

ABSTRACT
AMP-activated protein kinase (AMPK) and the homologous yeast SNF1 complex are key regulators of energy metabolism that counteract nutrient deficiency and ATP depletion by phosphorylating multiple enzymes and transcription factors that maintain energetic homeostasis. AMPK/SNF1 also promotes longevity in several model organisms, including yeast. Here we investigate the role of yeast SNF1 in mediating the extension of chronological life span (CLS) by caloric restriction (CR). We find that SNF1 activity is required throughout the transition of log phase to stationary phase (diauxic shift) for effective CLS extension. CR expands the period of maximal SNF1 activation beyond the diauxic shift, as indicated by Sak1-dependent T210 phosphorylation of the Snf1 catalytic α-subunit. A concomitant increase in ADP is consistent with SNF1 activation by ADP in vivo. Downstream of SNF1, the Cat8 and Adr1 transcription factors are required for full CR-induced CLS extension, implicating an alternative carbon source utilization for acetyl coenzyme A (acetyl-CoA) production and gluconeogenesis. Indeed, CR increased acetyl-CoA levels during the diauxic shift, along with expression of both acetyl-CoA synthetase genes ACS1 and ACS2. We conclude that CR maximizes Snf1 activity throughout and beyond the diauxic shift, thus optimizing the coordination of nucleocytosolic acetyl-CoA production with massive reorganization of the transcriptome and respiratory metabolism.
KEYWORDS: Snf1, AMPK, aging, yeast, caloric restriction, ADP, diauxic shift, acetyl-CoA
INTRODUCTION
Although each eukaryotic species suffers from its own set of age-related maladies, characteristics of the aging process at a cellular level are well conserved, including mitochondrial dysfunction, inefficient turnover of damaged proteins and organelles, accumulated reactive oxygen species (ROS) damage, and gradual breakdown of chromatin structure (reviewed in reference 1). Given the variety of the cellular components involved, it is astounding that any single intervention can alleviate the deterioration of all these processes. Caloric restriction (CR), however, slows the onset of aging-related pathologies and extends life span in almost every model organism tested, ranging from yeast to mammals (reviewed in reference 2). Some mechanistic commonalities for CR have emerged, including upregulation of cellular autophagy, increased mitochondrial respiration, protective oxidative stress responses, and decreased protein synthesis (reviewed in reference 3). The majority of these processes are regulated by a series of highly conserved nutrient signaling pathways, including the AMP-activated protein kinase (AMPK) signaling pathway (4).
AMPK is the eukaryotic cell's primary energy sensor, regulating metabolism and other downstream processes while interacting with other signaling pathways (5). Experimental evidence for AMPK as a mediator of the CR longevity response is mounting. In Caenorhabditis elegans, AMPK is activated and required for life span extension under certain CR regimens (6, 7). In Drosophila, overexpressing the upstream AMPK-activating kinase LKB1 extends life span (8), but metformin-induced AMPK activation does not (9). Whether AMPK directly mediates life span extension by CR in mouse remains an unsettled issue (10), but AMPK's effects on other known longevity pathways support its role as a CR nexus. In mice, AMPK signaling inhibits target of rapamycin (TOR) activity directly by phosphorylating the Raptor subunit (11) and indirectly by phosphorylating the upstream kinase tuberous sclerosis protein 2 (TSC2) (12). The FOXO family of transcription factors, downstream effectors of reduced insulin/insulin-like growth factor (IGF) signaling (rIIS)-mediated life span extension, are also direct targets of AMPK phosphorylation in C. elegans and mice (6, 13), but it is not clear how these phosphorylation events drive the effects on life span, if at all.
In the budding yeast Saccharomyces cerevisiae, the analogous AMPK complex is known as SNF1 (14). As in multicellular eukaryotes, downstream targets of SNF1 are involved in the regulation of multiple cellular processes, including metabolism, cell growth and division, autophagy, and adaptive stress responses (15, 16). Given this broad degree of conservation, we have utilized the yeast chronological life span (CLS) model of postmitotic aging to gain a better understanding of the role that AMPK/SNF1 signaling plays in CR-mediated life span extension at the cellular level. CLS is a measurement of cell survival during prolonged incubation in stationary phase (17) and is distinct from replicative life span (RLS), which is defined by the number of successful cell divisions before senescence. Here we demonstrate that SNF1 activity is required throughout the transition from fermentative to respiratory metabolism (termed the diauxic shift) to maximize CLS and to mediate the extension of CLS under CR conditions. CR significantly broadens the period of SNF1 activity during the transition, as measured by elevated T210 phosphorylation of the catalytic Snf1 α-subunit. Furthermore, the prolonged Snf1 phosphorylation correlates with CR-induced elevation of cellular ADP, consistent with ADP being a key metabolite that directly regulates SNF1 in vivo (18). We go on to identify the transcription factors Cat8 and Adr1 as key downstream SNF1 targets required for CLS extension by CR. Cells lacking Cat8 and Adr1 were unable to fully activate target gene transcription in response to CR during the diauxic shift, thus blocking CLS extension. A model of CR-mediated yeast CLS extension centered on SNF1 is presented.
RESULTS
Snf1 mediates maximum CLS extension by CR.
For the yeast chronological aging system, CR typically involves reducing the starting glucose concentration of a liquid culture at the time of inoculation and allowing the cells to enter stationary phase (19). Exponentially growing yeast cells preferentially ferment glucose into ethanol and acetate. As glucose is depleted, cells then reorganize their transcriptional and metabolic programs to favor the catabolism of secreted ethanol and acetate as nonfermentable carbon sources for energy and biomass production (20). This programmed transition is called the diauxic shift and involves a nutrient (glucose) depletion-triggered signaling cascade featuring activation of SNF1 (16), a heterotrimeric kinase complex consisting of a catalytic α-subunit (Snf1), a regulatory γ-subunit (Snf4), and one of three possible scaffolding β-subunits (Gal83, Sip1, or Sip2) (21) (Fig. 1A). Cells lacking Snf1 are defective in glycogen accumulation and do not properly enter stationary phase (22), resulting in a dramatically shortened CLS (23).
FIG 1.

Snf1 is required for normal chronological life span and life span extension by glucose and methionine restriction. (A) The yeast SNF1 complex shares a conserved heterotrimeric structure with mammalian AMPK consisting of one catalytic α-subunit (Snf1), a regulatory γ-subunit (Snf4), and one of three possible β-subunits (Sip1, Sip2, or Gal83). Activity of the complex requires phosphorylation of T210 within the activation loop of Snf1. KD, kinase domain of Snf1. (B) Semiquantitative CLS assay of WT (BY4741) grown under nonrestricted (NR, 2% glucose) or calorie-restricted (CR, 0.5% glucose) growth conditions. (C) CLS of the snf1Δ mutant (JS1394) under NR and CR conditions. (D) CLS of BY4741 in normal (125-mg/liter) or restricted (5-mg/liter) concentrations of methionine. (E) CLS of BY4741 supplemented with 25 mM isonicotinamide (INAM). (F) CLS of snf1Δ mutant under normal or restricted methionine conditions. (G) CLS of snf1Δ with or without INAM supplementation.
Expression profiling of cells grown to stationary phase under CR conditions revealed that the genes involved in acetate utilization were upregulated compared to those in nonrestricted (NR) yeast cells (24). Since Snf1 was known to be important for alternative carbon source utilization (25), we hypothesized that it was mediating the CLS extension induced by CR. To test this idea, we first deleted SNF1 and aged the mutant in synthetic complete (SC) medium with NR (2.0%) or CR (0.5%) initial glucose concentrations. CR extended the CLS of a wild-type (WT) strain (BY4741) (Fig. 1B) but had little effect on the short CLS of the snf1Δ mutant (Fig. 1C). Larger colonies that appeared after day 13 were derived from cells that underwent a phenomenon known as adaptive regrowth (or “gasping”), whereby spontaneous mutants that have a survival advantage occur within the population (26). SNF1 was also required for the extension of CLS induced by methionine restriction (Fig. 1D and F), another dietary regimen that extends the life span of organisms ranging from yeast to mice (reviewed in reference 27). However, the CLS-extending compound isonicotinamide (INAM) clearly extended CLS of the snf1Δ mutant (Fig. 1E and G), consistent with INAM changing the stationary-phase gene expression profile differently from CR (24) and indicating that INAM extends CLS through a more Snf1-independent mechanism.
To further investigate the role of Snf1 in CLS and dietary restriction, we focused on the glucose model of CR. It was possible that poor stationary-phase viability of the snf1Δ mutant was simply masking the beneficial effect of CR on CLS rather than CR functioning through SNF1 signaling. To address this idea, we took advantage of a specialized Snf1 mutant protein (Snf1-as) that functions normally but is specifically inactivated by the cell-permeable kinase inhibitor 2NM-PP1 (28). Importantly, this compound has no effect on the normal Snf1 protein. This system allowed for inactivation of Snf1-as after cells had already progressed through the diauxic shift. Based on growth curves, both the WT and snf1-as strains from NR and CR cultures entered the diauxic shift approximately 12 h after inoculation and stopped doubling by 24 h (Fig. 2A), indicating that they had progressed through the diauxic shift. 2NM-PP1 was added to the WT and snf1-as cultures either at the time of inoculation (0 h), at the onset of the diauxic shift (12 h), or after the shift (24 h) as indicated schematically in Fig. 2B. CLS was then quantitatively tracked for the first 10 days, which avoided the later appearance of adaptive regrowth. As expected, adding 2NM-PP1 had no effect on CLS of the WT SNF1 strain (Fig. 2C and D). However, 2NM-PP1 significantly shortened the CLS of the snf1-as mutant under NR and CR conditions regardless of when it was added to the cultures (Fig. 2E and F), thus phenocopying the snf1Δ effect on CLS. We conclude that Snf1 activity is required throughout and after the diauxic shift to sustain cell viability during stationary phase and to mediate the beneficial effect of CR on CLS.
FIG 2.

Inhibiting Snf1 after the diauxic shift shortens CLS. (A) Growth curve with SNF1 control strain (PY855) and mutant snf1-as strain (PY856) showing the cessation of cell growth within 24 h after inoculation. 2NM-PP1 was not added to the cultures. (B) Schematic diagram of timing for cell growth and addition of either dimethyl sulfoxide (DMSO) or 2NM-PP1 at various times either at the time of inoculation or at the indicated hours after inoculation. The percent viability was tracked starting at day 2. (C) Quantitative CLS assay with NR medium for the WT SNF1 control strain (PY855) treated with 2NM-PP1 at the time of inoculation (0 h) or after inoculation at 12 and 24 h. (D) Quantitative CLS assay with CR medium for strain PY855. (E) Quantitative CLS assay with NR medium for the mutant snf1-as strain (PY856). (F) CLS assay with CR medium for PY856. Error bars indicate standard deviations from 3 biological replicates.
CR promotes Snf1 activation during the diauxic shift.
Glucose depletion from yeast cultures induces Snf1 activation, which is indicated biochemically by phosphorylation of threonine 210 on the activation loop (29). T210 phosphorylation is analogous to phosphorylation of T172 in the human AMPK α-subunit and cross-recognized by an antibody specific for phospho-T172 (30). We hypothesized that one of the mechanisms by which CR extended CLS was through hyperactivation of Snf1. To test this idea, Snf1 T210 phosphorylation (T210-P) was initially tracked by Western blotting through a growth time course in NR or CR medium up to 28 h following inoculation. T210-P levels relative to total hemagglutinin (HA)-tagged Snf1 were extremely low in exponentially growing NR and CR cells (Fig. 3A and B, 10 h), indicating that simply reducing glucose from 2% to 0.5% was insufficient to activate Snf1 in exponentially growing cells. T210-P appeared earlier for the CR condition (16 h versus 22 h for NR), most likely because glucose expired more quickly in the CR cultures (Fig. 3A and B). This result also suggested that cells in CR cultures entered the diauxic shift ∼4 h earlier than NR cultures. By 28 h, there was significantly higher T210-P in CR cells, perhaps due to reduced phosphorylation in NR cells (Fig. 3B). Indeed, extending the time course to 48 h revealed a continual decline in T210-P after the peak 24-h time point under the NR condition, which was significantly delayed under the CR condition (Fig. 3C and D, 32- and 40-h time points). CR clearly expanded the period of time that Snf1 remained highly active during the transition into stationary phase, both earlier and later than under the NR condition.
FIG 3.

CR expands the time period of Snf1 T210 phosphorylation during growth in batch cultures. (A) Representative Western blot of whole-cell extracts using an antibody directed against human AMPKα phosphorylated on T172, which cross-reacts with yeast Snf1 phosphorylated at T210. Log phase is indicated by the 10-h time points. Snf1-HA is detected with an anti-HA antibody. (B) Quantitation of Snf1 T210-P levels relative to total Snf1-3HA from 3 biological replicates. (C) Representative Western blot for a time course extended to 48 h. (D) Quantitation of Snf1 T210-P levels in the extended time course from 3 biological replicates. (E) Quantitation of intracellular ADP levels from the extended time course samples. (F) Quantitation of intracellular ATP levels from the extended time course. Error bars represent standard deviations. Asterisks indicate P values <0.05 in the bar graphs.
We next addressed the mechanism of expanded Snf1 T210-P under the CR condition. Unlike mammalian AMPK, which is activated either by AMP or by ADP (31, 32), the yeast SNF1 complex is activated only by ADP (18). In vitro, ADP binds to the Snf4 γ-subunit and inhibits dephosphorylation of Snf1 T210 by Glc7 (18). AMP and ATP do not affect Snf1 T210-P or its catalytic activity (18). Therefore, we hypothesized that the expanded period of highly phosphorylated Snf1 was related to the intracellular ADP concentration. To assess this possibility, WT cells were grown under NR and CR conditions, and intracellular ADP concentration was measured across a time course similar to that in the above-described Western blot assays. ADP dropped significantly after the diauxic shift in NR cells (Fig. 3E, 24 h), correlating with the start of a steady decline in T210-P (Fig. 3C and D). CR, on the other hand, prevented the decline in ADP through the time course (Fig. 3E), consistent with the expansion of T210-P (Fig. 3C and D). Interestingly, ATP levels also remained elevated in CR-grown cells as was previously observed (33). Since ATP does not impact SNF1 activation, this result is consistent with the notion that SNF1 responds to the absolute ADP concentration rather than to the ADP/ATP ratio (18).
Upstream kinases have differential effects on CLS.
Having established that CR promotes extended Snf1 T210-P during CLS assays, we next asked whether a specific upstream kinase was responsible for the CR-induced life span extension. Snf1 is activated by any of 3 upstream kinases: Sak1, Elm1, and Tos3 (30, 34, 35). Although some studies found these kinases equally capable of phosphorylating Snf1 and functionally semiredundant under glucose depletion (34), others indicated variability in their ability to activate Snf1 under different stresses (36, 37). We quantitatively measured CLS for individual sak1Δ, elm1Δ, or tos3Δ mutants under NR and CR conditions and observed different phenotypes for each. The CLS of the tos3Δ mutant was indistinguishable from that of the WT in both types of medium (Fig. 4A and D). The elm1Δ mutant was longer lived than the WT under the NR condition, but the CLS was still fully extended by CR (Fig. 4B and D). In contrast, CLS of the sak1Δ mutant was slightly reduced compared to that of the WT under the NR condition, but the CR effect was blocked (Fig. 4C and D). This result strongly implicated Sak1 as the critical upstream Snf1 kinase for mediating CR effects on CLS. Next, the effect of each kinase mutant on Snf1 T210-P was examined by Western blotting. As shown in Fig. 4E, T210-P signal was generally reduced in each mutant compared to the WT, consistent with the previously observed redundancy among the three kinases. However, despite the overall reduction in Snf1 phosphorylation, CR still maintained T210-P into stationary phase (40 h) for the elm1Δ and tos3Δ mutants but not in the sak1Δ mutant. These results suggest that Sak1 is required for CR to maintain active Snf1 beyond the diauxic shift, which is important for CLS extension.
FIG 4.

Effects of deleting TOS3, ELM1, or SAK1 on CLS. (A) CLS of the WT strain BY4741 and the tos3Δ mutant SY841 in NR and CR media. (B) CLS of BY4741 and the elm1Δ mutant SY844 in NR and CR media. (C) CLS of BY4741 and the sak1Δ mutant SY848 in NR and CR media. (D) Mean CLS in days, SEMs, and 95% CI calculated from OASIS. (E) Western blots of Snf1 T210 phosphorylation or total Snf1-HA levels in WT (MSY1238), tos3Δ (JS1521), elm1Δ (JS1517), and sak1Δ (JS1519) strains. Cultures were grown in SC medium with either NR or CR glucose concentrations at the time of inoculation. Cells were harvested at the 9 (log phase)-, 16-, 24-, and 40-h time points.
Cat8 and Adr1 mediate CR effects on CLS downstream of Snf1.
Sak1 is the primary kinase for SNF1 complexes containing the Gal83 β-subunit (36). Upon activation, Gal83-containing complexes localize primarily to the nucleus (38), resulting in the altered expression of hundreds of genes through direct and indirect regulation of a series of transcriptional activators and repressors (16). This turned our attention to identifying key downstream effectors of Snf1 required for CR-induced CLS extension. A top candidate was Cat8, a transcription factor that activates genes involved in the consumption of ethanol and acetate via the glyoxylate and gluconeogenesis pathways (39, 40). Furthermore, Snf1 directly activates the Cat8 protein by phosphorylation (41, 42) and is required for activation of CAT8 gene expression (43). Some Cat8-regulated genes are coactivated by another transcription factor, Adr1 (44, 45). To determine if Cat8, Adr1, or both were important for CR-induced life span extension, the CLS of WT, cat8Δ, adr1Δ, and cat8Δ adr1Δ strains was tested. Initial semiquantitative spot assays strongly suggested that the CLS of cat8Δ and cat8Δ adr1Δ mutants was less responsive than that of the adr1Δ mutant and WT to extension by CR (Fig. 5A to D). Quantitative assays confirmed this, showing that CLS of the adr1Δ mutant was extended by CR to the same extent as the WT control strain, while CLS of the cat8Δ mutant was only partially extended (Fig. 5E to G). The CR effect was slightly more attenuated in the cat8Δ adr1Δ double mutant (Fig. 5E to G), indicating that Adr1 makes only a minor contribution to CLS in the absence of Cat8. Importantly, loss of Cat8 and/or Adr1 did not shorten CLS under NR growth conditions (Fig. 5E and G), strongly suggesting that other transcription factors can compensate for their loss in order to maintain cell survival under NR conditions. Under CR conditions, however, Cat8 and Adr1 together become critical for the extended CLS.
FIG 5.

CAT8 and ADR1 are together required for maximum CLS extension by CR. (A to D) Semiquantitative CLS assays showing relative cell survival over time for WT (BY4741), adr1Δ (SY826), cat8Δ (SY825), and cat8Δ adr1Δ (SY767) strains grown under NR or CR conditions. (E and F) Quantitative CLS assay for the same strains as described for panels A to D, grown in 2% glucose (NR) cultures (E) or in 0.5% glucose (CR) cultures (F). (G) Mean CLS in days, SEM, and 95% CI calculated from OASIS.
CR amplifies expression of specific Cat8/Adr1 targets.
The dependence of CLS extension on CAT8 and ADR1 suggested that expression of their target genes could be enhanced by the CR growth condition. Previous studies implicated acetate and acetic acid utilization as important for proper CLS maintenance (24, 46), so we focused quantitative reverse transcription (qRT)-PCR analysis on the expression of several Cat8/Adr1 target genes involved in the conversion of acetate into acetyl coenzyme A (acetyl-CoA). ACH1 encodes a mitochondrial enzyme that produces acetyl-CoA by transferring the CoA moiety of succinyl-CoA onto acetate (47, 48). ACS1, on the other hand, encodes a nucleocytoplasmic acetyl-CoA synthetase that utilizes acetate and free CoA as the substrates (49). Both are glucose repressed and activated during the diauxic shift (49, 50). ACH1 was activated during the diauxic shift, as expected, but there was not much difference between the NR and CR conditions other than the earlier activation in CR cultures (Fig. 6A, 14 h). Furthermore, ACH1 expression was only modestly attenuated in the cat8Δ adr1Δ mutant, suggesting the existence of compensatory transcription factors. In stark contrast, activation of ACS1 expression was strongly amplified by CR in a completely Cat8/Adr1-dependent manner (Fig. 6B). ACS1 and other genes with this pattern of expression, such as ADY2 (data not shown), likely work together to extend CLS during CR. For example, the acetate permease gene ADY2 (51) directly feeds acetate into the system for acetyl-CoA production.
FIG 6.

Effects of CR on acetyl-CoA and expression of genes involved in acetyl-CoA production. (A) Quantitative RT-PCR of ACH1 expression relative to control during a time course with WT (BY4741) and cat8Δ adr1Δ (JS767) strains. Strains were inoculated into NR (2% glucose) or CR (0.5% glucose) cultures. (B and C) Quantitative RT-PCR of ACS1 expression (B) and ACS2 expression (C) across the same time course. (D) Quantitation of acetyl-CoA from whole-cell extracts of BY4741 and JS767 derived from a time course of NR and CR cultures from log phase (10 h) to stationary phase (40 h). Asterisks indicate a significant difference between the WT and mutant strains for each time point (P < 0.01).
We also tested mRNA expression of ACS2, which encodes another nucleocytoplasmic acetyl-CoA synthetase that is not a Cat8/Adr1 target gene and has the opposite expression pattern of ACS1 (49). As expected, ACS2 expression decreased during the diauxic shift under the NR condition and was unaffected by deletion of CAT8 and ADR1 (Fig. 6C). Under the CR condition, however, ACS2 expression remained elevated past the shift in both strains. This was surprising and strongly implied that both acetyl-CoA synthetase isoforms were active during and beyond the diauxic shift under CR conditions. So, we next measured acetyl-CoA levels in each strain through a time course spanning log phase to stationary phase (Fig. 6D). As expected from an earlier study (52), acetyl-CoA in the WT strain decreased during the diauxic shift under the NR condition (Fig. 6D). However, CR prevented the post-log-phase reduction and instead increased acetyl-CoA beyond the log-phase concentration (Fig. 6D). CR also prevented acetyl-CoA depletion in the cat8Δ adr1Δ mutant, although the increase in acetyl-CoA over log-phase levels was lost (Fig. 6D). Taken together, these results suggest that both Acs1 and Acs2 contribute to CR-induced maintenance of elevated acetyl-CoA during the diauxic shift and the stationary phase, consistent with the amplified expression of Cat8/Adr1 target genes.
DISCUSSION
Yeast CLS is defined as the number of days that nondividing cells remain viable and is measured by growing liquid cultures into stationary phase and then monitoring cell viability over time. To mimic CR, glucose is reduced in the starting culture at the time of inoculation. Somehow, reduced glucose availability during log phase confers physiological changes that extend through the diauxic shift and into stationary phase. Since Snf1 activity is regulated by glucose concentration and required for the proper transition into stationary phase, we hypothesized that Snf1 was a key factor in mediating CR-induced CLS extension. We initially thought that reducing glucose to 0.5% might be sufficient to activate Snf1 during log phase, but this turned out to be incorrect. Instead, Snf1 was activated earlier in time course experiments because there was less glucose to be consumed in culture, driving cells into the diauxic shift earlier. This is why most of the Snf1-induced activities that we tested in this study occurred earlier under the CR condition. The striking result was that even though CR induced an earlier diauxic shift and Snf1 activation (T210 phosphorylation), the window of time that Snf1 remained active was also extended, suggesting that CR induces an overall expansion of the diauxic shift. This expansion likely optimizes the transcription profile and physiology necessary for long-term cell survival.
ADP and Snf1 activation during CR.
The AMPK complex in multicellular organisms is regulated by adenylate nucleotides at multiple levels, including allosteric activation by AMP, which serves as an indicator for low cellular energy pools (5), and AMP or ADP binding to the γ-subunit, which promotes activation by preventing T172 dephosphorylation (32). The yeast SNF1 complex differs in that AMP does not allosterically activate the enzyme. Instead, SNF1 is activated only by ADP, which binds to the Snf4 subunit to prevent Glc7-mediated dephosphorylation of T210 on the Snf1 subunit, at least in vitro (18, 53). Our results support a model whereby a relatively high cellular ADP concentration helps maximize Snf1 activation late into the diauxic shift and early stationary phase during growth under CR conditions, when ADP is usually being diminished under the NR condition (Fig. 3E; also see Fig. 7). Additional factors could be involved in prolonged SNF1 activation by CR, including further reduction of Glc7/Reg1 phosphatase activity or prolonged kinase activity from Sak1. While we cannot rule out contributions from unknown metabolites potentially stimulating SNF1 activity in vivo, ADP makes the most sense based on what is known. The mechanism of how CR maintains elevated ADP levels late into the diauxic shift remains unknown. It could be directly related to the high ATP levels that are also maintained into stationary phase by CR conditions (33), which we have also observed in our CLS system (Fig. 3F). ATP has no measurable effect on SNF1 activity in vitro, again suggesting that the absolute ADP concentration may be the critical factor, rather than the ADP/ATP ratio (18).
FIG 7.
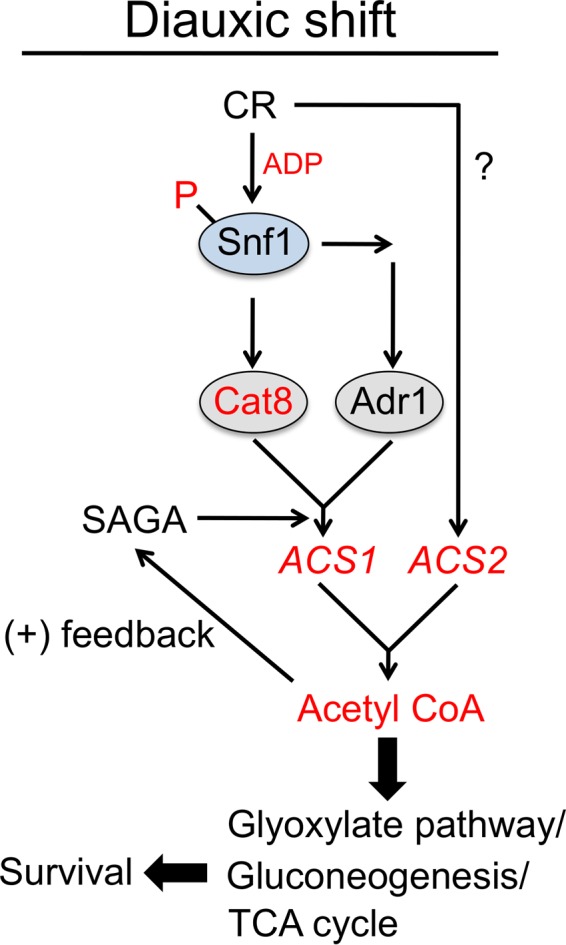
Model for CR-induced optimization of acetyl-CoA production during the diauxic shift to extend yeast CLS. CR maximizes Snf1 T210 phosphorylation in part by maintaining elevated ADP levels. Snf1 activates Cat8 and Adr1 transcription factors, which turn on multiple genes, including the acetyl-CoA synthetase gene ACS1. CR also maintains high ACS2 expression during the diauxic shift but independently of Cat8/Adr1. Elevated nucleocytoplasmic acetyl-CoA promotes histone acetyltransferase activity of the SAGA transcriptional coactivator complex, which amplifies expression of ACS1 and a subset of other Cat8/Adr1 target genes, resulting in a positive-feedback loop that drives sufficient acetyl-CoA production. Carbon skeletons are shunted through the glyoxylate cycle and gluconeogenesis pathway, supporting biomass formation during the early stages of the diauxic shift and storage of carbohydrates for extended cell survival. The full TCA cycle and respiration become more important later. Circles indicate proteins, and italics indicate genes. Red indicates components upregulated or enriched by CR (compared to NR) during or after the diauxic shift.
Elevated ADP and ATP fit very well with the enhancement of cellular respiration that occurs under CR conditions. Furthermore, growth on nonfermentable carbon sources strongly activates Snf1 and extends CLS (19). However, Cat8 and Adr1 are not required for the elevated ADP and ATP under CR conditions (data not shown), yet they are still required for robust CLS extension. This suggests that elevated ADP and Snf1 activation are not sufficient for CLS extension, and numerous downstream factors such as Cat8 and Adr1 that drive changes in transcription and likely other processes are also required.
CR-induced CLS extension is mediated by Cat8.
Longevity experiments in this study point to the transcriptional activator Cat8 as a key factor in the extension of CLS by CR. Phosphorylation of Cat8 by Snf1 occurs when glucose is depleted at the onset of the diauxic shift and correlates with transcription of its target genes (42). Cat8 is a Zn-finger cluster transcription factor that binds to a carbon source-responsive element (CSRE) in the promoter of its target genes and recruits coactivators, such as SAGA, to activate transcription (16). Both the ACH1 and the ASC1 target genes tested in this study contain a CSRE in their promoter, but only ACS1 showed a strong Cat8/Adr1-dependent transcriptional response to CR, indicating that they are differentially regulated despite both having a Cat8 binding site. The CSREs in different promoters have significant variation, with some elements considered stronger activation sequences than others. Consistently, ACS1 has a “strong” CSRE and ACH1 has a comparatively “weak” element (54). Furthermore, although Adr1 associates with the ACH1 promoter as measured by chromatin immunoprecipitation (ChIP), deleting ADR1 was previously shown to cause upregulation of ACH1 transcription (44). Strong CSREs also tend to be in genes that encode factors important early in the diauxic shift (54), such as ACS1 and ADY2. Among Cat8-activated genes are other transcription factors, such as Sip4, that turn on cellular processes more important later in the diauxic shift (54). Maintaining high Cat8 levels could make late diauxic shift processes such as respiration more efficient.
Efficient mitochondrial function has clearly been implicated in CLS extension by CR, such that a certain threshold of respiratory activity during exponential growth must be achieved in order for CR to extend CLS (55). Similarly, TOR inhibition was shown to increase the density of OXPHOS complexes in the mitochondria such that the mitochondria were preconditioned for respiration late in the diauxic shift and stationary phase (56). Extended Snf1 activation observed during CR could contribute to such a mechanism in the context of CR.
CR effects on acetyl-CoA during the diauxic shift.
Saccharomyces cerevisiae is a facultative anaerobe that prefers to generate ATP through glucose fermentation to ethanol even in the presence of oxygen. During growth on glucose-containing medium, most of the pyruvate generated by glycolysis is converted to acetaldehyde and then ethanol via alcohol dehydrogenases. Some of the acetaldehyde is converted to acetate by the cytosolic acetaldehyde dehydrogenase Ald6 (57) and then converted to acetyl-CoA by the nucleocytosolic acetyl-CoA synthetase Acs2 (58). Acetyl-CoA produced by this route is the major substrate for global histone acetylation carried out by histone acetyltransferases such as SAGA/Gcn5 and Sas2 (59). Its major role in maintaining acetyl-CoA also makes ACS2 essential when cells are grown in glucose (58). At the onset of the diauxic shift, however, ACS2 expression is repressed, while the other nucleocytosolic acetyl-CoA synthetase gene ACS1 is activated as a target of Cat8 (Fig. 6) (39, 49), presumably becoming a major source for acetyl-CoA production via acetate in the cytoplasm and nucleus.
Our results implicate increased ACS1 expression in CR-induced elevated acetyl-CoA levels, though we cannot rule out other sources. Ach1 may function to transfer acetyl groups from the mitochondria to the cytosol (60), forming cytosolic acetyl-CoA in the process to detoxify excess acetate and acetic acid (47), although ACH1 expression was not significantly enhanced by CR (Fig. 6A). Additionally, Snf1 activation inhibits acetyl-CoA carboxylase (ACC) (61), preventing the conversion of acetyl-CoA to malonyl-CoA, the first step in fatty acid biosynthesis. A more-effective inhibition of ACC via phosphorylation by Snf1 could therefore result in the elevation of acetyl-CoA levels (62), making the observed maintenance of acetyl-CoA under CR (Fig. 6D) more than a solely transcription-linked mechanism. However, loss of CAT8 and ADR1 prevented acetyl-CoA from accumulating above the log-phase level in CR cells (Fig. 6D), so the CAT8 ADR1-dependent hyperactivation of ACS1 expression could be responsible for this portion of the acetyl-CoA pool.
The correlation between activated Snf1 and acetyl-CoA maintenance in response to CR is consistent with previous results showing that Snf1 regulates acetyl-CoA homeostasis (62). Inappropriate nucleocytosolic acetyl-CoA production by Acs2 represses autophagy and shortens CLS under NR conditions (63), so sustained ACS2 expression and high acetyl-CoA induced by CR likely reflects optimized acetyl-CoA utilization. During the diauxic shift, elevated acetyl-CoA would contribute to CLS extension by driving transcription of multiple Cat8 target genes involved in the glyoxylate and gluconeogenesis pathways (Fig. 7), though there are certainly other possible targets and important transcription factors that could contribute. Part of the optimized acetyl-CoA utilization may therefore involve facilitating the activation of genes that produce more acetyl-CoA through targeted histone acetylation. Acetyl-CoA is the acetyl donor for histone acetylation by transcriptional coactivators such as the Gcn5-containing SAGA complex (64), a known coactivator for Cat8 and Adr1 (65, 66). Furthermore, the increase in acetyl-CoA and parallel activation of the glyoxylate and gluconeogenesis pathways will also provide carbon skeletons for biomass during diauxic growth and accumulation of trehalose, a key storage carbohydrate that functions in long-term yeast cell survival (46, 67). Once cells pass the diauxic shift and enter stationary phase, the elevated acetyl-CoA likely pushes flux toward the tricarboxylic acid (TCA) cycle and oxidative phosphorylation, as indicated by the maintenance of ATP pools into stationary phase (Fig. 3F) (33). Enhanced SNF1-mediated respiration by CR is also consistent with the activation of the Snf1 pathway that occurs in mutants that increase replicative life span through enhanced turnover of Mig1 (68), suggesting that SNF1 activation is a common feature of both CLS and RLS extension. Consistent with this result, deletion of SNF1 or the SIP2 β-subunit shortens RLS (69, 70). Interestingly, Snf1 can also negatively impact RLS when Sip2 is acetylated, although this works independently of caloric restriction (71). More work is clearly needed to dissect the SNF1-signaling pathway in more detail, which could have important implications toward AMPK in higher eukaryotes.
Our results in yeast imply that in multicellular eukaryotes, CR not only promotes AMPK activation but also temporally optimizes the signaling cascade from top to bottom. Metazoans do not ferment pyruvate into ethanol but rather reduce the pyruvate to lactate. The common process between them is therefore the ultimate use of the carbon skeletons from either ethanol or lactate for gluconeogenesis. Indeed, CR increases the activity of gluconeogenic enzymes in mouse liver and increases the acetyl-CoA level in old mice (72), consistent with our results in yeast.
MATERIALS AND METHODS
Yeast strains and media.
The wild-type (WT) S. cerevisiae lab strain used in this study was BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) (73). Strains containing additional gene deletions were either obtained from the yeast knockout collection or generated in the BY4741 background via PCR (74). Synthetic complete (SC) growth medium was used for all experiments (75), with glucose added to a final concentration of 2.0% (nonrestricted [NR]) or 0.5% (calorie restricted [CR]). To restrict methionine, SC medium contained 2% glucose and 5 mg/liter methionine instead of the normal 125-mg/liter concentration. Where indicated, isonicotinamide (INAM) was added to a final concentration of 25 mM. All cultures were grown at 30°C. The HA-tagged Snf1 strain (MSY1238) was kindly provided by Martin Schmidt (53). The snf1-as strain (PY856) was kindly provided by Karen Arndt and the 2NM-PP1 inhibitor by Kevan Shokat (28). All strains and genotypes are listed in Table 1.
TABLE 1.
Yeast strains
| Strain | Relevant genotype |
|---|---|
| BY4741a | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 |
| JS1394b | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 snf1Δ::kanMX |
| SY848c | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 sak1Δ::kanMX |
| SY844c | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 elm1Δ::kanMX |
| SY841c | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 tos3Δ::kanMX |
| SY825c | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 cat8Δ::kanMX |
| SY826c | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 adr1Δ::kanMX |
| SY767b | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 cat8Δ::kanMX adr1Δ::kanMX |
| MSY1238d | MATa his3Δ1 leu2Δ1 ura3-52 SNF1-3HA |
| JS1517 | MATa his3Δ1 leu2Δ1 ura3-52 SNF1-3HA elm1ΔkanMX4 |
| JS1519 | MATa his3Δ1 leu2Δ1 ura3-52 SNF1-3HA sak1ΔkanMX4 |
| JS1521 | MATa his3Δ1 leu2Δ1 ura3-52 SNF1-3HA tos3ΔkanMX4 |
| PY855e | MATa his3Δ200 ura3-52 snf1Δ10 [pSNF1-316 = CEN/ARS URA3 HA-SNF1] |
| PY856e | MATa his3Δ200 ura3-52 snf1Δ10 [pSNF1-I132G-316 = CEN/ARS URA3 HA-snf1-as] |
Chronological life span assays.
Semiquantitative CLS (spot) assays were performed as previously described (19). For the quantitative version of this assay, 2.5 μl of 1:10, 1:100, and 1:1,000 dilutions of the cultures was spotted onto yeast extract-peptone-dextrose (YPD) plates, which were then incubated at 30°C for 18 to 24 h to allow for microcolony formation. Images of the dilution spots were captured on a Nikon Eclipse E400 tetrad dissection microscope at a magnification of ×30, and the microcolonies were counted. At the end of each experiment, percent viability was calculated for each time point by normalizing to the day 2 totals. Standard deviation error bars were determined from at least 3 biological replicates. Mean life spans (days), standard errors of the means (SEM), and 95th percentile confidence intervals of the means (95% CI) were calculated using OASIS, an online program for Kaplan-Meier survival curve analysis (76).
Cultures for protein, nucleotide, mRNA, and acetyl-CoA quantification.
Overnight 10-ml cultures were started from single colonies and grown at 30°C in 15-ml glass culture tubes with loose-fitting metal caps on a New Brunswick Scientific roller drum. A spectrophotometer was used to determine the optical density at 600 nm (OD600) of each overnight culture, and appropriate aliquots were inoculated into 75 ml SC medium (either NR or CR) to achieve a starting OD600 of 0.015 in 250-ml Erlenmeyer flasks. This was considered the starting point for each time course. Cultures were grown at 30°C in a New Brunswick water bath shaker.
Western blot analysis.
At indicated time points, medium containing 5 OD units (∼1.4 × 108 cells) was harvested from the 75-ml cultures and transferred to 5-ml glass tubes, which were then submerged in a boiling water bath and rotated for 15 s to allow for more-even heat dispersal. After boiling for 3 min, which preserves the Snf1 phosphorylation state (77), samples were cooled at room temperature for 5 min, followed by transfer to 15-ml conical tubes and pelleting at 1,800 × g in an Eppendorf 5810R centrifuge, at 4°C for 5 min. Supernatants were discarded, and cell pellets were resuspended in 1 ml of ice-cold 20% trichloroacetic acid and transferred to a 1.5-ml microcentrifuge tube. Samples were then pelleted at 14,000 rpm in a microcentrifuge at 4°C for 1 min, and the supernatant was discarded. Whole-cell extracts were prepped by a combination of vortexing with glass beads and protein precipitation in 20% trichloroacetic acid.
Ten microliters of whole-cell extract was separated on 8% SDS-polyacrylamide gels in a Bio-Rad Mini-Protean apparatus at 130 V. A Bio-Rad Trans-Blot semidry apparatus was used at 25 V for 1 h to transfer protein from the gels to Millipore Immobilon-P membranes. Membranes were blocked for 1 h at room temperature with either 5% bovine serum albumin (BSA; for the P-AMPKα T172 antibody; Cell Signaling Technology) or 5% nonfat milk (for the anti-HA antibody; Promega) in 1× TBST (1× Tris-buffered saline–0.05% Tween 20) and then washed at room temperature with 1× TBST (5 times for 5 min each). Membranes were then incubated with diluted (1:2,000) primary antibody in either the BSA or nonfat milk blocking buffer overnight at 4°C. Membranes were washed again, and horseradish peroxidase (HRP)-conjugated secondary antibody (1:5,000) in the appropriate blocking buffer was added. The secondary incubations were performed at room temperature for 1 h, and the membranes were washed again. Proteins were detected using the enhanced chemiluminescence (ECL) method. ImageJ software was used to quantify the protein signal intensities from scanned images of the Western blot X-ray films. Snf1-P relative to Snf1-HA was calculated from three independent biological replicates. P values were calculated using the Student t test.
qRT-PCR analysis.
Total RNA was harvested from triplicate 50-ml cultures at the indicated time points using the hot acidic phenol method (78). cDNA was synthesized from 1 μg total RNA using a High Capacity cDNA reverse transcriptase kit (Applied Biosystems) in accordance with the manufacturer's instructions and then quantitated by PCRs using SYBR Hi-ROX Mastermix (Bioline) and a StepOnePlus real-time PCR system (Applied Biosystems). Transcript levels were normalized to levels of TFC1 mRNA (79). All DNA primers used for RT-PCR are listed in Table 2.
TABLE 2.
Oligonucleotides
| Oligonucleotide | Use | Sequence |
|---|---|---|
| JS2022 | TFC1 RT-PCR | AATGTACCAAAGCCACCACCC |
| JS2023 | TFC1 RT-PCR | ACCGCCTGGAGTGTCTGATT |
| JS2399 | ACS1 RT-PCR | TGATGACGCGCTAAGAGAGA |
| JS2400 | ACS1 RT-PCR | AACGGGTGTGCATGGATAGT |
| JS2409 | ACH1 RT-PCR | GTTAACACGGCTACGCCTTC |
| JS2410 | ACH1 RT-PCR | ATCGCAACAACCTTTTCAGG |
| JS2415 | ACS2 RT-PCR | CTTGGGTACCGCCTCAATAA |
| JS2416 | ACS2 RT-PCR | ATTTCGGCTTCACCTACACG |
| JS2511 | CAT8 HA tag | GATAATGTATCTGATTTATTCCAATGGCAAAACGCCAAACGGATCCCCGGGTTAATTAA |
| JS2512 | CAT8 HA tagging | GGATTCCGTTTTGAATATATTACACTATGAAATAAAGAAGAATTCGAGCTCGTTTAAAC |
| JS2525 | ADY2 RT-PCR | GGTTAGCTCCTGCTCCAGTG |
| JS2526 | ADY2 RT-PCR | ACATAGCACAACCGACGACA |
| JS2630 | CAT8 RT-PCR | GCATGGAATGGACAATGTG |
| JS2631 | CAT8 RT-PCR | GACCGATCCAAGTCATCGTT |
Quantification of intracellular adenosine nucleotides.
To extract nucleotides from yeast cells, an amount equal to 2.5 OD600 units of cells (∼7 × 107 cells) was transferred at the indicated time points from 50-ml cultures into 1.5-ml microcentrifuge tubes and pelleted at 1,500 rpm and 4°C. Cells were washed in 1 ml ice-cold Tris-EDTA (TE; 10 mM Tris, 1 mM EDTA, pH 8.0) and suspended in 250 μl LETS buffer (10 mM Tris [pH 8], 100 mM LiCl, 10 mM EDTA, 0.5% SDS). A 250-μl volume of 1:1 phenol-chloroform was added, and the samples were vortexed. The samples were then pelleted at 14,000 rpm in a microcentrifuge at 4°C, and the aqueous supernatants were transferred to a fresh microcentrifuge tube and diluted 1:10 with ice-cold TE buffer. To determine the ATP content of each sample, 4 μl of ATP buffer (30 mM KPO4 [pH 7.3], 40 mM MgSO4, 8% Tween 20) was added to 8 μl of sample. After incubation at room temperature for 10 min, 4 μl of AXP buffer (30 mM KPO4 [pH 7.3], 40 mM MgSO4) was added, and the samples were incubated for an additional 30 min at 30°C. A 10-μl volume of each sample was then used to determine ATP content using the ATP Determination kit (Invitrogen) in accordance with the manufacturer's instruction.
To quantify intracellular ADP, all ADP in the samples was first converted to ATP during the 30°C incubation by the addition of 4 μl ADP buffer (30 mM KPO4 [pH 7.3], 40 mM MgSO4, 0.4 mM phosphoenolpyruvate, 0.0224 mg/ml pyruvate kinase) in place of AXP buffer. The ATP Determination kit (Invitrogen) was then used to determine the total concentration of ATP (both initial ATP and converted ADP) in each sample. Final intracellular ADP concentrations were then calculated by deducting each sample's previously determined ATP content.
Quantification of intracellular acetyl-CoA.
To quantify intracellular acetyl-CoA, 10 OD600 units of cells was transferred from the 75-ml cultures described above into 15-ml conical tubes at various time points. Sodium azide was added to each sample to a final concentration of 10 mM. The cells were then spun down for 5 min at 1,800 × g at 4°C in a tabletop centrifuge. The supernatant was removed, and the pellet was resuspended in 250 μl of 10% perchloric acid. The cells were then lysed with the addition of glass beads and vortexing. The lysed samples were then spun down at 800 × g for 5 min at 4°C, and 150 μl of the supernatants was transferred to new 1.5-ml microcentrifuge tubes. The lysate was neutralized to pH 6 to 8 with 50 μl of 3 M KHCO3, and the resulting precipitate was spun down for 3 min at 14,000 rpm in the microcentrifuge. A 125-μl volume of supernatant was then transferred to a new microcentrifuge tube and kept on ice. The concentration of acetyl-CoA in each sample was immediately measured using a Sigma acetyl-CoA assay kit in accordance with the manufacturer instructions.
ACKNOWLEDGMENTS
We thank Martin Schmidt and Rhonda McCartney for strains and plasmids, as well as extensive advice with Snf1 protocols. We also thank Flora Rutaganira and Kevan Shokat for 2NM-PP1 and Margaret Shirra and Karen Arndt for the snf1-as strain. We thank Ben Tu and Zheng Kuang for advice on acetyl-CoA measurements, David Auble and Martin Schmidt for critically reading the manuscript, and Smith lab members for helpful discussions.
This work was supported in part by funding from the University of Virginia School of Medicine and the Department of Biochemistry and Molecular Genetics, as well as NIH grant GM075240.
REFERENCES
- 1.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. 2013. The hallmarks of aging. Cell 153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taormina G, Mirisola MG. 2014. Calorie restriction in mammals and simple model organisms. Biomed Res Int 2014:308690. doi: 10.1155/2014/308690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruetenik A, Barrientos A. 2015. Dietary restriction, mitochondrial function and aging: from yeast to humans. Biochim Biophys Acta 1847:1434–1447. doi: 10.1016/j.bbabio.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finkel T. 2015. The metabolic regulation of aging. Nat Med 21:1416–1423. doi: 10.1038/nm.3998. [DOI] [PubMed] [Google Scholar]
- 5.Hardie DG, Schaffer BE, Brunet A. 2016. AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol 26:190–201. doi: 10.1016/j.tcb.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP, Brunet A. 2007. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr Biol 17:1646–1656. doi: 10.1016/j.cub.2007.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. 2007. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab 6:280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 8.Funakoshi M, Tsuda M, Muramatsu K, Hatsuda H, Morishita S, Aigaki T. 2011. A gain-of-function screen identifies wdb and lkb1 as lifespan-extending genes in Drosophila. Biochem Biophys Res Commun 405:667–672. doi: 10.1016/j.bbrc.2011.01.090. [DOI] [PubMed] [Google Scholar]
- 9.Slack C, Foley A, Partridge L. 2012. Activation of AMPK by the putative dietary restriction mimetic metformin is insufficient to extend lifespan in Drosophila. PLoS One 7:e47699. doi: 10.1371/journal.pone.0047699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Canto C, Auwerx J. 2011. Calorie restriction: is AMPK a key sensor and effector? Physiology 26:214–224. doi: 10.1152/physiol.00010.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. 2008. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inoki K, Zhu T, Guan KL. 2003. TSC2 mediates cellular energy response to control cell growth and survival. Cell 115:577–590. doi: 10.1016/S0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 13.Nakashima K, Yakabe Y. 2007. AMPK activation stimulates myofibrillar protein degradation and expression of atrophy-related ubiquitin ligases by increasing FOXO transcription factors in C2C12 myotubes. Biosci Biotechnol Biochem 71:1650–1656. doi: 10.1271/bbb.70057. [DOI] [PubMed] [Google Scholar]
- 14.Woods A, Cheung PC, Smith FC, Davison MD, Scott J, Beri RK, Carling D. 1996. Characterization of AMP-activated protein kinase beta and gamma subunits. Assembly of the heterotrimeric complex in vitro. J Biol Chem 271:10282–10290. [DOI] [PubMed] [Google Scholar]
- 15.Burkewitz K, Zhang Y, Mair WB. 2014. AMPK at the nexus of energetics and aging. Cell Metab 20:10–25. doi: 10.1016/j.cmet.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conrad M, Schothorst J, Kankipati HN, Van Zeebroeck G, Rubio-Texeira M, Thevelein JM. 2014. Nutrient sensing and signaling in the yeast Saccharomyces cerevisiae. FEMS Microbiol Rev 38:254–299. doi: 10.1111/1574-6976.12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fabrizio P, Longo VD. 2003. The chronological life span of Saccharomyces cerevisiae. Aging Cell 2:73–81. doi: 10.1046/j.1474-9728.2003.00033.x. [DOI] [PubMed] [Google Scholar]
- 18.Mayer FV, Heath R, Underwood E, Sanders MJ, Carmena D, McCartney RR, Leiper FC, Xiao B, Jing C, Walker PA, Haire LF, Ogrodowicz R, Martin SR, Schmidt MC, Gamblin SJ, Carling D. 2011. ADP regulates SNF1, the Saccharomyces cerevisiae homolog of AMP-activated protein kinase. Cell Metab 14:707–714. doi: 10.1016/j.cmet.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith DL Jr, McClure JM, Matecic M, Smith JS. 2007. Calorie restriction extends the chronological lifespan of Saccharomyces cerevisiae independently of the Sirtuins. Aging Cell 6:649–662. doi: 10.1111/j.1474-9726.2007.00326.x. [DOI] [PubMed] [Google Scholar]
- 20.Gray JV, Petsko GA, Johnston GC, Ringe D, Singer RA, Werner-Washburne M. 2004. “Sleeping beauty”: quiescence in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 68:187–206. doi: 10.1128/MMBR.68.2.187-206.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amodeo GA, Rudolph MJ, Tong L. 2007. Crystal structure of the heterotrimer core of Saccharomyces cerevisiae AMPK homologue SNF1. Nature 449:492–495. doi: 10.1038/nature06127. [DOI] [PubMed] [Google Scholar]
- 22.Thompson-Jaeger S, Francois J, Gaughran JP, Tatchell K. 1991. Deletion of SNF1 affects the nutrient response of yeast and resembles mutations which activate the adenylate cyclase pathway. Genetics 129:697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weinberger M, Mesquita A, Caroll T, Marks L, Yang H, Zhang Z, Ludovico P, Burhans WC. 2010. Growth signaling promotes chronological aging in budding yeast by inducing superoxide anions that inhibit quiescence. Aging 2:709–726. doi: 10.18632/aging.100215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wierman MB, Matecic M, Valsakumar V, Li M, Smith DL Jr, Bekiranov S, Smith JS. 2015. Functional genomic analysis reveals overlapping and distinct features of chronologically long-lived yeast populations. Aging 7:177–194. doi: 10.18632/aging.100729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Celenza JL, Carlson M. 1984. Cloning and genetic mapping of SNF1, a gene required for expression of glucose-repressible genes in Saccharomyces cerevisiae. Mol Cell Biol 4:49–53. doi: 10.1128/MCB.4.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fabrizio P, Battistella L, Vardavas R, Gattazzo C, Liou LL, Diaspro A, Dossen JW, Gralla EB, Longo VD. 2004. Superoxide is a mediator of an altruistic aging program in Saccharomyces cerevisiae. J Cell Biol 166:1055–1067. doi: 10.1083/jcb.200404002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McIsaac RS, Lewis KN, Gibney PA, Buffenstein R. 2016. From yeast to human: exploring the comparative biology of methionine restriction in extending eukaryotic life span. Ann N Y Acad Sci 1363:155–170. doi: 10.1111/nyas.13032. [DOI] [PubMed] [Google Scholar]
- 28.Shirra MK, McCartney RR, Zhang C, Shokat KM, Schmidt MC, Arndt KM. 2008. A chemical genomics study identifies Snf1 as a repressor of GCN4 translation. J Biol Chem 283:35889–35898. doi: 10.1074/jbc.M805325200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCartney RR, Schmidt MC. 2001. Regulation of Snf1 kinase. Activation requires phosphorylation of threonine 210 by an upstream kinase as well as a distinct step mediated by the Snf4 subunit. J Biol Chem 276:36460–36466. [DOI] [PubMed] [Google Scholar]
- 30.Sutherland CM, Hawley SA, McCartney RR, Leech A, Stark MJ, Schmidt MC, Hardie DG. 2003. Elm1p is one of three upstream kinases for the Saccharomyces cerevisiae SNF1 complex. Curr Biol 13:1299–1305. doi: 10.1016/S0960-9822(03)00459-7. [DOI] [PubMed] [Google Scholar]
- 31.Auciello FR, Ross FA, Ikematsu N, Hardie DG. 2014. Oxidative stress activates AMPK in cultured cells primarily by increasing cellular AMP and/or ADP. FEBS Lett 588:3361–3366. doi: 10.1016/j.febslet.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, Saiu P, Howell SA, Aasland R, Martin SR, Carling D, Gamblin SJ. 2011. Structure of mammalian AMPK and its regulation by ADP. Nature 472:230–233. doi: 10.1038/nature09932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi JS, Lee CK. 2013. Maintenance of cellular ATP level by caloric restriction correlates chronological survival of budding yeast. Biochem Biophys Res Commun 439:126–131. doi: 10.1016/j.bbrc.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 34.Hong SP, Leiper FC, Woods A, Carling D, Carlson M. 2003. Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc Natl Acad Sci U S A 100:8839–8843. doi: 10.1073/pnas.1533136100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nath N, McCartney RR, Schmidt MC. 2003. Yeast Pak1 kinase associates with and activates Snf1. Mol Cell Biol 23:3909–3917. doi: 10.1128/MCB.23.11.3909-3917.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hedbacker K, Hong SP, Carlson M. 2004. Pak1 protein kinase regulates activation and nuclear localization of Snf1-Gal83 protein kinase. Mol Cell Biol 24:8255–8263. doi: 10.1128/MCB.24.18.8255-8263.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCartney RR, Rubenstein EM, Schmidt MC. 2005. Snf1 kinase complexes with different beta subunits display stress-dependent preferences for the three Snf1-activating kinases. Curr Genet 47:335–344. doi: 10.1007/s00294-005-0576-2. [DOI] [PubMed] [Google Scholar]
- 38.Vincent O, Townley R, Kuchin S, Carlson M. 2001. Subcellular localization of the Snf1 kinase is regulated by specific beta subunits and a novel glucose signaling mechanism. Genes Dev 15:1104–1114. doi: 10.1101/gad.879301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haurie V, Perrot M, Mini T, Jeno P, Sagliocco F, Boucherie H. 2001. The transcriptional activator Cat8p provides a major contribution to the reprogramming of carbon metabolism during the diauxic shift in Saccharomyces cerevisiae. J Biol Chem 276:76–85. doi: 10.1074/jbc.M008752200. [DOI] [PubMed] [Google Scholar]
- 40.Hedges D, Proft M, Entian KD. 1995. CAT8, a new zinc cluster-encoding gene necessary for derepression of gluconeogenic enzymes in the yeast Saccharomyces cerevisiae. Mol Cell Biol 15:1915–1922. doi: 10.1128/MCB.15.4.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Charbon G, Breunig KD, Wattiez R, Vandenhaute J, Noel-Georis I. 2004. Key role of Ser562/661 in Snf1-dependent regulation of Cat8p in Saccharomyces cerevisiae and Kluyveromyces lactis. Mol Cell Biol 24:4083–4091. doi: 10.1128/MCB.24.10.4083-4091.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Randez-Gil F, Bojunga N, Proft M, Entian KD. 1997. Glucose derepression of gluconeogenic enzymes in Saccharomyces cerevisiae correlates with phosphorylation of the gene activator Cat8p. Mol Cell Biol 17:2502–2510. doi: 10.1128/MCB.17.5.2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rahner A, Scholer A, Martens E, Gollwitzer B, Schuller HJ. 1996. Dual influence of the yeast Cat1p (Snf1p) protein kinase on carbon source-dependent transcriptional activation of gluconeogenic genes by the regulatory gene CAT8. Nucleic Acids Res 24:2331–2337. doi: 10.1093/nar/24.12.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tachibana C, Yoo JY, Tagne JB, Kacherovsky N, Lee TI, Young ET. 2005. Combined global localization analysis and transcriptome data identify genes that are directly coregulated by Adr1 and Cat8. Mol Cell Biol 25:2138–2146. doi: 10.1128/MCB.25.6.2138-2146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Young ET, Dombek KM, Tachibana C, Ideker T. 2003. Multiple pathways are co-regulated by the protein kinase Snf1 and the transcription factors Adr1 and Cat8. J Biol Chem 278:26146–26158. doi: 10.1074/jbc.M301981200. [DOI] [PubMed] [Google Scholar]
- 46.Hu J, Wei M, Mirzaei H, Madia F, Mirisola M, Amparo C, Chagoury S, Kennedy B, Longo VD. 2014. Tor-Sch9 deficiency activates catabolism of the ketone body-like acetic acid to promote trehalose accumulation and longevity. Aging Cell 13:457–467. doi: 10.1111/acel.12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fleck CB, Brock M. 2009. Re-characterisation of Saccharomyces cerevisiae Ach1p: fungal CoA-transferases are involved in acetic acid detoxification. Fungal Genet Biol 46:473–485. doi: 10.1016/j.fgb.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 48.Lee FJ, Lin LW, Smith JA. 1996. Acetyl-CoA hydrolase involved in acetate utilization in Saccharomyces cerevisiae. Biochim Biophys Acta 1297:105–109. doi: 10.1016/0167-4838(96)00109-4. [DOI] [PubMed] [Google Scholar]
- 49.de Jong-Gubbels P, van den Berg MA, Steensma HY, van Dijken JP, Pronk JT. 1997. The Saccharomyces cerevisiae acetyl-coenzyme A synthetase encoded by the ACS1 gene, but not the ACS2-encoded enzyme, is subject to glucose catabolite inactivation. FEMS Microbiol Lett 153:75–81. doi: 10.1111/j.1574-6968.1997.tb10466.x. [DOI] [PubMed] [Google Scholar]
- 50.Lee FJ, Lin LW, Smith JA. 1990. A glucose-repressible gene encodes acetyl-CoA hydrolase from Saccharomyces cerevisiae. J Biol Chem 265:7413–7418. [PubMed] [Google Scholar]
- 51.Paiva S, Devaux F, Barbosa S, Jacq C, Casal M. 2004. Ady2p is essential for the acetate permease activity in the yeast Saccharomyces cerevisiae. Yeast 21:201–210. doi: 10.1002/yea.1056. [DOI] [PubMed] [Google Scholar]
- 52.Cai L, Sutter BM, Li B, Tu BP. 2011. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell 42:426–437. doi: 10.1016/j.molcel.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rubenstein EM, McCartney RR, Zhang C, Shokat KM, Shirra MK, Arndt KM, Schmidt MC. 2008. Access denied: Snf1 activation loop phosphorylation is controlled by availability of the phosphorylated threonine 210 to the PP1 phosphatase. J Biol Chem 283:222–230. doi: 10.1074/jbc.M707957200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roth S, Kumme J, Schuller HJ. 2004. Transcriptional activators Cat8 and Sip4 discriminate between sequence variants of the carbon source-responsive promoter element in the yeast Saccharomyces cerevisiae. Curr Genet 45:121–128. doi: 10.1007/s00294-003-0476-2. [DOI] [PubMed] [Google Scholar]
- 55.Ocampo A, Liu J, Schroeder EA, Shadel GS, Barrientos A. 2012. Mitochondrial respiratory thresholds regulate yeast chronological life span and its extension by caloric restriction. Cell Metab 16:55–67. doi: 10.1016/j.cmet.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pan Y, Shadel GS. 2009. Extension of chronological life span by reduced TOR signaling requires down-regulation of Sch9p and involves increased mitochondrial OXPHOS complex density. Aging 1:131–145. doi: 10.18632/aging.100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meaden PG, Dickinson FM, Mifsud A, Tessier W, Westwater J, Bussey H, Midgley M. 1997. The ALD6 gene of Saccharomyces cerevisiae encodes a cytosolic, Mg2+-activated acetaldehyde dehydrogenase. Yeast 13:1319–1327. doi:. [DOI] [PubMed] [Google Scholar]
- 58.Van den Berg MA, Steensma HY. 1995. ACS2, a Saccharomyces cerevisiae gene encoding acetyl-coenzyme A synthetase, essential for growth on glucose. Eur J Biochem 231:704–713. doi: 10.1111/j.1432-1033.1995.tb20751.x. [DOI] [PubMed] [Google Scholar]
- 59.Takahashi H, McCaffery JM, Irizarry RA, Boeke JD. 2006. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol Cell 23:207–217. doi: 10.1016/j.molcel.2006.05.040. [DOI] [PubMed] [Google Scholar]
- 60.Chen Y, Zhang Y, Siewers V, Nielsen J. 2015. Ach1 is involved in shuttling mitochondrial acetyl units for cytosolic C2 provision in Saccharomyces cerevisiae lacking pyruvate decarboxylase. FEMS Yeast Res 15(3):pii:fov015. doi: 10.1093/femsyr/fov015. [DOI] [PubMed] [Google Scholar]
- 61.Woods A, Munday MR, Scott J, Yang X, Carlson M, Carling D. 1994. Yeast SNF1 is functionally related to mammalian AMP-activated protein kinase and regulates acetyl-CoA carboxylase in vivo. J Biol Chem 269:19509–19515. [PubMed] [Google Scholar]
- 62.Zhang M, Galdieri L, Vancura A. 2013. The yeast AMPK homolog SNF1 regulates acetyl coenzyme A homeostasis and histone acetylation. Mol Cell Biol 33:4701–4717. doi: 10.1128/MCB.00198-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marino G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, Schroeder S, Pendl T, Harger A, Niso-Santano M, Zamzami N, Scoazec M, Durand S, Enot DP, Fernandez AF, Martins I, Kepp O, Senovilla L, Bauvy C, Morselli E, Vacchelli E, Bennetzen M, Magnes C, Sinner F, Pieber T, Lopez-Otin C, Maiuri MC, Codogno P, Andersen JS, Hill JA, Madeo F, Kroemer G. 2014. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell 53:710–725. doi: 10.1016/j.molcel.2014.01.016. [DOI] [PubMed] [Google Scholar]
- 64.Grant PA, Duggan L, Cote J, Roberts SM, Brownell JE, Candau R, Ohba R, Owen-Hughes T, Allis CD, Winston F, Berger SL, Workman JL. 1997. Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes Dev 11:1640–1650. doi: 10.1101/gad.11.13.1640. [DOI] [PubMed] [Google Scholar]
- 65.Abate G, Bastonini E, Braun KA, Verdone L, Young ET, Caserta M. 2012. Snf1/AMPK regulates Gcn5 occupancy, H3 acetylation and chromatin remodelling at S cerevisiae ADY2 promoter. Biochim Biophys Acta 1819:419–427. doi: 10.1016/j.bbagrm.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Biddick RK, Law GL, Chin KK, Young ET. 2008. The transcriptional coactivators SAGA, SWI/SNF, and mediator make distinct contributions to activation of glucose-repressed genes. J Biol Chem 283:33101–33109. doi: 10.1074/jbc.M805258200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kyryakov P, Beach A, Richard VR, Burstein MT, Leonov A, Levy S, Titorenko VI. 2012. Caloric restriction extends yeast chronological lifespan by altering a pattern of age-related changes in trehalose concentration. Front Physiol 3:256. doi: 10.3389/fphys.2012.00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yao Y, Tsuchiyama S, Yang C, Bulteau AL, He C, Robison B, Tsuchiya M, Miller D, Briones V, Tar K, Potrero A, Friguet B, Kennedy BK, Schmidt M. 2015. Proteasomes, Sir2, and Hxk2 form an interconnected aging network that impinges on the AMPK/Snf1-regulated transcriptional repressor Mig1. PLoS Genet 11:e1004968. doi: 10.1371/journal.pgen.1004968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiao R, Postnikoff S, Harkness TA, Arnason TG. 2015. The SNF1 kinase ubiquitin-associated domain restrains its activation, activity, and the yeast life span. J Biol Chem 290:15393–15404. doi: 10.1074/jbc.M115.647032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lin SS, Manchester JK, Gordon JI. 2003. Sip2, an N-myristoylated beta subunit of Snf1 kinase, regulates aging in Saccharomyces cerevisiae by affecting cellular histone kinase activity, recombination at rDNA loci, and silencing. J Biol Chem 278:13390–13397. doi: 10.1074/jbc.M212818200. [DOI] [PubMed] [Google Scholar]
- 71.Lu JY, Lin YY, Sheu JC, Wu JT, Lee FJ, Chen Y, Lin MI, Chiang FT, Tai TY, Berger SL, Zhao Y, Tsai KS, Zhu H, Chuang LM, Boeke JD. 2011. Acetylation of yeast AMPK controls intrinsic aging independently of caloric restriction. Cell 146:969–979. doi: 10.1016/j.cell.2011.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hagopian K, Ramsey JJ, Weindruch R. 2003. Caloric restriction increases gluconeogenic and transaminase enzyme activities in mouse liver. Exp Gerontol 38:267–278. doi: 10.1016/S0531-5565(02)00202-4. [DOI] [PubMed] [Google Scholar]
- 73.Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14:115–132. doi:. [DOI] [PubMed] [Google Scholar]
- 74.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M'Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Veronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, Johnston M, Davis RW. 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 75.Matecic M, Smith DL, Pan X, Maqani N, Bekiranov S, Boeke JD, Smith JS. 2010. A microarray-based genetic screen for yeast chronological aging factors. PLoS Genet 6:e1000921. doi: 10.1371/journal.pgen.1000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Han SK, Lee D, Lee H, Kim D, Son HG, Yang JS, Lee SV, Kim S. 2016. OASIS 2: online application for survival analysis 2 with features for the analysis of maximal lifespan and healthspan in aging research. Oncotarget 7:56147–56152. doi: 10.18632/oncotarget.11269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Orlova M, Barrett L, Kuchin S. 2008. Detection of endogenous Snf1 and its activation state: application to Saccharomyces and Candida species. Yeast 25:745–754. doi: 10.1002/yea.1628. [DOI] [PubMed] [Google Scholar]
- 78.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (ed). 2000. Current protocols in molecular biology. John Wiley & Sons, Inc., New York, NY. [Google Scholar]
- 79.Teste MA, Duquenne M, Francois JM, Parrou JL. 2009. Validation of reference genes for quantitative expression analysis by real-time RT-PCR in Saccharomyces cerevisiae. BMC Mol Biol 10:99. doi: 10.1186/1471-2199-10-99. [DOI] [PMC free article] [PubMed] [Google Scholar]