

ABSTRACT
In bacteria, replication forks assembled at a replication origin travel to the terminus, often a few megabases away. They may encounter obstacles that trigger replisome disassembly, rendering replication restart from abandoned forks crucial for cell viability. During the past 25 years, the genes that encode replication restart proteins have been identified and genetically characterized. In parallel, the enzymes were purified and analyzed in vitro, where they can catalyze replication initiation in a sequence-independent manner from fork-like DNA structures. This work also revealed a close link between replication and homologous recombination, as replication restart from recombination intermediates is an essential step of DNA double-strand break repair in bacteria and, conversely, arrested replication forks can be acted upon by recombination proteins and converted into various recombination substrates. In this review, we summarize this intense period of research that led to the characterization of the ubiquitous replication restart protein PriA and its partners, to the definition of several replication restart pathways in vivo, and to the description of tight links between replication and homologous recombination, responsible for the importance of replication restart in the maintenance of genome stability.
KEYWORDS: DNA replication, homologous recombination
INTRODUCTION
Replication of a circular bacterial chromosome normally initiates at a unique origin. For bidirectional DNA replication, two replication forks are established that replicate the DNA in opposite directions until they meet in the terminus region. Replication fork progression involves the coordinated action of two complexes, the primosome to open the DNA duplex and synthesize primers and the replisome to catalyze the concerted DNA synthesis of both DNA strands. The two DNA strands at the replication fork are antiparallel, and DNA synthesis occurs only in the 5′-to-3′ direction. Therefore, to synthesize both strands in a concerted and semiconservative fashion, one strand is synthesized mainly continuously (the leading strand) and the other is synthesized discontinuously (the lagging strand). The fragments generated on the discontinuous strand are 1 to 2 kb in length and are called Okazaki fragments (OF). In Escherichia coli, the primosome is composed of the DnaB helicase that opens the parental strands and the DnaG primase that interacts transiently with DnaB and synthesizes the RNA primers at the onset of each OF synthesis. DnaB is a hexameric helicase that encircles single-stranded DNA (ssDNA) and translocates on the lagging-strand template in a 5′-to-3′ direction. The DNA polymerase III holoenzyme (Pol III HE) synthesizes both nascent DNA strands, and its action is stimulated by interactions with DnaB and with the ssDNA binding protein (SSB) that covers the lagging-strand template (1).
The first committed step in the assembly of a replication fork is DnaB loading. In bacteria, two pathways for DnaB loading have been described, a sequence-specific, DnaA-dependent pathway at the chromosome origin (oriC) (2) and a sequence-independent pathway that rescues inactivated or broken and repaired replication forks requiring the replication restart proteins PriA, PriB, PriC, DnaT, and possibly Rep. In both processes, after identification of the proper DNA sequence or structure, exposure of a region of ssDNA by displacement of DNA-bound SSB allows DnaC to load the DnaB helicase onto the DNA to initiate replisome assembly. Depending on where DnaB is loaded, DnaC will require interactions either between DnaA and DnaB or between combinations of the replication restart proteins to facilitate the process.
Because of the remarkable stability of replisomes assembled from purified components on DNA in vitro, it was long thought that in vivo replication forks assembled at the origin could progress toward the terminus unimpeded. However, in the 1990s, the preprimosomal proteins were identified as proteins that promote replication initiation independently of DnaA. Originally identified as proteins required for the conversion of phage ssDNA into a duplex and called n (or factor Y), n′, n″, and i replication proteins, the preprimosomal proteins were rebaptized PriA, PriB, PriC, and DnaT when it was realized that they play a role in chromosome replication (but are not required for replication initiation at oriC) (3, 4). Their action required the recognition of a specific ssDNA region of the circular ϕX174 genome that is able to adopt a special secondary structure (hairpin) called the primosome assembly site (PAS) (5). The order of assembly and the stoichiometries of final “preprimosome” were determined as PriA (two or three monomers), PriB (two dimers), DnaT (one trimer), PriC (one monomer), and the DnaB helicase loaded by DnaC (one hexamer) (6–8). The preprimosome becomes the primosome in the presence of the primase DnaG. PriC is the only nonessential protein for preprimosome assembly. Its presence stimulated preprimosome assembly, and PriC was considered to be a stability factor in these reports (7, 8). The observation that PriA promoted oriC-independent replication in vivo (9) and that cells lacking the PriA protein suffered severe growth defects (10) suggested a role for primosome assembly in E. coli chromosome replication. Work in several laboratories led to the conclusion that the replisome may need to be reloaded in a PriA-dependent way during chromosome replication and that failure to do so has severe consequences (11, 12).
The essential role of PriA for the recombinational repair of DNA double-strand breaks (DSBs) indicated that homologous recombination triggers replication restart (10, 13–15). Conversely, arrested replication forks were shown to be targeted by recombination enzymes and cause chromosome rearrangements (16, 17). Another demonstration of the close relationship between replication restart and recombination is that the poorly partitioned nucleoids observed in a subpopulation of priA mutant cells is caused by the homologous recombination machinery (18). The discovery of links between replication and recombination gave rise to the notion that replication fork restart plays an important role in genome stability, which started an era of strong interest and intense studies.
The viability defects caused by the inactivation of homologous recombination functions are far less dramatic than the inactivation of replication restart. Therefore, it is clear from the genetic data that the replication restart proteins are mainly required to reload a replisome at abandoned replication forks. Accordingly, PriA is required for the viability of cells in which replication arrest is increased but does not trigger fork breakage, for example, in gyrase and topoisomerase IV mutants where forks are arrested by the accumulation of positive supercoils (19, 20). An early estimate of replication restart frequency was based on the percentage of partially replicated chromosomes in a dnaC2(Ts) mutant as determined by flow cytometry. That study suggested that the range could be around 18 to 25% of the E. coli cells in a population (21). In contrast, direct measurements of helicase stability in the same dnaC2(Ts) mutant by single-molecule microscopy showed that both helicase complexes disassembled within 20 min in 86% of the cells studied (22). The reasons for the discrepancy between these population and single-cell studies are unknown. In the same single-molecule microscopy study, measurements of the lifetimes of DNA complexes suggest that restart could be as frequent as five times per generation in Bacillus subtilis, as in E. coli (22). This result highlights the importance of replication restart and shows clearly that replication restart most often does not involve homologous recombination, as this frequency would then be incompatible with the viability of recombination mutants. In this review, we will describe next the PriA-dependent replication restart process and PriA partners in vivo and in vitro and then various reactions that eventually take place prior to PriA-dependent replication restart.
GENETICS OF PriA AND ITS PARTNERS
Three pathways of replication restart were originally proposed on the basis of the patterns of synthetic lethality between pairs of null mutants. The three pathways are outlined in Fig. 1. They are referred to as PriA-PriB-DnaT, PriA-PriC-DnaT, and PriC. (As described below, we now feel that a more parsimonious interpretation of the current evidence would remove the Rep helicase from the PriA-independent pathway, where it has been previously placed [15], and call this pathway just PriC, as in Fig. 1.) priA and dnaT null mutants are deficient for both PriA-PriB-DnaT and PriA-PriC-DnaT pathways and rely solely on the PriC pathway. They have the most extreme phenotypes, showing poor cell growth/viability, high basal levels of SOS expression, defects in nucleoid morphology (a partitioning-defective phenotype), sensitivity to UV irradiation, and recombination deficiency (23–26) (Fig. 2). The priA dnaT double mutant is viable and has phenotypes similar to those of the single mutants (24). This shows that PriA and DnaT are not required for the PriC pathway and that PriA and DnaT are often needed, but not at each replication round, or the mutants would not be viable. priB and priC null mutants individually have little effect on cellular physiology, while the priBC double mutant is inviable (27). This result led to the proposals of a PriA-independent PriC pathway (28) and that the PriAB-DnaT and PriAC-DnaT pathways are formally equal. Experimentally, however, one can detect differences between the two PriA-dependent pathways. Inactivation of priB or priC does not have the same consequences in strains that have an additional mutation that increases the frequency of replication arrest. For example, inactivation of priB is five times more deleterious than inactivation of priC in a holD mutant (defective for a polymerase III subunit) and leads to rich medium sensitivity in a gyrB(Ts) mutant at semipermissive temperature (defective for gyrase), whereas inactivation of priC has no effect. These results led to the proposal that the PriA-PriB-DnaT pathway is the major PriA pathway in vivo (20, 29).
FIG 1.
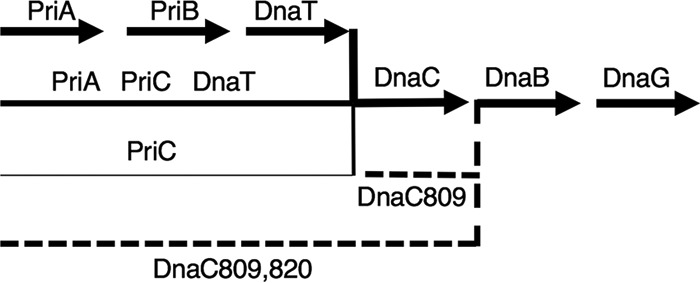
Replication restart pathways. Schematic representation of pathways for replication restart as deduced from genetic evidence. The starting substrates are abandoned forks or D loops formed by homologous recombination at dsDNA ends. The product of the reaction is a DNA replication fork with DnaB and DnaG loaded and ready to interact with the DNA Pol III HE. The PriA-PriB-DnaT pathway is the major pathway in vivo and the only one fully reconstituted in vitro. The existence of a PriA-PriC-DnaT pathway is deduced from the weak viability of the priA and dnaT mutants, in contrast to the full viability of a priB mutant and the nonviability of a priB priC double mutant. This pathway could not be reconstituted in vitro, where PriC alone is sufficient for primosome assembly. This model, however, is the only one that accounts for the phenotypes listed above. The PriC pathway was deduced from the colethality of a priC mutation with each of the priA and priB mutations. It was originally proposed to involve Rep because of the lethality of rep priA and rep priB, but this observation may rather result from replication blockage by DNA-bound RNA polymerases in the rep mutant and a need for PriA and PriB in mutants where forks are frequently arrested. Nevertheless, replication restart can be catalyzed by PriC and Rep in vitro by using substrates where DnaB loading is blocked by a 5′ lagging-strand end (20). Finally, DnaC809,820 suppresses all replication restart defects, suggesting that it allows DnaB loading at forks on its own in vivo. In contrast, DnaC809 is unable to restore priA viability in the rep or holD context, where replication fork arrest is frequent, or in a priC context. The latter observation suggested a role for PriC in the DnaC809 pathway. However, in vivo and in vitro results were contradictory, as DnaC809 could load DnaB on an SSB-covered substrate without the help of any other protein. Some unknown protein might prevent PriC and DnaC809 from acting alone in vivo, leading to the existence of the PriA-PriC-DnaT and PriC-DnaC809 pathways. Solid lines represent pathways available in wild-type cells (their thickness represents the relative amount of use), and dashed lines represents suppressor pathways.
FIG 2.
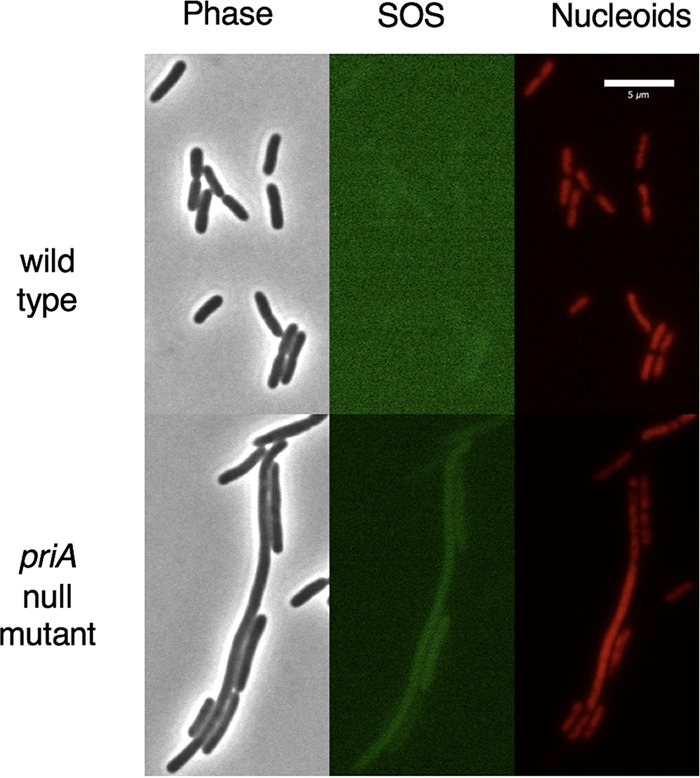
Micrographs showing the growth of two strains of bacteria on an agarose pad made of minimal medium. The two strains contain a hupA-mCherry protein fusion to visualize the nucleoids and the sulAp-green fluorescent protein promoter fusion to visualize the amount of SOS expression (106, 107). The priA null mutation is priA2::kan. The lighter green color of the wild-type SOS image is due to increased contrast enhancement to show what little, if any, SOS expression there is in those cells. The priA null mutant shows an example of a long cell with high levels of SOS expression and a poorly partitioning nucleoid and examples of middle-sized cells having both high and low levels of SOS expression and nucleoids that look wild type or Par−. Such cells represent 16% of the total cells in a priA mutant culture in minimal medium. Also shown are examples of short, wild-type-size cells having very low SOS expression and wild-type nucleoids, which make up the remaining 84%.
Mutations in priA causing defects in the PriA-PriB-DnaT and the PriA-PriC-DnaT pathways can be suppressed by different missense mutations in dnaC (27, 28). dnaC809 is an example of one type of suppressor. It suppresses the priA mutant growth defects in an otherwise wild-type context. However, it does not suppress the lethality of the priA priC, priA rep, and priA holD double mutations (28, 29). The first two observations were originally interpreted as replication restart occurring in a priA dnaC809 mutant via a PriC-Rep replication restart pathway. It was later proposed that the efficiency of replication restart by DnaC809 could be too low for the viability of priA rep and priA holD mutants because mutations in rep and holD increase the frequency of replication fork arrest (29). In contrast, the priC mutation does not increase replication fork arrest and the lack of suppression by dnaC809 supports the idea that all replication restart pathways are inactivated in the priA priC and priB priC double mutants (Fig. 1). A second type of suppressor was selected in a priBC dnaC809 triple mutant. This suppressor is a second mutation in dnaC809, called dnaC809,820, and it makes the suppression of priA mutants independent of PriC, Rep, and HolD. This suggested that dnaC809,820 may be a more efficient dnaC suppressor mutation than dnaC809 (28, 29). A third type was found in a priB rep double mutant; it was called dnaC824, and it only partially suppressed priA null mutant phenotypes (30).
The PriC pathway is a minor pathway of replication restart in E. coli. It was proposed on the basis of the lethality of the priA priC and priB priC mutations. As explained above, it was also originally proposed to include Rep because of the lethality of each priC and rep mutation in a priA dnaC809 context (28). However, Rep possesses another function in E. coli, which is to remove DNA-bound proteins from the path of replication forks. Consequently, rep inactivation increases the frequency of replication fork arrest, which can account for the lethality of the rep priA mutation and the lack of rescue by the dnaC809 suppressor mutation (31–33). It should be noted that the priC mutant does not show any of the rep mutant phenotypes besides its colethality with priA (28, 34). Conversely, the only other phenotype of the priC mutant is colethality with certain dnaA(Ts) (dnaA46 and dnaA508) mutations at permissive temperatures. This colethality is not observed in the rep mutant (and dnaA46) and suggests a role for PriC during replication initiation, assuming that DnaA does not play a role in replication restart (35, 36).
BIOCHEMISTRY OF PriA AND ITS PARTNERS
Several studies have explored the biochemical details of the restart reaction. The PriA protein was shown to have two activities, a 3′-5′ helicase activity that is not required for primosome assembly in vitro or for replication restart in vivo and a primosome assembly activity (37). Using PAS as the substrate for loading, it was revealed that PriA would bind first. This binding was then stabilized by the addition of PriB, and the addition of DnaT allows for the loading of DnaB by DnaC (8). High levels of DnaT can render this reaction PriB independent (6). PriA was then shown to recognize and promote primosome assembly at displacement (D) loops, an early recombination intermediate made by RecA (Fig. 3) (38, 39). Finally, the binding of preprimosomal proteins was tested on three types of Y structures resembling a replication fork with only a 3′ leading-strand end at the point of the fork, only a 5′ lagging-strand end, or both (Fig. 3). The structure with a 3′ leading end is an isomer of a D loop. PriA was shown to preferentially recognize forks with such a nascent 3′ end, while PriC recognizes Y structures with a gap in the leading-strand template (40). Surprisingly, the recognition of different structures by these two proteins in vitro is at odds with a redundant function in vivo, unless each of these structures can be derived from the other one. PriA recognition of DNA 3′ ends at forks is mediated by its N-terminal domain (41). PriA binds PriB in the helicase domain, while PriB has overlapping binding sites for DnaT and ssDNA (42). Weak interactions and competing binding sites trigger a dynamic process for the formation of the PriA-PriB-DnaT complex (42). This provides a molecular handle for DnaC to recognize the proper substrate to load DnaB (42, 43). PriC alone provides a platform to load DnaB onto a substrate in vitro (40, 42, 44). Although no helicase function is required for primosome assembly in vitro when the lagging-strand template is single stranded, the presence of a 5′ DNA end at the tip of the fork renders a 3′-5′ helicase activity necessary for the reaction. This activity can be provided in vitro by PriA or Rep, whose role in the reaction may thus be to unwind a region of lagging-strand duplex so that DnaC can have a region of ssDNA to load DnaB (45). Note that, in vivo, a Y structure with a gap in both strands is not possible since the cDNA strands would anneal. The study of physical interactions between the replication restart proteins and their multisubunit complexes with DNA offers more in-depth insight into these sophisticated reactions (Table 1). To this end, several groups have reported functional and/or physical interactions between E. coli PriA and PriB (6, 42, 43), PriA and DnaT (6), PriB and DnaT (42), PriC and DnaB (46), PriC and Rep (45), and Rep and DnaB (47) and between Klebsiella pneumoniae PriC and DnaT (48).
FIG 3.
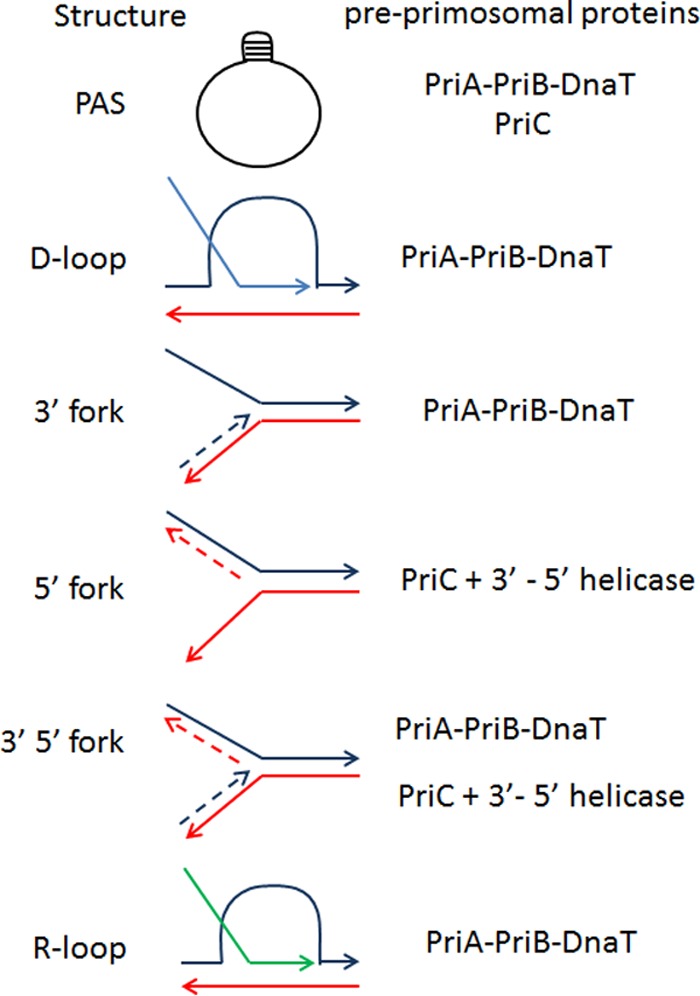
Schematic representation of the DNA molecules recognized and acted upon by preprimosomal proteins in vitro and presumably in vivo. From top to bottom, ϕX174 PAS, D loop resulting from the RecA-mediated invasion of a parental molecule (dark blue and red lines) by a homologous DNA RecA filament (light blue line), Y structure mimicking a replication fork with a fully synthesized leading-strand end and a gap in the lagging strand, Y structure mimicking a replication fork with a fully synthesized lagging-strand end and a gap in the leading strand, Y structure mimicking a replication fork with fully synthesized leading- and lagging-strand ends (in Y structures, parental strands are shown as full lines and newly synthesized strands are shown as dashed lines), and R loop (the RNA molecule is drawn as a green line). The preprimosomal proteins shown to assemble a primosome in these structures are indicated on the right. PriC action on PAS was deduced from the increased activity of the PriA-PriB-DnaT proteins in its presence. The 3′-5′ helicase function of PriA is required for its action on Y molecules with a dsDNA lagging strand.
TABLE 1.
Genes that encode replication restart proteins and activities, functions, and interactions of their products
| Gene | Protein activity in vitro | Protein function(s) in vivo | Interaction partner(s) |
|---|---|---|---|
| dnaA | Site-specific DNA binding protein at oriC | Targets replication initiation to oriC | DnaB, DnaC |
| dnaB | 5′-3′ helicase | Replicative helicase | Pol III clamp loader, DnaC, DnaG, Rep |
| dnaC | DnaB helicase loader | DnaB helicase loader during chromosome replication | DnaB, DnaA |
| dnaG | Primase | Primer synthesis during chromosome replication | DnaB |
| dnaT | Part of PriA-PriB-DnaT preprimosome complex | Essential for PriA-PriB-DnaT and PriA-PriC-DnaT replication restart pathways | PriA, PriB |
| priA | Structure-specific DNA binding protein (PAS, D loop, Y structure), 3′-5′ helicase | Targets replication restart to PAS, inactivated forks, and recombination intermediates | PriB, DnaT, SSB |
| priB | Part of PriA-PriB-DnaT preprimosome complex | Essential for the PriA-PriB-DnaT replication restart pathway | PriA, DnaT |
| priC | Preprimosome protein that promotes DnaB loading by itself | Preprimosome protein of PriC and PriA-PriC-DnaT replication restart pathways | SSB DnaB |
| rep | 3′-5′ helicase, removes proteins from DNA, can act together with PriC at a 5′ fork | Removes proteins from path of replication forks | ϕX174 protein A, M13 gpII, DnaB, PriC |
| ssb | ssDNA binding protein | Protects ssDNA, targets proteins to forks | PriA, PriC, Pol III, several other proteins |
It is clear that all substrates considered to be important in the restart process have regions of ssDNA that are likely to be coated with SSB in vivo. In vitro study has shown that DnaC809 can load DnaB on a DNA substrate bound with SSB, whereas DnaC cannot, suggesting that an important role of the preprimosomal proteins is to overcome the SSB barrier to DnaB loading (49). It has been shown that SSB can interact with many different replication and recombination proteins through its C terminus, including PriA and PriC (44, 50–52). These interactions were proposed to be important for SSB to recruit these proteins to replication forks and to play a role in primosome assembly. It is also known that SSB can bind ssDNA in two modes, one SSB tetramer binding either 35 (SSB35) or 65 (SSB65) nucleotides. SSB35 is associated with DNA replication, and SSB65 is associated with DNA repair. Single-molecule experiments have shown that both PriA and PriC promote shifting of the SSB binding mode from SSB65 to SSB35 (44, 53). It was proposed that PriA and PriC might thus release a small region of ssDNA where DnaC can load DnaB.
Other studies have revealed structures of PriA, PriB, and PriC by themselves and in the absence of DNA. A full-length structure of PriA from K. pneumoniae with 88% identity with E. coli PriA and the ability to complement PriA mutants of E. coli revealed a structure with six tightly clustered domains (53). The first domain has been crystallized before from the E. coli protein and shown to be able to bind the 3′-OH group of ssDNA (41). The next is a winged helix domain that is capable of binding double-stranded DNA (dsDNA). Then come two domains that comprise the ATPase activity. Encoded between these two larger domains is a cysteine-rich region (CRR) that binds two Zn2+ atoms. The CRR domain is thought to be important for both helicase activity and protein-protein interactions (6, 54). Lastly, the C terminus stabilizes the N-terminal DNA binding domain and the ATPase domains. It also has the ability to bind ssDNA. A model has been put forth that has PriA interacting with the DNA at the point of a replication fork with the 3′ end of the newly synthesized strand of DNA, with a small amount of duplex DNA ahead of the fork (yet to be unwound), and with a region of ssDNA on the lagging-strand template where DnaB is to be loaded. The CRR domain associated with helicase activity is poised to peel away the 5′ end of the strand of DNA annealed to the lagging-strand template. An X-ray crystal structure of E. coli PriB revealed an OB fold that is strikingly similar to SSB, although these two proteins bind ssDNA in different ways (55). This study also revealed residues important for the binding of PriA and ssDNA. A nuclear magnetic resonance structure of PriC protein from Cronobacter sakazakii (41% identical to E. coli PriC) revealed a compact alpha-helical structure that brings together residues identified as involved with the binding of ssDNA, SSB, and possibly DnaB (46).
REPLICATION RESTART IN BACTERIA OTHER THAN E. COLI
PriA is a ubiquitous protein in bacteria with a conserved action, even though it acts with different partner proteins in different bacteria (56–58). In B. subtilis, the three proteins required for PriA-dependent replication restart, DnaB, DnaI, and DnaD, also act at the replication origin (56–59). The helicase loader is a two-protein complex, DnaB-DnaI, and interestingly, helicase loading is catalyzed in E. coli and B. subtilis by two different molecular mechanisms. In E. coli, the hexameric helicase is preassembled in solution as a ring that is opened and reclosed around ssDNA during loading, while in B. subtilis, monomers are directly assembled onto ssDNA to form the hexameric ring structure (56–58). The third protein, DnaD, may play a structural role in the process (60). In Helicobacter pylori, the only replication restart protein identified is PriA (reviewed in reference 61). Like all bacteria that lack homologues of the E. coli DnaC and B. subtilis DnaI helicase loaders, H. pylori encodes a protein called DciA, which represents a third class of helicase loader (62). Replication restart proteins have been characterized genetically and biochemically in several bacterial species. For example, PriA and PriB were studied in Neisseria gonorrhoeae, a bacterium that lacks PriC (63, 64). PriB from K. pneumoniae was shown to be very similar to the E. coli enzyme, in contrast to N. gonorrhoeae PriB (65). The Deinococcus radiodurans PriA protein lacks the helicase activity (66). The function and structure of Gram-positive DnaD protein have been explored in Staphylococcus aureus (67 and references therein). The priA gene is absent from certain intracellular symbionts, and this sometimes coincides with the absence of the recA gene (68).
REACTIONS THAT CAN TAKE PLACE PRIOR TO PriA-DEPENDENT RESTART
Replication restart from a D loop made by RecBCD- and RecA-dependent strand invasion.
Several conditions where PriA is required for viability are linked to the occurrence of chromosomal DSBs and the coupling of DSB repair with PriA-dependent replication restart. Replication restart from a D loop formed by homologous recombination involves the same PriA partners as replication restart from replication fork structures (Fig. 3). PriA and PriB, together with DnaT, target the D loop and allow DnaC-catalyzed loading of DnaB on the strand that will become the lagging-strand template, which triggers the assembly of a replisome and replication initiation (69). As for replication initiation in vitro from other structures, PriC had only a minor stimulatory effect on the reaction. Break-induced DNA replication occurs in bacteria, phages, and eukaryotic cells (70). The coupling of DSB repair and replication restart ensures that replication is restarted after a broken or reversed fork is reintegrated into the chromosome by homologous recombination (Fig. 4, one-ended recombinational repair). On the other hand, owing to the potent exonuclease V action of RecBCD, DSB repair in bacteria is accompanied by extensive DNA degradation. Consequently, two D loops are independently formed at the two dsDNA ends of a DSB, and DSB repair in bacteria involves the merging of two PriA-dependent replication forks assembled at these D loops (71, 72) (Fig. 4, ends-in replication). Finally, insertion of a linear DNA creates oppositely oriented replication forks that can eventually copy the entire molecule (Fig. 4, ends-out replication) (73, 74).
FIG 4.

Replication restart following the repair of different substrates by RecBCD, RecA-mediated homologous recombination. On the left, one-ended homologous recombination at a broken replication fork restores unidirectional replication. In the middle, homologous recombination at a dsDNA break installs two converging replication forks. On the right, homologous recombination at the two ends of a linear molecule installs two diverging replication forks. Lines are DNA strands. Green arrows indicate the sites of replication restart in D loops. Adapted from reference 73.
Replication fork reversal.
Although bacteria are fully equipped to restart replication from abandoned replication forks, in some cases, replication forks are processed prior to restart. The reaction of replication fork reversal involves annealing of the newly synthesized leading- and lagging-strand ends at a blocked fork. This forms a dsDNA end adjacent to a Holliday junction (Fig. 5). The dsDNA end can be either degraded by RecBCD or recombined by RecBCD and RecA. Both reactions result in the formation of a PriA substrate that allows replication restart. Replication fork reversal was observed (i) in several E. coli replication mutants in which different replisome components are affected (75), (ii) in the rep mutant that lacks the main helicase responsible for clearing DNA-bound proteins (mainly RNA polymerases) from the path of replication forks (76), (iii) in the priA mutant, suggesting that it occurs in cells that fail to restart replication after spontaneous arrest (77), (iv) in HU-treated cells and in a ribonucleotide reductase mutant (nrdA) (78, 79), (v) after accumulation of topoisomerase I-DNA covalent complexes (80), (vi) in UV-irradiated cells (81), (vii) after encounter of replication forks with an oppositely oriented highly transcribed sequence (82), and (viii) in bacteria other than E. coli, such as in Pseudomonas syringae at low temperature (83).
FIG 5.
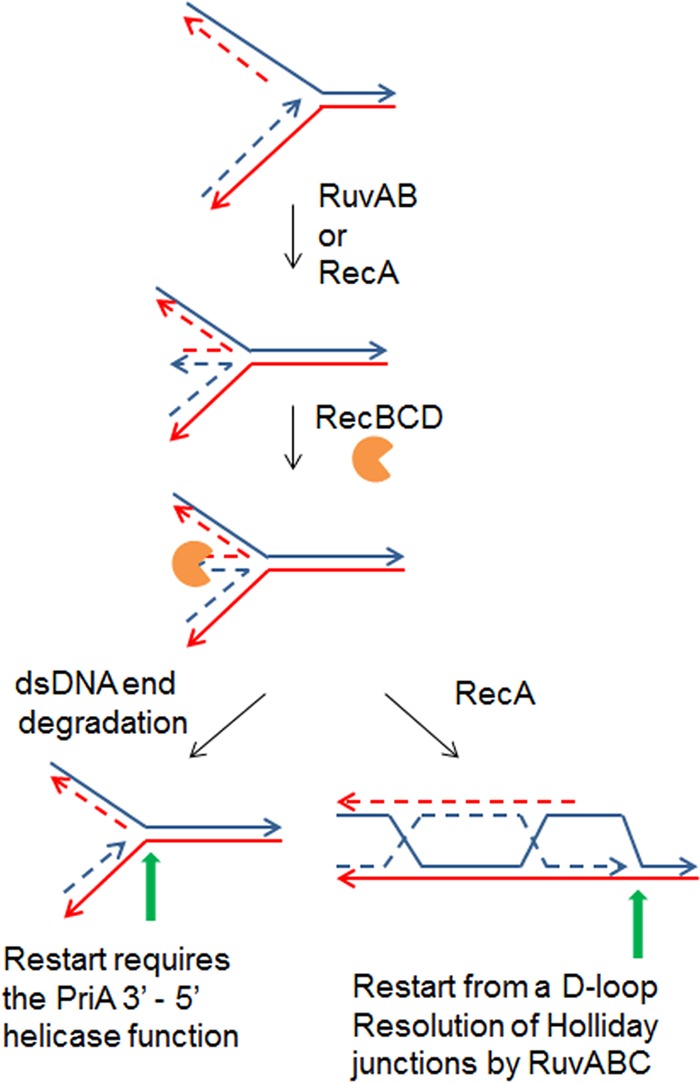
Replication restart after replication fork reversal. In a first step, the arrested fork is reversed by annealing of the leading- and lagging-strand ends, a reaction catalyzed by RuvAB or RecA. A dsDNA end is generated and acted upon by RecBCD. Either it is entirely degraded, resulting in a fork with full leading- and lagging-strand ends, which could account for the requirement for the PriA helicase function to unwind the lagging-strand end for DnaB loading, or the dsDNA end is recombined with the template DNA, creating a D loop that can be restarted by the PriA-PriB-DnaT pathway. Solid lines represent template DNA, dashed lines represent newly synthesized DNA, indented circles represent RecBCD, and green arrows represent sites of PriA binding for replication restart.
PriA is required for the viability of cells that undergo replication fork reversal (28, 29, 76). After replication fork reversal, the dsDNA end can be either degraded or recombined by RecBCD. As described above, homologous recombination renders all proteins of the PriA-PriB-DnaT pathway essential for restart. Although PriA helicase activity is not required for replication restart after homologous recombination (10), the growth of rep and holD mutants is impaired by inactivation of this activity (28, 29). This requirement could result from full degradation by RecBCD of the dsDNA end at some reversed forks, producing a fully duplex fork (with full leading- and lagging-strand ends and no gap) and thus rendering 5′ unwinding by the PriA helicase activity essential for primosome assembly (Fig. 5). Finally, PriA is required in UV-irradiated cells for oriC-independent replication (9) and for replication fork resumption after UV irradiation (84). The requirement for PriA may relate to the occurrence of replication fork reversal in UV-irradiated cells (81).
Replication restart allows the action of the UvrD helicase at protein-blocked forks.
Ter-Tus are physiological replication arrest sites present in the terminus region of the chromosome. They arrest replication forks coming from one direction and are positioned to form a replication fork trap ensuring that replication terminates in the region opposite to the origin (85). Interestingly, homologous recombination proteins and the UvrD helicase were shown to be required for replication restart at forks blocked by ectopic Ter-Tus complexes (86, 87). It was shown that a following round of oriC-initiated replication results in “rear ending” at the first blocked forks and the formation of dsDNA ends that need to be recombined for viability. The UvrD helicase was also required for viability and was proposed to dislodge the Tus-Ter complex from DNA. However, the requirement for homologous recombination proteins in a UvrD+ context implies that the UvrD helicase cannot directly clear Tus-blocked forks. It was proposed that in vivo replication restart from the D loop formed by homologous recombination allows UvrD to gain access to the fork, to clear DNA-bound proteins, and thus allow replication restart (87). It implies that PriA-dependent replication forks initiated from a recombination intermediate may differ from forks initiated at the chromosome origin, in this particular case, by the presence of UvrD. Furthermore, in the rep mutant, UvrD is essential for the removal of transcriptional obstacles from the path of replication forks (31, 32, 88). However, the presence of UvrD does not prevent replication fork reversal in this mutant. The data indicate that UvrD cannot clear blocked forks directly but goes in after fork reversal and resetting, acting at PriA-dependent restarting forks. Finally, the same phenomenon was also observed upon replication blockage by an oppositely oriented highly transcribed sequence (inverted rrn), where two accessory helicases are required to remove the obstacle, and they also act after replication fork reversal (31, 82).
Replication fork restart from R loops and during phage Mu integration.
In addition to recombination intermediates and abandoned forks, PriA can initiate replication from R loops in vivo (Fig. 3). R loops are formed in the E. coli chromosome in cells that lack the enzymes that degrade or unwind them, i.e., rnh (RNase H) and recG mutants, and are used in these mutants for oriC-DnaA-independent replication (9, 89–91). Finally, PriA and DnaT are essential for phage Mu integration into the E. coli chromosome by replicative and nonreplicative pathways (92, 93). During in vitro replicative transposition, PriA and its partners act after the transpososome is released by a specific complex. Interestingly, in vitro, the choice of the PriA pathway depends on whether the substrate is deproteinized (PriA-PriB) or whether the complex that releases the transpososome is still present (PriA-PriC) (93).
Overreplication in the RecG mutant.
The RecG helicase can unwind various DNA structures in vitro, leading to the proposal that RecG has multiple functions in vivo (reviewed in reference 94). In vitro and in vivo, in addition to targeting Holliday junctions, RecG acts at R loops, D loops, and replication forks. It is known that DNA replication initiation is increased at these three different types of substrates in a recG mutant. This suggests that one role of RecG in vivo is to counteract these oriC-independent methods of initiation of DNA replication (89, 95, 96). Furthermore, suppressors of the recG mutant's hypersensitivity to DNA-damaging agents lie in the priA gene and inactivate the helicase activity of PriA. This further suggests that that PriA and RecG have targets in common in vivo (97). In early 2000, it was proposed that RecG promotes replication across UV lesions by catalyzing fork reversal. However, when this model was revisited, it was shown that recG inactivation either had the opposite effect, enhancing replication rather than decreasing it (95), or no effect on replication restart in UV-irradiated cells (81, 98). It was recently proposed that RecG prevents overreplication initiation by directing PriA binding to blocked forks or to D loops formed by homologous recombination (71, 99, 100). Action of RecG and PriA on the same forks would allow RecG to determine the direction of replication restart by PriA and, conversely, allow PriA to prevent replication fork reversal by RecG (71, 99). Note that RecG has the same preferential DNA target as PriC (Y structure with a 5′ lagging-strand end and a gap in the leading strand), but whether RecG affects PriC action in vitro, accounting for the in vivo versus in vitro discrepancies, has not be explored.
Replication restart-associated genomic instability.
Although PriA-dependent replication restart may occur less than once per generation, these events are important because of their consequences for genome stability. A holD point mutant, affected in one of the Pol III HE subunits and isolated by screening for hyperrecombination mutants, undergoes replication fork reversal (101, 102). Recombination-dependent replication occurs in stationary phase, where it leads to Pol IV (DinB)-dependent mutagenesis (reviewed in reference 103). Stationary-phase DinB-dependent mutagenesis was largely increased in the recG mutant (104), possibly because of abnormal replication initiations from recombination intermediates in the absence of RecG. A dnaB(Ts) mutation strongly stimulates tandem repeat recombination, and, as for stationary-phase mutagenesis, in addition to all DSB repair proteins, DinB and RecG play a role in dnaB(Ts)-promoted instability, presumably through the stabilization of D loops used for replication restart (105). These few examples illustrate how, because of the action of recombination proteins at blocked forks, replication arrests do not need to be frequent to have dramatic consequences for genome stability.
CONCLUSION
The actors of replication restart in bacteria have now been identified and characterized both genetically and biochemically. The amount of knowledge accumulated to date on their crucial role in vivo and on their functioning as molecular machines in vitro is impressive. Nevertheless, several questions remain open. For example, why does PriC require either a mutated DnaC protein (such as DnaC809) or PriA and DnaT to function in vivo, whereas it acts alone in vitro? How can different substrates for PriA and PriC in vitro allow redundant functions in vivo? Why is transcription identified as the main cause of replication arrest in wild-type cells, although replication and transcription have evolved together and bacteria are equipped to deal with such obstacles? What is the activity altered in some dnaA(Ts) mutants that is required for viability in priC mutants at the permissive temperature? The E. coli chromosome is entirely replicated in about 40 min, which implies that each ∼2,320-kb chromosome arm is replicated with an average speed close to 1 kb/s. Is replication restart so rapid that replication arrests have a negligible effect on the average replication speed, or is replication progression even more rapid than we believe?
ACKNOWLEDGMENTS
We thank Andrei Kuzminov for reading the manuscript and offering many helpful comments and suggestions. We also acknowledge other researchers in this field who contributed to our understanding of these processes whose papers were not cited because of space limitation.
S.J.S. was supported by GM098885 from the National Institutes of Health. B.M. was supported by Agence National de la Recherche ANR 11 BSV5 006 01.
REFERENCES
- 1.McHenry CS. 2011. DNA replicases from a bacterial perspective. Annu Rev Biochem 80:403–436. doi: 10.1146/annurev-biochem-061208-091655. [DOI] [PubMed] [Google Scholar]
- 2.Masai H, Nomura N, Arai K. 1990. The ABC-primosome. A novel priming system employing dnaA, dnaB, dnaC, and primase on a hairpin containing a dnaA box sequence. J Biol Chem 265:15134–15144. [PubMed] [Google Scholar]
- 3.Nurse P, DiGate RJ, Zavitz KH, Marians KJ. 1990. Molecular cloning and DNA sequence analysis of Escherichia coli priA, the gene encoding the primosomal protein replication factor Y. Proc Natl Acad Sci U S A 87:4615–4619. doi: 10.1073/pnas.87.12.4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zavitz KH, Digate RJ, Marians KJ. 1991. The PriB and PriC replication proteins of Escherichia coli. Genes, DNA sequence, overexpression, and purification. J Biol Chem 266:13988–13995. [PubMed] [Google Scholar]
- 5.Greenbaum JH, Marians KJ. 1985. Mutational analysis of primosome assembly sites. Evidence for alternative DNA structures. J Biol Chem 260:12266–12272. [PubMed] [Google Scholar]
- 6.Liu J, Nurse P, Marians KJ. 1996. The ordered assembly of the phiX174-type primosome. III. PriB facilitates complex formation between PriA and DnaT. J Biol Chem 271:15656–15661. [DOI] [PubMed] [Google Scholar]
- 7.Ng JY, Marians KJ. 1996. The ordered assembly of the phiX174-type primosome. I. Isolation and identification of intermediate protein-DNA complexes. J Biol Chem 271:15642–15648. [DOI] [PubMed] [Google Scholar]
- 8.Ng JY, Marians KJ. 1996. The ordered assembly of the phiX174-type primosome. II. Preservation of primosome composition from assembly through replication. J Biol Chem 271:15649–15655. [DOI] [PubMed] [Google Scholar]
- 9.Masai H, Asai T, Kubota Y, Arai K, Kogoma T. 1994. Escherichia coli PriA protein is essential for inducible and constitutive stable DNA replication. EMBO J 13:5338–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sandler SJ, Samra HS, Clark AJ. 1996. Differential suppression of priA2::kan phenotypes in Escherichia coli K-12 by mutations in priA, lexA, and dnaC. Genetics 143:5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gabbai CB, Marians KJ. 2010. Recruitment to stalled replication forks of the PriA DNA helicase and replisome-loading activities is essential for survival. DNA Repair (Amst) 9:202–209. doi: 10.1016/j.dnarep.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sandler SJ, Marians KJ. 2000. Role of PriA in replication fork reactivation in Escherichia coli. J Bacteriol 182:9–13. doi: 10.1128/JB.182.1.9-13.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kogoma T, Cadwell GW, Barnard KG, Asai T. 1996. The DNA replication priming protein, PriA, is required for homologous recombination and double-strand break repair. J Bacteriol 178:1258–1264. doi: 10.1128/jb.178.5.1258-1264.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuzminov A, Stahl FW. 1999. Double-strand end repair via the RecBC pathway in Escherichia coli primes DNA replication. Genes Dev 13:345–356. doi: 10.1101/gad.13.3.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Motamedi MR, Szigety SK, Rosenberg SM. 1999. Double-strand-break repair recombination in Escherichia coli: physical evidence for a DNA replication mechanism in vivo. Genes Dev 13:2889–2903. doi: 10.1101/gad.13.21.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Michel B, Flores MJ, Viguera E, Grompone G, Seigneur M, Bidnenko V. 2001. Rescue of arrested replication forks by homologous recombination. Proc Natl Acad Sci U S A 98:8181–8188. doi: 10.1073/pnas.111008798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Michel B, Grompone G, Flores MJ, Bidnenko V. 2004. Multiple pathways process stalled replication forks. Proc Natl Acad Sci U S A 101:12783–12788. doi: 10.1073/pnas.0401586101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCool JD, Sandler SJ. 2001. Effects of mutations involving cell division, recombination, and chromosome dimer resolution on a priA2:: kan mutant. Proc Nat Acad Sci USA 98:8203–8210. doi: 10.1073/pnas.121007698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grompone G, Bidnenko V, Ehrlich SD, Michel B. 2004. PriA is essential for viability of the Escherichia coli topoisomerase IV parE10(Ts) mutant. J Bacteriol 186:1197–1199. doi: 10.1128/JB.186.4.1197-1199.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grompone G, Ehrlich SD, Michel B. 2003. Replication restart in gyrB Escherichia coli mutants. Mol Microbiol 48:845–854. doi: 10.1046/j.1365-2958.2003.03480.x. [DOI] [PubMed] [Google Scholar]
- 21.Maisnier-Patin S, Nordstrom K, Dasgupta S. 2001. Replication arrests during a single round of replication of the Escherichia coli chromosome in the absence of DnaC activity. Mol Microbiol 42:1371–1382. doi: 10.1046/j.1365-2958.2001.02718.x. [DOI] [PubMed] [Google Scholar]
- 22.Mangiameli S, Merrikh CN, Wiggins PA, Merrikh H. 2017. Transcription leads to pervasive replisome instability in bacteria. Elife 6:e19848. doi: 10.7554/eLife.19848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee EH, Kornberg A. 1991. Replication deficiencies in priA mutants of Escherichia coli lacking the primosomal replication n′ protein. Proc Natl Acad Sci U S A 88:3029–3032. doi: 10.1073/pnas.88.8.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCool JD, Ford CC, Sandler SJ. 2004. A dnaT mutant with phenotypes similar to those of a priA2::kan mutant in Escherichia coli K-12. Genetics 167:569–578. doi: 10.1534/genetics.103.025296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nurse P, Zavitz KH, Marians KJ. 1991. Inactivation of the Escherichia coli PriA DNA replication protein induces the SOS response. J Bacteriol 173:6686–6693. doi: 10.1128/jb.173.21.6686-6693.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sandler SJ. 1996. Overlapping functions for recF and priA in cell viability and UV-inducible SOS expression are distinguished by dnaC809 in Escherichia coli K-12. Mol Microbiol 19:871–880. doi: 10.1046/j.1365-2958.1996.429959.x. [DOI] [PubMed] [Google Scholar]
- 27.Sandler SJ, Marians KJ, Zavitz KH, Coutu J, Parent MA, Clark AJ. 1999. dnaC mutations suppress defects in DNA replication- and recombination-associated functions in priB and priC double mutants in Escherichia coli K-12. Mol Microbiol 34:91–101. doi: 10.1046/j.1365-2958.1999.01576.x. [DOI] [PubMed] [Google Scholar]
- 28.Sandler SJ. 2000. Multiple genetic pathways for restarting DNA replication forks in Escherichia coli K-12. Genetics 155:487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flores MJ, Ehrlich SD, Michel B. 2002. Primosome assembly requirement for replication restart in the Escherichia coli holDG10 replication mutant. Mol Microbiol 44:783–792. doi: 10.1046/j.1365-2958.2002.02913.x. [DOI] [PubMed] [Google Scholar]
- 30.Boonsombat R, Yeh SP, Milne A, Sandler SJ. 2006. A novel dnaC mutation that suppresses priB rep mutant phenotypes in Escherichia coli K-12. Mol Microbiol 60:973–983. doi: 10.1111/j.1365-2958.2006.05147.x. [DOI] [PubMed] [Google Scholar]
- 31.Boubakri H, de Septenville AL, Viguera E, Michel B. 2010. The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J 29:145–157. doi: 10.1038/emboj.2009.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guy CP, Atkinson J, Gupta MK, Mahdi AA, Gwynn EJ, Rudolph CJ, Moon PB, van Knippenberg IC, Cadman CJ, Dillingham MS, Lloyd RG, McGlynn P. 2009. Rep provides a second motor at the replisome to promote duplication of protein-bound DNA. Mol Cell 36:654–666. doi: 10.1016/j.molcel.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGlynn P. 2011. Helicases that underpin replication of protein-bound DNA in Escherichia coli. Biochem Soc Trans 39:606–610. doi: 10.1042/BST0390606. [DOI] [PubMed] [Google Scholar]
- 34.Atkinson J, Gupta MK, Rudolph CJ, Bell H, Lloyd RG, McGlynn P. 2011. Localization of an accessory helicase at the replisome is critical in sustaining efficient genome duplication. Nucleic Acids Res 39:949–957. doi: 10.1093/nar/gkq889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hinds T, Sandler SJ. 2004. Allele specific synthetic lethality between priC and dnaAts alleles at the permissive temperature of 30 degrees C in E. coli K-12. BMC Microbiol 4:47. doi: 10.1186/1471-2180-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Michel B, Ehrlich SD, Uzest M. 1997. DNA double-strand breaks caused by replication arrest. EMBO J 16:430–438. doi: 10.1093/emboj/16.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zavitz KH, Marians KJ. 1992. ATPase-deficient mutants of the Escherichia coli DNA replication protein PriA are capable of catalyzing the assembly of active primosomes. J Biol Chem 267:6933–6940. [PubMed] [Google Scholar]
- 38.Liu J, Marians KJ. 1999. PriA-directed assembly of a primosome on D loop DNA. J Biol Chem 274:25033–25041. doi: 10.1074/jbc.274.35.25033. [DOI] [PubMed] [Google Scholar]
- 39.McGlynn P, AlDeib AA, Liu J, Marians KJ, Lloyd RG. 1997. The DNA replication protein PriA and the recombination protein RecG bind D-loops. J Mol Biol 270:212–221. doi: 10.1006/jmbi.1997.1120. [DOI] [PubMed] [Google Scholar]
- 40.Heller RC, Marians KJ. 2005. The disposition of nascent strands at stalled replication forks dictates the pathway of replisome loading during restart. Mol Cell 17:733–743. doi: 10.1016/j.molcel.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 41.Sasaki K, Ose T, Okamoto N, Maenaka K, Tanaka T, Masai H, Saito M, Shirai T, Kohda D. 2007. Structural basis of the 3′-end recognition of a leading strand in stalled replication forks by PriA. EMBO J 26:2584–2593. doi: 10.1038/sj.emboj.7601697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lopper M, Boonsombat R, Sandler SJ, Keck JL. 2007. A hand-off mechanism for primosome assembly in replication restart. Mol Cell 26:781–793. doi: 10.1016/j.molcel.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cadman CJ, Lopper M, Moon PB, Keck JL, McGlynn P. 2005. PriB stimulates PriA helicase via an interaction with single-stranded DNA. J Biol Chem 280:39693–39700. doi: 10.1074/jbc.M508521200. [DOI] [PubMed] [Google Scholar]
- 44.Wessel SR, Marceau AH, Massoni SC, Zhou R, Ha T, Sandler SJ, Keck JL. 2013. PriC-mediated DNA replication restart requires PriC complex formation with the single-stranded DNA-binding protein. J Biol Chem 288:17569–17578. doi: 10.1074/jbc.M113.478156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heller RC, Marians KJ. 2005. Unwinding of the nascent lagging strand by Rep and PriA enables the direct restart of stalled replication forks. J Biol Chem 280:34143–34151. doi: 10.1074/jbc.M507224200. [DOI] [PubMed] [Google Scholar]
- 46.Wessel SR, Cornilescu CC, Cornilescu G, Metz A, Leroux M, Hu K, Sandler SJ, Markley JL, Keck JL. 2016. Structure and function of the PriC DNA replication restart protein. J Biol Chem 291:18384–18396. doi: 10.1074/jbc.M116.738781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Atkinson J, Gupta MK, McGlynn P. 2011. Interaction of Rep and DnaB on DNA. Nucleic Acids Res 39:1351–1359. doi: 10.1093/nar/gkq975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang CC, Huang CY. 2016. DnaT is a PriC-binding protein. Biochem Biophys Res Commun 477:988–992. doi: 10.1016/j.bbrc.2016.07.016. [DOI] [PubMed] [Google Scholar]
- 49.Xu LW, Marians KJ. 2000. Purification and characterization of DnaC810, a primosomal protein capable of bypassing PriA function. J Biol Chem 275:8196–8205. doi: 10.1074/jbc.275.11.8196. [DOI] [PubMed] [Google Scholar]
- 50.Cadman CJ, McGlynn P. 2004. PriA helicase and SSB interact physically and functionally. Nucleic Acids Res 32:6378–6387. doi: 10.1093/nar/gkh980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kozlov AG, Jezewska MJ, Bujalowski W, Lohman TM. 2010. Binding specificity of Escherichia coli single-stranded DNA binding protein for the chi subunit of DNA pol III holoenzyme and PriA helicase. Biochemistry 49:3555–3566. doi: 10.1021/bi100069s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lecointe F, Serena C, Velten M, Costes A, McGovern S, Meile JC, Errington J, Ehrlich SD, Noirot P, Polard P. 2007. Anticipating chromosomal replication fork arrest: SSB targets repair DNA helicases to active forks. EMBO J 26:4239–4251. doi: 10.1038/sj.emboj.7601848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhattacharyya B, George NP, Thurmes TM, Zhou R, Jani N, Wessel SR, Sandler SJ, Ha T, Keck JL. 2014. Structural mechanisms of PriA-mediated DNA replication restart. Proc Natl Acad Sci U S A 111:1373–1378. doi: 10.1073/pnas.1318001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zavitz KH, Marians KJ. 1993. Helicase-deficient cysteine to glycine substitution mutants of Escherichia coli replication protein PriA retain single-stranded DNA-dependent ATPase activity. Zn2+ stimulation of mutant PriA helicase and primosome assembly activities. J Biol Chem 268:4337–4346. [PubMed] [Google Scholar]
- 55.Huang CY, Hsu CH, Sun YJ, Wu HN, Hsiao CD. 2006. Complexed crystal structure of replication restart primosome protein PriB reveals a novel single-stranded DNA-binding mode. Nucleic Acids Res 34:3878–3886. doi: 10.1093/nar/gkl536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bruand C, Farache M, McGovern S, Ehrlich SD, Polard P. 2001. DnaB, DnaD and DnaI proteins are components of the Bacillus subtilis replication restart primosome. Mol Microbiol 42:245–255. doi: 10.1046/j.1365-2958.2001.02631.x. [DOI] [PubMed] [Google Scholar]
- 57.Polard P, Marsin S, McGovern S, Velten M, Wigley DB, Ehrlich SD, Bruand C. 2002. Restart of DNA replication in Gram-positive bacteria: functional characterisation of the Bacillus subtilis PriA initiator. Nucleic Acid Res 30:1593–1605. doi: 10.1093/nar/30.7.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Velten M, McGovern S, Marsin S, Ehrlich SD, Noirot P, Polard P. 2003. A two-protein strategy for the functional loading of a cellular replicative DNA helicase. Mol Cell 11:1009–1020. doi: 10.1016/S1097-2765(03)00130-8. [DOI] [PubMed] [Google Scholar]
- 59.Marsin S, McGovern S, Ehrlich SD, Bruand C, Polard P. 2001. Early steps of Bacillus subtilis primosome assembly. J Biol Chem 276:45818–45825. doi: 10.1074/jbc.M101996200. [DOI] [PubMed] [Google Scholar]
- 60.Bruand C, Velten M, McGovern S, Marsin S, Serena C, Ehrlich SD, Polard P. 2005. Functional interplay between the Bacillus subtilis DnaD and DnaB proteins essential for initiation and re-initiation of DNA replication. Mol Microbiol 55:1138–1150. doi: 10.1111/j.1365-2958.2004.04451.x. [DOI] [PubMed] [Google Scholar]
- 61.Nitharwal RG, Verma V, Dasgupta S, Dhar SK. 2011. Helicobacter pylori chromosomal DNA replication: current status and future perspectives. FEBS Lett 585:7–17. doi: 10.1016/j.febslet.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 62.Brézellec P, Vallet-Gely I, Possoz C, Quevillon-Cheruel S, Ferat JL. 2016. DciA is an ancestral replicative helicase operator essential for bacterial replication initiation. Nat Commun 7:13271. doi: 10.1038/ncomms13271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dong J, George NP, Duckett KL, DeBeer MA, Lopper ME. 2010. The crystal structure of Neisseria gonorrhoeae PriB reveals mechanistic differences among bacterial DNA replication restart pathways. Nucleic Acids Res 38:499–509. doi: 10.1093/nar/gkp1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kline KA, Seifert HS. 2005. Mutation of the priA gene of Neisseria gonorrhoeae affects DNA transformation and DNA repair. J Bacteriol 187:5347–5355. doi: 10.1128/JB.187.15.5347-5355.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Berg L, Lopper ME. 2011. The priB gene of Klebsiella pneumoniae encodes a 104-amino acid protein that is similar in structure and function to Escherichia coli PriB. PLoS One 6:e24494. doi: 10.1371/journal.pone.0024494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lopper ME, Boone J, Morrow C. 2015. Deinococcus radiodurans PriA is a pseudohelicase. PLoS One 10:e0133419. doi: 10.1371/journal.pone.0133419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang YH, Lien Y, Huang CC, Huang CY. 2016. Characterization of Staphylococcus aureus primosomal DnaD protein: highly conserved C-terminal region is crucial for ssDNA and PriA helicase binding but not for DnaA protein-binding and self-tetramerization. PLoS One 11:e0157593. doi: 10.1371/journal.pone.0157593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klasson L, Andersson SG. 2006. Strong asymmetric mutation bias in endosymbiont genomes coincide [sic] with loss of genes for replication restart pathways. Mol Biol Evol 23:1031–1039. doi: 10.1093/molbev/msj107. [DOI] [PubMed] [Google Scholar]
- 69.Xu L, Marians KJ. 2003. PriA mediates DNA replication pathway choice at recombination intermediates. Mol Cell 11:817–826. doi: 10.1016/S1097-2765(03)00061-3. [DOI] [PubMed] [Google Scholar]
- 70.Anand RP, Lovett ST, Haber JE. 2013. Break-induced DNA replication. Cold Spring Harb Perspect Biol 5:a010397. doi: 10.1101/cshperspect.a010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Azeroglu B, Mawer JS, Cockram CA, White MA, Hasan AM, Filatenkova M, Leach DR. 2016. RecG directs DNA synthesis during double-strand break repair. PLoS Genet 12:e1005799. doi: 10.1371/journal.pgen.1005799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mawer JS, Leach DR. 2014. Branch migration prevents DNA loss during double-strand break repair. PLoS Genet 10:e1004485. doi: 10.1371/journal.pgen.1004485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Michel B, Leach D. 5 November 2012, posting date. Homologous recombination—enzymes and pathways. EcoSal Plus doi: 10.1128/ecosalplus.7.2.7. [DOI] [PubMed] [Google Scholar]
- 74.Smith GR. 1991. Conjugational recombination in E. coli: myths and mechanisms. Cell 64:19–27. doi: 10.1016/0092-8674(91)90205-D. [DOI] [PubMed] [Google Scholar]
- 75.Michel B, Boubakri H, Baharoglu Z, Lemasson M, Lestini R. 2007. Recombination proteins and rescue of arrested replication forks. DNA Repair (Amst) 6:967–980. doi: 10.1016/j.dnarep.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 76.Seigneur M, Bidnenko V, Ehrlich SD, Michel B. 1998. RuvAB acts at arrested replication forks. Cell 95:419–430. doi: 10.1016/S0092-8674(00)81772-9. [DOI] [PubMed] [Google Scholar]
- 77.Grompone G, Ehrlich D, Michel B. 2004. Cells defective for replication restart undergo replication fork reversal. EMBO Rep 5:607–612. doi: 10.1038/sj.embor.7400167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guarino E, Jimenez-Sanchez A, Guzman EC. 2007. Defective ribonucleoside diphosphate reductase impairs replication fork progression in Escherichia coli. J Bacteriol 189:3496–3501. doi: 10.1128/JB.01632-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guarino E, Salguero I, Jimenez-Sanchez A, Guzman EC. 2007. Double-strand break generation under deoxyribonucleotide starvation in Escherichia coli. J Bacteriol 189:5782–5786. doi: 10.1128/JB.00411-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sutherland JH, Tse-Dinh YC. 2010. Analysis of RuvABC and RecG involvement in the Escherichia coli response to the covalent topoisomerase-DNA complex. J Bacteriol 192:4445–4451. doi: 10.1128/JB.00350-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Khan SR, Kuzminov A. 2012. Replication forks stalled at ultraviolet lesions are rescued via RecA and RuvABC protein-catalyzed disintegration in Escherichia coli. J Biol Chem 287:6250–6265. doi: 10.1074/jbc.M111.322990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.De Septenville AL, Duigou S, Boubakri H, Michel B. 2012. Replication fork reversal after replication-transcription collision. PLoS Genet 8:e1002622. doi: 10.1371/journal.pgen.1002622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sinha AK, Pavankumar TL, Kamisetty S, Mittal P, Ray MK. 2013. Replication arrest is a major threat to growth at low temperature in Antarctic Pseudomonas syringae Lz4W. Mol Microbiol 89:792–810. doi: 10.1111/mmi.12315. [DOI] [PubMed] [Google Scholar]
- 84.Rangarajan S, Woodgate R, Goodman MF. 2002. Replication restart in UV-irradiated Escherichia coli involving pols II, III, V, PriA, RecA and RecFOR proteins. Mol Microbiol 43:617–628. doi: 10.1046/j.1365-2958.2002.02747.x. [DOI] [PubMed] [Google Scholar]
- 85.Neylon C, Kralicek AV, Hill TM, Dixon NE. 2005. Replication termination in Escherichia coli: structure and antihelicase activity of the Tus-Ter complex. Microbiol Mol Biol Rev 69:501–526. doi: 10.1128/MMBR.69.3.501-526.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bidnenko V, Ehrlich SD, Michel B. 2002. Replication fork collapse at replication terminator sequences. EMBO J 21:3898–3907. doi: 10.1093/emboj/cdf369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bidnenko V, Lestini R, Michel B. 2006. The Escherichia coli UvrD helicase is essential for Tus removal during recombination-dependent replication restart from Ter sites. Mol Microbiol 62:382–396. doi: 10.1111/j.1365-2958.2006.05382.x. [DOI] [PubMed] [Google Scholar]
- 88.Baharoglu Z, Lestini R, Duigou S, Michel B. 2010. RNA polymerase mutations that facilitate replication progression in the rep uvrD recF mutant lacking two accessory replicative helicases. Mol Microbiol 77:324–336. doi: 10.1111/j.1365-2958.2010.07208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hong XK, Cadwell GW, Kogoma T. 1995. Escherichia coli RecG and RecA proteins in R-loop formation. EMBO J 14:2385–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kogoma T. 1997. Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol Mol Biol Rev 61:212–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sandler SJ. 2005. Requirements for replication restart proteins during constitutive stable DNA replication in Escherichia coli K-12. Genetics 169:1799–1806. doi: 10.1534/genetics.104.036962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jang S, Sandler SJ, Harshey RM. 2012. Mu insertions are repaired by the double-strand break repair pathway of Escherichia coli. PLoS Genet 8:e1002642. doi: 10.1371/journal.pgen.1002642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.North SH, Nakai H. 2005. Host factors that promote transpososome disassembly and the PriA-PriC pathway for restart primosome assembly. Mol Microbiol 56:1601–1616. doi: 10.1111/j.1365-2958.2005.04639.x. [DOI] [PubMed] [Google Scholar]
- 94.Lloyd RG, Rudolph CJ. 2016. 25 years on and no end in sight: a perspective on the role of RecG protein. Curr Genet 62:827–840. doi: 10.1007/s00294-016-0589-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rudolph CJ, Upton AL, Harris L, Lloyd RG. 2009. Pathological replication in cells lacking RecG DNA translocase. Mol Microbiol 73:352–366. doi: 10.1111/j.1365-2958.2009.06773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rudolph CJ, Upton AL, Stockum A, Nieduszynski CA, Lloyd RG. 2013. Avoiding chromosome pathology when replication forks collide. Nature 500:608–611. doi: 10.1038/nature12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Al-Deib AA, Mahdi AA, Lloyd RG. 1996. Modulation of recombination and DNA repair by the RecG and PriA helicases of Escherichia coli K-12. J Bacteriol 178:6782–6789. doi: 10.1128/jb.178.23.6782-6789.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Donaldson JR, Courcelle CT, Courcelle J. 2004. RuvAB and RecG are not essential for the recovery of DNA synthesis following UV-induced DNA damage in Escherichia coli. Genetics 166:1631–1640. doi: 10.1534/genetics.166.4.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Azeroglu B, Leach DR. 2 February 2017. RecG controls DNA amplification at double-strand breaks and arrested replication forks. FEBS Lett doi: 10.1002/1873-3468.12583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tanaka T, Masai H. 2006. Stabilization of a stalled replication fork by concerted actions of two helicases. J Biol Chem 281:3484–3493. doi: 10.1074/jbc.M510979200. [DOI] [PubMed] [Google Scholar]
- 101.Baharoglu Z, Petranovic M, Flores MJ, Michel B. 2006. RuvAB is essential for replication forks reversal in certain replication mutants. EMBO J 25:596–604. doi: 10.1038/sj.emboj.7600941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Flores MJ, Bierne H, Ehrlich SD, Michel B. 2001. Impairment of lagging strand synthesis triggers the formation of a RuvABC substrate at replication forks. EMBO J 20:619–629. doi: 10.1093/emboj/20.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Williams AB, Foster PL. 5 November 2012, posting date. Stress-induced mutagenesis. EcoSal Plus doi: 10.1128/ecosalplus.7.2.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Layton JC, Foster PL. 2003. Error-prone DNA polymerase IV is controlled by the stress-response sigma factor, RpoS, in Escherichia coli. Mol Microbiol 50:549–561. doi: 10.1046/j.1365-2958.2003.03704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lovett ST. 2006. Replication arrest-stimulated recombination: dependence on the RecA paralog, RadA/Sms and translesion polymerase, DinB. DNA Repair (Amst) 5:1421–1427. doi: 10.1016/j.dnarep.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 106.Marceau AH, Bahng S, Massoni SC, George NP, Sandler SJ, Marians KJ, Keck JL. 2011. Structure of the SSB-DNA polymerase III interface and its role in DNA replication. EMBO J 30:4236–4247. doi: 10.1038/emboj.2011.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.McCool JD, Long E, Petrosino JF, Sandler HA, Rosenberg SM, Sandler SJ. 2004. Measurement of SOS expression in individual Escherichia coli K-12 cells using fluorescence microscopy. Mol Microbiol 53:1343–1357. doi: 10.1111/j.1365-2958.2004.04225.x. [DOI] [PubMed] [Google Scholar]