

Abstract
Switchgrass (Panicum virgatum L.) is a cellulosic biofuel feedstock and their effects on bacterial communities in deep soils remain poorly understood. To reveal the responses of bacterial communities to long-term switchgrass cultivation through the soil profile, we examined the shift of soil microbial communities with depth profiles of 0–60 cm in five-year switchgrass cultivation and fallow plots. The Illumina sequencing of the 16S rRNA gene showed that switchgrass cultivation significantly increased microbial OTU richness, rather than microbial Shannon diversity; however, there was no significant difference in the structure of microbial communities between switchgrass cultivation and fallow soils. Both switchgrass cultivation and fallow soils exhibited significant negative vertical spatial decay of microbial similarity, indicating that more vertical depth distant soils had more dissimilar communities. Specifically, switchgrass cultivation soils showed more beta-diversity variations across soil depth profile. Through network analysis, more connections and closer relationships of microbial taxa were observed in soils under switchgrass cultivation, suggesting that microbial co-occurrence patterns were substantially influenced by switchgrass cultivation. Overall, our study suggested that five-year switchgrass cultivation could generated more beta-diversity variations across soil depth and more complex inter-relationships of microbial taxa, although did not significantly shape the structure of soil microbial community.
Introduction
Switchgrass (Panicum virgatum L.) is a perennial C-4 grass with high photosynthetic efficiency and biomass production potential1. It has received considerable attentions during the last several decades since it is recognized as a promising crop for biofuel production by the US Department of Energy (DOE) Herbaceous Energy Crops Program (HECP)2–4. The widespread of this perennial biofuel crops could shift the land use towards the renewable, biomass-based energy systems, and influence the soil ecosystems subsequently5. Particularly, soil microbes can respond rapidly to the environmental changes caused by plant6, 7. The plant could regulate soil microbial community structure via root exudates8. Switchgrass can release up to 20% of fixed carbon to the rhizosphere through exudation9. Although agronomic knowledge of switchgrass has grown increasingly2, 4, however, their influence on soil microbial community still remain uncovered.
Soil microbes play fundamental roles in soil biogeochemical processes of the carbon, nitrogen, and inorganic element cycles10. The vast majority of researches on soil microbial communities have focused on the top 15 cm of the soil column or less, therefore our understanding of soil microbes is limited to surface horizons11, 12. The microbial biomass often exhibits exponential decreases with depth and is greatest in surface soil, while there is still a large population of microbes in the subsoil (below 15 cm) because of the large volume throughout the depth of soil profile11–14. Due to the proximity to parent material, the deeper microbes might play potential important roles in soil formation processes, ecosystem biochemistry and pollutant degradation, as well as in maintaining the quality of groundwater15–17. The sub-surface horizons can harbor a large amount of organic C during long-term turnover, associating with the activity of microbes in subsoil18, 19. Previous studies have showed that microbial communities could significantly change with soil depths, and the microbial diversity of microorganisms typically decreases with depth11, 20. However, the characterization and spatial variability of microbial communities at deeper soils remains poorly understood under the influence of Switchgrass growing.
Microorganisms form complex interaction webs within a specific ecological niche, and understanding the interactions among microorganisms is important to explore the complexity of functional processes21. Co-occurrence network analysis could provide comprehensive perspective into the complex microbial interactions, such as commensalism, competition and predation22. However, most of the previous studies on the microbial community structures have been conducted based on the technologies of clone library analysis, community-level physiological profiles, phospholipid fatty acids (PLFAs), terminal restriction fragment length polymorphism (T-RFLP) and denaturing gradient gel electrophoresis (DGGE). These methods provided limited information to comprehensively resolve the phylogenetic responses of microbial communities to environmental changes due to the small number of sequences analyzed23. Recently the next-generation sequencing technologies have made broad and deep surveys of microbial communities taking full advantage of the network analysis approaches24. It could grab rare species and describe the overall microbial community diversity25. Therefore, network analysis have been applied to explore the microbial co-occurrence patterns in diverse environments including marine water26, soil22, 27 and activated sludge28. While, we still have only a limited understanding of the effects of Switchgrass growing on the co-occurrence patterns of microbial communities.
In present study, we conducted a metagenomic analysis of soil microbial communities via high-throughput sequencing of the 16S rRNA gene to examine their shifts with depth profiles and switchgrass cultivation. Soil samples were obtained from a five-year switchgrass experimental area in the Guanzhong plain of Shaanxi Province. The objective was to investigate the responses of bacterial communities to long-term switchgrass cultivation within the soil profile. Specifically, we addressed the following detailed questions: (i) Do switchgrass cultivations change the structure of bacterial community throughout the depth of soil profile? (ii) How the bacterial communities shift with the soil depths? (iii) What are the co-occurrence patterns between bacterial taxa responding to switchgrass cultivation?
Results
Distribution of bacterial taxa
A data set of 1,116,764 quality sequences was produced from the soil samples in switchgrass cultivation and fallow plots with four soil depth profiles (Table 1), including 0–10 cm (Layer 1), 10–20 cm (Layer 2), 20–30 cm (Layer 3) and 30–60 cm (Layer 4). The mean number of sequences per sample (n = 24) was 46,532 (max = 58,071, min = 29,126, SD = 8941). The total OTU number was 12,547 defined by 97% sequence similarity. These OTUs were assigned to 64 phyla, 171 classes, 269 orders, 336 families and 526 genera. Of the OTUs, 99.5% (12,489 OTUs) were Bacteria and only 0.4% (54 OTUs) were affiliated with the Archaea domain. Five bacterial phyla including Proteobacteria, Acidobacteria, Bacteroidetes, Firmicutes and Gemmatimonadetes were predominant (relative abundance >5%), and accounted for 74.5% of the total sequences; and in addition, Planctomycetes, Actinobacteria, Verrucomicrobia, Chlorobi and Nitrospirae were present in most soil samples at low relative abundances (Fig. 1). At class level, Gammaproteobacteria (13.8%), Alphaproteobacteria (9.3%), Betaproteobacteria (6.7%) and Deltaproteobacteria (6.3%) were the dominant Proteobacteria with an overall relative abundance of 37.6%; Acidobacteria-6, Clostridia, Bacteroidia and Chloracidobacteria were also predominant groups, accounted for 22.0% of the total sequences in the soil samples (Supplementary Fig. S1). In general, Archaea accounted for 2.1% of all the sequences, and the the dominant taxa was Candidatus Nitrososphaera (phylum Crenarchaeota).
Table 1.
Microbial alpha-diversity characteristics for the fallow and switchgrass soils in four layers.
| Soil samples | No. of high quality reads | Diversity Index | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Observed OTU97 | Chao1 | Shannon | Coverage | ||||||||
| Average | SD* | Average | SD | Average | SD | Average | SD | Average | SD | ||
| Fallow | Layer 1 | 48925 | 7455 | 3786 | 770 | 5038.84 | 1423.12 | 10.11 | 0.44 | 0.95 | 0.02 |
| Layer 2 | 46956 | 10201 | 3480 | 857 | 4696.74 | 1529.37 | 9.64 | 0.99 | 0.96 | 0.02 | |
| Layer 3 | 43865 | 12645 | 3802 | 369 | 4924.32 | 1293.83 | 10.26 | 0.05 | 0.96 | 0.02 | |
| Layer 4 | 35819 | 5319 | 3005 | 413 | 3459.12 | 460.01 | 9.31 | 0.71 | 0.97 | 0.00 | |
| Switchgrass | Layer 1 | 51365 | 8028 | 4307 | 192 | 5866.10 | 653.67 | 10.41 | 0.15 | 0.95 | 0.01 |
| Layer 2 | 45093 | 13879 | 4018 | 871 | 5686.97 | 1263.94 | 10.09 | 0.72 | 0.95 | 0.01 | |
| Layer 3 | 47804 | 4410 | 4101 | 275 | 5538.59 | 451.94 | 10.35 | 0.14 | 0.95 | 0.01 | |
| Layer 4 | 52428 | 3250 | 3590 | 565 | 5050.41 | 678.51 | 8.69 | 1.35 | 0.95 | 0.01 | |
*Standard deviation.
Figure 1.
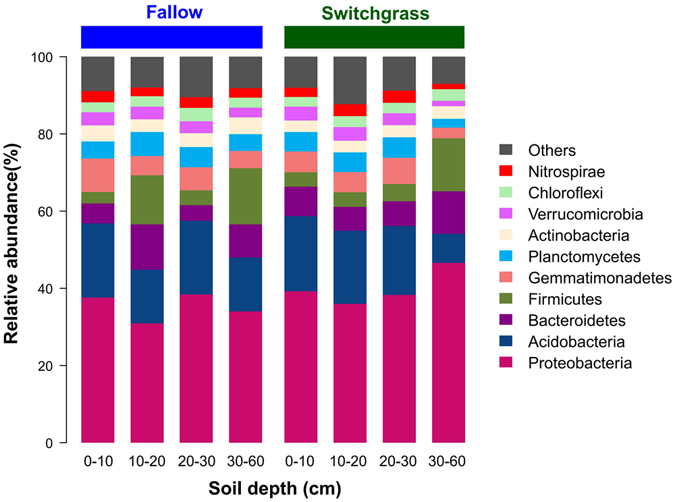
Relative abundances of bacterial phyla at 0–60 cm depth in switchgrass and fallow soils.
Assembly patterns of bacterial community
Microbial alpha-diversity was measured using the observed OTU richness and Shannon–Wiener index. The observed OTU richness was significantly higher in switchgrass cultivation soil samples than that in fallow soils, tested by Wilcoxon rank-sum test (P < 0.05; Supplementary Fig. S2). While, Shannon index did not significantly different between these two groups. On the other hand, we found Shannon index significantly decreased with soil depth in switchgrass cultivation plots (P < 0.05); whereas, this trend was not significant in fallow plots (Fig. 2). And the observed OTUs richness did not show significantly change with soil depth in either switchgrass cultivation or fallow plots.
Figure 2.

Changes in microbial diversity (Shannon index) with soil depth in switchgrass and fallow plots, estimated via linear regression.
The CAP analysis based on Bray–Curtis distance (Fig. 3A), demonstrated that bacterial community varied with depth, which were confirmed by ANOSIM (P < 0.05). The Canonical discriminant analysis (CDA) of the predominant microbial taxa (relative abundance >0.5%) at genus levels revealed taxonomic associations with soil depth (Fig. 3B). Different layers of soil profiles distinguished specific microbial taxa. In layer 1, Aquicella, Kaistobacter, Sphingomonas and Gemmata were the abundant genera; Steroidobacter and Candidatus Nitrososphaera were dominant in soils of layer 2; Lysobacter, Pirellula, Nitrospira and Planctomyces were dominant in layer 3; Halomonas, Shewanella and Ruminococcus were abundant genera in soils of layer 4.
Figure 3.

Microbial distribution patterns varied with soil depth in switchgrass and fallow plots. (A) Constrained analysis of the principal coordinates (CAP) of microbial communities following four soil layers of different plots (Layer 1: 0–10 cm, Layer 2: 10–20 cm, Layer 3: 20–30 cm and Layer 4: 30–60 cm) based on the Bray–Curtis distance. (B) Canonical discriminant analysis (CDA) comparing soil layers against microbial taxa loadings based on genera with relative abundance levels >0.5%. Arrows represent the degree of correlation between each taxon and each layer as a measure of the predictive discrimination of each layer.
There was no significant difference in the structure of microbial communities between switchgrass cultivation and fallow soils, either in integrate soil profiles (ANOSIM P = 0.113; PERMANOVA P = 0.203) or each single layer. While some significant taxonomic differences between these two groups soils were examined by Wilcoxon rank-sum test (P < 0.05) based on the top 1000 most abundant OTUs (Supplementary Figs S3 and S4). For example, Novosphingobium, Fluviicola, Flavobacterium, Alcanivorax, Shewanella and Sorangium were significantly higher in relative abundance in soils with switchgrass cultivation; whereas, the abundance of families Rhodospirillaceae and Gaiellaceae, and the genera Gemmata and Pilimelia increased significantly in fallow soils.
Vertical spatial variations of bacterial community
To investigate the vertical spatial variations of bacterial community, we estimated the relationships between soil depth profiles and bacterial community similarities based on Bray–Curtis distance (Fig. 4). The significant negative vertical spatial decay of bacterial community relationships were found in linear regression for both switchgrass cultivation and fallow soils, indicating that more vertical depth distant soils had more dissimilar communities. In particular, the switchgrass cultivation soils had a steeper slope, indicating that there were more beta-diversity variations with the increased vertical depth under switchgrass cultivation.
Figure 4.
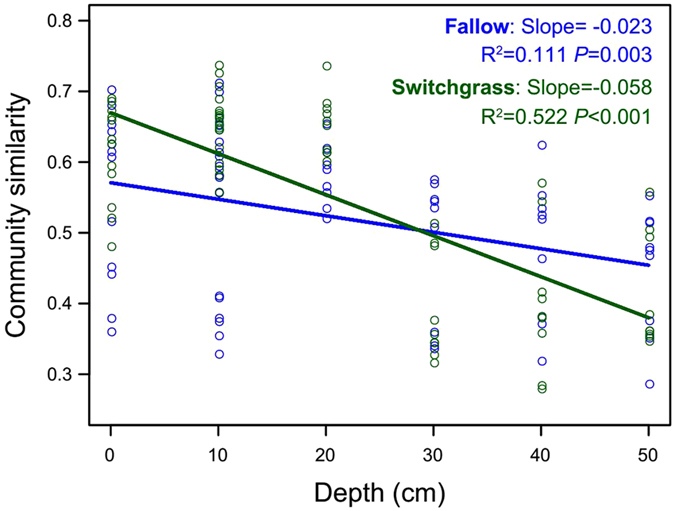
The vertical spatial variations of bacterial community similarity in switchgrass and fallow soils, estimated the relationships between soil depth profiles and microbial community similarities based on Bray–Curtis distance. The lines denote the least-squares linear regressions across soil depth.
To further explore vertical spatial variations of the dominated bacterial taxa, we estimated the correlations between relative abundance of these taxa and soil depths via Pearson coefficient (Supplementary Table S1 and Table 2). In fallow soils, phylum Crenarchaeota and classes Gemmatimonadetes, Thaumarchaeota, Saprospirae and Cytophagia were significantly and negatively correlated with soil depth; and classes Gammaproteobacteria were positively correlated with soil depth. For switchgrass cultivation soils, as soil depth increased, the relative abundances of phyla Acidobacteria, Verrucomicrobia and Armatimonadetes were significantly decreased in switchgrass cultivation soils; while, the abundance of Firmicutes and Cyanobacteria significantly increased. At class level, Gammaproteobacteria, Clostridia and Bacteroidia were significantly and positively correlated with soil depth; and Acidobacteria-6, Betaproteobacteria, Chloracidobacteria, Pedosphaerae, Cytophagia and Saprospirae were negatively correlated with soil depth. Additionally, the significant taxa were more in switchgrass cultivation soils than in fallow soils, confirmed more beta-diversity variations under switchgrass cultivation.
Table 2.
The vertical spatial variations of the dominated microbial taxa at class level in the fallow and switchgrass soils, correlations between relative abundance of these taxa and soil depths were estimated via Pearson coefficient.
| Class | Fallow | Switchgrass | ||
|---|---|---|---|---|
| Pearson coefficient | P value | Pearson coefficient | P value | |
| Gammaproteobacteria | 0.598 | 0.040 | 0.603 | 0.038 |
| Alphaproteobacteria | −0.372 | 0.233 | −0.393 | 0.207 |
| Acidobacteria-6 | −0.186 | 0.562 | −0.747 | 0.005 |
| Betaproteobacteria | −0.541 | 0.069 | −0.627 | 0.029 |
| Clostridia | 0.272 | 0.393 | 0.632 | 0.028 |
| Deltaproteobacteria | −0.505 | 0.094 | −0.476 | 0.118 |
| Bacteroidia | 0.197 | 0.539 | 0.593 | 0.042 |
| Chloracidobacteria | −0.264 | 0.407 | −0.699 | 0.011 |
| Planctomycetia | −0.057 | 0.861 | −0.490 | 0.105 |
| Nitrospira | −0.085 | 0.792 | −0.384 | 0.218 |
| Gemmatimonadetes | −0.678 | 0.015 | −0.468 | 0.125 |
| Pedosphaerae | −0.269 | 0.398 | −0.636 | 0.026 |
| Thaumarchaeota | −0.708 | 0.010 | −0.271 | 0.394 |
| Cytophagia | −0.629 | 0.028 | −0.671 | 0.017 |
| PRR-12 | 0.401 | 0.196 | −0.331 | 0.294 |
| Saprospirae | −0.757 | 0.004 | −0.767 | 0.004 |
| Gemm-1 | 0.411 | 0.184 | −0.084 | 0.794 |
| Anaerolineae | 0.101 | 0.755 | 0.255 | 0.423 |
| iii1-8 | −0.728 | 0.007 | −0.494 | 0.102 |
| Actinobacteria | 0.389 | 0.212 | 0.536 | 0.073 |
Co-occurrence network analysis
The soil microbial networks were generated for switchgrass cultivation and fallow soils, respectively (Fig. 5). The topological properties were calculated to describe the complex pattern of inter-relationships among nodes, and to distinguish differences in taxa correlations between these two group soils (Table 3). Specifically, the structural properties of the switchgrass network were greater than the fallow network, indicating more connection and closer relationships of microbial taxa under switchgrass cultivation.
Figure 5.
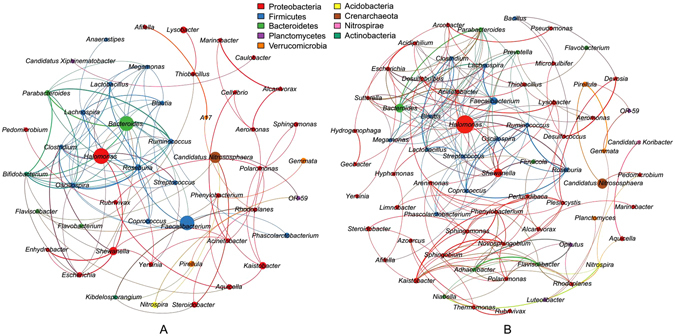
Network of co-occurring microbial genera based on correlation analysis for fallow (A) and switchgrass cultivation (B) soils. A connection stands for a strong (Spearman’s ρ > 0.6) and significant (P < 0.01) correlation. The size of each node is proportional to the relative abundance; the thickness of each connection between two nodes (edge) is proportional to the value of Spearman’s correlation coefficients. The nodes were colored by phylum.
Table 3.
Topological properties of co-occurring networks obtained from switchgrass cultivation and fallow soils.
| Nodes | Edges | Modularity (MD) | Clustering coefficient (CC) | Average path length (APL) | Network diameter (ND) | Average degree (AD) | |
|---|---|---|---|---|---|---|---|
| Fallow | 49 | 113 | 0.456 | 0.565 | 2.608 | 4.764 | 2.306 |
| Switchgrass | 69 | 204 | 0.577 | 0.592 | 5.081 | 11.537 | 2.957 |
Based on betweenness centrality scores, the top five genera identified as keystone taxa were Arenimonas, Clostridium, Thiobacillus, Lysobacter and Nitrospira in fallow network; Shewanella, Acinetobacter, Rhodoplanes, Aeromonas and Bacteroides were keystone taxa in switchgrass network. The keystone taxa differed greatly between these two networks. Furthermore, betweenness centrality of the switchgrass network was much stronger than that of the fallow network (P < 0.05, Wilcoxon rank-sum test; Supplementary Fig. S5), which could confirm that switchgrass network have more complex inter-relationships of microbial taxa.
Discussion
As a high photosynthetic efficiency and biofuel production potential perennial C-4 grass, the widespread planting of switchgrass might provide great economic value. However, whether switchgrass cultivation influences the soil ecosystems, particularly in the deep soils profiles, still remain uncovered. The present study aimed to reveal the responses of microbial communities to long-term switchgrass cultivation within the soil profiles of 0–60 cm. Our results showed that switchgrass cultivations did not significantly change the structure of soil microbial community, but generated more beta-diversity variations across soil depth and more complex inter-relationships of microbial taxa.
Plant could regulate soil microbial community structure through the root architecture, exudates, and mucilage8. The rhizodeposits from plant roots appear to be a major driving force in the regulation of microbial diversity and activity29–31. Previous study revealed that switchgrass could enrich specific microbial species in the rhizosphere, which were able to utilize root exudates5. However, we did not observe significant difference in bacterial communities between switchgrass cultivation and fallow soils. It might be explained that the affected zones of roots are small, and plants might not be enough to influence the whole soil ecosystems. In our study, the soils were obtained from a five-year switchgrass cultivation area. Thus, our results suggest that long-term switchgrass cultivations could not significantly change the structure of soil microbial communities. In other context, switchgrass cultivation caused some specific taxonomic differences compared with the fallow soils. The enriched taxa in soils with switchgrass cultivation were mainly affiliated with Proteobacteria, Bacteroidetes and Acidobacteria (Supplementary Fig. S3). Previous study reported that Proteobacteria and Acidobacteria were the dominant members in the switchgrass rhizosphere soils5. Particularly, Proteobacteria were active utilizers of fresh photosynthate; while, Acidobacteria preferred to complex organic matter, rather than simple root-derived dissolved organic carbon5. Soil Bacteroidetes were typically copiotrophic and were most abundant in nutrient rich soils, including rhizosphere soils32. Additionally, we found that genera Novosphingobium, Fluviicola and Flavobacterium were enriched under switchgrass cultivation. Novosphingobium and Flavobacterium were the dominant root exudate utilizers in switchgrass rhizosphere reportedly32. Fluviicola was isolated as an endophytic bacterium through addition of plant extract to nutrient media33. Root exudates are a key factor in shaping microbiome, and the ability to utilize root exudates is an important trait that allows microorganisms to be competitive in the rhizosphere5. Additionally, soil microbiome–plant feedback mechanisms are closely associated with ecosystem function and primary productivity in terrestrial habitats34, 35. Switchgrass has been reported to require much less fertilizer input and to generate high yields compared to many other crops36, 37. The enriched microbial taxa in switchgrass cultivation soils were selectively assembled, and might be benefit of plant growth and health. These beneficial microbes might support nutrients for the high annual biomass production of switchgrass, which usually referred to as plant growth promoting rhizobacteria (PGPR). In our study, the switchgrass cultivation enriched bacteria belonged to Flavobacterium, Xanthomonadaceae and Pseudomonadaceae were reported as PGPR38, 39.
Previous work demonstrated that the diversity of microorganisms typically decreases with soil depth12, 14. In present study, we only found that microbial Shannon diversity significantly decreased with soil depth in switchgrass cultivation plots, while not in fallow plots. Switchgrass cultivation might provide nutrients via root exudates, which might different across the soil profiles due to the length of root. This could be supported by another work of our lab, which was conducted in the same experimental area (manuscript submitted). My colleagues found that soil organic carbon was found significantly higher in switchgrass cultivation soils than that in fallow soils through soil layers (Supplementary Fig. S6). This could also explain that the microbial richness was significantly higher under switchgrass cultivation.
Soil depth had a highly significant effect on the structure of microbial communities, especially in the switchgrass cultivation plots. Both switchgrass cultivation and fallow soils exhibited significant negative vertical spatial decay of microbial community similarity relationships, and the switchgrass cultivation soils had a steeper slope (Fig. 4). This indicated that more vertical depth distant soils had more dissimilar communities, and switchgrass cultivation generated more beta-diversity variations across soil depth. Switchgrass cultivation could provide different kinds of nutrients via root exudates, resulting in the complex environmental heterogeneity throughout the soil depth. Higher amplitude of variation in environmental conditions could explain the high variations in beta-diversity40. Previous researches showed that the subsoil microbial communities were distinct from topsoil communities11, 12, 17. In present study, the relative abundance of Firmicutes, Cyanobacteria, Gammaproteobacteria and Bacteroidia increased with soil depth. Some observed changes was similar to other studies11, 20, 41. Firmicutes can survive in extreme environments, and Cyanobacteria generally occur in harsh desert environments42. Gammaproteobacteria were likely to promote plant and root growth by fixing nitrogen and producing growth hormones43. On the other hand, the relative abundance of Acidobacteria, Verrucomicrobia, Crenarchaeota, Betaproteobacteria and Gemmatimonadetes decreased as soil depth increases. Previous works reported that Acidobacteria was negatively correlated with pH, which was increased with soil depths14, 44, 45. Crenarchaeota, dominated by class Thaumarchaeota in our study, is widespread speculation of driving the autotrophic nitrification46. Soil Verrucomicrobia were oligotrophic and able to grow under conditions of low C availability47. While ecological niches inhabited by Crenarchaeota and Verrucomicrobia remain largely undetermined12.
Although the entire soil microbial communities were not significantly changed, the microbial inter-relationships were substantially influenced by switchgrass cultivation. Through co-occurrence network analysis, we found that structural properties of the switchgrass network were greater than the fallow network, indicating more connection and closer relationships of microbial taxa under switchgrass cultivation. Comparing network-level topological features can provide us with insight into variations in the co-occurrence patterns between different communities48. Additionally, betweenness centrality of the switchgrass network was much stronger than that of the fallow network, which could confirmed more complex inter-relationships of microbial taxa under switchgrass cultivation. Discerning the modules maintaining the connectivity in network, betweenness centrality represents the potential of an individual node influence on the interactions of other nodes in the network, and has been used to define the keystone species in the ecosystems49–52. High betweenness centrality value indicates a core and central location of this node in the network, whereas low betweenness centrality value indicates a more peripheral location48. Switchgrass could secrete root exudates to the soil ecosystems, including sugars, amino acids and other organic acids53, which can be easily utilized by complex microbial communities. This might be supported by higher values of soil organic carbon under switchgrass cultivation (Supplementary Fig. S6). For microorganisms, wide niches can support the coexistence of species within the communities54. In this case, plants could supply carbon (C) to soil generating intense microbial activities and interactions55. In previous study, rhizosphere networks for wild oat were more complex than those in surrounding soils, indicating the rhizosphere has a greater potential for interactions and niche-sharing56. Roots might promote the development of niches populated by dominant taxa, which would concurrently yield greater interactions, greater co-variations due to shared niches, and overall result in more complex co-occurrence patterns over time. Conversely, the complex microbial interactions including cooperative or syntrophic interactions among PGPRs might also be benefit for plant growth and health. Microorganisms can communicate with each other through various signal molecules57. Specially, rhizosphere microorganisms are more competent at producing signal molecules58, which might enhance the microbial feedback with plants.
Conclusion
Overall, our results showed that soil depth had a highly significant effect on the bacterial communities. Both switchgrass cultivation and fallow soils exhibited the significant negative vertical spatial decay of bacterial similarity relationships. Some dominated taxa regularly changed across soil profiles. However, five-year switchgrass cultivations did not significantly change the structure of soil bacterial community, but generated more beta-diversity variations across soil depth. Furthermore, the bacterial co-occurrence patterns were substantially influenced by switchgrass cultivation. More connection and closer relationships of bacterial taxa were observed in soils under switchgrass cultivation. In future works, more complete information of microbial taxonomic and functional data should be integrated to better understand of the microbial ecology of the soil profile and their response to long-term switchgrass cultivation.
Materials
Study area and soil sampling
The switchgrass experiment was carried out over the period 2011–2015 in an experimental area of Northwest A&F University, located in the Guanzhong plain of Shaanxi Province (Fig. 1). The soil series was a clay loam. Switchgrass (cultivars Cave-in-rock and Sunburst) plots were established in September 2011, where winter wheat was cultivated before. Switchgrass was sown into the plots at a seeding rate of 11.2 kg pure live seed ha−1 and fertilized with 56 kg N ha−1. The fallow plots were adjacent to the switchgrass plots. Both plots were rain fed and no irrigation. After planting, no weed control and no additional fertilizers were applied. The research plots for switchgrass and fallow were 5 × 6 m and replicated three times.
Soil samples were randomly collected from the field in each switchgrass and fallow plots on October 15, 2015. Soil cores were collected with a core sampler at four depths (0–10, 10–20, 20–30 and 30–60 cm). In total, twenty-four soil samples (two plots × four depths × three replicates) were collected, transported to the laboratory in sterile plastic bags on dry ice, and then stored at −80 °C for microbial analyses.
DNA extraction and purification
Community DNA was extracted from 0.5 g of soil samples using the MP FastDNA®SPIN Kit for soil (MP Biochemicals, Solon, OH, USA) according to the manufacturer protocol. The V4 hypervariable regions of the 16S rRNA gene was amplified using primers 515 F (5′-GTG CCA GCM GCC GCG GTA A-3′) and 806 R (5′-GGA CTA CHV GGG TWT CTA AT-3′), with the forward primer modified to contain a unique 6 nt barcode at the 5′ end. All PCR reactions were performed with 30 μl system with 15 μL of Phusion® High-Fidelity PCR Master Mix (New England Biolabs), 0.2 μM of forward and reverse primers and about 10 ng template DNA. The thermal cycling conditions as following: initial denaturation at 98 °C for 1 min, followed by 30 cycles of denaturation at 98 °C for 10 s, annealing at 50 °C for 30 s, and extension at 72 °C for 60 s, and an extension step at 72 °C for 5 min after cycling was complete. All samples were amplified in triplicate, and no-template controls were included in all steps of the process. Triplicate PCR amplicons were pooled together and then mixed with the same volume of 1 × loading buffer (containing SYB green). They were detected by electrophoresis in a 2% (w/v) agarose gel. PCR products with bright bands were mixed in equal density ratios and purified with GeneJET Gel Extraction Kit (Thermo Scientific, MA, USA). The purified PCR amplicons were sequenced using the Illumina HiSeq 2500 platform at Novogene Bioinformatics Technology Co., Ltd. (Beijing, China).
Sequence analysis of the 16S rRNA amplicons
Paired-end reads were merged using FLASH (V1.2.7, http://ccb.jhu.edu/software/FLASH/), and filtered according to the literature59. The acquired sequences were chimera detected and removed using USEARCH software based on the UCHIME algorithm60. The sequences were assigned to each sample with the unique barcodes. Sequence analysis was performed by the UPARSE software package using the UPARSE-OTU and UPARSE-OTUref algorithms. Operational taxonomic units (OTUs) were clustered at the 97% similarity level61. Singletons were removed from downstream analyses. The representative sequences for each OTU were assigned to their taxonomic group using the RDP classifier at an 80% confidence threshold59.
Data analyses
Alpha and beta diversity were calculated based on 29126 reads per sample (minimum number of sequences required to normalize the differences in sequencing depth) using QIIME (http://qiime.org/index.html), with multiple indices (observed species and Shannon-Wiener index) and the Bray-Curtis distance between samples. Constrained analysis of principal coordinates (CAP) based on Bray-Curtis distance was performed to investigate the relationship between microbial community composition and soil depth under switchgrass and fallow plots. Canonical discriminant analysis (CDA) was used to identify the taxa associated with different soil layers based on genera with relative abundance levels >0.5%. ANOSIM62 and permutational multivariate analysis of variance (PERMANOVA)63 were performed to determine whether samples from each groups contained significant differences in their species diversity. The vertical spatial decay of microbial similarity was calculated as the linear least-squares regression relationships between soil depth and the microbial similarity (based on 1 – dissimilarity of the Bray-Curtis distance metric).
Network was used to explore co-occurrence patterns of microbial taxa within switchgrass and fallow soils. The genera with relative abundances above 0.05% were selected. A Spearman’s correlation between two genera was considered statistically robust if the Spearman’s correlation coefficient (ρ) was >0.6 and the P-value was <0.0122. All the robust correlations identified from pairwise comparison of the genera abundance form a correlation network where each node represents one genus, and each edge stands for a strong and significant correlation between the nodes. To describe the topology of the resulting networks, a set of measures (number of nodes and edges, average path length, network diameter, average degree, clustering coefficient and modularity) was calculated using igraph64 packages in R environment and networks were visualized using the interactive platform Gephi65–67. The betweenness centrality values of each node were estimated. This topological feature indicated the relevance of a node as capable of holding together communicating nodes, were used to define the keystone species49, 52.
All statistics analyses were performed in R environment (http://www.r-project.org) unless otherwise indicated.
Electronic supplementary material
Acknowledgements
This study was supported by the National Natural Science Foundation of China (31402128), the Natural Science Foundation of Shaanxi Province of China (2016JQ3012), PhD Research Start-up Funds (Z111020312) from Northwest A&F University and the Fundamental Research Funds for the Central Universities (2014YB007) of Northwest A&F University.
Author Contributions
S.H., M.L. and T.H. conceived and designed the experiment, S.H., L.G., F.M. and M.Y. performed experiments, and S.J. did the data analysis. S.H. worte the manuscript. All authors have read the manuscript and agree with its content.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-03778-7
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Tianming Hu, Email: hutianming@126.com.
Mingxiu Long, Email: longmingxiu@nwsuaf.edu.cn.
References
- 1.Bouton JH. Molecular breeding of switchgrass for use as a biofuel crop. Current opinion in genetics & development. 2007;17:553–558. doi: 10.1016/j.gde.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 2.Parrish DJ, Fike JH. The biology and agronomy of switchgrass for biofuels. BPTS. 2005;24:423–459. doi: 10.1080/07352680500316433. [DOI] [Google Scholar]
- 3.Sanderson MA, Adler PR, Boateng AA, Casler MD, Sarath G. Switchgrass as a biofuels feedstock in the USA. Can J Plant Sci. 2006;86:1315. doi: 10.4141/P06-136. [DOI] [Google Scholar]
- 4.Wright L, Turhollow A. Switchgrass selection as a “model” bioenergy crop: a history of the process. Biomass Bioenerg. 2010;34:851–868. doi: 10.1016/j.biombioe.2010.01.030. [DOI] [Google Scholar]
- 5.Mao Y, Li X, Smyth EM, Yannarell AC, Mackie RI. Enrichment of specific bacterial and eukaryotic microbes in the rhizosphere of switchgrass (Panicum virgatum L.) through root exudates. Environmental microbiology reports. 2014;6:293–306. doi: 10.1111/1758-2229.12152. [DOI] [PubMed] [Google Scholar]
- 6.Mao Y, Yannarell AC, Davis SC, Mackie RI. Impact of different bioenergy crops on N‐cycling bacterial and archaeal communities in soil. Environ Microbiol. 2013;15:928–942. doi: 10.1111/j.1462-2920.2012.02844.x. [DOI] [PubMed] [Google Scholar]
- 7.Yannarell AC, Busby RR, Denight ML, Gebhart DL, Taylor SJ. Soil bacteria and fungi respond on different spatial scales to invasion by the legume Lespedeza cuneata. Frontiers in microbiology. 2011;2:127. doi: 10.3389/fmicb.2011.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh BK, Millard P, Whiteley AS, Murrell JC. Unravelling rhizosphere–microbial interactions: opportunities and limitations. Trends Microbiol. 2004;12:386–393. doi: 10.1016/j.tim.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen C. Rhizodeposition of organic C by plants: mechanisms and controls. Agronomie. 2003;23:375–396. doi: 10.1051/agro:2003011. [DOI] [Google Scholar]
- 10.Falkowski PG, Fenchel T, Delong EF. The microbial engines that drive Earth’s biogeochemical cycles. Science. 2008;320:1034–1039. doi: 10.1126/science.1153213. [DOI] [PubMed] [Google Scholar]
- 11.Li C, Yan K, Tang L, Jia Z, Li Y. Change in deep soil microbial communities due to long-term fertilization. Soil Biol Biochem. 2014;75:264–272. doi: 10.1016/j.soilbio.2014.04.023. [DOI] [Google Scholar]
- 12.Eilers KG, Debenport S, Anderson S, Fierer N. Digging deeper to find unique microbial communities: the strong effect of depth on the structure of bacterial and archaeal communities in soil. Soil Biol Biochem. 2012;50:58–65. doi: 10.1016/j.soilbio.2012.03.011. [DOI] [Google Scholar]
- 13.Hartmann M, Lee S, Hallam SJ, Mohn WW. Bacterial, archaeal and eukaryal community structures throughout soil horizons of harvested and naturally disturbed forest stands. Environ Microbiol. 2009;11:3045–3062. doi: 10.1111/j.1462-2920.2009.02008.x. [DOI] [PubMed] [Google Scholar]
- 14.Will C, et al. Horizon-specific bacterial community composition of German grassland soils, as revealed by pyrosequencing-based analysis of 16S rRNA genes. Appl Environ Microb. 2010;76:6751–6759. doi: 10.1128/AEM.01063-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buss H, et al. The coupling of biological iron cycling and mineral weathering during saprolite formation, Luquillo Mountains, Puerto Rico. Geobiology. 2005;3:247–260. doi: 10.1111/j.1472-4669.2006.00058.x. [DOI] [Google Scholar]
- 16.Madsen, E. L. Impacts of agricultural practices on subsurface microbial ecology. Advances in agronomy (USA) (1995).
- 17.Fierer N, Schimel JP, Holden PA. Variations in microbial community composition through two soil depth profiles. Soil Biol Biochem. 2003;35:167–176. doi: 10.1016/S0038-0717(02)00251-1. [DOI] [Google Scholar]
- 18.Rumpel C, Kögel-Knabner I. Deep soil organic matter—a key but poorly understood component of terrestrial C cycle. Plant Soil. 2011;338:143–158. doi: 10.1007/s11104-010-0391-5. [DOI] [Google Scholar]
- 19.Fierer N, Chadwick OA, Trumbore SE. Production of CO2 in soil profiles of a California annual grassland. Ecosystems. 2005;8:412–429. doi: 10.1007/s10021-003-0151-y. [DOI] [Google Scholar]
- 20.Li C-h, Tang L-s, Jia Z-j, Li Y. Profile Changes in the Soil Microbial Community When Desert Becomes Oasis. PloS one. 2015;10:e0139626. doi: 10.1371/journal.pone.0139626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faust K, Raes J. Microbial interactions: from networks to models. Nat Rev Microbiol. 2012;10:538–550. doi: 10.1038/nrmicro2832. [DOI] [PubMed] [Google Scholar]
- 22.Barberán A, Bates ST, Casamayor EO, Fierer N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012;6:343–351. doi: 10.1038/ismej.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Y, et al. Successful bioremediation of an aged and heavily contaminated soil using a microbial/plant combination strategy. J Hazard Mater. 2014;264:430–438. doi: 10.1016/j.jhazmat.2013.10.071. [DOI] [PubMed] [Google Scholar]
- 24.Berry D, Widder S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front Microbiol. 2014;5:219. doi: 10.3389/fmicb.2014.00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liao X, et al. Pyrosequencing analysis of bacterial communities in drinking water biofilters receiving influents of different types. Process Biochem. 2013;48:703–707. doi: 10.1016/j.procbio.2013.02.033. [DOI] [Google Scholar]
- 26.Beman JM, Steele JA, Fuhrman JA. Co-occurrence patterns for abundant marine archaeal and bacterial lineages in the deep chlorophyll maximum of coastal California. ISME J. 2011;5:1077–1085. doi: 10.1038/ismej.2010.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiao S, et al. Bacterial communities in oil contaminated soils: Biogeography and co-occurrence patterns. Soil Biol Biochem. 2016;98:64–73. doi: 10.1016/j.soilbio.2016.04.005. [DOI] [Google Scholar]
- 28.Ju F, Xia Y, Guo F, Wang Z, Zhang T. Taxonomic relatedness shapes bacterial assembly in activated sludge of globally distributed wastewater treatment plants. Environ Microbiol. 2014;16:2421–2432. doi: 10.1111/1462-2920.12355. [DOI] [PubMed] [Google Scholar]
- 29.Lundberg DS, et al. Defining the core Arabidopsis thaliana root microbiome. Nature. 2012;488:86–90. doi: 10.1038/nature11237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bulgarelli D, et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature. 2012;488:91–95. doi: 10.1038/nature11336. [DOI] [PubMed] [Google Scholar]
- 31.Mendes LW, Kuramae EE, Navarrete AA, van Veen JA, Tsai SM. Taxonomical and functional microbial community selection in soybean rhizosphere. ISME J. 2014;8:1577–1587. doi: 10.1038/ismej.2014.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fierer N, Bradford MA, Jackson RB. Toward an ecological classification of soil bacteria. Ecology. 2007;88:1354–1364. doi: 10.1890/05-1839. [DOI] [PubMed] [Google Scholar]
- 33.Eevers N, et al. Optimization of isolation and cultivation of bacterial endophytes through addition of plant extract to nutrient media. Microbial biotechnology. 2015;8:707–715. doi: 10.1111/1751-7915.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bagchi R, et al. Pathogens and insect herbivores drive rainforest plant diversity and composition. Nature. 2014;506:85–88. doi: 10.1038/nature12911. [DOI] [PubMed] [Google Scholar]
- 35.Wagg C, Bender SF, Widmer F, van der Heijden MG. Soil biodiversity and soil community composition determine ecosystem multifunctionality. Proc Natl Acad Sci USA. 2014;111:5266–5270. doi: 10.1073/pnas.1320054111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tilman D, Hill J, Lehman C. Carbon-negative biofuels from low-input high-diversity grassland biomass. Science. 2006;314:1598–1600. doi: 10.1126/science.1133306. [DOI] [PubMed] [Google Scholar]
- 37.Schmer MR, Vogel KP, Mitchell RB, Perrin RK. Net energy of cellulosic ethanol from switchgrass. Proc Natl Acad Sci USA. 2008;105:464–469. doi: 10.1073/pnas.0704767105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhattacharyya P, Jha D. Plant growth-promoting rhizobacteria (PGPR): emergence in agriculture. World J Microb Biot. 2012;28:1327–1350. doi: 10.1007/s11274-011-0979-9. [DOI] [PubMed] [Google Scholar]
- 39.Gray E, Smith D. Intracellular and extracellular PGPR: commonalities and distinctions in the plant–bacterium signaling processes. Soil Biol Biochem. 2005;37:395–412. doi: 10.1016/j.soilbio.2004.08.030. [DOI] [Google Scholar]
- 40.Dini-Andreote F, et al. Dynamics of bacterial community succession in a salt marsh chronosequence: evidences for temporal niche partitioning. ISME J. 2014;8:1989–2001. doi: 10.1038/ismej.2014.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hansel CM, Fendorf S, Jardine PM, Francis CA. Changes in bacterial and archaeal community structure and functional diversity along a geochemically variable soil profile. Appl Environ Microb. 2008;74:1620–1633. doi: 10.1128/AEM.01787-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Evans R, Johansen J. Microbiotic crusts and ecosystem processes. Crit Rev Plant Sci. 1999;18:183–225. doi: 10.1016/S0735-2689(99)00384-6. [DOI] [Google Scholar]
- 43.Kuklinsky‐Sobral J, et al. Isolation and characterization of soybean‐associated bacteria and their potential for plant growth promotion. Environ Microbiol. 2004;6:1244–1251. doi: 10.1111/j.1462-2920.2004.00658.x. [DOI] [PubMed] [Google Scholar]
- 44.Jones RT, et al. A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses. ISME J. 2009;3:442–453. doi: 10.1038/ismej.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fierer N, Jackson RB. The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci USA. 2006;103:626–631. doi: 10.1073/pnas.0507535103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leininger S, et al. Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature. 2006;442:806–809. doi: 10.1038/nature04983. [DOI] [PubMed] [Google Scholar]
- 47.Bergmann GT, et al. The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biol Biochem. 2011;43:1450–1455. doi: 10.1016/j.soilbio.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma, B. et al. Geographic patterns of co-occurrence network topological features for soil microbiota at continental scale in eastern China. ISME J (2016). [DOI] [PMC free article] [PubMed]
- 49.González AMM, Dalsgaard B, Olesen JM. Centrality measures and the importance of generalist species in pollination networks. Ecol Complex. 2010;7:36–43. doi: 10.1016/j.ecocom.2009.03.008. [DOI] [Google Scholar]
- 50.Banerjee, S. et al. Determinants of bacterial communities in Canadian agroforestry systems. Environ Microbiol (2015). [DOI] [PubMed]
- 51.Greenblum S, Turnbaugh PJ, Borenstein E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc Natl Acad Sci USA. 2012;109:594–599. doi: 10.1073/pnas.1116053109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vick-Majors TJ, Priscu JC, Amaral-Zettler LA. Modular community structure suggests metabolic plasticity during the transition to polar night in ice-covered Antarctic lakes. ISME J. 2014;8:778–789. doi: 10.1038/ismej.2013.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rovira AD. Plant root exudates. The Botanical Review. 1969;35:35–57. doi: 10.1007/BF02859887. [DOI] [Google Scholar]
- 54.Stegen JC, Lin X, Konopka AE, Fredrickson JK. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 2012;6:1653–1664. doi: 10.1038/ismej.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Philippot L, Raaijmakers JM, Lemanceau P, Van Der Putten WH. Going back to the roots: the microbial ecology of the rhizosphere. Nat Rev Microbiol. 2013;11:789–799. doi: 10.1038/nrmicro3109. [DOI] [PubMed] [Google Scholar]
- 56.Shi S, et al. The interconnected rhizosphere: High network complexity dominates rhizosphere assemblages. Ecol Lett. 2016;19:926–936. doi: 10.1111/ele.12630. [DOI] [PubMed] [Google Scholar]
- 57.Keller L, Surette MG. Communication in bacteria: an ecological and evolutionary perspective. Nat Rev Microbiol. 2006;4:249–258. doi: 10.1038/nrmicro1383. [DOI] [PubMed] [Google Scholar]
- 58.Elasri M, et al. Acyl-homoserine lactone production is more common among plant-associated Pseudomonas spp. than among soilborne Pseudomonas spp. Appl Environ Microb. 2001;67:1198–1209. doi: 10.1128/AEM.67.3.1198-1209.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Caporaso JG, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA. 2011;108:4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 62.Clarke KR. Non‐parametric multivariate analyses of changes in community structure. Aust J Ecol. 1993;18:117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x. [DOI] [Google Scholar]
- 63.Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46. [Google Scholar]
- 64.Csardi G, Nepusz T. The igraph software package for complex network research. InterJournal, Complex Systems. 2006;1695:1–9. [Google Scholar]
- 65.Bastian M, Heymann S, Jacomy M. Gephi: an open source software for exploring and manipulating networks. ICWSM. 2009;8:361–362. [Google Scholar]
- 66.Newman ME. Modularity and community structure in networks. Proc Natl Acad Sci USA. 2006;103:8577–8582. doi: 10.1073/pnas.0601602103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Newman ME. The structure and function of complex networks. Siam Rev. 2003;45:167–256. doi: 10.1137/S003614450342480. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.