

ABSTRACT
We analyzed the within-household evolution of two household-associated Escherichia coli strains from pandemic clonal group ST131-H30, using isolates recovered from five individuals within two families, each of which had a distinct strain. Family 1's strain was represented by a urine isolate from the index patient (older sister) with recurrent cystitis and a blood isolate from her younger sister with fatal urosepsis. Family 2's strain was represented by a urine isolate from the index patient (father) with pyelonephritis and renal abscesses, blood and kidney drainage isolates from the daughter with emphysematous pyelonephritis, and urine and fecal isolates from the mother with cystitis. Collectively, the several variants of each family's strain had accumulated a total of 8 (family 1) and 39 (family 2) point mutations; no two isolates were identical. Of the 47 total mutations, 36 resulted in amino acid changes or truncation of coded proteins. Fourteen such mutations (39%) targeted genes encoding transcriptional regulators, and 9 (25%) involved DNA-binding transcription factors (TFs), which significantly exceeded the relative contribution of TF genes to the isolates' genomes (∼6%). At least one-half of the transcriptional regulator mutations were inactivating, based on phenotypic and/or transcriptional analysis. In particular, inactivating mutations in the global regulator LrhA (repressor of type 1 fimbriae and flagella) occurred in the blood isolates from both households and increased the virulence of E. coli strains in a murine sepsis model. The results indicate that E. coli undergoes adaptive evolution between and/or within hosts, generating subpopulations with distinctive phenotypes and virulence potential.
IMPORTANCE The clonal evolution of bacterial strains associated with interhost transmission is poorly understood. We characterized the genome sequences of clonal descendants of two Escherichia coli strains, recovered at different time points from multiple individuals within two households who had different types of urinary tract infection. We found evidence that the E. coli strains underwent extensive mutational diversification between and within these individuals, driven disproportionately by inactivation of transcriptional regulators. In urosepsis isolates, the mutations observed in the global regulator LrhA increased bacterial virulence in a murine sepsis model. Our findings help in understanding the adaptive dynamics and strategies of E. coli during short-term natural evolution.
KEYWORDS: E. coli, adaptive mutation, clonal evolution, sepsis, transcriptional regulator, urinary tract infection
INTRODUCTION
In bacteria, adaptive diversification, the fundamental phenomenon underlying evolution, is driven by gene transfer or mutation. Studies involving the experimental evolution of bacterial strains have provided great insights into possible molecular mechanisms of adaptive diversification (1–4). In natural populations, however, adaptive diversification of same-strain (clonal) descendants remains poorly understood, with most of the available evidence coming from serial clinical isolates obtained over extended periods from patients with chronic lung infections (5, 6). Here we used a novel approach by comparing derivatives of a single Escherichia coli strain isolated from patients with different clinical syndromes who were first-degree relatives, had contact with one another, and at some time had lived together. This allowed for real-life assessment of genomic difference resulting from bacterial strain transmission between, and/or persistence within, different hosts.
E. coli is the leading cause of urinary tract infection (UTI) and bacterial sepsis in humans (7–9). To understand the mechanisms involved in clonal divergence of natural E. coli isolates, closely related strains must be compared. One of the most important extraintestinal pathogenic E. coli (ExPEC) lineages is sequence type 131 (ST131), with its multidrug-resistant subgroup H30 (10, 11). Several case reports have described different types of infections due to E. coli ST131-H30 in members of the same household (12–14). Pulsed-field gel electrophoresis profiles indicated that the isolates represent clonal derivatives of the same strain; however, different levels of virulence in the mouse sepsis model suggested that those isolates underwent clonal evolution affecting their pathogenic potential (15). Recent whole-genome sequencing of some of these same-household strains confirmed their clonal relatedness but also pointed to some level of genetic diversification (16). However, the nature or functional significance of the genomic variations between the isolates has not been addressed.
Adaptive diversification of bacteria occurs both in sympatric (same, enclosed) environments and in allopatric (different, compartmentalized) environments (2, 17–19). Adaptation of bacteria to either fluctuating or completely novel conditions generally requires a rapid shift in the expression of genes for different metabolic pathways or surface-bound organelles. Mutations affecting regulatory loci are likely to produce a rapid pleiotropic phenotypic effect on the expression of multiple genes and, hence, might be more beneficial for adaptation in a novel environment. Indeed, several experimental (1, 18, 19) and observational (5, 20, 21) studies described the occurrence of mutations in regulatory sequences. For example, the anti-sigma factor MucA, involved in the repression of biosynthesis of the exopolysaccharide alginate capsule in Pseudomonas aeruginosa, was found to be a hot spot for mutations in bacteria from chronically infected patients with cystic fibrosis (22, 23). In turn, LacI, the transcriptional repressor of transport and metabolism of lactose, was targeted frequently by mutations in E. coli strains evolving in various artificial lactose-containing environments in experimental studies (1, 24).
Here, we show that same-household strains undergo between- or within-host clonal diversification that is adaptive and is driven to a significant extent by mutations in transcriptional regulator genes, with such mutations often leading to gene inactivation and, in some instances, changes in virulence.
RESULTS
Close relatedness of the within-household strains.
Single nucleotide polymorphisms (SNPs) were extracted from genomic sequence data of seven E. coli isolates from two independent households. As a reference against which the detected within-household mutations could be compared to determine their origin, from our collection of 47 sequenced E. coli ST131-H30 isolates we selected and analyzed similarly the 2 nonhousehold strains that were phylogenetically closest to each of the household clusters (16) (Fig. 1).
FIG 1.

Genetic diversification of clonally related E. coli strains isolated from patients sharing the same household. The trees are based on accumulated SNPs as determined from the assembled genome sequences of household 1 and the draft genome sequences of household 2 isolates. The lengths of branches are proportional to the numbers of accumulated SNPs (also shown in italics). CD358 and JJ2555 are the closest strains in our H30 collection to the household 1 and household 2 isolates, respectively. The number of SNPs differentiating CD358 and JJ2555 from each other and the household isolates was determined for core genes only. The index patient isolate in each household cluster is shown in red.
The household 1 isolates included E. coli strains JJ1887, from the urine of the index patient (older sister) with recurrent cystitis, and JJ1886, from the bloodstream of her younger sister with fatal urosepsis (12). The household 2 isolates included strain JJ2546, from the urine of the index patient (father) with pyelonephritis/renal abscesses, and two isolates from his daughter (JJ2547, from kidney drainage; JJ2548, from blood), who developed emphysematous pyelonephritis and nonlethal urosepsis within 2 weeks after visiting her father in his hospital room. Two other isolates were obtained 6 years later from the daughter's mother when she developed cystitis, with strain JJ2963 isolated from urine and strain JJ2974 concurrently isolated from feces.
Based on SNPs across the whole genome, the two isolates from household 1 differed by 8 SNPs, while the branch connecting them to the closest epidemiologically unrelated E. coli strain from our collection of ST131-H30 isolates, strain CD358, contained 376 SNPs for the core genome only (Fig. 1). The isolates from household 2 were slightly more diverse, differing from each other by a total of 38 SNPs, with a pairwise difference ranging from 3 SNPs (between the mother's fecal and urine isolates) to 27 SNPs (between the mother's and father's urine isolates) (Fig. 1). Again, however, the branch connecting the household 2 clade to the nearest epidemiologically unrelated strain in our collection, strain JJ2555, contained 458 SNPs; moreover, the branches between the two present clades of household isolates contained a total of 1,695 SNPs.
Thus, although all of the present household isolates belonged to the same sub-ST clonal group of E. coli, ST131-H30, and differed from one another by multiple SNPs, within-household isolates were much more closely related to each other than to the nearest reference collection strain and, especially, to isolates from the other study household. This suggests strongly that these within-household isolates are clonal derivatives of a single strain per household rather than an admixture of independently acquired strains from the ST131-H30 clonal group. Although each strain appears to evolve while circulating within the corresponding household, the actual host-to-host dynamics and the timeline of strain transmission are unknown and could involve any household member as an index carrier, including independent transmission from an unknown member or even a pet.
Limited information on the mutational rates of closely related E. coli strains is available. An increased mutational rate in both pathogenic and commensal bacteria has been linked to the existence within the clonal population of so-called mutators that are frequently deficient in a postreplicative repair system (25, 26). Screening of the present within-household isolates for the mutator phenotype using a rifampin resistance assay showed that the frequency of rifampin-resistant mutants in overnight cultures of the bacteria remained at the same low level as in the control E. coli K-12 strain MG1655 (27), not exceeding 2 mutants per 108 cells (see Table S1 in the supplemental material).
Genome comparison of urine and blood source isolates from household 1.
The above-mentioned 8 SNPs that differentiated household 1 blood isolate JJ1886 from urine isolate JJ1887 were all located in different chromosomal genes. Of these SNPs, one resulted in a premature stop codon, five were nonsynonymous (i.e., amino acid replacement) mutations, and two were synonymous (i.e., silent) mutations (Table 1). According to the phylogeny (16) (Fig. 1), 5 of 8 SNPs occurred in JJ1886 (the blood isolate), including a premature stop codon mutation in rpoS, nonsynonymous mutations in lacI, lrhA, and ydjF, and a synonymous mutation in ompN. The 3 SNPs that occurred in JJ1887 (the urine isolate) included nonsynonymous mutations in cspE and dapA and a synonymous mutation in gadA. Though the parsimony-based phylogenetic analysis of household isolates and other ST131-H30 strains provides a plausible scenario of within-household diversification (Fig. 1), the absolute hierarchy of the mutational events cannot be determined, and the occurrence of reverse mutations that would affect the presented pattern of accumulated mutations cannot be excluded.
TABLE 1.
Genetic differences between household 1 strains JJ1886 and JJ1887a
| Locus tag | Chromosomal position | Genetic locusb | Function(s) | Genetic change (amino acid change) | Presence of genetic change in strain: |
|
|---|---|---|---|---|---|---|
| JJ1886 | JJ1887 | |||||
| P423_01815 | 400099–401058 | lacI | LacI transcription factor | G890A (G297E) | + | |
| P423_03060 | 666606–666815 | cspE | Cold shock protein CspE | G140A (G47D) | + | |
| P423_03335 | 717032–717919 | dapA | Dihydrodipicolinate synthetase | G85A (V29M) | + | |
| P423_07890 | 1590530–1591663 | ompN | Outer membrane porin | C372T (SYN) | + | |
| P423_09420 | 1916277–1917035 | ydjF | DeoR-family transcription factor | G292A (A98T) | + | |
| P423_12780 | 2598783–2599721 | lrhA | LrhA transcription factor | C565T (L189F) | + | |
| P423_15025 | 3056283–3057116 | rpoS | RNA polymerase sigma factor RpoS | C140A (S47stop) | + | |
| P423_19575 | 4002705–4004105 | gadA | Glutamate decarboxylase | C717T (SYN) | + | |
| Plasmid | NA | pJJ1886_4 | Conjugative plasmid | Absence | + | |
| Plasmid | NA | pJJ1887_5 | R-factor plasmid | Absence | + | |
| PAI | 4907731–4949587 | PAI | Iron acquisition system (Fec), antigen 43 adhesin (Ag43), hemolysin expression-modulating protein (Hha) | Duplication | + | |
| P423_04930 | 1027744–1027875 | NA | Phage hypothetical protein | IS66 insertion | + | |
| NA | 1447788–c | NA | Intergenic phage region | IS66 insertion | + | |
| NA | NA | NA | NA | IS66 insertion upstream of PAI duplicate | + | |
| NA | NA | NA | NA | IS66 insertion downstream of PAI duplicate | + | |
PAI, pathogenicity-associated island; NA, not applicable; SYN, synonymous; IS66, insertion sequence 66; +, genetic change present. Locus tags and chromosomal positions are given as in the reference JJ1886 genome.
For those genes without a designated name in the JJ1886 annotation, the homolog designation available from annotations of other E. coli strains was used. Genes encoding transcriptional regulators are shown in bold.
This chromosomal insertion of IS66 is not present in the originally published sequence of the JJ1886 genome.
Apart from these 8 SNPs, two additional genetic differences distinguished JJ1887 and JJ1886 (Table 1). First, both JJ1886 and JJ1887 carried five plasmids; four of these were identical among the isolates, but each isolate carried one unique plasmid. The JJ1887-specific plasmid (pJJ1887_5) is a unique IncF-type plasmid that is 130,603 bp long and carries, among other genes, an antibiotic resistance cassette encoding resistance to tetracycline, gentamicin, and trimethoprim-sulfamethoxazole. The JJ1886-specific plasmid (pJJ1886_4) is a unique IncP-type plasmid that is 55,956 bp long. Its only functionally annotated genes are those encoding the type IV secretion system that is part of a conjugation apparatus (NCBI accession number NC_022650).
Second, the chromosome of JJ1886 had a duplicated copy of an ∼42-kb pathogenicity-associated island (PAI) that was present in JJ1887 as a single copy (see Fig. S1 in the supplemental material). This PAI carried the iron acquisition fec operon and the agn43 gene, which encodes the urovirulence-associated antigen 43 (Ag43) adhesin (see below). The duplicated region was flanked by transposase-encoding insertion sequences (IS66, ∼2.5 kbp), suggesting a mobile-element-associated duplication (Fig. S1). In JJ1886, additional copies of the same IS66 element were inserted in two other regions, within separate prophages (Table 1). The remainder of the JJ1886 chromosome had the same architecture as that of JJ1887.
Thus, JJ1886 and JJ1887 underwent recent clonal evolution via gene loss, duplications, and point mutations, with most SNPs being structural in nature, i.e., affecting the amino acid composition or length of the encoded proteins.
Structural mutations preferentially target genes encoding transcriptional regulators.
Among the genes targeted by structural mutations in the household 1 strains, two of those mutated in JJ1886, lacI and lrhA, encode well-characterized transcription factors (TFs), i.e., the repressor of the lactose degradation operon, LacI, and the global regulator LrhA. Screening for TFs using the P2TF database (http://www.p2tf.org/) revealed that, based on a predicted helix-turn-helix DNA-binding domain, another JJ1886 mutated gene, ydjF, also encodes a TF, from the DeoR family of regulators. Overall, when the P2TF database was used to screen the entire well-annotated JJ1886 chromosome for TFs (with correction for the known PAI duplication), 299 (6.0%) of 4,979 total genes in JJ1886 were predicted to encode TFs. Thus, the fact that 3 of 6 genes (50%) with structural mutations encode TFs suggested that the structural alterations specifically target such proteins (P < 0.005). Moreover, as indicated by genome annotation and KEGG functional categorization (http://www.genome.jp/kegg/), two other genes in JJ1886 that contain structural mutations encode proteins that function as transcriptional regulators: RpoS, a stress response sigma factor, and CspE, a cold shock response protein that serves as an RNA chaperone and transcription antiterminator (28, 29). In contrast, both of the observed synonymous mutations occurred in genes that encode proteins with a nonregulatory function: outer membrane protein OmpN and glutamate decarboxylase GadA (Table 1).
Point mutations in transcriptional regulators are inactivating.
We examined the functional impact of the observed mutations that involved transcriptional regulators with relatively well-characterized phenotypic effects, RpoS, LacI and LrhA, all of which were targeted in blood isolate JJ1886 from household 1.
RpoS controls the expression of numerous genes that promote bacterial survival under a variety of stress conditions, including oxidative stress (29–31). A strain's oxidative stress response can be tested by exposing bacteria to hydrogen peroxide (32). As mentioned above, the SNP in rpoS in JJ1886 results in a premature stop codon, truncating the RpoS protein at 46 of 330 amino acids. When JJ1887 and JJ1886 were exposed to 2.5 mM hydrogen peroxide for 1 h, survival was significantly greater for JJ1887 than JJ1886 (56.6% ± 13% versus 13.8% ± 3%; P < 0.05) (Fig. 2A, left panel). Complementation of JJ1886 with a low-copy-number plasmid expressing wild-type rpoS from JJ1887 (rpoSJJ1887), but not with rpoS from JJ1886 (rpoSJJ1886) or with the empty vector, improved the survival of JJ1886 to nearly the same level as that of JJ1887 (Fig. 2A, right panel). Thus, as expected, the premature stop codon in rpoS of blood isolate JJ1886 resulted in a phenotype corresponding with inactivation of the RpoS transcriptional regulator. (Of note, inactivating mutations in RpoS also occur frequently during in vitro passage and storage of E. coli isolates [see Discussion].)
FIG 2.
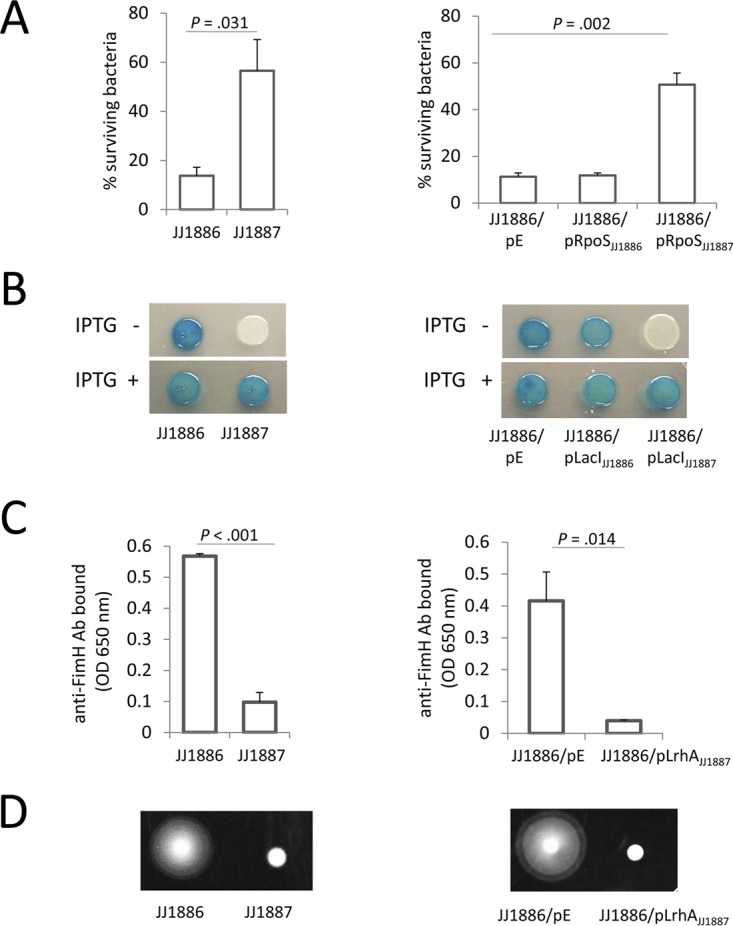
Phenotypic characteristics of wild-type and gene-complemented E. coli strains. (A) Sensitivity of bacteria to oxidative stress, as measured by a hydrogen peroxide survival assay. Data are means ± standard errors of the means (SEM) (n = 3). (B) Beta-galactosidase (LacZ) activity of bacteria grown for 6 h on X-Gal LB agar in the absence and presence of IPTG. (C) Surface expression of type-1 fimbriae, as detected by enzyme-linked immunosorbent assay (ELISA) using an anti-FimH antibody. (D) Motility of bacteria grown on LB 0.2% agar for 6 h. For the gene complementation study, JJ1886 bacteria were transformed with a control (empty) plasmid (pE; pACYC184) or pACYC184 carrying rpoS, lacI, or lrhA from JJ1886 or JJ1887 (pRpoSJJ1886, pRpoSJJ1887, pLacIJJ1886, pLacIJJ1887, and pLrhAJJ1887, respectively).
We next examined the phenotypic effect of the point amino acid substitution in the repressor of the lactose degradation operon, LacI, that occurs in JJ1886 (G297E). When JJ1886 and JJ1887 were streaked onto X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) agar in the absence of the LacI inhibitor IPTG (isopropyl-β-d-thiogalactopyranoside), only JJ1886 displayed LacZ activity, indicating that the lac operon is constitutively active in JJ1886 but not JJ1887 (Fig. 2B, left panel). Addition of IPTG dramatically increased LacZ activity in JJ1887, with no effect in JJ1886 (Fig. 2B, left panel). Transcomplementation of JJ1886 with lacIJJ1887 but not with lacIJJ1886 or an empty vector plasmid restored LacZ repression in the absence of IPTG (Fig. 2B, right panel). Thus, the G297E mutation in LacIJJ1886 is inactivating. Indeed, examination of the LacI crystal structure revealed that the mutated LacI residue 297 is located in the repressor's regulatory domain, in close proximity to the binding pocket of LacI inhibitors (see Fig. S2 in the supplemental material).
LrhA is a global regulator of E. coli genes that, among other effects, represses expression of mannose-specific type 1 fimbriae and flagella (33). Whereas cells of neither JJ1886 nor JJ1887 were fimbriated when grown on Luria-Bertani (LB) agar (see Fig. S3 in the supplemental material), after two consecutive passages in LB broth JJ1886 demonstrated significantly higher expression levels of type 1 fimbriae than did JJ1887 (Fig. 2C, left panel; Fig. S3). This suggests that JJ1886 is more efficient than JJ1887 in turning on fimbrial expression. Also, when grown in soft agar, motility was greater for JJ1886 than JJ1887 (Fig. 2D, left panel), suggesting higher expression levels of flagella by this blood isolate. Transformation of JJ1886 with a plasmid expressing wild-type lrhA from JJ1887 suppressed both fimbriation and motility (Fig. 2C and D, right panels). Taken together, these results suggested that the L189F mutation in LrhA of JJ1886 is inactivating.
Thus, similar to the truncating mutation in RpoS in JJ1886, nonsynonymous mutations in TFs LacI and LrhA in blood isolate JJ1886 appear to result in loss of function of these regulatory proteins.
Significant transcriptional profile differences between the blood and cystitis strains.
Gene expression in the household 1 isolates was tested using bacteria grown in two different media, i.e., LB broth (which is nutrient rich) and filter-sterilized human urine.
For both strains, more genes were upregulated in urine than in broth: 1,116 versus 242, respectively, in JJ1886 and 1,282 versus 198, respectively, in JJ1887 (P < 0.005 for each comparison) (see Fig. S4 and Data Set S1 in the supplemental material). For growth in urine, the most highly upregulated genes in both JJ1886 and JJ1887 were those encoding an inhibitor of glucose transporter, sgrT (P423_00375), the mhp gene cluster responsible for catabolism of aromatic compounds as a carbon source, and different genes encoding iron-sulfur (Fe-S) cluster proteins (e.g., sufA and ytfE). Interestingly, both strains also demonstrated significant upregulation in urine of the iron acquisition fec operon, which is located in the PAI that is present in a single copy in JJ1887 and is duplicated in JJ1886 (Fig. 3).
FIG 3.

Gene expression pattern of a 42-kb pathogenicity island (PAI) in two sisters' strains in urine and broth. Gene expression was determined by transcriptome analysis of strains grown in urine (upper x axis) or LB broth (lower x axis) and is measured in reads per kilobase per million mapped reads (RPKM). Selected open reading frames (ORFs) with increased expression in urine and/or broth are labeled. Average data for three biological replicates are shown.
JJ1886 and JJ1887 differed in the expression levels of certain genes in urine. As expected from the PAI duplication in JJ1886, the corresponding mRNA reads were on average twice as abundant in JJ1886 as in JJ1887 (notably excluding the ang43 leader RNA, which is known to regulate Ag43 adhesin expression [34]). Beside these PAI-associated genes, genes with higher expression in JJ1886 than in JJ1887 included those within the lac operon, within three different operons encoding flagella (flhCD, flgBCDEFG, and fliC), and within a large locus corresponding to the lambdoid mEp460-like prophage (Fig. 4; see also Data Set S2 in the supplemental material). Overall, however, in urine ∼3 times more genes were overexpressed in JJ1887 than in JJ1886 (216 versus 68 genes). The 216 genes overexpressed in JJ1887 included those of the acid response system (ybaS, gadBC, gadE, gadA, hdeAB, etc.) and other stress response genes (Fig. 4 and Data Set S2).
FIG 4.

Differential gene expression between two sisters' strains in urine. The gene expressions of sepsis isolate JJ1886 and cystitis isolate JJ1887 during growth in urine were determined by whole-transcriptome analysis. Fold differences (log10) in expression levels are shown. Values above the x axis are for genes differentially upregulated in JJ1886; values below the x axis are for genes differentially upregulated in JJ1887. Only ORFs are shown. Genes controlled by RpoS, LrhA, and LacI are labeled in yellow, red, and green, respectively (as determined by gene complementation studies). Overlapping control by multiple regulators is shown in blue (LrhA and RpoS) and orange (LacI and LrhA). Average data for three biological replicates are shown.
In contrast to what was observed in urine, in LB broth more genes were overexpressed in blood isolate JJ1886 than in urine isolate JJ1887 (297 versus 223 genes, respectively; P < 0.005) (Data Set S2). Notably, after lacI loci, these included type 1 fimbrial genes, which were well expressed in broth in both strains but to a significantly greater extent in JJ1886 than JJ1887. Importantly, in contrast to constitutively overexpressed lacI loci in JJ1886 (regardless of the medium conditions), the fimbrial genes, similar to flagellar genes, were inversely expressed in LB and urine yet in each instance represented the most highly overexpressed traits in blood isolate JJ1886 (Data Set S2).
Among the genes overexpressed in JJ1887 relative to JJ1886 in LB again were multiple acid response genes (Data Set S2) and cspA, encoding the major cold shock protein, CspA (Data Set S2). CspA, which itself is a transcriptional regulator, was shown to be downregulated by CspE in rich medium (28). As noted above, JJ1887 has a nonsynonymous mutation in cspE, suggesting that the resulting G47D amino acid substitution has an inactivating effect on CspE's regulatory function.
For the plasmids common to JJ1886 and JJ1887, only relatively minor gene expression differences were observed between the two strains (Table S2), whether in urine or LB broth.
Thus, a large number of genes were differentially expressed between blood isolate JJ1886 and urine isolate JJ1887 in either urine or rich medium, presumably as the result of the observed genetic differences, most of which affected transcriptional regulators.
Inactivation of transcriptional regulators is responsible for most gene expression differences.
We tested how complementation of mutations in rpoS, lacI, and lrhA in JJ1886 affects the differences in transcriptional profiles between the blood and urine isolates. This comparison was done only with growth in urine.
Regarding rpoS, complementation of JJ1886 with wild-type rpoSJJ1887 increased the expression of 71 of the 216 genes (33%) that were underexpressed in JJ1886 relative to JJ1887, which accounted collectively for 25% of all differentially expressed genes among the household 1 isolates (Fig. 4, yellow). In contrast, none of the genes that were overexpressed in JJ1886 relative to JJ1887 appeared to be affected by the rpoS mutation in JJ1886. Regarding lacI, complementation of JJ1886 with wild-type lacIJJ1887 resulted in suppression of the lac operon to the level of JJ1887 (see Fig. S5A in the supplemental material) but had no major impact on expression of JJ1886 genes outside the lac operon (with less than 1% of all differentially expressed genes between the isolates being affected by lacI complementation; see Data Set S3 in the supplemental material). Finally, regarding lrhA, complementation of JJ1886 with wild-type lrhAJJ1887 resulted in repression of flagellar genes, including flhDC, encoding master regulators of flagellar biosynthesis, and fliC, encoding the major flagellar structural subunit (Fig. 4 and Data Set S3). The lrhA complementation also resulted in the downregulation of most genes in the lambdoid mEp460-like prophage locus that were overexpressed in urine in JJ1886 relative to JJ1887. In total, complementation with lrhA affected 9.5% of all differentially expressed genes among the household 1 isolates (Fig. 4; see also Fig. S5B and Data Set S3 in the supplemental material). Overall, the altered expression of 97 of the 284 genes that were differentially expressed between JJ1886 and JJ1887 in urine was mitigated by complementation of JJ1886 with wild-type copies of the three transcriptional regulator-encoding genes.
We also took advantage of the fact that the pJJ1887_5 plasmid (which is specific to JJ1887 and carries a multidrug resistance cassette) is lost spontaneously from ∼20% of JJ1887 cells during passage in the absence of antibiotics. For this, we compared the transcriptional profiles, in urine, of wild-type and plasmid-cured JJ1887 variants. Only 4 of the genes differentially expressed between JJ1886 and JJ1887 were affected by the plasmid loss (see Data Set S4 in the supplemental material).
Overall, the differential transcriptional profiles of the clonal derivatives JJ1886 and JJ1887 correlated closely with the observed inactivating mutations in the transcriptional regulators of blood isolate JJ1886.
LrhA activity suppresses E. coli virulence in a murine sepsis model.
As previously shown using a subcutaneous sepsis model, blood (sepsis) isolate JJ1886 demonstrated greater systemic virulence in mice than urine (cystitis) isolate JJ1887 (15). We used the same experimental approach to compare three recombinant derivatives of JJ1886, each complemented with a wild-type copy of rpoS, lacI, or lrhA from JJ1887.
Whereas mice injected with JJ1886 transformants complemented with wild-type copies of rpoS or lacI (from JJ1887) showed the same rapid lethality as mice injected with JJ1886 transformed with the empty vector, mice injected with a JJ1886 transformant complemented with lrhAJJ1887 showed significantly decreased illness scores relative to control mice (P < 0.001) (Fig. 5) and had 50% reduced mortality at 3 days postinfection. Thus, inactivation of LrhA appears to be responsible, at least partially, for the increased mortality and enhanced severity of infection observed with JJ1886 in the murine sepsis model, which parallels the clinical phenotypes observed with these isolates in their source patients.
FIG 5.
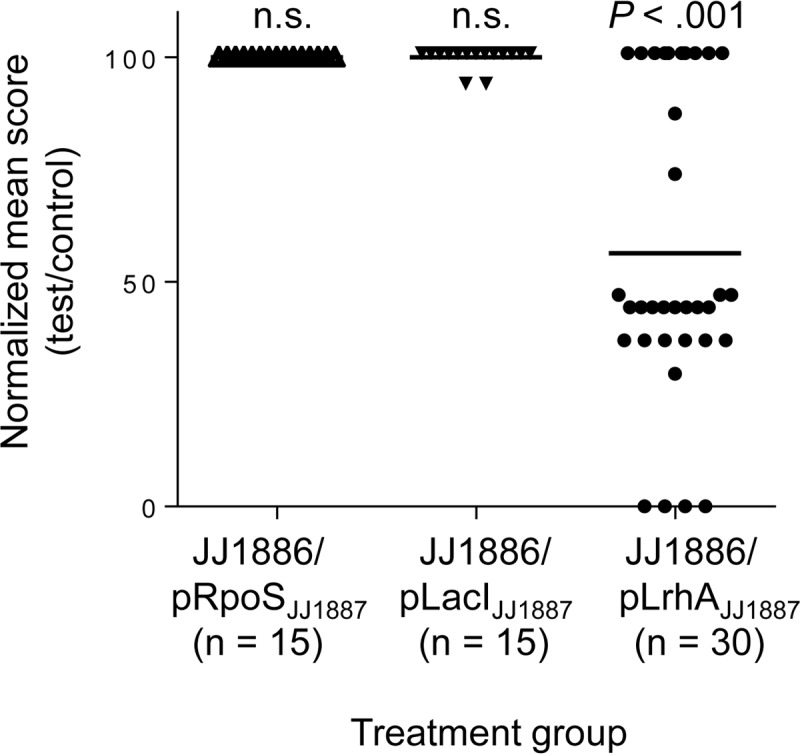
Effects of gene complementation on JJ1886 pathogenicity in mice. Mice were inoculated with JJ1886 bacteria transformed with an empty plasmid (pE; pACYC184) or pACYC184 carrying the rpoS, lacI, or lrhA gene from strain JJ1887 (pRpoSJJ1887, pLacIJJ1887, or pLrhAJJ1887). The percent change in the clinical status of each individual mouse (averaged over 3 days postchallenge) in each treatment group relative to the mean value for the control group (pE) is shown (normalized mean score). n, number of mice tested. Horizontal bars indicate the group means. Data shown are combined from three independent experiments. Significance of the difference in mean score between treatment groups and control was evaluated using simple linear regression (for pRpoSJJ1887 and pLacIJJ1887, using data obtained in one independent experiment each) or by multilevel mixed-effects linear regression, to adjust for random effects (for pLrhAJJ1887, using data from two independent experiments); n.s., not significant.
Transcriptional regulators are targeted by mutations also in a second set of clonally evolved strains.
We analyzed the nature of genetic variations in the five isolates from household 2 (Fig. 1). Unlike the household 1 isolates, for the household 2 isolates only draft genome sequences were available (16).
A total of 38 SNPs and a single 6-bp insertion were identified between the five household 2 isolates (Fig. 1 and Table 2). Of the SNPs, 29 were nonsynonymous, 5 were synonymous, and 4 were intergenic (Table 2). Of the 30 total structural changes (29 SNPs, one insertion), 6 mutations (19.4%)—including 5 nonsynonymous SNPs (in rbsR, malT, fhlA, ntrC, and yebC) and the 6-bp insertion (in lrhA)—affected genes encoding known or putative TFs in other strains (Table 2). As with the household 1 isolates, the proportion of mutations targeting TFs was significantly higher than expected for a random distribution based on the proportion of TFs among all protein-coding genes in JJ1886 (i.e., 2% versus 0.06%; P < 0.05). Also, 3 additional genes with nonsynonymous changes, cspE, degS, and rapZ, encoded other types of transcriptional regulators (28, 35, 36), while none of the 5 genes with synonymous changes did so. Notably, 2 of 4 intergenic SNPs also were found upstream of genes encoding transcriptional regulators: one directly in the promoter of the DNA-binding nucleoid protein Dps (37, 38) and the other in the 5′ untranslated region (UTR) of cspA, encoding the major cold shock protein CspA, which functions as an RNA chaperone (39) (Table 2; see also Table S3).
TABLE 2.
Genetic differences between household 2 E. coli strainsc
| Genea | Gene product | Mutationb | Presence of mutation in strain |
||||
|---|---|---|---|---|---|---|---|
| Daughter's |
Mother's |
||||||
| Father's JJ2546 (urine) | JJ2547 (kidney drainage) | JJ2548 (blood) | JJ2963 (urine) | JJ2974 (feces) | |||
| P423_00080 | Arylsulfatase | G32A (G11D) | + | + | |||
| degS | Peptidase RseP (regulator of sigma E) | C259A (H87N) | + | ||||
| ykgC | Pyridine nucleotide-disulfide oxidoreductase | A445G (T149A) | + | + | |||
| nfsB | Dihydropteridine reductase | G73T (E25stop) | + | ||||
| ybdK | Gamma-glutamyl:cysteine ligase | G759A (M253I) | + | ||||
| cspE | Cold shock protein CspE (RNA chaperone) | G157T (E53stop) | + | ||||
| lipA | Lipoyl synthase | T683C (I228T) | + | + | |||
| dps | Stationary-phase protection protein Dps | G127T (intergenic) | + | ||||
| ycgJ | Hypothetical protein | C121T (intergenic) | + | ||||
| dnaC | DNA replication protein DnaC (phage) | C333A (SYN) | + | + | |||
| ydbH | Hypothetical protein YdbH | G1937A (W646stop) | + | + | |||
| yebC | Putative transcriptional regulator | C239T (A80V) | + | + | |||
| yfaL | Autotransporter | C1431T (SYN) | + | ||||
| lrhA | Transcriptional regulator LrhA | A41_T42ins 6 bp (D14_L15ins LD) | + | ||||
| P423_12990 | Peptidase S10 | C476T (P159L) | + | ||||
| fadJ | Fatty acid oxidation protein subunit alpha | G1863A (SYN) | + | ||||
| P423_13360 | Membrane protein | T129A (SYN) | + | + | |||
| crr | PTS glucose transporter subunit IIA (EIIA_Glc) | A260T (E87V) | + | ||||
| fhlA | Sigma 54-dependent transcriptional regulator | G878A (W293stop) | + | + | |||
| sdaC | Serine/threonine transporter | A25G (I9V) | + | ||||
| mltA | Murein transglycosylase A | C206T (S69L) | + | ||||
| P423_16465 | 9-O-Acetyl-N-acetylneuraminic acid deacetylase | A351C (L117F) | + | ||||
| P423_16480 | N-Acetylneuraminic acid mutarotase | T407C (I136T) | + | + | |||
| P423_16705 | Polysialic acid transporter | C44T (intergenic) | + | + | |||
| rapZ | GlmZ (sRNA)-inactivating NTPase | C1486T (N188S) | + | + | |||
| malT | Transcriptional regulator | A1004C (E335A) | + | ||||
| ftsY | Signal recognition particle-docking protein | C1486T (R496stop) | + | + | |||
| P423_19450 | Oligopeptidase A | A303G (SYN) | + | ||||
| cspA | Cold shock protein upstream CspA | C74A (intergenic) | + | ||||
| glyS | Glycyl-tRNA synthetase (subunit B) | C1933T (R645C) | + | ||||
| gltS | Sodium/glutamate symport carrier protein GltS | C848T (L283P) | + | ||||
| frvA | Fructose-specific PTS (EIIA_Frc) | C371T (A124V) | + | + | |||
| ilvN | Acetolactate synthase | A35C (E12A) | + | + | |||
| rbsR | Transcriptional regulator RbsR | G748T (V253F) | + | ||||
| P423_21035 | Arylsulfatase | C1292T (T431I) | + | + | |||
| P423_21850 | Hypothetical protein | T765G (I255M) | + | + | + | ||
| ntrC | Sigma 54-dependent transcriptional regulator | G817A (A273T) | + | + | |||
| bsmA | Biofilm stress and motility protein BsmA | A133G (I45V) | + | ||||
| P423_24355 | Restriction endonuclease | C1223T (P408L) | + | ||||
Gene number or name as in reference JJ1886 genome (NCBI accession number NC_022648). For those genes without a designated name in the JJ1886 annotation, the homolog designation available from annotations of other E. coli strains was used. Genes encoding transcriptional regulators are shown in bold.
The position of an intergenic mutation is shown as the distance from a start codon of the gene. Amino acid changes are given in parentheses.
SYN, synonymous; ins, insertion; PTS, phosphotransferase system; +, mutation is present.
Two genes were mutated in isolates from both households. Like the household 1 blood isolate JJ1886, the household 2 daughter's blood isolate JJ2548 carried a mutation in lrhA, but in this case the 6-bp insertion resulted in a two-amino-acid insertion between residues 14 and 15 of mature LrhA. This insertional mutation apparently disabled LrhA function because, as with JJ1886, strain JJ2548 showed increased motility at baseline that was repressed by introduction of a plasmid carrying wild-type lrhAJJ1887 (see Fig. S6A in the supplemental material). Also, like the household 1 urine isolate JJ1887, the household 2 father's urine isolate JJ2546 had a mutation in the cold shock protein gene cspE that resulted in a premature stop codon and truncation of the 69-amino-acid protein after residue 53, in contrast to the nonsynonymous change in cspE in JJ1887 (G47D).
Besides cspE, mutations in the household 2 isolates resulted in premature stop codons in fhlA, ydbH, ftsY, and nfsB. Of these, fhlA, which encodes the TF FhlA (40), was mutated in both the fecal and urine isolates from the mother. Also, although the repressor gene lacI was not altered in any of the household 2 isolates (as it was in household 1 blood isolate JJ1886), the household 2 daughter's blood isolate JJ2548 harbored a nonsynonymous mutation in rbsR, which encodes the repressor of the rbs operon that controls transport and metabolism of d-ribose, another alternative carbon source for E. coli (41). Interestingly, transcriptome sequencing (RNA-seq) analysis of the household 2 isolates when grown in urine indicated significantly higher expression of the rbs operon in the daughter's blood isolate JJ2548 than in the father's urine isolate JJ2546 (Fig. S6B), suggesting that in JJ2548 the RbsR repressor may be at least partially inactivated.
Thus, genome analysis of a second set of clonally evolved strains from a different household revealed a substantial level of structural mutations that again targeted genes encoding TFs and other transcriptional regulators, with some obvious or potentially inactivating effects and some commonality of targeted genes across the two households.
Natural origin of the mutational changes.
The presence of the observed mutations in multiple independently obtained isolates provides strong evidence that the mutations occurred naturally, i.e., before rather than after bacterial isolation. Among the household 2 isolates, 14 mutations were found in both the mother's urine and fecal isolates (JJ2963 and JJ2974) (Fig. 1 and Table 2), including 3 truncating mutations in ydbH, ftsY, and the TF gene fhlA and 2 nonsynonymous mutations in other transcriptional regulator-encoding genes (rapZ and ntrC). Also, a nonsynonymous SNP in the P423_21850 locus was found in 3 of the household's isolates (i.e., the father's isolate and both of the mother's isolates, collected 6 years apart), while a nonsynonymous mutation in a putative TF, YebC, was found in both of the daughter's isolates.
Thus, nearly one-half of the mutations found in the household 2 isolates are almost certainly naturally occurring, including some of the truncating and nonsynonymous mutations in genes coding for transcriptional regulators.
DISCUSSION
Here, we demonstrate that within-household evolution of extraintestinal E. coli strains was associated with clonal divergence, driven by positive selection. We found that single-strain derivatives isolated from patients within a given household accumulated functional genomic changes, most commonly mutations altering the structure of encoded proteins but also including plasmid loss or gain and PAI duplication. Transcriptional regulators appear to be a preferred target for such structural alterations. Strong experimental evidence indicated that for many mutations their loss-of-function effect, including inactivation of the global regulator LrhA in sepsis strains, correlated with increased illness severity and mortality in the murine sepsis model.
Previous studies have shown that transcriptional regulators comprise <10% of the E. coli genome (42, 43). This group of genes includes genes that encode TFs, sigma factors, RNA chaperones, nucleoid proteins, and other regulators (e.g., regulatory RNAs) that act at different stages of transcription (42, 44, 45). TFs, which are DNA-binding proteins that usually target the regulated gene's operator region, constitute the largest group of regulators, being encoded by ∼300 genes (42, 46, 47). This is fully consistent with the number of TFs (299, of a total of 4,979 genes) that we identified by comparing the P2TF database (48) with the single fully assembled and annotated genome from this study, that of sepsis isolate JJ1886, from household 1. Considering that transcriptional regulator genes comprise only a small fraction of protein-encoding genes overall, the observed high mutation rate among TFs and other regulatory genes indicates that such genes were targeted preferentially by positive selection and, thus, that the corresponding mutations likely are adaptive.
Of the observed 9 mutations in TFs and 5 mutations in other transcriptional regulators, 5 and 3 mutations, respectively, were inactivating based on phenotypic assays, gene expression analysis, or the truncating nature of the mutations. Experimental loss of RpoS function was demonstrated previously to be advantageous under nutrient-limiting conditions, by redirecting cellular resources from a stress response to a hunger response (49). A similar role was proposed also for natural RpoS-inactivating mutations (50). However, in the in-host environment, stress and nutritional competition appear to be equally important, in that different types of mutations, i.e., loss-of-function mutations and those affecting the expression level of RpoS, are selected (51–53). Interestingly, virulence analysis of different clonally related E. coli isolates from the same patient with distinct RpoS expression levels showed that a lower level of RpoS was strongly associated with better growth in vitro and greater virulence in the murine septicemia model (51). This suggests that rpoS inactivation could confer a fitness advantage and thus be positively selected in the host.
However, rpoS inactivation occurs frequently during laboratory passage and storage of strains (32), so the possibility of in vitro artifacts must be considered. Here, the mutations that we identified in the household 1 isolates occurred in only one isolate each. Thus, it is impossible to be fully confident that they were not acquired in the laboratory. However, in the household 2 isolates, a total of 16 mutations, including 3 of 6 affecting various TFs, were positioned on internal branches of the phylogenetic tree, i.e., were shared by independent isolates. This confirms their independent and thus presumably natural origin. Also, we found at least one example in which a mutation in LrhA (R17G) was found in independent isolates from the same patient (from blood and urine specimens [see Table S4 in the supplemental material]), supporting a natural origin for this mutation.
Interestingly, the position of the R17G LrhA mutation is in close proximity to the 14/LD/15 LrhA insertion in the blood isolate from household 2 (JJ2548), which was experimentally confirmed to have an inactivating effect on LrhA function. However, no crystal structure of E. coli LrhA—or of LrhA from any other species—is currently available, and although some insights might be obtained from homology models (e.g., those available in the UniProt database), any predictions regarding how these amino acid residues are involved in LrhA function would be only speculative. That said, these residues' effect could be direct or mediated through a distal conformational effect on the DNA-binding groove (as in case of the 14/LD/15 amino acid insertion and the R17G mutation in the putative DNA-binding domain) or interference with the ligand interaction properties (as for the L189F mutation localizing in the putative ligand-binding domain).
Additionally, the fact that, in strains from both households, lrhA and cspE were inactivated selectively in isolates of a particular origin (blood and urine, respectively) suggests that these and possibly some of the other observed mutations could have a pathoadaptive effect specific for a particular clinical syndrome or extraintestinal body compartment. Because LrhA is a global negative regulator of flagella and type 1 fimbriae (33, 54), its inactivation causes a switch from “off” to “on” of these phase-variable traits, under appropriate inducing conditions. The ability to produce flagella and type 1 fimbriae was shown to play a critical role in the extraintestinal virulence of E. coli (55–57). In a murine model of UTI, flagellum-mediated motility allowed E. coli ascension to the upper urinary tract and correlated strongly with systemic dissemination (58, 59). Also, highly motile E. coli strains are isolated more commonly from patients with urosepsis than from those with uncomplicated UTI (60).
Mannose-specific type 1 fimbriae were also shown to be critical in the establishment of bladder and renal colonization in a murine model (61–63). In our study, the mutational inactivation of LrhA in blood isolate JJ1886, besides causing overexpression of flagella and type 1 fimbriae, resulted also in upregulation of early and late loci of lambdoid prophage. Increased induction of latent phage during infection could increase the lysis of bacterial cells and thus be a factor that contributes to sepsis. Our attempt to isolate the phage from in vitro-cultured JJ1886 failed. Therefore, alternative technical approaches, e.g., proteomics, need to be considered in future studies to fully validate the observed gene expression profile.
In contrast to lrhA, the possible pathoadaptive effects of cspE mutations in cystitis isolates are unclear. Interestingly, however, CspA, which is repressed by CspE, was the most upregulated protein in E. coli harvested from voided urine from UTI patients or experimentally infected mice (64, 65), suggesting that inactivation of CspE may be adaptive for this particular niche.
Among other notable mutations, LacI inactivation might be pathoadaptive for urinary isolates, given the evidence that loss of LacI function might be advantageous in an environment with low or inconsistent concentrations of lactose-like sugars (1). Under such conditions, and with glucose depletion, lacI mutants are better able than wild-type strains to rapidly switch their carbon metabolism from glucose to lactose. Interestingly, lacZ was highly upregulated during and was critical for E. coli intracellular persistence in the bladder epithelium (66), suggesting that in that environment bacteria might consume lactose or noninducing galactoside substrates.
Importantly, of the three mutation-inactivated TFs that were tested here, LrhA was the only one for which complementation with a functional gene copy affected survival in a murine septicemia model. This result suggested that transcriptional reprogramming due to LrhA inactivation might be critical in determining the augmented virulence of the isolate in its original human host (12). However, an important contributory role of the source patient's underlying health status cannot be excluded.
Nevertheless, similar increases in virulence due to inactivation of transcriptional regulator have been reported for methicillin-susceptible Staphylococcus aureus in colonized individuals during progression from nasal carriage to fatal bloodstream infections (67). In contrast, the loss-of-function SNPs that appeared in virulence loci in Pseudomonas aeruginosa during long-term adaptation in chronic airway infections resulted in decreased bacterial virulence (5).
Loss-of-function mutations in virulence genes have also been proposed to be responsible for attenuated pathogenicity in E. coli associated with asymptomatic bacteriuria (ABU) in humans (68, 69). Such inactivating mutations, together with frequent gene deletion, are considered signatures of reductive evolution in such ABU strains, which evolved from uropathogenic E. coli (70). Thus, although in the present isolates the occurrence in transcriptional regulators of loss-of-function mutations represents, from a gene function perspective, reductive evolution of these bacteria, it also provides evidence that loss-of-function mutations could be pathoadaptive.
In any environmental niche, including the virulence habitats of bacteria, to be successful an organism must improve its fitness and thereby compete more effectively and achieve increased numbers over time. Therefore, an adaptation might result in increased or decreased virulence of an etiological agent, depending on the accompanying host damage, and thus, prediction of clinical outcome of infection may not be straightforward.
Apart from point mutations, support for the hypothesis that some of the present clinical isolates underwent pathoadaptive evolution in the course of clonal diversification comes from the fact that sepsis isolate JJ1886 has a duplicated 42-kb PAI. This region comprises different virulence-associated loci, in particular, genes involved in iron uptake (fec locus) that here were highly expressed in urine. High expression of this and other iron uptake systems was also demonstrated in E. coli harvested directly from patients' urine (64, 71), suggesting their pathoadaptive significance.
Although various plausible hypotheses could explain how the mutations discussed above could be selected in extraintestinal virulence compartments, the human gut itself is a highly dynamic and compartmentalized environment, with oscillating nutrient composition and other physiological conditions (72, 73). This could allow adaptive diversification within resident strains via many different mechanisms, including gene inactivation. Indeed, the household 2 mother's late-occurring fecal isolate JJ2973 acquired a total of 15 different mutations in comparison to the father's (index patient) isolate from 6 years earlier, including mutations in transcriptional regulators and truncating mutations. Hence, adaptive clonal diversification likely is not limited to clinical isolates but reflects the diversity of the host's fecal population. In a murine model of gut colonization, commensal E. coli strains exhibited an exceptionally high mutation rate of 10−5, with similar mutations emerging continuously across different hosts (74). Thus, particular subpopulations of fecal bacteria could be better preadapted for spread into the urinary tract or, from there, invasion of and survival in the bloodstream.
One might expect that mutation of one regulatory network could be accompanied by, or even lead to, a cascade of changes in other networks, some possibly compensatory (75). This could result in the accumulation of multiple adaptive mutations in the same strain, as observed here. It is also possible that transmission from one host to another, besides creating bottleneck effects, could intensify the pressure for adaptive diversification because of differences in the gut environment due to between-host differences in diet, immunity status, or microbiota composition. Over time, the clonal diversity of same-strain descendants within a given host could reach a high-level equilibrium that is maintained by frequency-dependent or other types of positive selection, at least over the short term (76).
Though the actual dynamics and sequence of transmission within any household are unknown, phylogenetic reconstruction of strain relatedness allowed us to demonstrate that adaptive diversification of bacteria could be happening in nature over a relatively short time scale, similar to that shown under experimental evolution conditions. Mutational inactivation of regulatory genes appears to be a major component of this phenomenon, at least within the emergent pandemic pathogen ST131-H30. A full understanding of the exact nature of the forces driving same-strain clonal diversification between hosts or within a given host, however, will require multiple sampling of the bacterial population from the same or different compartments. Moreover, a combined analysis of genetic data from the bacteria and the host (e.g., host cell gene expression during infection or carriage) could provide even deeper insight into selection pressures for bacterial adaptation and thus better validation of the adaptive potential of the observed genetic changes.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains used in this study are listed in Table S4 in the supplemental material. Wild-type strains were grown in LB broth or sterile human urine (E. V. Sokurenko laboratory collection) at 37°C without shaking. Human urine was collected from at least 3 healthy volunteers, pooled, and filter sterilized. Recombinant plasmid-transformed bacteria (see below) were cultured in the presence of chloramphenicol, unless stated otherwise.
Bacterial phenotype characteristics.
Lactose degradation-positive or -negative phenotype was tested by growing bacteria on LB agar containing X-Gal (160 μg/ml) in the presence or absence of IPTG as a LacI inhibitor. Bacterial motility was tested by spotting 5-μl aliquots of overnight LB broth onto 0.2% agar LB plates, followed by 6 h of incubation at 37°C. Hydrogen peroxide sensitivity was tested as described previously (32), with minor modifications. Briefly, 10-μl aliquots of overnight LB cultures were transferred to 1 ml phosphate-buffered saline (PBS) or PBS containing 2.5 mM hydrogen peroxide, followed by 1 h of incubation at 37°C without shaking. Dilutions of bacterial samples were plated on LB agar for enumeration. The frequency of mutations conferring resistance to the antibiotic rifampin was determined as described previously (25).
For analysis of type 1 fimbria expression, bacteria were grown aerobically on LB agar and in LB broth without shaking. Bacteria were washed and resuspended in PBS (optical density [OD], 2), added to microtiter plate wells, and incubated for 1 h at 37°C. Wells were then washed with PBS and quenched with 0.2% bovine serum albumin (BSA) in PBS for 15 to 20 min. Adherent bacteria were probed with a 1:1,000 diluted rabbit anti-FimH pilin domain serum (77), followed by a secondary anti-rabbit horseradish peroxide-conjugated antibody (Bio-Rad). After extensive washing, the color reaction was developed by adding 3,3′,5,5′-tetramethylbenzidine (TMB) to the wells, and absorbance was read at 650 nm. Alternatively, expression of type 1 fimbriae was assayed by flow cytometry (78). Briefly, bacterial cells were fixed with 4% paraformaldehyde for 20 min at room temperature, washed with PBS, and incubated with 0.2% bovine serum albumin for 30 min. Cells (∼108) were then labeled with 50 μg/ml anti-FimH monoclonal antibody (MAb21 [79]) in the presence of 52 mM α-methyl-d-mannopyranoside for 1 h, followed by incubation with 1:500 diluted Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen). Propidium iodide (PI; final concentration, 40 μM) was added for the last 30 min of secondary antibody incubation. After washing, fluorescence of 100,000 events per sample was measured using FACScan (Becton Dickinson). The fluorescence was calculated for PI-positive cells.
Gene amplification and cloning.
For gene cloning, lacI, rpoS, and lrhA were amplified from genomic DNA of JJ1886 and JJ1887 by PCR using the primers listed in Table S5 in the supplemental material. PCR fragments were then digested with XbaI/XhoI (New England BioLabs) and ligated with T4 ligase (New England BioLabs) into a modified pACYC184 vector (pACYC184.ex) containing an XhoI site just downstream of the XbaI site. The resulting plasmid constructs are listed in Table S4, posted at the URL above.
Genome alignment and analysis.
The fully assembled reference genomes for JJ1886 (NC_022648 [80]) and JJ1887 (NZ_CP014316 [81]) were aligned to each other using Progressive Mauve (82) and inspected for large-scale rearrangements, inversions, and duplications. Candidate sequence variations were reported directly from the alignment and confirmed by mapping raw Illumina reads obtained for those genomes (11, 16) to the assembled reference sequences using Bowtie 2 (83). The alignments were refined using SAMtools (84).
For SNP analysis, raw Illumina sequencing reads (16) were aligned to the reference genome (JJ1886 or JJ1887) using the BWA-MEM algorithm (85). The alignments were refined with SAMtools (84), and variants were called using BCFtools (http://www.htslib.org). Alignments were further refined to mark PCR duplicates, perform local realignment around indels, and recalibrate read base quality scores with Picard Tools (http://broadinstitute.github.io/picard) and the Broad Genome Analysis Toolkit per the GATK best practices guide (86). Variant calls were generated using the GATK Haplotype Caller, and low-confidence calls were filtered. Annotation for variant calls was accomplished using SnpEff (87). All identified DNA changes were confirmed by PCR and Sanger sequencing (for primer sequences, see Table S5). Instances in which the assembled sequence conflicted with the raw sequencing data and PCR results were considered to represent assembly errors (Table 1, IS66 insertion).
RNA-seq.
Bacteria from LB agar plates were inoculated into LB broth or filter-sterilized human urine and were cultured overnight at 37°C without shaking, followed by a second overnight passage through the same medium. The cultures were then mixed with RNA protect reagent (Qiagen), and total bacterial RNA was extracted using the RNeasy minikit (Qiagen). Residual genomic DNA was removed by DNase I (Roche) digestion. The RNA samples were rRNA depleted using the RiboZero-rRNA removal kit (for Gram-negative bacteria; Epicenter). cDNA libraries were prepared using the Truseq stranded mRNA kit (Illumina) and, after normalization, underwent sequencing on an Illumina MiSeq platform using the MiSeq 150 cycle kit (Illumina). FASTQ files were analyzed using Rockhopper bacterial RNA software (http://cs.wellesley.edu/~btjaden/Rockhopper/ [88]) with the JJ1886 chromosome (NC_022648) and/or plasmids (NC_022649-51 and NC_022661-62) used as reference. On average, 5 to 6 million high-quality (i.e., Q of >30, where Q is the Phred quality score defining the base call accuracy and a Q score of 30 to a base is equivalent to the probability of an incorrect base call 1 in 1,000 times) reads were obtained for each sample, of which ∼90% aligned to the reference genome. Gene expression was computed by Rockhopper and was expressed as reads per kilobase per million mapped reads (RPKM), except that instead of the total number of reads, the upper quartile of total reads was applied. For differential gene expression, only reads aligned to annotated regions were analyzed. Three biological replicates were performed for wild-type JJ1886 and JJ1887 and two for any other tested bacteria. The negative binominal distribution test implemented in Rockhopper was used for determination of statistical significance (q value), with q values of <0.01 considered to be statistically significant (88).
Murine sepsis model.
All mouse work in this study was carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health under protocol no. 120603, which was reviewed and approved by the Minneapolis VA Medical Center Institutional Animal Care and Use Committee.
The systemic virulence levels of bacterial strains were compared using an established murine model of sepsis (89). Briefly, Swiss-Webster mice (mean weight, ∼21.6 g) were injected subcutaneously with approximately 108 CFU of log-phase bacteria suspended in PBS and were observed twice daily over the following 3 days. The health status was scored on a 5-step scale (1, healthy; 2, minimally ill; 3, moderately ill; 4, severely ill; 5, dead). With the worst score on a given day used as that day's score, the mean of the three daily health status scores was calculated for each mouse. Mice were euthanized if observed in stage 4 illnesses or at the end of the 3-day observation period, whichever came first. Mice euthanized on day 1 or 2 received a score of 5 for the subsequent day(s).
Statistical analysis.
Fisher's exact test (two-tailed) was used to compare categorical variables (GraphPad Prism 6). Continuous variables were compared using Student's t test, unless stated otherwise (GraphPad Prism 6). P values of <0.05 were considered statistically significant.
Accession number(s).
The raw RNA-seq reads data were deposited at the NCBI Sequence Read Archive (SRA) under BioProject PRJNA350853.
Supplementary Material
ACKNOWLEDGMENTS
We thank Steve Moseley (University of Washington, Seattle) for critical reading of the manuscript and Brian Johnston (Veterans Affairs Medical Center, Minneapolis) and Anahit Hovhannisyan (University of Washington) for technical assistance.
This work was supported by the National Institutes of Health (5R01AI106007-03 to E.V.S. and 5R21AI117654-02 to L.B.P. and J.R.J.). This material is also based in part on work supported by the Office of Research and Development, Medical Research Service, Department of Veterans Affairs (1 I01 CX000920-01 to J.R.J.).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00036-17.
REFERENCES
- 1.Quan S, Ray JC, Kwota Z, Duong T, Balazsi G, Cooper TF, Monds RD. 2012. Adaptive evolution of the lactose utilization network in experimentally evolved populations of Escherichia coli. PLoS Genet 8:e1002444. doi: 10.1371/journal.pgen.1002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang L, Spira B, Zhou Z, Feng L, Maharjan RP, Li X, Li F, McKenzie C, Reeves PR, Ferenci T. 2010. Divergence involving global regulatory gene mutations in an Escherichia coli population evolving under phosphate limitation. Genome Biol Evol 2:478–487. doi: 10.1093/gbe/evq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zambrano MM, Siegele DA, Almiron M, Tormo A, Kolter R. 1993. Microbial competition: Escherichia coli mutants that take over stationary phase cultures. Science 259:1757–1760. doi: 10.1126/science.7681219. [DOI] [PubMed] [Google Scholar]
- 4.Kinnersley M, Wenger J, Kroll E, Adams J, Sherlock G, Rosenzweig F. 2014. Ex uno plures: clonal reinforcement drives evolution of a simple microbial community. PLoS Genet 10:e1004430. doi: 10.1371/journal.pgen.1004430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, D'Argenio DA, Miller SI, Ramsey BW, Speert DP, Moskowitz SM, Burns JL, Kaul R, Olson MV. 2006. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A 103:8487–8492. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang L, Jelsbak L, Marvig RL, Damkiaer S, Workman CT, Rau MH, Hansen SK, Folkesson A, Johansen HK, Ciofu O, Hoiby N, Sommer MO, Molin S. 2011. Evolutionary dynamics of bacteria in a human host environment. Proc Natl Acad Sci U S A 108:7481–7486. doi: 10.1073/pnas.1018249108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vincent JL, Rello J, Marshall J, Silva E, Anzueto A, Martin CD, Moreno R, Lipman J, Gomersall C, Sakr Y, Reinhart K. 2009. International study of the prevalence and outcomes of infection in intensive care units. JAMA 302:2323–2329. doi: 10.1001/jama.2009.1754. [DOI] [PubMed] [Google Scholar]
- 8.Elixhauser A, Friedman B, Stranges E. 2006. Septicemia in U.S. hospitals, 2009: statistical brief #122. https://www.hcup-us.ahrq.gov/reports/statbriefs/sb122.pdf. [PubMed]
- 9.Foxman B. 2014. Urinary tract infection syndromes: occurrence, recurrence, bacteriology, risk factors, and disease burden. Infect Dis Clin North Am 28:1–13. doi: 10.1016/j.idc.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 10.Johnson JR, Tchesnokova V, Johnston B, Clabots C, Roberts PL, Billig M, Riddell K, Rogers P, Qin X, Butler-Wu S, Price LB, Aziz M, Nicolas-Chanoine MH, Debroy C, Robicsek A, Hansen G, Urban C, Platell J, Trott DJ, Zhanel G, Weissman SJ, Cookson BT, Fang FC, Limaye AP, Scholes D, Chattopadhyay S, Hooper DC, Sokurenko EV. 2013. Abrupt emergence of a single dominant multidrug-resistant strain of Escherichia coli. J Infect Dis 207:919–928. doi: 10.1093/infdis/jis933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Price LB, Johnson JR, Aziz M, Clabots C, Johnston B, Tchesnokova V, Nordstrom L, Billig M, Chattopadhyay S, Stegger M, Andersen PS, Pearson T, Riddell K, Rogers P, Scholes D, Kahl B, Keim P, Sokurenko EV. 2013. The epidemic of extended-spectrum-beta-lactamase-producing Escherichia coli ST131 is driven by a single highly pathogenic subclone, H30-Rx. mBio 4:e00377-13. doi: 10.1128/mBio.00377-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Owens RC Jr, Johnson JR, Stogsdill P, Yarmus L, Lolans K, Quinn J. 2011. Community transmission in the United States of a CTX-M-15-producing sequence type ST131 Escherichia coli strain resulting in death. J Clin Microbiol 49:3406–3408. doi: 10.1128/JCM.00993-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ender PT, Gajanana D, Johnston B, Clabots C, Tamarkin FJ, Johnson JR. 2009. Transmission of an extended-spectrum-beta-lactamase-producing Escherichia coli (sequence type ST131) strain between a father and daughter resulting in septic shock and emphysematous pyelonephritis. J Clin Microbiol 47:3780–3782. doi: 10.1128/JCM.01361-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson JR, Anderson JT, Clabots C, Johnston B, Cooperstock M. 2010. Within-household sharing of a fluoroquinolone-resistant Escherichia coli sequence type ST131 strain causing pediatric osteoarticular infection. Pediatr Infect Dis J 29:473–475. doi: 10.1097/INF.0b013e3181c89bd7. [DOI] [PubMed] [Google Scholar]
- 15.Johnson JR, Porter SB, Zhanel G, Kuskowski MA, Denamur E. 2012. Virulence of Escherichia coli clinical isolates in a murine sepsis model in relation to sequence type ST131 status, fluoroquinolone resistance, and virulence genotype. Infect Immun 80:1554–1562. doi: 10.1128/IAI.06388-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson JR, Davis G, Clabots C, Johnston BD, Porter S, DebRoy C, Pomputius W, Ender PT, Cooperstock M, Slater BS, Banerjee R, Miller S, Kisiela DI, Sokurenko EV, Aziz M, Price LB. 2016. Household clustering of Escherichia coli sequence type 131 clinical and fecal isolates according to whole genome sequence analysis. Open Forum Infect Dis 3:ofw129. doi: 10.1093/ofid/ofw129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maharjan R, Seeto S, Notley-McRobb L, Ferenci T. 2006. Clonal adaptive radiation in a constant environment. Science 313:514–517. doi: 10.1126/science.1129865. [DOI] [PubMed] [Google Scholar]
- 18.Saxer G, Krepps MD, Merkley ED, Ansong C, Deatherage Kaiser BL, Valovska MT, Ristic N, Yeh PT, Prakash VP, Leiser OP, Nakhleh L, Gibbons HS, Kreuzer HW, Shamoo Y. 2014. Mutations in global regulators lead to metabolic selection during adaptation to complex environments. PLoS Genet 10:e1004872. doi: 10.1371/journal.pgen.1004872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spencer CC, Bertrand M, Travisano M, Doebeli M. 2007. Adaptive diversification in genes that regulate resource use in Escherichia coli. PLoS Genet 3:e15. doi: 10.1371/journal.pgen.0030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carter MQ, Parker CT, Louie JW, Huynh S, Fagerquist CK, Mandrell RE. 2012. RcsB contributes to the distinct stress fitness among Escherichia coli O157:H7 curli variants of the 1993 hamburger-associated outbreak strains. Appl Environ Microbiol 78:7706–7719. doi: 10.1128/AEM.02157-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Das S, Lindemann C, Young BC, Muller J, Osterreich B, Ternette N, Winkler AC, Paprotka K, Reinhardt R, Forstner KU, Allen E, Flaxman A, Yamaguchi Y, Rollier CS, van Diemen P, Blattner S, Remmele CW, Selle M, Dittrich M, Muller T, Vogel J, Ohlsen K, Crook DW, Massey R, Wilson DJ, Rudel T, Wyllie DH, Fraunholz MJ. 2016. Natural mutations in a Staphylococcus aureus virulence regulator attenuate cytotoxicity but permit bacteremia and abscess formation. Proc Natl Acad Sci U S A 113:E3101–E3110. doi: 10.1073/pnas.1520255113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bragonzi A, Wiehlmann L, Klockgether J, Cramer N, Worlitzsch D, Doring G, Tummler B. 2006. Sequence diversity of the mucABD locus in Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Microbiology 152:3261–3269. doi: 10.1099/mic.0.29175-0. [DOI] [PubMed] [Google Scholar]
- 23.Martin DW, Schurr MJ, Mudd MH, Govan JR, Holloway BW, Deretic V. 1993. Mechanism of conversion to mucoidy in Pseudomonas aeruginosa infecting cystic fibrosis patients. Proc Natl Acad Sci U S A 90:8377–8381. doi: 10.1073/pnas.90.18.8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooper TF, Lenski RE. 2010. Experimental evolution with E. coli in diverse resource environments. I. Fluctuating environments promote divergence of replicate populations. BMC Evol Biol 10:11. doi: 10.1186/1471-2148-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LeClerc JE, Li B, Payne WL, Cebula TA. 1996. High mutation frequencies among Escherichia coli and Salmonella pathogens. Science 274:1208–1211. doi: 10.1126/science.274.5290.1208. [DOI] [PubMed] [Google Scholar]
- 26.Matic I, Radman M, Taddei F, Picard B, Doit C, Bingen E, Denamur E, Elion J. 1997. Highly variable mutation rates in commensal and pathogenic Escherichia coli. Science 277:1833–1834. doi: 10.1126/science.277.5333.1833. [DOI] [PubMed] [Google Scholar]
- 27.Blattner FR, Plunkett G III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 28.Bae W, Phadtare S, Severinov K, Inouye M. 1999. Characterization of Escherichia coli cspE, whose product negatively regulates transcription of cspA, the gene for the major cold shock protein. Mol Microbiol 31:1429–1441. doi: 10.1046/j.1365-2958.1999.01284.x. [DOI] [PubMed] [Google Scholar]
- 29.Battesti A, Majdalani N, Gottesman S. 2011. The RpoS-mediated general stress response in Escherichia coli. Annu Rev Microbiol 65:189–213. doi: 10.1146/annurev-micro-090110-102946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eisenstark A, Calcutt MJ, Becker-Hapak M, Ivanova A. 1996. Role of Escherichia coli rpoS and associated genes in defense against oxidative damage. Free Radic Biol Med 21:975–993. doi: 10.1016/S0891-5849(96)00154-2. [DOI] [PubMed] [Google Scholar]
- 31.Hengge-Aronis R, Klein W, Lange R, Rimmele M, Boos W. 1991. Trehalose synthesis genes are controlled by the putative sigma factor encoded by rpoS and are involved in stationary-phase thermotolerance in Escherichia coli. J Bacteriol 173:7918–7924. doi: 10.1128/jb.173.24.7918-7924.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hryckowian AJ, Welch RA. 2013. RpoS contributes to phagocyte oxidase-mediated stress resistance during urinary tract infection by Escherichia coli CFT073. mBio 4:e00023-13. doi: 10.1128/mBio.00023-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lehnen D, Blumer C, Polen T, Wackwitz B, Wendisch VF, Unden G. 2002. LrhA as a new transcriptional key regulator of flagella, motility and chemotaxis genes in Escherichia coli. Mol Microbiol 45:521–532. doi: 10.1046/j.1365-2958.2002.03032.x. [DOI] [PubMed] [Google Scholar]
- 34.Wallecha A, Oreh H, van der Woude MW, deHaseth PL. 2014. Control of gene expression at a bacterial leader RNA, the agn43 gene encoding outer membrane protein Ag43 of Escherichia coli. J Bacteriol 196:2728–2735. doi: 10.1128/JB.01680-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alba BM, Zhong HJ, Pelayo JC, Gross CA. 2001. degS (hhoB) is an essential Escherichia coli gene whose indispensable function is to provide sigma (E) activity. Mol Microbiol 40:1323–1333. doi: 10.1046/j.1365-2958.2001.02475.x. [DOI] [PubMed] [Google Scholar]
- 36.Gopel Y, Papenfort K, Reichenbach B, Vogel J, Gorke B. 2013. Targeted decay of a regulatory small RNA by an adaptor protein for RNase E and counteraction by an anti-adaptor RNA. Genes Dev 27:552–564. doi: 10.1101/gad.210112.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Purtov YA, Glazunova OA, Antipov SS, Pokusaeva VO, Fesenko EE, Preobrazhenskaya EV, Shavkunov KS, Tutukina MN, Lukyanov VI, Ozoline ON. 2014. Promoter islands as a platform for interaction with nucleoid proteins and transcription factors. J Bioinform Comput Biol 12:1441006. doi: 10.1142/S0219720014410066. [DOI] [PubMed] [Google Scholar]
- 38.Jeong KC, Baumler DJ, Kaspar CW. 2006. dps expression in Escherichia coli O157:H7 requires an extended -10 region and is affected by the cAMP receptor protein. Biochim Biophys Acta 1759:51–59. doi: 10.1016/j.bbaexp.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 39.Jiang W, Fang L, Inouye M. 1996. The role of the 5′-end untranslated region of the mRNA for CspA, the major cold-shock protein of Escherichia coli, in cold-shock adaptation. J Bacteriol 178:4919–4925. doi: 10.1128/jb.178.16.4919-4925.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schlensog V, Bock A. 1990. Identification and sequence analysis of the gene encoding the transcriptional activator of the formate hydrogenlyase system of Escherichia coli. Mol Microbiol 4:1319–1327. doi: 10.1111/j.1365-2958.1990.tb00711.x. [DOI] [PubMed] [Google Scholar]
- 41.Mauzy CA, Hermodson MA. 1992. Structural and functional analyses of the repressor, RbsR, of the ribose operon of Escherichia coli. Protein Sci 1:831–842. doi: 10.1002/pro.5560010701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ishihama A. 2010. Prokaryotic genome regulation: multifactor promoters, multitarget regulators and hierarchic networks. FEMS Microbiol Rev 34:628–645. doi: 10.1111/j.1574-6976.2010.00227.x. [DOI] [PubMed] [Google Scholar]
- 43.Thieffry D, Huerta AM, Perez-Rueda E, Collado-Vides J. 1998. From specific gene regulation to genomic networks: a global analysis of transcriptional regulation in Escherichia coli. Bioessays 20:433–440. doi:. [DOI] [PubMed] [Google Scholar]
- 44.Stulke J. 2002. Control of transcription termination in bacteria by RNA-binding proteins that modulate RNA structures. Arch Microbiol 177:433–440. doi: 10.1007/s00203-002-0407-5. [DOI] [PubMed] [Google Scholar]
- 45.Browning DF, Busby SJ. 2004. The regulation of bacterial transcription initiation. Nat Rev Microbiol 2:57–65. doi: 10.1038/nrmicro787. [DOI] [PubMed] [Google Scholar]
- 46.Perez-Rueda E, Collado-Vides J. 2000. The repertoire of DNA-binding transcriptional regulators in Escherichia coli K-12. Nucleic Acids Res 28:1838–1847. doi: 10.1093/nar/28.8.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Charoensawan V, Wilson D, Teichmann SA. 2010. Genomic repertoires of DNA-binding transcription factors across the tree of life. Nucleic Acids Res 38:7364–7377. doi: 10.1093/nar/gkq617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ortet P, De Luca G, Whitworth DE, Barakat M. 2012. P2TF: a comprehensive resource for analysis of prokaryotic transcription factors. BMC Genomics 13:628. doi: 10.1186/1471-2164-13-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Notley-McRobb L, King T, Ferenci T. 2002. rpoS mutations and loss of general stress resistance in Escherichia coli populations as a consequence of conflict between competing stress responses. J Bacteriol 184:806–811. doi: 10.1128/JB.184.3.806-811.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferenci T. 2003. What is driving the acquisition of mutS and rpoS polymorphisms in Escherichia coli? Trends Microbiol 11:457–461. doi: 10.1016/j.tim.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 51.Levert M, Zamfir O, Clermont O, Bouvet O, Lespinats S, Hipeaux MC, Branger C, Picard B, Saint-Ruf C, Norel F, Balliau T, Zivy M, Le Nagard H, Cruveiller S, Chane-Woon-Ming B, Nilsson S, Gudelj I, Phan K, Ferenci T, Tenaillon O, Denamur E. 2010. Molecular and evolutionary bases of within-patient genotypic and phenotypic diversity in Escherichia coli extraintestinal infections. PLoS Pathog 6:e1001125. doi: 10.1371/journal.ppat.1001125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Price EP, Sarovich DS, Mayo M, Tuanyok A, Drees KP, Kaestli M, Beckstrom-Sternberg SM, Babic-Sternberg JS, Kidd TJ, Bell SC, Keim P, Pearson T, Currie BJ. 2013. Within-host evolution of Burkholderia pseudomallei over a twelve-year chronic carriage infection. mBio 4:e00388-13. doi: 10.1128/mBio.00388-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dong T, Chiang SM, Joyce C, Yu R, Schellhorn HE. 2009. Polymorphism and selection of rpoS in pathogenic Escherichia coli. BMC Microbiol 9:118. doi: 10.1186/1471-2180-9-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blumer C, Kleefeld A, Lehnen D, Heintz M, Dobrindt U, Nagy G, Michaelis K, Emody L, Polen T, Rachel R, Wendisch VF, Unden G. 2005. Regulation of type 1 fimbriae synthesis and biofilm formation by the transcriptional regulator LrhA of Escherichia coli. Microbiology 151:3287–3298. doi: 10.1099/mic.0.28098-0. [DOI] [PubMed] [Google Scholar]
- 55.Wiles TJ, Kulesus RR, Mulvey MA. 2008. Origins and virulence mechanisms of uropathogenic Escherichia coli. Exp Mol Pathol 85:11–19. doi: 10.1016/j.yexmp.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adlerberth I, Hanson LA, Svanborg C, Svennerholm AM, Nordgren S, Wold AE. 1995. Adhesins of Escherichia coli associated with extra-intestinal pathogenicity confer binding to colonic epithelial cells. Microb Pathog 18:373–385. doi: 10.1006/mpat.1995.0034. [DOI] [PubMed] [Google Scholar]
- 57.Schierack P, Walk N, Ewers C, Wilking H, Steinruck H, Filter M, Wieler LH. 2008. ExPEC-typical virulence-associated genes correlate with successful colonization by intestinal E. coli in a small piglet group. Environ Microbiol 10:1742–1751. doi: 10.1111/j.1462-2920.2008.01595.x. [DOI] [PubMed] [Google Scholar]
- 58.Lane MC, Alteri CJ, Smith SN, Mobley HL. 2007. Expression of flagella is coincident with uropathogenic Escherichia coli ascension to the upper urinary tract. Proc Natl Acad Sci U S A 104:16669–16674. doi: 10.1073/pnas.0607898104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lane MC, Lockatell V, Monterosso G, Lamphier D, Weinert J, Hebel JR, Johnson DE, Mobley HL. 2005. Role of motility in the colonization of uropathogenic Escherichia coli in the urinary tract. Infect Immun 73:7644–7656. doi: 10.1128/IAI.73.11.7644-7656.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kao CY, Lin WH, Tseng CC, Wu AB, Wang MC, Wu JJ. 2014. The complex interplay among bacterial motility and virulence factors in different Escherichia coli infections. Eur J Clin Microbiol Infect Dis 33:2157–2162. doi: 10.1007/s10096-014-2171-2. [DOI] [PubMed] [Google Scholar]
- 61.Melican K, Sandoval RM, Kader A, Josefsson L, Tanner GA, Molitoris BA, Richter-Dahlfors A. 2011. Uropathogenic Escherichia coli P and type 1 fimbriae act in synergy in a living host to facilitate renal colonization leading to nephron obstruction. PLoS Pathog 7:e1001298. doi: 10.1371/journal.ppat.1001298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Connell I, Agace W, Klemm P, Schembri M, Marild S, Svanborg C. 1996. Type 1 fimbrial expression enhances Escherichia coli virulence for the urinary tract. Proc Natl Acad Sci U S A 93:9827–9832. doi: 10.1073/pnas.93.18.9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rosen DA, Hooton TM, Stamm WE, Humphrey PA, Hultgren SJ. 2007. Detection of intracellular bacterial communities in human urinary tract infection. PLoS Med 4:e329. doi: 10.1371/journal.pmed.0040329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hagan EC, Lloyd AL, Rasko DA, Faerber GJ, Mobley HL. 2010. Escherichia coli global gene expression in urine from women with urinary tract infection. PLoS Pathog 6:e1001187. doi: 10.1371/journal.ppat.1001187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Snyder JA, Haugen BJ, Buckles EL, Lockatell CV, Johnson DE, Donnenberg MS, Welch RA, Mobley HL. 2004. Transcriptome of uropathogenic Escherichia coli during urinary tract infection. Infect Immun 72:6373–6381. doi: 10.1128/IAI.72.11.6373-6381.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Conover MS, Hadjifrangiskou M, Palermo JJ, Hibbing ME, Dodson KW, Hultgren SJ. 2016. Metabolic requirements of Escherichia coli in intracellular bacterial communities during urinary tract infection pathogenesis. mBio 7:e00104–16. doi: 10.1128/mBio.00104-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Young BC, Golubchik T, Batty EM, Fung R, Larner-Svensson H, Votintseva AA, Miller RR, Godwin H, Knox K, Everitt RG, Iqbal Z, Rimmer AJ, Cule M, Ip CL, Didelot X, Harding RM, Donnelly P, Peto TE, Crook DW, Bowden R, Wilson DJ. 2012. Evolutionary dynamics of Staphylococcus aureus during progression from carriage to disease. Proc Natl Acad Sci U S A 109:4550–4555. doi: 10.1073/pnas.1113219109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klemm P, Roos V, Ulett GC, Svanborg C, Schembri MA. 2006. Molecular characterization of the Escherichia coli asymptomatic bacteriuria strain 83972: the taming of a pathogen. Infect Immun 74:781–785. doi: 10.1128/IAI.74.1.781-785.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Salvador E, Wagenlehner F, Kohler CD, Mellmann A, Hacker J, Svanborg C, Dobrindt U. 2012. Comparison of asymptomatic bacteriuria Escherichia coli isolates from healthy individuals versus those from hospital patients shows that long-term bladder colonization selects for attenuated virulence phenotypes. Infect Immun 80:668–678. doi: 10.1128/IAI.06191-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dobrindt U, Wullt B, Svanborg C. 2016. Asymptomatic bacteriuria as a model to study the coevolution of hosts and bacteria. Pathogens 5:E21. doi: 10.3390/pathogens5010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Subashchandrabose S, Hazen TH, Brumbaugh AR, Himpsl SD, Smith SN, Ernst RD, Rasko DA, Mobley HL. 2014. Host-specific induction of Escherichia coli fitness genes during human urinary tract infection. Proc Natl Acad Sci U S A 111:18327–18332. doi: 10.1073/pnas.1415959112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Albenberg L, Esipova TV, Judge CP, Bittinger K, Chen J, Laughlin A, Grunberg S, Baldassano RN, Lewis JD, Li H, Thom SR, Bushman FD, Vinogradov SA, Wu GD. 2014. Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology 147:1055-63.e8. doi: 10.1053/j.gastro.2014.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li H, Limenitakis JP, Fuhrer T, Geuking MB, Lawson MA, Wyss M, Brugiroux S, Keller I, Macpherson JA, Rupp S, Stolp B, Stein JV, Stecher B, Sauer U, McCoy KD, Macpherson AJ. 2015. The outer mucus layer hosts a distinct intestinal microbial niche. Nat Commun 6:8292. doi: 10.1038/ncomms9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lourenco M, Ramiro RS, Guleresi D, Barroso-Batista J, Xavier KB, Gordo I, Sousa A. 2016. A mutational hotspot and strong selection contribute to the order of mutations selected for during Escherichia coli adaptation to the gut. PLoS Genet 12:e1006420. doi: 10.1371/journal.pgen.1006420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stoebel DM, Hokamp K, Last MS, Dorman CJ. 2009. Compensatory evolution of gene regulation in response to stress by Escherichia coli lacking RpoS. PLoS Genet 5:e1000671. doi: 10.1371/journal.pgen.1000671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Golubchik T, Batty EM, Miller RR, Farr H, Young BC, Larner-Svensson H, Fung R, Godwin H, Knox K, Votintseva A, Everitt RG, Street T, Cule M, Ip CL, Didelot X, Peto TE, Harding RM, Wilson DJ, Crook DW, Bowden R. 2013. Within-host evolution of Staphylococcus aureus during asymptomatic carriage. PLoS One 8:e61319. doi: 10.1371/journal.pone.0061319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tchesnokova V, Aprikian P, Kisiela D, Gowey S, Korotkova N, Thomas W, Sokurenko E. 2011. Type 1 fimbrial adhesin FimH elicits an immune response that enhances cell adhesion of Escherichia coli. Infect Immun 79:3895–3904. doi: 10.1128/IAI.05169-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kisiela D, Laskowska A, Sapeta A, Kuczkowski M, Wieliczko A, Ugorski M. 2006. Functional characterization of the FimH adhesin from Salmonella enterica serovar Enteritidis. Microbiology 152:1337–1346. doi: 10.1099/mic.0.28588-0. [DOI] [PubMed] [Google Scholar]
- 79.Tchesnokova V, Aprikian P, Yakovenko O, Larock C, Kidd B, Vogel V, Thomas W, Sokurenko E. 2008. Integrin-like allosteric properties of the catch bond-forming FimH adhesin of Escherichia coli. J Biol Chem 283:7823–7833. doi: 10.1074/jbc.M707804200. [DOI] [PubMed] [Google Scholar]
- 80.Andersen PS, Stegger M, Aziz M, Contente-Cuomo T, Gibbons HS, Keim P, Sokurenko EV, Johnson JR, Price LB. 2013. Complete genome sequence of the epidemic and highly virulent CTX-M-15-producing H30-Rx subclone of Escherichia coli ST131. Genome Announc 1(6):e00988-13. doi: 10.1128/genomeA.00988-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Johnson TJ, Aziz M, Liu CM, Sokurenko E, Kisiela DI, Paul S, Andersen P, Johnson JR, Price LB. 2016. Complete genome sequence of a CTX-M-15-producing Escherichia coli strain from the H30Rx subclone of sequence type 131 from a patient with recurrent urinary tract infections, closely related to a lethal urosepsis isolate from the patient's sister. Genome Announc 4(3):e00334-16. doi: 10.1128/genomeA.00334-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Darling AC, Mau B, Blattner FR, Perna NT. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Broad Institute of Harvard and MIT https://archive.org/stream/arxiv-1303.3997/1303.3997_djvu.txt.
- 86.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA. 2013. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43:11.10.1–33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tjaden B. 2015. De novo assembly of bacterial transcriptomes from RNA-seq data. Genome Biol 16:1. doi: 10.1186/s13059-014-0572-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Johnson JR, Clermont O, Menard M, Kuskowski MA, Picard B, Denamur E. 2006. Experimental mouse lethality of Escherichia coli isolates, in relation to accessory traits, phylogenetic group, and ecological source. J Infect Dis 194:1141–1150. doi: 10.1086/507305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.