

ABSTRACT
Chemotaxis is the movement of cells in response to gradients of diverse chemical cues. Motile bacteria utilize a conserved chemotaxis signal transduction system to bias their motility and navigate through a gradient. A central regulator of chemotaxis is the histidine kinase CheA. This cytoplasmic protein interacts with membrane-bound receptors, which assemble into large polar arrays, to propagate the signal. In the alphaproteobacterium Azospirillum brasilense, Che1 controls transient increases in swimming speed during chemotaxis, but it also biases the cell length at division. However, the exact underlying molecular mechanisms for Che1-dependent control of multiple cellular behaviors are not known. Here, we identify specific domains of the CheA1 histidine kinase implicated in modulating each of these functions. We show that CheA1 is produced in two isoforms: a membrane-anchored isoform produced as a fusion with a conserved seven-transmembrane domain of unknown function (TMX) at the N terminus and a soluble isoform similar to prototypical CheA. Site-directed and deletion mutagenesis combined with behavioral assays confirm the role of CheA1 in chemotaxis and implicate the TMX domain in mediating changes in cell length. Fluorescence microscopy further reveals that the membrane-anchored isoform is distributed around the cell surface while the soluble isoform localizes at the cell poles. Together, the data provide a mechanism for the role of Che1 in controlling multiple unrelated cellular behaviors via acquisition of a new domain in CheA1 and production of distinct functional isoforms.
IMPORTANCE Chemotaxis provides a significant competitive advantage to bacteria in the environment, and this function has been transferred laterally multiple times, with evidence of functional divergence in different genomic contexts. The molecular principles that underlie functional diversification of chemotaxis in various genomic contexts are unknown. Here, we provide a molecular mechanism by which a single CheA protein controls two unrelated functions: chemotaxis and cell length. Acquisition of this multifunctionality is seemingly a recent evolutionary event. The findings illustrate a mechanism by which chemotaxis function may be co-opted to regulate additional cellular functions.
KEYWORDS: Azospirillum, chemotaxis, signal transduction
INTRODUCTION
Chemotaxis, the directed movement of motile bacteria in gradients of diverse chemoeffectors, promotes colonization of various niches and the establishment of plant-microbe associations (1, 2). The histidine kinase CheA is a central regulator of chemotaxis. In the Escherichia coli model, when chemical signals bind to chemoreceptors, CheA is autophosphorylated. This phospho-CheA then promotes changes in swimming direction via phosphotransfer to CheY, yielding phospho-CheY with an increased affinity to flagellar motors. A chemotaxis-specific phosphatase, CheZ, acts on phospho-CheY to terminate the signal. Phospho-CheA also plays a role in receptor adaptation to background conditions. Methylation levels of the chemoreceptors are altered through either a phosphotransfer from phospho-CheA to the methylesterase CheB or by the addition of methyl groups via the methyltransferase CheR (3, 4). E. coli possesses a single chemotaxis system, and the functional CheA is a homodimer, with each monomer containing five domains denoted P1 to P5 (Fig. 1A) (5). The ATP binding domain of CheA (P4) phosphorylates at the conserved histidine residue in the N-terminal phosphorylation domain (P1) (Fig. 1A). P2 is a docking site for interactions with CheY and CheB, which are both activated through a phosphorylation event from phospho-CheA; P3 serves as a dimerization domain. The P5 domain closely resembles and also interacts with the adaptor protein CheW, which couples chemoreceptors to CheA (6). Together, CheA, through its P5 domains, and CheW interact with the cytoplasmic tips of the trimers of receptor dimers to form a ring-shaped baseplate (7, 8). Each baseplate interacts with neighboring baseplates, making chemotaxis signal transduction a cooperative process that both amplifies and propagates the chemotaxis signal (7–9).
FIG 1.
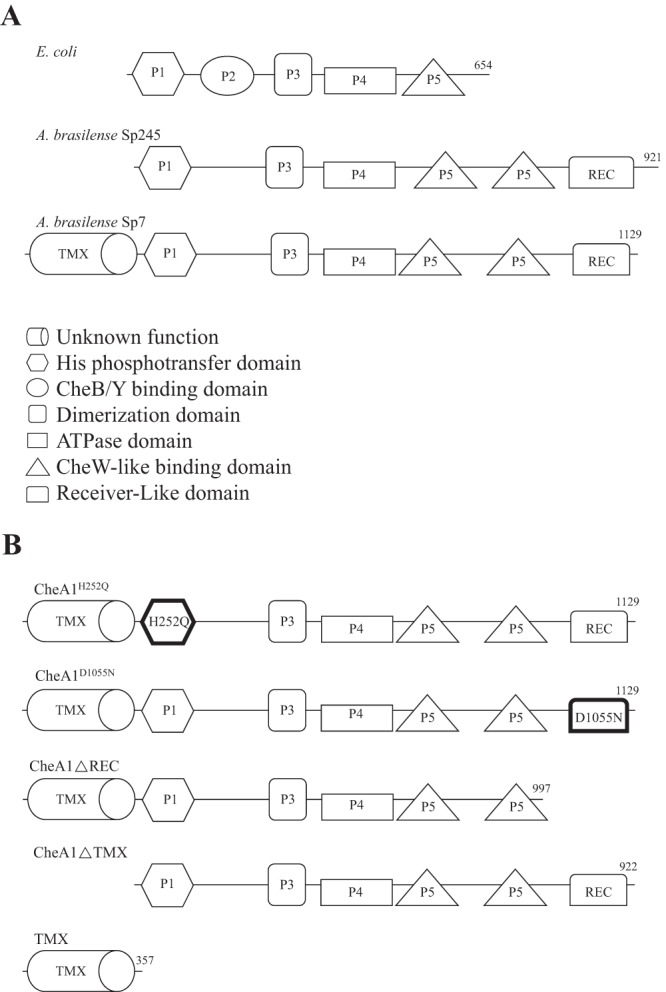
(A) Domain architecture of CheA orthologs in E. coli and A. brasilense strains Sp7 and Sp245. Top, the E. coli CheA contains a histidine phosphotransfer domain which harbors the conserved His residue (P1) followed by a CheB/Y binding domain (P2). The dimerization (dimer) domain (P3) precedes the ATPase domain (P4), which is necessary to phosphorylate the His residue. The binding domain (P5) resides in the C-terminal region. Middle, A. brasilense strain Sp245 CheA1 possesses the P1, P3, P4, and P5 domains and also a receiver-like (REC) domain. Bottom, A. brasilense strain Sp7 CheA1 possesses domains similar to those of strain Sp245, but it also possesses an N-terminal seven-transmembrane region of unknown function (TMX). Numbers at the C termini denote the total number of amino acids present. (B) Schematic representation of A. brasilense strain Sp7 CheA1 variants used in this study. Variants with single-residue replacements (CheA1H252Q and CheA1D1055N) are in boldface.
Compared to the case for E. coli chemotaxis, the complete genome sequences of most motile bacteria indicate the presence of more than one chemotaxis (Che) operon. In addition to chemotaxis, some chemotaxis systems are implicated in the regulation of cellular functions other than flagellar motility: some systems regulate the extension-retraction cycle of type IV pili to promote twitching (10), others regulate enzymatic activities implicated in the transition from a vegetative to a sessile lifestyle (11), and some exert control of developmental programs (12–14). Chemotaxis systems that regulate alternative cellular functions often lack a CheY homolog and instead possess enzymes or transcription factors that function to mediate the signaling output (15). Chemotaxis systems that control type IV pili activities are characterized by CheA homologs which possess multiple P1 domains at their N terminus (15). Therefore, comparative analysis of the architecture of chemotaxis systems can provide clues as to the putative function of some chemotaxis operons.
The alphaproteobacterium Azospirillum brasilense's genome encodes four signal transduction pathways. The Che1 pathway controls transient changes in the swimming speed that accompany the simultaneous suppression of changes in the swimming direction during a response to a chemoattractant (16). These changes in swimming velocity modulate the propensity of cells to clump by cell-cell interactions (16–18). However, deletion of che1 or of the gene for the histidine kinase of the Che1 pathway, cheA1, has a minor effect on chemotaxis (17). An additional Che pathway, which has a more dominant role in controlling chemotaxis, has recently been identified (19). In addition to regulating transient increases in swimming speed, Che1 is implicated in regulating cell length: mutations that render cheA1, che1, or cheY1 nonfunctional also result in cells of relatively shorter lengths than the wild type, while mutations that abolish the function of enzymes required for chemotaxis receptor adaptation, CheB1 and CheR1, result in cells significantly longer than the wild type, with these effects most evident in dividing cells (17). These data thus suggest that Che1 regulates two distinct functions, i.e., transient changes in swimming speed upon chemo-stimulation and cell length, but the underlying mechanisms are not known (16). The A. brasilense Che1 pathway comprises all homologs of the E. coli chemotaxis operon, including a CheY1 homolog that functions to regulate transient increases in swimming velocity but excluding CheZ (16). Che1 also includes a CheA1 homolog with an additional N-terminal domain of unknown function (20). In this study, we aimed to identify the domain(s) of CheA1 that contributes to these distinct functions, namely, chemotaxis and cell length regulation. We provide experimental evidence that the N-terminal domain of CheA1, which we name TMX, is dispensable for chemotaxis but is associated with the function of CheA1 in cell length regulation. We also show that CheA1 is produced in two isoforms: (i) a soluble isoform lacking the N-terminal TMX which localizes as small foci at the cell poles and (ii) a larger, membrane-anchored, full-length protein that is distributed throughout the cell surface. Together, our data suggest a molecular mechanism by which the A. brasilense Che1 pathway regulates unrelated cellular functions via the production of functionally and spatially distinct populations of CheA1.
RESULTS
Domain architecture reveals unique N-terminal fusion to CheA1.
To determine how CheA1 from A. brasilense may regulate multiple cellular functions, we first analyzed its domain architecture. Compared to the canonical E. coli CheA, the A. brasilense CheA1 lacks a P2 domain, contains a second P5 domain, and carries a receiver (REC) domain at the C terminus (SM00448) (Fig. 1A) (21). Strikingly, the A. brasilense strain Sp7 CheA1 also possesses an additional N-terminal domain, which we refer to here as TMX, predicted to be closely related to a polytopic hemolysin III-related (HlyIII) protein of unknown function (Fig. 1A). This particular CheA domain appears to be unique to strain Sp7; the CheA1 homolog from the closely related A. brasilense strain Sp245 lacks the N-terminal TMX domain but is otherwise similar to the CheA1 of strain Sp7 (Fig. 1A). TMX found at the N terminus of CheA1 from A. brasilense Sp7 is thus a unique feature of this protein in this particular strain.
The presence of the N-terminal TMX domain is associated with the CheA1-dependent cell length phenotype but is dispensable for chemotaxis.
The different architectures of CheA1 in two closely related strains of A. brasilense prompted us to repeat the behavioral studies performed in strain Sp7 on strain Sp245. We constructed a derivative of strain Sp245 lacking the CheA1 homolog (ΔcheA1Sp245) and analyzed chemotaxis and cell length phenotypes. We found that the ΔcheA1Sp245 mutant was defective in chemotaxis: the average swarm diameter on a soft agar plate was 4.1 ± 0.03 mm for the wild type and 1.8 ± 0.02 mm for the ΔcheA1Sp245 mutant. We also observed that the doubling times of Sp245 and the ΔcheA1Sp245 mutant were different (6.7 ± 0.9 h for the wild type and 8.1 ± 0.8 h for the ΔcheA1Sp245 mutant), suggesting that the difference in the chemotaxis ring diameter is likely the result of differences in doubling times, since growth is required to observe chemotaxis in this assay. To further evaluate potential chemotaxis defects, we analyzed the swimming pattern of free-swimming cells and found that the ΔcheA1Sp245 mutant strain was able to reverse swimming direction at a rate similar to that for its parent (data not shown). This suggests that the CheA1 of strain Sp245 is not the major CheA homolog regulating all chemotaxis responses, which is similar to the CheA1 in strain Sp7 (17). Interestingly, mutating cheA1Sp245 had no effect on cell length: the average length of cells in a population of the wild-type strain Sp245 and its ΔcheA1Sp245 mutant was 2.6 ± 0.3 μm. Therefore, the phenotype of the ΔcheA1Sp245 strain is similar to that of its Sp7 counterpart in that it has no major role in chemotaxis. However, it is distinct from the Sp7 ΔcheA1 phenotype in that mutating cheA1 in strain Sp245 does not cause a defect in cell length. These observations suggest that the CheA1-dependent cell length phenotype observed in strain Sp7 is linked to the presence of the TMX domain.
To further characterize the roles of TMX and other CheA1 domains in the control of cell length and chemotaxis, we generated cheA1 mutants carrying either site-specific mutations of conserved residues or in-frame domain deletions. Specifically, we analyzed CheA1H252Q, a variant with a mutation in the putative autophosphorylation histidine residue previously implicated in CheA1 function during chemotaxis (16), as well as CheA1D1055N, which is mutated in the conserved putative phosphorylatable Asp1055 residue in the C-terminal REC domain of the protein (Fig. 1B). We also constructed and analyzed a strain expressing only the TMX domain, as well as strains expressing CheA1ΔTMX or CheA1ΔREC, which carry in-frame deletions of the TMX domain or the REC domain, respectively (Fig. 1B). As previously described (17), the ΔcheA1 mutant strain carrying an empty vector produced significantly shorter cells than the Sp7 wild-type strain. However, this defect was complemented by introducing a medium-copy-number plasmid carrying the full-length cheA1 gene (pBBRCheA1) (Fig. 2). Expressing CheA1H252Q or CheA1ΔREC from a broad-host-range plasmid fully restored cell length to the ΔcheA1 mutant strain, indicating that CheA1 autophosphorylation and its REC domain play no role in cell length control. In contrast, expressing a CheA1 variant in which TMX was deleted in frame (CheA1ΔTMX) failed to restore the cell length phenotype to the ΔcheA1 mutant strain (Fig. 2). Surprisingly, expressing CheA1D1055Nor TMX alone in the ΔcheA1 strain not only restored but further increased the cell length (Fig. 2).
FIG 2.
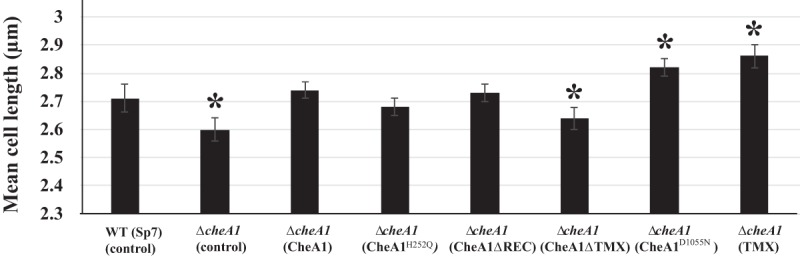
Roles of different domains of CheA1 in cell length regulation. *, statistically significant difference in average cell lengths relative to that of the wild-type strain, at a P value of <0.05 (n = 50) by Student's t test.
The effect of TMX on cell length could possibly result from altered growth rates of the strains carrying these vectors. To address this, doubling times of strains expressing these CheA1 constructs were determined. The wild-type Sp7(pBBR) (control) strain doubled faster (2.6 ± 0.4 h) than any of the ΔcheA1 mutant derivatives, but all ΔcheA1 mutant derivatives had doubling times which were not significantly different from each other. The doubling times were 4.7 ± 0.3 h for the ΔcheA1(pBBR) mutant, 5.6 ± 0.5 h for the ΔcheA1(pBBRCheA1) mutant, 4.8 ± 0.5 h for the ΔcheA1(pBBRCheA1H252Q) mutant, 4.6 ± 0.2 h for the ΔcheA1(pBBRCheA1ΔREC) mutant, 4.8 ± 0.5 h for the ΔcheA1(pBBRCheA1ΔTMX) mutant, 5.3 ± 0.7 h for the ΔcheA1(pBBRCheA1D1055N) mutant, and 4.6 ± 0.2 h for the ΔcheA1(pBBRTMX) mutant. These data suggest that the cell length phenotype, which is different in the strains expressing these constructs (Fig. 2), is not directly the result of impaired growth. We also considered the possibility that membrane-bound CheA1 variants could have indirect effects on cell length due to altered protein expression levels from the medium-copy-number plasmid, including protein degradation. To test this, we used a polyclonal antibody raised against soluble CheA1 (CheA1ΔTMX) and Western blots to compare levels of protein expression in the wild type and the ΔcheA1 mutant strains expressing membrane-bound CheA1 variants (Fig. 3). As expected, the anti-CheA1ΔTMX antibodies recognized a single large band, at the predicted molecular weight, in all strains except the ΔcheA1 mutant control. Comparing the CheA1 band intensity across the samples revealed that, relative to the wild-type Sp7 strain carrying an empty vector control, the ΔcheA1 mutant derivatives expressed CheA1 and its variants at levels that were comparable between them and represented about twice the levels of CheA1 detected in the wild type. Specifically, CheA1, CheA1H252Q, CheA1D1055N, and CheA1ΔREC expression levels were on average 1.8, 1.9, 2.0, and 2.2 times the wild-type levels, respectively. Based on these data, we hypothesize that TMX may also be produced at about twice the native levels under these conditions. Despite the observation that the expression of CheA1, or its variants, from the pBBR vector yielded about twice the amount of protein produced under native conditions, we did not see any evidence of degradation. Furthermore, all strains expressed similar CheA1 levels, but they each produced different cell length phenotypes, with expression of CheA1 or CheA1H252Q restoring the wild-type cell length phenotype while expression of CheA1D1055N or CheA1ΔREC caused an increase beyond the wild-type cell length (Fig. 2). These data argue against altered protein expression levels causing the cell length phenotypes and rather suggest that it is the presence of TMX in CheA1 of A. brasilense strain Sp7 that is associated with the effect on cell length.
FIG 3.
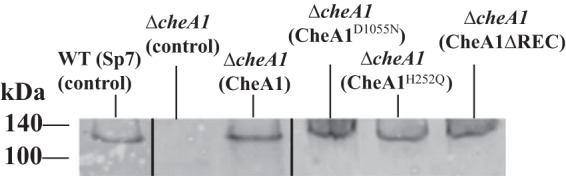
Detection of CheA1 in whole-cell lysates of A. brasilense wild-type strain Sp7 and its ΔcheA1 mutant derivatives expressing CheA1 and its variants from a plasmid by Western blotting. The mutant derivatives were expressing CheA1, variants with single-amino-acid residue replacements, or truncated variants of the protein from a medium-copy-number broad-host-range plasmid. A polyclonal antibody raised against CheA1ΔTMX was affinity purified as described in Materials and Methods prior to use. Each well is identified with the strain and with the parental or variant protein expressed from the broad-host-range vector in parentheses. The black lines indicate junctions separating lanes that were spliced from the original image in order to show samples run on the same gel in a single row.
We then assessed TMX's role in chemotaxis through soft agar assays of the wild-type, ΔcheA1 mutant, and mutant derivatives (Fig. 4A). Chemotaxis was fully restored when CheA1 or CheA1ΔTMX was expressed from the medium-copy-vector in the ΔcheA1 mutant background (Fig. 4A). However, a plasmid expressing only the TMX domain was unable to restore chemotaxis and, in fact, appeared to inhibit chemotaxis. In A. brasilense strain Sp7, CheA1 controls transient increases in free-swimming cell speed in response to attractants, such as oxygen. Therefore, we also analyzed the same CheA1 alleles in a temporal gradient assay for aerotaxis (Fig. 4B). Similar to the response of the wild-type strain carrying an empty vector, expressing CheA1 or CheA1ΔTMX in the ΔcheA1 mutant strain resulted in statistically significant increases in the swimming speed in response to a switch from anaerobiosis to air (Fig. 4B). As expected, a ΔcheA1 mutant strain carrying an empty vector lacked this response. A similar defect was also observed when the TMX domain was expressed alone from a plasmid in this mutant (Fig. 4B). Together, the results suggest that TMX present within CheA1 in A. brasilense strain Sp7 is dispensable for chemotaxis.
FIG 4.
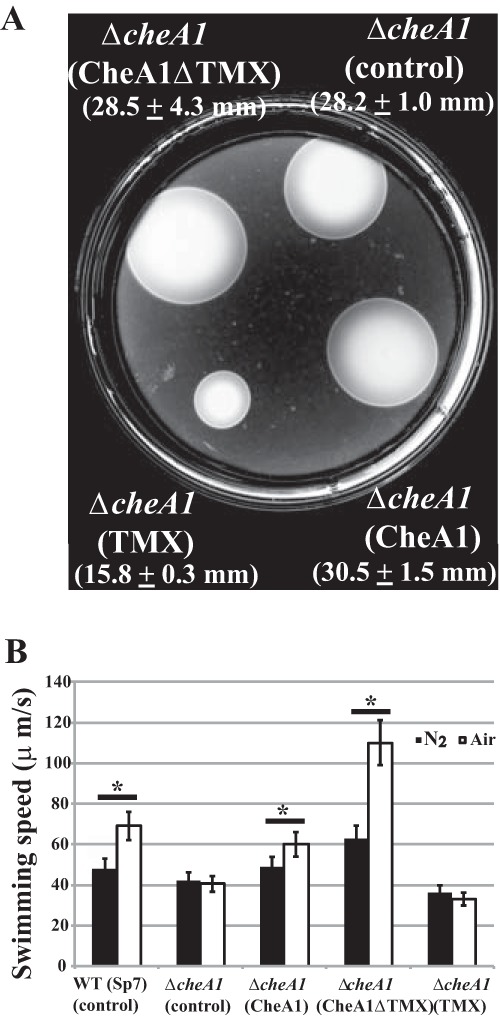
Roles of unique domains of A. brasilense strain Sp7 CheA1 in chemotaxis. (A) Chemotaxis of the ΔcheA1 mutant strain expressing CheA1 or truncated versions of this protein from a broad-host-range plasmid was analyzed in the soft agar plate assay supplemented with appropriate antibiotics. The chemotaxis of the wild-type strain Sp7 carrying an empty vector control [Sp7(pBBR)]; not shown) was similar to that of the ΔcheA1(pBBRCheA1) strain, and the average chemotaxis ring diameter was 30.2 ± 2.9 mm. The picture was taken at 48 h postinoculation. The average chemotaxis ring diameters obtained from three replicates are shown. (B) The temporal gradient assay for aerotaxis was used to measure transient increases in cell swimming speed upon switching free-swimming cells from an anaerobic atmosphere (nitrogen gas) to air. Each strain is identified, with the parental or variant protein it expresses from the broad-host-range vector in parentheses. *, statistically significant difference in the swimming speed between before and after switching to air (P < 0.05 by Student's t test).
CheA1 is produced as two isoforms.
The results above were puzzling regarding how distinct domains of CheA1 could function in chemotaxis or cell length regulation with no obvious functional overlap. Further, the presence of the predicted polytopic TMX domain at the N terminus of CheA1 suggested that CheA1 from A. brasilense may be anchored to the membrane, a proposition which, if confirmed, would challenge existing paradigms regarding the subcellular assembly of chemotaxis signaling complexes, namely, that CheA is a soluble protein which interacts with chemoreceptors at cell poles (3, 22). To address this issue, we analyzed whole-cell extracts of the wild-type strain and the ΔcheA1 mutant derivative using a Western blot and affinity-purified anti-CheA1 antibodies raised against soluble CheA1 (CheA1ΔTMX). This analysis revealed the presence of two bands present exclusively in the wild-type lysates and absent from the ΔcheA1 mutant lysates (Fig. 5A). The molecular mass of the full-length CheA1 from strain Sp7 is predicted to be 120 kDa, which we detected as a thick band on whole-cell extracts. In addition, a very faint band that migrated at around 100 kDa was also detected in the whole-cell lysates of the wild-type strain. Similar Western blot analyses of whole-cell lysates of several Azospirillum strains, for which the genome is sequenced and not predicted to encode a membrane-anchored CheA1, revealed only one unique cross-reacting band at about 100 kDa (data not shown). Given the specific detection of this large band in Sp7 with antibodies raised against the soluble CheA1ΔTMX and the fact that the N-terminal TMX domain is predicted to encode a protein with seven membrane-spanning segments, we hypothesize that the high-molecular-weight band detected in whole-cell lysates of the wild type corresponds to a full-length CheA1 containing the TMX domain and the smaller band corresponds to a soluble CheA1 without the TMX domain. We used cell fractionation to further investigate the subcellular localization of CheA1 (Fig. 5B). A broad band at around 120 kDa, likely corresponding to full-length CheA1, was found exclusively in the membrane fraction of strain Sp7 and was absent in the soluble fraction of this strain or in the lysates of the ΔcheA1 mutant treated under similar conditions. Analysis of the proteins produced in the soluble fractions of the wild-type strain indicated the presence of a single band corresponding to a product at about 98 kDa that was absent from both the membrane fraction and the ΔcheA1 lysates (Fig. 5B). The predicted molecular mass of a soluble CheA1 produced without the N-terminal TMX domain is 98 kDa, suggesting the presence of a soluble CheA1 in A. brasilense strain Sp7. We note that the samples corresponding to the soluble fraction analyzed here were also more concentrated than the whole-cell lysates or the membrane fraction (Fig. 5), underscoring the low concentration in cells. Based on this observation, we surmise that a soluble CheA1 isoform is present in the cytoplasm of A. brasilense at relatively low concentrations compared to the membrane-bound isoform. To determine the relative abundance of bands detected in the Western blots, we used densitometry analysis and found that the thicker band migrating at 120 kDa was about 11-fold more intense than the smaller band. Together, these data indicate that CheA1 from A. brasilense strain Sp7 is produced in two isoforms: a membrane-bound form corresponding to a full-length CheA1 anchored to the membrane via the polytopic TMX domain and a soluble form corresponding to CheA1 lacking the TMX domain and that most resembles the architecture of CheA homologs from other organisms.
FIG 5.
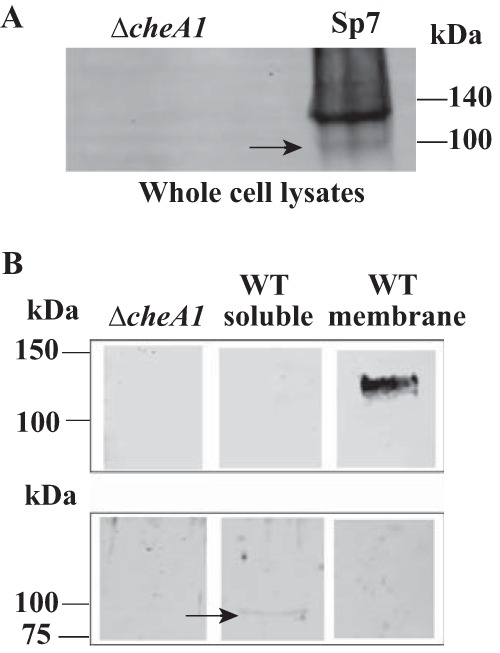
Detection of CheA1 in different subcellular fractions of A. brasilense wild-type strain Sp7 and its ΔcheA1 mutant derivative by Western blotting. (A) Whole-cell lysates of the wild type and the ΔcheA1 mutant were probed with affinity-purified polyclonal antibodies raised against a recombinantly produced soluble CheA1ΔTMX from A. brasilense Sp7, as described in Materials and Methods. (B) Different subcellular fractions collected from the wild-type strain Sp7 were probed with affinity-purified polyclonal antibodies raised against a recombinantly produced soluble CheA1ΔTMX from A. brasilense Sp7. Note that different protocols were used for sample preparation and SDS-PAGE for the Western blots shown in panels A and B (see Materials and Methods). In panel A, an anti-CheAΔTMX antibody, which was further purified against whole proteins extracted from the Δche1 mutant, was used. These samples were run on a 7.5% gel and transferred to a 0.45-μm hydrophobic PVDF transfer membrane. In panel B, an anti-CheAΔTMX antibody which had not been further purified against whole proteins extracted from the Δche1 mutant was used. The membrane fraction was resuspended in a solubilizing buffer (1× PBS with 2% Triton X-100 and 1 mM PMSF) and run on an 8 to 16% gradient gel before being transferred to a nitrocellulose membrane. For the cytosolic fraction, these samples were concentrated via acetone precipitation before being loaded in a 10% gel and transferred to a nitrocellulose membrane. The numbers on the left indicate the positions of the molecular mass markers run on the same gels. The arrows point to the soluble, faint band corresponding to CheA1ΔTMX.
The CheA1 isoforms localize to the cell surface and to the cell poles.
To corroborate the cell fractionation data, we constructed and expressed, from broad-host-range plasmids, fluorescently tagged versions of the full-length CheA1 (CheA1-yellow fluorescent protein [YFP]), of CheA1ΔTMX (CheA1ΔTMX-YFP), and of isolated TMX (TMX-YFP). The fluorescent constructs were expressed in both the wild-type strain Sp7 and the ΔcheA1 mutant backgrounds (Fig. 6A). The constructs were functional in that they restored the wild-type phenotype in cell length regulation and chemotaxis to a ΔcheA1 mutant strain (see Fig. S1 in the supplemental material). In both backgrounds, CheA1-YFP was diffuse throughout the cells with occasional foci, both polar and nonpolar, that were relatively dim and seen best when expressed in the ΔcheA1 strain background. In contrast, CheA1ΔTMX-YFP was present as punctate foci at the cell poles in both strain backgrounds (Fig. 6). The localization of TMX-YFP, expressed in either the Sp7 or ΔcheA1 strain background, was diffuse throughout the cell, although occasional punctate foci were observed, though they did not appear to localize in any particular region of the cell surface (Fig. 6A). The localization of CheA1ΔTMX-YFP at the cell poles was similar to the localization of a chemotaxis receptor Tlp1-YFP, which was previously shown to function in chemotaxis (23). Quantitative evaluation of the distribution of fluorescence in the cell population expressing CheA1-YFP, CheA1ΔTMX-YFP, and TMX-YFP corroborated these observations (Fig. 6B). In addition, cells expressing CheA1-YFP displayed localization patterns more consistent with those seen in cells expressing TMX-YFP, presumably corresponding to the higher level of membrane-anchored CheA1-YFP relative to that of the soluble, polar-localized CheA1ΔTMX isoform. Together, these data are consistent with the hypothesis that the soluble isoform of CheA1 localizes as polar clusters while the full-length CheA1 localizes independently to the cell surface via the N-terminal TMX domain.
FIG 6.
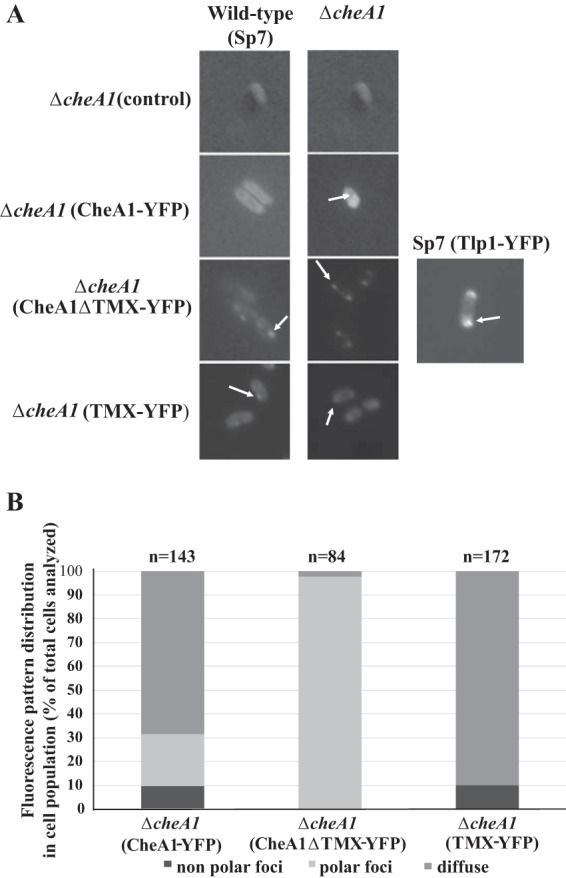
Fluorescence microscopy of YFP-tagged chemotaxis proteins from A. brasilense wild-type strain Sp7 and its ΔcheA1 mutant derivative. (A) Representative fluorescence microscopy images. The fluorescent constructs were expressed from the native cheA1 promoter on a broad-host-range plasmid, pRH005. For expression of Tlp1-YFP, the fusion was expressed from the lac promoter present on the pRH005 vector backbone. Arrows indicate punctate foci in cells. (B) Quantitation of the distribution of fluorescence in the population of cells analyzed. Each strain is identified, with the variant protein fusion to YFP expressed from the broad-host-range vector in parentheses. The total numbers (n) of cells analyzed are shown on the graph. The cells included in the analysis were imaged from at least 3 independent cultures.
DISCUSSION
Here we provide experimental evidence supporting a model for the multifunctional role of CheA1 from A. brasilense Sp7 in regulating chemotaxis and cell length at division (16, 17). In all Bacteria and Archaea analyzed to date, the chemotaxis receptors assemble together with cytoplasmic chemotaxis proteins, including CheA homologs, at the cell poles, where they form complex signaling arrays which function to amplify and propagate signals (3, 7, 8, 22). The soluble CheA1 isoform from the Sp7 strain studied here localized at the cell poles, consistent with its function in chemotaxis. The full-length, membrane-anchored CheA1 was distributed throughout the cells, with no apparent distinct localization. Given its localization on the cell surface, the function of the membrane-anchored CheA1, while unknown, is expected to be independent of the chemotaxis signaling function of the protein, consistent with results obtained here. This hypothesis is further supported by the observation that chemotaxis receptor mutants that were shown to signal (e.g., Tlp1 [24] and AerC [25]) and localize at the cell poles (AerC [25]) in a Che1-dependent manner do not display any cell length phenotype, similar to those produced as a result of mutations in cheA1 (17).
Our results suggest that the N-terminal TMX domain of CheA1 from strain Sp7 plays a role in modulating cell length. Deletion of the sequence corresponding to TMX from CheA1 yielded alleles unable to restore the wild-type cell length phenotype to the ΔcheA1 mutant, but expression of TMX alone rescued this phenotype. TMX is a conserved, single-domain protein of unknown function that is homologous to a hemolysin III-related (HlyIII) protein in Bacillus cereus (26, 27). Some evidence, obtained using recombinantly expressed HlyIII proteins in the heterologous E. coli background, suggested that HlyIII-related proteins function as hemolysins (28), but their presence in all bacteria makes this hypothesis unlikely. The function of HlyIII proteins has not been conclusively determined (29), but they were shown to be distant homologs of adipoR receptors found in yeasts and humans (29), the function of which also remains to be established. The data obtained here suggest that TMX, as shown here fused to the A. brasilense CheA1 protein, may have a function that ultimately alters how cells control cell length, but a direct role for TMX as a cell length regulator has yet to be established. Whether this is a conserved function of HlyIII-related homologs also remains to be determined.
Interestingly, expression of TMX alone or a mutation replacing the conserved aspartate residue within the CheA1 REC domain (D1055) yielded strains that were even longer than the wild-type strain. This is unexpected, and it also suggests possible features for TMX function. First, the deletion of the REC domain in CheA1ΔREC did not have this effect on the cell length phenotype, suggesting that it is not a direct function of the REC domain. The cell length phenotype of the CheA1D1055N mutant supports our findings that CheA1 is produced as a full-length, membrane-anchored isoform, and it further suggests that phosphorylation of the conserved aspartate residue within the REC domain has a regulatory role in the TMX output. This is consistent with the previously described functions of REC domains as regulatory modules in other hybrid histidine CheA homologs (30, 31). For example, mutating the REC domain of CheA3, which is involved in controlling cyst development in the closely related Rhodospirillum centenum, causes a loss of regulation of cyst formation, as those mutant derivatives produce more cysts than wild-type cells do (10, 31). One interpretation of our data in light of these previous findings is that phosphorylation of D1055 causes a conformational change within the full-length, membrane-anchored CheA1, which ultimately alters the ability of TMX to interact with the target protein(s), thereby affecting the cell length phenotype. The effect of expressing TMX alone, from a plasmid, on cell length is also consistent with interfering with protein-protein interactions that TMX may be engaged in for its function. This hypothesis is also consistent with previous data showing that other mutations within Che1 proteins (e.g., CheY1) that interact with CheA1 also affect cell length bias (17). Interactions of Che1 proteins with CheA1 are expected to depend on canonical CheA1 domains, which are present in both the soluble and the membrane-anchored CheA1 isoforms. Together, the data thus suggest that the function of TMX in regulating cell length in A. brasilense Sp7 may depend on interactions with other proteins.
The existence of CheA isoforms in chemotactic bacteria is not a new concept; E. coli produces CheA in two isoforms by utilizing a second translational start site (32, 33). What is unique about the CheA1 isoforms in A. brasilense strain Sp7 is that each isoform contributes to the regulation of two unrelated cellular functions. How the two CheA1 isoforms in A. brasilense are produced remains to be determined. Unlike in E. coli, there is no evidence for the presence of a ribosome binding site that would permit initiation of translation of the soluble CheA1 isoform (20, 32). Other possible mechanisms include transcriptional regulation from an alternative promoter as well as posttranscriptional and/or posttranslational processing. Further experimental investigation is required to establish the exact molecular mechanism involved in the production of these two isoforms.
Our data indicate that the insertion of the polytopic TMX domain at the N terminus of CheA1 of A. brasilense strain Sp7 is a recent event. None of the other Azospirillum species strains sequenced to date shows evidence of a similar domain insertion (34–39). In fact, the draft genome of an Sp7 strain generated by a team of South Korean scientists also lacks the TMX fusion to CheA1 (NCBI BioProject PRJNA293508). A. brasilense genomes are organized as 7 replicons: one chromosome and 6 large plasmids named p1 through p6 (34–39). The DNA regions coding for CheA1 and for TMX are both located on the largest plasmid, p1, in all A. brasilense strains with available genome sequences, but they are not adjacent and are separated by several kilobases, precluding a simple frameshifting or slippage mutation between adjacent genes. Genome plasticity and extensive genomic rearrangements are evidenced in the Azospirillum genomes sequenced to date, including those of several strains of A. brasilense (34–38). These include the lack of synteny between replicons of strains from the same species, the abundance and density of insertion sequence IS elements in all genomes, and evidence of plasmid loss or rearrangements and of repeated phage infections. The recent TMX acquisition by CheA1 of strain Sp7 is also supported by the lack of functional divergence between CheA1 proteins of different A. brasilense strains. Our results show that the minor role of the soluble CheA1 in A. brasilense Sp7 chemotaxis (16) is conserved in strain Sp245, in which the fusion event between TMX and CheA1 has not occurred. Comparative genome analysis of diverse bacteria shows that most domain acquisition events that generate novel multidomain protein architectures occur at the N or C termini of proteins (40). The insertion of TMX at the N terminus of CheA1 from strain Sp7 is consistent with these observations, but the mechanism involved is unknown. Analyses of protein domain fusion or insertion events in completely sequenced genomes of closely related organisms have been previously used to predict protein-protein interactions (41). However, we have no evidence that TMX and CheA1 functionally interact. Our results here show that the domain acquisition event that fused TMX to CheA1 resulted in both protein functions being maintained through the production of two CheA1 isoforms. While we do not know how TMX exerts its effect on cell length, this function is not obviously related to the control of the motility pattern mediated by CheA1. Mechanistic insight into how TMX functions to regulate cell length in A. brasilense should provide conclusive experimental evidence regarding a possible functional relationship.
MATERIALS AND METHODS
Strains, media, and chemicals.
The bacterial strains used in this study are listed in Table 1. A. brasilense strain Sp7 (ATCC 29145) (42) and mutant derivatives were grown at 28°C on minimal medium for A. brasilense (MMAB) plates supplemented with malate (10 mM final concentration) or with both malate (5 mM) and fructose (5 mM) for cell size measurements (see below), since these were determined to be the best substrates to observe cell size differences (43). MMAB is either supplemented with a nitrogen source (ammonium chloride at 18.7 mM), with cells incubated with shaking (200 rpm), or without nitrogen supplementation for growth under nitrogen fixation conditions, without shaking to limit oxygen diffusion. The ΔcheA1 and cheA1H252Q derivatives were described previously (16, 17) (Table 1).
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Genotype or relevant characteristic(s) | Reference or source |
|---|---|---|
| Escherichia coli strains | ||
| Top10 | Cloning strain | Invitrogen |
| OMNImax | Cloning strain | Invitrogen |
| S17-1 | thi endA recA hsdR RP4-2Tc::Mu-Km::Tn7 | 25 |
| Azospirillum brasilense strains | ||
| Sp7 | Parental strain, wild type | ATCC (ATCC 29145) |
| Sp245 | Parental strain, wild type | 20 |
| ΔcheA1 mutant | Δ(cheA1)::gusA-Km (Km) | 17 |
| Δche1 mutant | Δ(cheA1-cheR1)::Cm (Cm) | 17 |
| ΔcheA1Sp245 mutant | ΔcheA1:Gm insertion in Sp245 | This work |
| Plasmids | ||
| pDONR 221 | Cloning vector | Invitrogen |
| Topo 2.1 | Cloning vector | Thermo Fisher |
| pBBR1MCS3 | Cloning vector | 23 |
| pBSKII(+) | Cloning vector | Stratagene |
| pRH005 | Gateway destination vector for C-terminal YFP fusion with protein of interest | 27 |
| pDONRTMX | pDONR221 carrying TMX | This work |
| pDONRCheA1 | pDONR221 carrying CheA1 | This work |
| pDONRCheA1ΔTMX | pDONR221 carrying CheA1ΔTMX | This work |
| pDONRTlp1 | pDONR221 carrying Tlp1 | This work |
| pBBRCheA1 | pBBR1MCS3 expressing CheA1 | 17 |
| pBBRCheA1ΔTMX | pBBR1MCS3 expressing CheA1 with the TMX domain deleted | This work |
| pBBRCheA1ΔREC | pBBR1MCS3 expressing CheA1 with the REC domain deleted | This work |
| pBBRTMX | pBBR1MCS3 expressing the TMX domain of CheA1 | This work |
| pRHCheA1 | pRH005 expressing CheA1 | This work |
| pRHTMX | pRH005 expressing TMX domain | This work |
| pRHCheA1ΔTMX | pRH005 expressing CheA1 with the TMX domain deleted | This work |
| pRHTlp1 | pRH005 expressing Tlp1 | This work |
| pKGmob-Gm | Mobilizable suicide plasmid | Lab strain |
| pKGmob-cheA1 | pKGmob-Gm expressing Sp245 CheA1 | This work |
| pBBRCheA1H252Q | pBBR1MCS3 expressing CheA1H252Q | Lab strain |
| pBBRCheA1D1055N | pBBR1MCS3 expressing CheA1D1055N | This work |
| pBSKCheA1 | pBSKII(+) carrying CheA1 | This work |
| pBSKCheA1ΔTMX | pBSKII(+) carrying CheA1 with the TMX domain deleted | This work |
Behavioral assays.
For comparison of chemotaxis responses in the soft agar assay, bacteria were inoculated into MMAB solidified with 0.3% (wt/vol) agar (soft agar plates) and supplemented with 18.7 mM ammonium chloride and 10 mM malate. Plates were inoculated with culture in the mid-log phase of growth (optical density at 600 nm [OD600], 0.4 to 0.6) and grown at 28°C. Chemotactic rings were measured after 48 h of incubation. The temporal gradient assays for aerotaxis and computerized motion analysis were performed as previously described (24). Briefly, free-swimming cells were incubated on a slide placed inside a microchamber with an atmosphere of nitrogen gas (anaerobiosis) or air, which represents an attractant signal for the cells. Videos (10 s in length) for each time point were recorded, and swimming speed was analyzed through these videos using CellTrack software. The swimming speed of free-swimming cells was determined from a minimum of 75 cells tracked in 3 independent experiments. The swimming speeds denote averages calculated from the tracks of free-swimming cells 1 min prior to and 1 min after a switch from an atmosphere of N2 to air.
Mutant construction.
To generate a cheA1 gene in which the sequence corresponding to the N-terminal TMX domain was deleted, CheA1ΔTMX, the full-length CheA1 with its putative promoter region, was amplified by PCR from genomic DNA using the CheOp-HindIII-F and CheOp-BamHI-Rev primers (Table 2). These primers include HindIII and BamHI restriction sites at their 5′ and 3′ termini, respectively, to facilitate cloning. The PCR fragment was digested with HindIII and BamHI and cloned into the pBSKII(+) vector digested with the same enzymes, yielding pBSKCheA1. An in-frame deletion of the TMX domain was obtained using pBSKCheA1 as a template in an inverse PCR strategy as described previously (44). The outward-facing primers used were CheA1ΔTMX-F2 and CheA1ΔTMX-Rev. These primers also included BglII restriction sites at their 5′ ends to facilitate recircularization after inverse PCR, generating pBSKCheA1ΔTMX. These primers allowed for an in-frame deletion of the TMX region from nucleotide 66 to 215 of the cheA1 sequence (GenBank accession number AAL47021.1). The cheA1D1055N mutant was generated using the QuikChange II site-directed mutagenesis kit (Stratagene), verified by sequencing, and then cloned into pCR 2.1 TOPO vector (Invitrogen). The putative cheA1 promoter was isolated from a plasmid containing full-length cheA1 using CheA1promXhoI-F and CheA1DNup HindIII-R, which amplify a region upstream of the cheA1 start codon. The promoter was then cloned into the pCR2.1 TOPO vector and sequenced for verification. The cheA1D1055N mutant and the cheA1 promoter were then isolated by using restriction enzymes (HindIII and XbaI for isolating the cheA1D1055N fragment and XhoI and HindIII for isolating the cheA1 promoter) and ligated into the pBBR1MCS3 cloning vector (45). The pBBRCheA1D1055N plasmid was then transformed into chemically competent E. coli S17-1 cells, followed by biparental conjugation into the A. brasilense ΔcheA1 mutant background, as described previously (46). An A. brasilense strain Sp245 mutant derivative in which cheA1 is inactivated was constructed as following. First, a 509-bp internal segment within the cheA1 from A. brasilense strain Sp245, cheA1Sp245, was amplified using Sp245cheA1-F and Sp245cheA1-R and then cloned into pCR 2.1 TOPO, sequenced for verification, cloned into the suicide vector pKGmob-cheA1 (Table 1), and introduced into E. coli chemically competent S17-1 cells (47, 48). Biparental mating was used to introduce the suicide vector carrying the internal cheA1Sp245 fragment into the wild-type Sp245 strain, as described above. Single recombination mutation events were selected and verified by PCR, and a representative was used as an A. brasilense Sp245 ΔcheA1Sp245 mutant derivative. To generate ΔcheA1(pBBRCheA1ΔREC), pBBRCheA1 was used as a template with the primers CheA1prom KpnI-F and CheA1ΔREC XhoI-R (Tables 1 and 2). The insert generated by PCR was digested with KpnI and XhoI, cloned into pBBR1MCS3, and digested with the same restriction enzymes. This vector was then transformed into E. coli S17-1 chemically competent cells and mated into the A. brasilense ΔcheA1 derivative via biparental mating as described above.
TABLE 2.
Primers used in this study
| Primer name | Sequencea |
|---|---|
| CheOp-HindIII-F | 5′-CCCAAGCTTCAGCGCGATGAACTGGTTGGACC |
| CheOp-BamHI-Rev | 5′-CGCGGATCCCGTAATCAGACGGTCCTGGTTAG |
| CheA1ΔTMX-F2 | 5′-GAAGATCTGCGGTCGCGCGGAATTC |
| CheA1ΔTMX-Rev | 5′-GAAGATCTGACCGCGTCCGCCGCACG |
| CheA1promXhoI-F | 5′-CCGCTCGAGCAGCGCGATGAACTGGTT |
| CheA1DNup HindIII-R | 5′-AAGCTTCACGGCGGTGACGTC |
| Sp245cheA1-F | 5′-GAATCACGTGACGTCGGA |
| Sp245cheA1-R | 5′-CGACGCCGCGGCCCGACA |
| CheA1prom KpnI-F | 5′-CGGGGTACCCAGCGCGATGAACTGGTTG |
| CheA1ΔREC XhoI-R | 5′-CTCGAGTCATGGAGCGCCCTCTCC |
| GWCheA1-F | 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTCAGCGCGATGAACTGGTT |
| GWCheA1-R | 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTGCGGCACCTTTCTGCTCG |
| GWCheA1TMX-R | 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTTCCACGTCGAGCAACGAC |
| Tlp1GWFwd | 5′-GGGGACAAGTTTGTACAAAAAGCAGGCTCCGGCATGCGGATCAGGGC |
| Tlp1GWRev | 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCCGGCCAGCCCACCGGGCGCCT |
Italics indicate restriction enzyme sequences.
Fluorescence tagging of CheA1 and its variants.
Gateway cloning (Invitrogen) and the pRH005 vector were used to construct all yellow fluorescent protein (YFP) fusions (49). The pRH005 vector allows cloning of any gene to generate products fused in frame with YFP at their C termini. All genes to be fused in frame with the YFP gene in pRH005 (cheA1, cheA1ΔTMX, tmX, and tlp1) were amplified by PCR using pBBRCheA1 or pBBRCheA1ΔTMX (cheA1, cheA1ΔTMX, and tmX) or Sp7 genomic DNA (tlp1) as a template along with the following primers: for amplification of cheA1, GWCheA1-F and GWCheA1-R; for amplification of cheA1ΔTMX, GWCheA1-F and GWCheA1-R; for amplification of tmX, GWCheA1-F and GWCheA1TMX-R; and for amplification of tlp1, Tlp1GWFwd and Tlp1GWRev (Table 2). The PCR products, flanked by gateway sites on 5′ and 3′ ends, were then introduced into pDONR221 (Invitrogen), via a BP Clonase reaction (Invitrogen) and transformed into E. coli OmniMax chemically competent cells following the manufacturer's recommendations (Invitrogen). The constructs were verified by sequencing prior to transfer into the pRH005 vector via an LR Clonase reaction, performed per the manufacturer's instructions (Invitrogen). Final constructs were introduced into wild-type A. brasilense strain Sp7 and ΔcheA1 mutant backgrounds via biparental conjugation, as described above.
Fluorescence Imaging.
All cells expressing fluorescently tagged proteins were prepared by growing them to low density (OD600 of 0.2 to 0.4) in 5 ml of MMAB supplemented with 10 mM malate. Cells from these cultures were then pelleted via centrifugation. Thirty microliters of concentrated cells was pipetted onto a 1% low-melting-point (LMP) agarose pad in 1× phosphate-buffered saline (PBS) with a coverslip and left undisturbed for 2 to 3 min. Images were acquired using the YFP or fluorescein isothiocyanate (FITC) filter on a Nikon ECLIPSE 80i fluorescence microscope with a Nikon CoolSnap HQ2-cooled charge-coupled device (CCD) camera.
Cell length measurements.
Cells to be analyzed were grown at 28°C to mid-log phase (OD600 of 0.4 to 0.6) in 5 ml of liquid MMAB supplemented with 18.5 mM ammonium chloride, 5 mM malate, and 5 mM fructose (17). Cells were concentrated by centrifugation, stained with 1 μM FM4-64 vital fluorescent dye (Invitrogen Molecular Probes), incubated for 5 min in the dark, centrifuged at maximum speed for 1 to 2 min, and pipetted on top of 1% LMP agarose prepared in 1× PBS. Images were acquired using the tetramethyl rhodamine isocyanate (TRITC) filter on a Nikon ECLIPSE 80i fluorescence microscope with a Nikon CoolSnap HQ2-cooled CCD camera. Cell length measurements in micrometers were obtained using the Nikon NIS-Elements BR program from at least 50 cells per sample in four different fields of view.
Doubling time.
Cells were grown at 28°C to mid-log phase (OD600 of 0.4 to 0.6) either in 5 ml of liquid MMAB supplemented with 18.5 mM ammonium chloride and 10 mM malate and with tetracycline (10 μg/ml) or in 5 ml of liquid TY with the same supplements. Forty microliters of the liquid culture was inoculated into 260 μl of fresh MMAB with tetracycline (10 μg/ml), and the resulting cultures underwent a growth curve on a microplate reader. The absorbance (OD600) of each sample was taken every 20 min for a total of 16 h, and the doubling time was calculated from the growth curve. All curves were determined in triplicate.
Cell fractionation.
Cultures were collected at log phase (OD600 of 0.4 to 0.6) via centrifugation at 10,000 × g for 10 min at 4°C, and the pellets were resuspended in 50 mM potassium phosphate buffer containing 2 mM MgCl2 and 1 mM proteinase inhibitor phenylmethylsulfonyl fluoride (PMSF) (pH 7.0). Cells were then disrupted by passage through a French press three times at a cell pressure of 25,000 lb/in2. The cell lysate was centrifuged (Fiberlite F13-14 × 50cy fixed angle rotor) at 10,000 × g for 20 min at 4°C, and the supernatant centrifuged at 110,000 × g for 120 min at 4°C (Ti 50.2 fixed-angle ultracentrifuge rotor). The pellet was suspended in the phosphate buffer once more and centrifuged again at 110,000 × g for 120 min at 4°C. The membrane pellet was solubilized in 1× PBS with 2% Triton X-100 and 1 mM PMSF. Protein concentrations were measured for each sample using the Bradford assay (50), and each sample was adjusted to the same protein concentration prior to loading into the SDS-polyacrylamide gel. Cell fractions were verified as membrane or cytosolic by differential staining with the hydrophobic molecular dye FM1-43, which is a lipophilic dye that fluoresces exclusively upon insertion into the cell membrane (Invitrogen Molecular Probes). The cell fractionated samples were mixed in a 1:1 ratio with the membrane-specific FM1-43 molecular dye (Invitrogen), incubated at room temperature for 15 min in the dark, and imaged by fluorescence microscopy using an FITC filter. Fluorescence intensity was compared between samples and with buffer controls exposed under similar conditions.
Western blotting.
For constructs containing the pBBR1MCS3 cloning vector, 50 ml of each sample were grown to mid-log phase (OD600 of 0.7 to 0.8) in liquid MMAB supplemented with 18.5 mM ammonium chloride, 10 mM malate, and tetracycline (10 μg/ml). Cells were pelleted and resuspended in 200 μl fresh radioimmunoprecipitation assay (RIPA) buffer. The cells were sonicated (Fisher Sonic Dismembrator model 100) for 30 s at a 3% amplitude of pulse, followed by a 1-min rest period, for a total of five cycles. Equal volumes of protein (15 μg) and 2× Laemmli buffer with reducing agent were placed on the heat block at 70°C for 8 min and loaded into the wells of a 7.5% Mini-Protean precast gel (Bio-Rad). The gel ran at 120 V for 90 min or until the dye front reached the reference line. The gel was then transferred to a 0.45-μm hydrophobic polyvinylidene difluoride (PVDF) transfer membrane (Immobilon) using a semidry transfer apparatus (Bio-Rad). The transfer ran at 15 V for 35 min, and then the membrane was air dried for 45 min. The membrane was washed in 1× Tris-buffered saline–Tween 20 (TBST) for 5 min at room temperature before being blocked for 1.5 h at room temperature in a 5% milk solution in 1× TBST. Afterwards, the membrane was rinsed for 5 min in 1× TBST. For detection of CheA1 and variants, an anti-CheA1ΔTMX polyclonal antibody was purified using a negative-affinity protocol. Briefly, 25 ml of ΔcheA1 cells was grown to mid-log phase and spun down. The pellet was resuspended in 0.1 M NaHCO3 buffer (pH 8.3) supplemented with 0.5 M NaCl. Cells were then disrupted by passage through a French press three times at a cell pressure of 25,000 lb/in2 and were centrifuged before the supernatant was collected. The whole-cell proteins were then immobilized onto activated Sepharose 4B beads during an overnight incubation of proteins and beads at 4°C. Unreacted proteins were washed with coupling buffer (0.1 M carbonate buffer, 0.5 M NaCl, pH 8.3), and unreacted groups were blocked by incubating the beads overnight at 4°C in 0.2 M glycine (pH 8.0). The beads were washed with coupling buffer, resuspended in 1× PBS, and loaded into a column. The column was equilibrated with 1× PBS before antiserum was passed through the column and collected. The flowthrough was reapplied to the column three times to remove any contaminants and to concentrate the antibody. We further purified this flowthrough by preparing a column containing immobilized whole proteins extracted from the Δche1 mutant, which lacked the entire A. brasilense Che1 operon (17), and passed the antiserum through the column. The resulting antibody was used at a 1:100 dilution, and a secondary antibody (Li-Cor donkey anti-rabbit IRDye 800CW) was applied at a 1:5,000 dilution for 1 h. Membranes were imaged on an Odyssey CLx infrared imaging system, and band intensities were quantified using ImageStudioLite software (4.0; Li-Cor). A minimum of four images from independent samples was used. For cell fractionated samples, a total volume of 100 ml of cells was grown to mid-log phase (OD600 of 0.7 to 0.8) in liquid MMAB supplemented with 18.5 mM ammonium chloride and 10 mM malate before being fractionated as described above. Equal volumes of the membrane pellet, which was resuspended in 1× PBS with 2% Triton X-100, 1 mM PMSF, and 2× Laemmli buffer with reducing agent, were boiled for 5 min prior to being loaded, at an amount of 31 μg, into an 8 to 16% Mini-Protean precast gradient gel (Bio-Rad). The gel was run at 200 V for 30 min before being transferred to a 0.2 μM nitrocellulose membrane using a semidry transfer apparatus (Bio-Rad). The transfer was performed at 15 V for 25 min, followed by membrane washes in 1× TBST for 5 min at room temperature. The membrane was blocked for 1 h at room temperature in a 5% milk solution in 1× TBST and rinsed for 5 min in 1× TBST. For the cytosolic fraction, the same protocol was followed except that the samples were loaded at a concentration of 20.5 μg and run on a 10% Mini-Protean precast gel (Bio-Rad). For detection of CheA1 in the cell fractionated samples, an anti-CheA1ΔTMX polyclonal antibody, raised against soluble CheA1ΔTMX from A. brasilense strain Sp7 but not further purified against whole proteins extracted from the Δche1 mutant, was used at a 1:4 dilution, and a secondary antibody (Li-Cor donkey anti-rabbit IRDye 800CW) was applied at a 1:10,000 dilution for 1 h. Membranes were imaged on an Odyssey CLx Infrared imaging system. For whole-cell extracts, the same protocol as for cells bearing the pBBR1MCS3 cloning vector was used. Cells were grown as described above, and then 15 μg was loaded into the wells of a 7.5% gel Mini-Protean precast gel (Bio-Rad) and run at 120 V for 90 min or until the dye front reached the reference line. The gel was then transferred to a 0.45-μm hydrophobic PVDF transfer membrane (Immobilon) using a semidry transfer apparatus (Bio-Rad). The transfer to the nitrocellulose membrane was performed at 15 V for 35 min, and the membrane was air dried for 45 min and then washed in 1× TBST for 5 min at room temperature. The membrane was blocked for 1 h at room temperature in a 5% milk solution in 1× TBST and rinsed for 5 min in 1× TBST. The same anti-CheA1ΔTMX polyclonal antibody, which was further affinity purified with immobilized whole protein extracts from the Δche1 strain (17) as described above, was used at a 1:100 dilution, and the secondary antibody (Li-Cor donkey anti-rabbit IRDye 800CW) was applied for 1 h at room temperature at a 1:5,000 dilution. Samples were imaged using an Odyssey CLx infrared imaging system, and band intensities were quantified using ImageStudioLite software.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Science Foundation (NSF) grants MCB-0919819 and MCB 1330344, awarded to G.A. We acknowledge support from the University of Tennessee College of Arts and Sciences and the BCMB department to attend the ASM Scientific Writing and Publishing Institute, which provided assistance with scientific writing development.
Any opinions, findings, conclusions, or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
We thank Anastasia Aksenova for valuable input and technical assistance.
We declare no conflict of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00189-17.
REFERENCES
- 1.Krell T, Lacal J, Munoz-Martinez F, Reyes-Darias JA, Cadirci BH, Garcia-Fontana C, Ramos JL. 2011. Diversity at its best: bacterial taxis. Environ Microbiol 13:1115–1124. doi: 10.1111/j.1462-2920.2010.02383.x. [DOI] [PubMed] [Google Scholar]
- 2.Alexandre G. 2010. Coupling metabolism and chemotaxis-dependent behaviours by energy taxis receptors. Microbiology 156:2283–2293. doi: 10.1099/mic.0.039214-0. [DOI] [PubMed] [Google Scholar]
- 3.Wadhams GH, Armitage JP. 2004. Making sense of it all: bacterial chemotaxis. Nat Rev Mol Cell Biol 5:1024–1037. doi: 10.1038/nrm1524. [DOI] [PubMed] [Google Scholar]
- 4.Levit MN, Stock JB. 2002. Receptor methylation controls the magnitude of stimulus-response coupling in bacterial chemotaxis. J Biol Chem 277:36760–36765. doi: 10.1074/jbc.M204325200. [DOI] [PubMed] [Google Scholar]
- 5.Surette MG, Levit M, Liu Y, Lukat G, Ninfa EG, Ninfa A, Stock JB. 1996. Dimerization is required for the activity of the protein histidine kinase CheA that mediates signal transduction in bacterial chemotaxis. J Biol Chem 271:939–945. doi: 10.1074/jbc.271.2.939. [DOI] [PubMed] [Google Scholar]
- 6.Bilwes AM, Alex LA, Crane BR, Simon MI. 1999. Structure of CheA, a signal-transducing histidine kinase. Cell 96:131–141. doi: 10.1016/S0092-8674(00)80966-6. [DOI] [PubMed] [Google Scholar]
- 7.Briegel A, Ladinsky MS, Oikonomou C, Jones CW, Harris MJ, Fowler DJ, Chang YW, Thompson LK, Armitage JP, Jensen GJ. 2014. Structure of bacterial cytoplasmic chemoreceptor arrays and implications for chemotactic signaling. eLife 3:e02151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Briegel A, Li X, Bilwes AM, Hughes KT, Jensen GJ, Crane BR. 2012. Bacterial chemoreceptor arrays are hexagonally packed trimers of receptor dimers networked by rings of kinase and coupling proteins. Proc Natl Acad Sci U S A 109:3766–3771. doi: 10.1073/pnas.1115719109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li M, Hazelbauer GL. 2011. Core unit of chemotaxis signaling complexes. Proc Natl Acad Sci U S A 108:9390–9395. doi: 10.1073/pnas.1104824108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun H, Zusman DR, Shi W. 2000. Type IV pilus of Myxococcus xanthus is a motility apparatus controlled by the frz chemosensory system. Curr Biol 10:1143–1146. doi: 10.1016/S0960-9822(00)00705-3. [DOI] [PubMed] [Google Scholar]
- 11.Güvener ZT, Harwood CS. 2007. Subcellular location characteristics of the Pseudomonas aeruginosa GGDEF protein, WspR, indicate that it produces cyclic-di-GMP in response to growth on surfaces. Mol Microbiol 66:1459–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirby JR, Zusman DR. 2003. Chemosensory regulation of developmental gene expression in Myxococcus xanthus. Proc Natl Acad Sci U S A 100:2008–2013. doi: 10.1073/pnas.0330944100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bible AN, Khalsa-Moyers GK, Mukherjee T, Green CS, Mishra P, Purcell A, Aksenova A, Hurst GB, Alexandre G. 2015. Metabolic adaptations of Azospirillum brasilense to oxygen stress by cell-to-cell clumping and flocculation. Appl Environ Microbiol 81:8346–8357. doi: 10.1128/AEM.02782-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berleman JE, Bauer CE. 2005. Involvement of a Che-like signal transduction cascade in regulating cyst cell development in Rhodospirillum centenum. Mol Microbiol 56:1457–1466. doi: 10.1111/j.1365-2958.2005.04646.x. [DOI] [PubMed] [Google Scholar]
- 15.Wuichet K, Zhulin IB. 2010. Origins and diversification of a complex signal transduction system in prokaryotes. Sci Signal 3:ra50. doi: 10.1126/scisignal.2000724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bible A, Russell MH, Alexandre G. 2012. The Azospirillum brasilense Che1 chemotaxis pathway controls swimming velocity, which affects transient cell-to-cell clumping. J Bacteriol 194:3343–3355. doi: 10.1128/JB.00310-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bible AN, Stephens BB, Ortega DR, Xie Z, Alexandre G. 2008. Function of a chemotaxis-like signal transduction pathway in modulating motility, cell clumping, and cell length in the alphaproteobacterium Azospirillum brasilense. J Bacteriol 190:6365–6375. doi: 10.1128/JB.00734-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qi X, Nellas RB, Byrn MW, Russell MH, Bible AN, Alexandre G, Shen T. 2013. Swimming motility plays a key role in the stochastic dynamics of cell clumping. Phys Biol 10:026005. doi: 10.1088/1478-3975/10/2/026005. [DOI] [PubMed] [Google Scholar]
- 19.Mukherjee T, Kumar D, Burriss N, Xie Z, Alexandre G. 27 May 2016. Azospirillum brasilense chemotaxis depends on two signaling pathways regulating distinct motility parameters. J Bacteriol doi: 10.1128/jb.00020-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hauwaerts D, Alexandre G, Das SK, Vanderleyden J, Zhulin IB. 2002. A major chemotaxis gene cluster in Azospirillum brasilense and relationships between chemotaxis operons in α-proteobacteria. FEMS Microbiol Lett 208:61–67. [DOI] [PubMed] [Google Scholar]
- 21.Ponting CP, Schultz J, Milpetz F, Bork P. 1999. SMART: identification and annotation of domains from signalling and extracellular protein sequences. Nucleic Acids Res 27:229–232. doi: 10.1093/nar/27.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kentner D, Sourjik V. 2006. Spatial organization of the bacterial chemotaxis system. Curr Opin Microbiol 9:619–624. doi: 10.1016/j.mib.2006.10.012. [DOI] [PubMed] [Google Scholar]
- 23.Greer-Phillips SE, Stephens BB, Alexandre G. 2004. An energy taxis transducer promotes root colonization by Azospirillum brasilense. J Bacteriol 186:6595–6604. doi: 10.1128/JB.186.19.6595-6604.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russell MH, Bible AN, Fang X, Gooding JR, Campagna SR, Gomelsky M, Alexandre G. 2013. Integration of the second messenger c-di-GMP into the chemotactic signaling pathway. mBio 4:e00001-13. doi: 10.1128/mBio.00001-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie Z, Ulrich LE, Zhulin IB, Alexandre G. 2010. PAS domain containing chemoreceptor couples dynamic changes in metabolism with chemotaxis. Proc Natl Acad Sci U A 107:2235–2240. doi: 10.1073/pnas.0910055107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baida GE, Kuzmin NP. 1995. Cloning and primary structure of a new hemolysin gene from Bacillus cereus. Biochim Biophys Acta 1264:151–154. doi: 10.1016/0167-4781(95)00150-F. [DOI] [PubMed] [Google Scholar]
- 27.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer ELL, Tate J, Punta M. 2014. Pfam: the protein families database. Nucleic Acids Res 42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Y-C, Chang M-C, Chuang Y-C, Jeang C-L. 2004. Characterization and virulence of hemolysin III from Vibrio vulnificus. Curr Microbiol 49:175–179. doi: 10.1007/s00284-004-4288-5. [DOI] [PubMed] [Google Scholar]
- 29.Tang YT, Hu T, Arterburn M, Boyle B, Bright JM, Emtage PC, Funk WD. 2005. PAQR proteins: a novel membrane receptor family defined by an ancient 7-transmembrane pass motif. J Mol Evol 61:372–380. doi: 10.1007/s00239-004-0375-2. [DOI] [PubMed] [Google Scholar]
- 30.Inclan YF, Laurent S, Zusman DR. 2008. The receiver domain of FrzE, a CheA-CheY fusion protein, regulates the CheA histidine kinase activity and downstream signalling to the A- and S-motility systems of Myxococcus xanthus. Mol Microbiol 68:1328–1339. doi: 10.1111/j.1365-2958.2008.06238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He K, Marden JN, Quardokus EM, Bauer CE. 2013. Phosphate flow between hybrid histidine kinases CheA(3) and CheS(3) controls Rhodospirillum centenum cyst formation. PLoS Genet 9:e1004002. doi: 10.1371/journal.pgen.1004002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kofoid EC, Parkinson JS. 1991. Tandem translation starts in the cheA locus of Escherichia coli. J Bacteriol 173:2116–2119. doi: 10.1128/jb.173.6.2116-2119.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith RA, Parkinson JS. 1980. Overlapping genes at the cheA locus of Escherichia coli. Proc Natl Acad Sci U S A 77:5370–5374. doi: 10.1073/pnas.77.9.5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wisniewski-Dyé F, Borziak K, Khalsa-Moyers G, Alexandre G, Sukharnikov LO, Wuichet K, Hurst GB, McDonald WH, Robertson JS, Barbe V, Calteau A, Rouy Z, Mangenot S, Prigent-Combaret C, Normand P, Boyer M, Siguier P, Dessaux Y, Elmerich C, Condemine G, Krishnen G, Kennedy I, Paterson AH, González V, Mavingui P, Zhulin IB. 2011. Azospirillum genomes reveal transition of bacteria from aquatic to terrestrial environments. PLoS Genet 7:e1002430. doi: 10.1371/journal.pgen.1002430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaneko T, Minamisawa K, Isawa T, Nakatsukasa H, Mitsui H, Kawaharada Y, Nakamura Y, Watanabe A, Kawashima K, Ono A, Shimizu Y, Takahashi C, Minami C, Fujishiro T, Kohara M, Katoh M, Nakazaki N, Nakayama S, Yamada M, Tabata S, Sato S. 2010. Complete genomic structure of the cultivated rice endophyte Azospirillum sp. B510. DNA Res 17:37–50. doi: 10.1093/dnares/dsp026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rivera D, Revale S, Molina R, Gualpa J, Puente M, Maroniche G, Paris G, Baker D, Clavijo B, McLay K, Spaepen S, Perticari A, Vazquez M, Wisniewski-Dyé F, Watkins C, Martinez-Abarca F, Vanderleyden J, Cassan F. 2014. Complete genome sequence of the model rhizosphere strain Azospirillum brasilense Az39, successfully applied in agriculture. Genome Announc 2:e00683-14. doi: 10.1128/genomeA.00683-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwak Y, Shin JH. 2015. First Azospirillum genome from aquatic environments: whole-genome sequence of Azospirillum thiophilum BV-S, a novel diazotroph harboring a capacity of sulfur-chemolithotrophy from a sulfide spring. Mar Genomics doi: 10.1016/j.margen.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 38.Wisniewski-Dyé F, Lozano L, Acosta-Cruz E, Borland S, Drogue B, Prigent-Combaret C, Rouy Z, Barbe V, Herrera AM, Gonzalez V, Mavingui P. 2012. Genome sequence of Azospirillum brasilense CBG497 and comparative analyses of Azospirillum core and accessory genomes provide insight into niche adaptation. Genes (Basel) 3:576–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin-Didonet CC, Chubatsu LS, Souza EM, Kleina M, Rego FG, Rigo LU, Yates MG, Pedrosa FO. 2000. Genome structure of the genus Azospirillum. J Bacteriol 182:4113–4116. doi: 10.1128/JB.182.14.4113-4116.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pasek S, Risler JL, Brezellec P. 2006. Gene fusion/fission is a major contributor to evolution of multi-domain bacterial proteins. Bioinformatics 22:1418–1423. doi: 10.1093/bioinformatics/btl135. [DOI] [PubMed] [Google Scholar]
- 41.Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D. 1999. Detecting protein function and protein-protein interactions from genome sequences. Science 285:751–753. doi: 10.1126/science.285.5428.751. [DOI] [PubMed] [Google Scholar]
- 42.Baldani VLD, de B Alvarez MA, Baldani JI xd, Bereiner J. 1986. Establishment of inoculated Azospirillum spp. in the rhizosphere and in roots of field grown wheat and sorghum. Plant Soil 90:35–46. doi: 10.1007/BF02277385. [DOI] [Google Scholar]
- 43.Vanstockem M, Michiels K, Vanderleyden J, Van Gool AP. 1987. Transposon mutagenesis of Azospirillum brasilense and Azospirillum lipoferum: physical analysis of Tn5 and Tn5-Mob insertion mutants. Appl Environ Microbiol 53:410–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tomic-Canic M, Bernerd F, Blumenberg M. 1996. A simple method to introduce internal deletions or mutations into any position of a target DNA sequence. Methods Mol Biol 57:249–257. [DOI] [PubMed] [Google Scholar]
- 45.Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM II, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176. doi: 10.1016/0378-1119(95)00584-1. [DOI] [PubMed] [Google Scholar]
- 46.Stephens BB, Loar SN, Alexandre G. 2006. Role of CheB and CheR in the complex chemotactic and aerotactic pathway of Azospirillum brasilense. J Bacteriol 188:4759–4768. doi: 10.1128/JB.00267-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simon R, Priefer U, Puhler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nat Biotechnol 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 48.Katzen F, Becker A, Ielmini MV, Oddo CG, Ielpi L. 1999. New mobilizable vectors suitable for gene replacement in gram-negative bacteria and their use in mapping of the 3′ end of the Xanthomonas campestris pv. campestris gum operon Appl Environ Microbiol 65:278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hallez R, Letesson JJ, Vandenhaute J, De Bolle X. 2007. Gateway-based destination vectors for functional analyses of bacterial ORFeomes: application to the Min system in Brucella abortus. Appl Environ Microbiol 73:1375–1379. doi: 10.1128/AEM.01873-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.