990100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stramiello M, Wagner JJ. D1/5 receptor-mediated enhancement of LTP requires PKA, Src family kinases, and NR2B-containing NMDARs. Neuropharmacology. 2008;55:871–877. doi: 10.1016/j.neuropharm.2008.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zausinger S, Hungerhuber E, Baethmann A, Reulen H, Schmid-Elsaesser R. Neurological impairment in rats after transient middle cerebral artery occlusion: a comparative study under various treatment paradigms. Brain research. 2000;863:94–105. doi: 10.1016/S0006-8993(00)02100-4. [DOI] [PubMed] [Google Scholar]
- 41.Kerchner GA, Nicoll RA. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nature reviews. Neuroscience. 2008;9:813–825. doi: 10.1038/nrn2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gu GJ, et al. Mechanism of ischemic tolerance induced by hyperbaric oxygen preconditioning involves upregulation of hypoxia-inducible factor-1alpha and erythropoietin in rats. Journal of applied physiology (Bethesda, Md. 1985) 2008;104:1185–1191. doi: 10.1152/japplphysiol.00323.2007. [DOI] [PubMed] [Google Scholar]
- 43.Tamburri A, Dudilot A, Licea S, Bourgeois C, Boehm J. NMDA-receptor activation but not ion flux is required for amyloid-beta induced synaptic depression. PloS one. 2013;8:e65350. doi: 10.1371/journal.pone.0065350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu F, Schafer DP, McCullough LD. TTC, fluoro-Jade B and NeuN staining confirm evolving phases of infarction induced by middle cerebral artery occlusion. Journal of neuroscience methods. 2009;179:1–8. doi: 10.1016/j.jneumeth.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buick R. When did oxygenic photosynthesis evolve? Philosophical transactions of the Royal Society of London. Series B, Biological sciences. 2008;363:2731–2743. doi: 10.1098/rstb.2008.0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swanson RA, et al. A semiautomated method for measuring brain infarct volume. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- 47.Raz L, et al. Role of Rac1 GTPase in NADPH oxidase activation and cognitive impairment following cerebral ischemia in the rat. PloS one. 2010;5:e12606. doi: 10.1371/journal.pone.0012606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vemuganti R, Dempsey RJ, Bowen KK. Inhibition of intercellular adhesion molecule-1 protein expression by antisense oligonucleotides is neuroprotective after transient middle cerebral artery occlusion in rat. Stroke; a journal of cerebral circulation. 2004;35:179–184. doi: 10.1161/01.STR.0000106479.53235.3E. [DOI] [PubMed] [Google Scholar]
- 49.Schmued LC, Stowers CC, Scallet AC, Xu LL. Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Research. 2005;1035:24–31. doi: 10.1016/j.brainres.2004.11.054. [DOI] [PubMed] [Google Scholar]
- 50.Ashwal S, Tone B, Tian HR, Cole DJ, Pearce WJ. Core and penumbral nitric oxide synthase activity during cerebral ischemia and reperfusion. Stroke; a journal of cerebral circulation. 1998;29:1037–1046. doi: 10.1161/01.STR.29.5.1037. [DOI] [PubMed] [Google Scholar]
- 51.Chen J, et al. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke; a journal of cerebral circulation. 2001;32:2682–2688. doi: 10.1161/hs1101.098367. [DOI] [PubMed] [Google Scholar]
- 52.Clifton GL, et al. Marked protection by moderate hypothermia after experimental traumatic brain injury. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1991;11:114–121. doi: 10.1038/jcbfm.1991.13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.