

ABSTRACT
The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor that is known as a mediator of toxic responses. Recently, it was shown that the AhR has dual functions. Besides being a transcription factor, it also possesses an intrinsic E3 ubiquitin ligase function that targets, e.g., the steroid receptors for proteasomal degradation. The aim of this study was to identify the molecular switch that determines whether the AhR acts as a transcription factor or an E3 ubiquitin ligase. To do this, we used the breast cancer cell line MCF7, which expresses a functional estrogen receptor alpha (ERα) signaling pathway. Our data suggest that aryl hydrocarbon receptor nuclear translocator (ARNT) plays an important role in the modulation of the dual functions of the AhR. ARNT knockdown dramatically impaired the transcriptional activation properties of the ligand-activated AhR but did not affect its E3 ubiquitin ligase function. The availability of ARNT itself is modulated by another basic helix-loop-helix (bHLH)–Per-ARNT-SIM (PAS) protein, the repressor of AhR function (AhRR). MCF7 cells overexpressing the AhRR showed lower ERα protein levels, reduced responsiveness to estradiol, and reduced growth rates. Importantly, when these cells were used to produce estrogen-dependent xenograft tumors in SCID mice, we also observed lower ERα protein levels and a reduced tumor mass, implying a tumor-suppressive-like function of the AhR in MCF7 xenograft tumors.
KEYWORDS: aryl hydrocarbon receptor, molecular switch, E3 ubiquitin ligase, transcription factor, aryl hydrocarbon receptor nuclear translocator, aryl hydrocarbon receptor repressor
INTRODUCTION
Basic helix-loop-helix (bHLH)–Per-aryl hydrocarbon receptor (AhR) nuclear translocator (ARNT)-SIM (PAS) proteins function as biological sensors that respond to physiological stimuli and environmental signals to mediate adaptive cellular responses. In general, these proteins form heterodimers that consist of a signal-induced subunit and an unregulated, ubiquitously expressed subunit (1). A member of the family of bHLH-PAS proteins is the AhR. Initially identified by Poland and Knutson (2) as a binding site for planar, nonhalogenated, and halogenated xenobiotics, in particular the highly toxic 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), the AhR was viewed for a long time as a mediator of xenobiotic-induced toxicity and carcinogenesis. However, mouse knockdown studies have revealed a role for the AhR beyond xenobiotic-induced toxicity and carcinogenesis. For example, the AhR has been shown to be involved in fetal liver development (3), immune cell modulation and differentiation (4–6), and gastrointestinal homeostasis (7, 8). The developmental defects observed in AhR−/− mice and the strong conservation of the AhR throughout evolution (for a review, see reference 9) clearly point toward an essential role of this receptor in cell proliferation and differentiation in both developmental and physiological processes.
The molecular events following the activation of the AhR by ligands such as TCDD are fairly well understood. In its latent form, the AhR resides in the cytoplasm bound to heat shock protein 90 and cochaperones. Upon ligand binding, the AhR translocates to the nucleus, where it dimerizes with its partner protein ARNT and binds to its cognate response elements, the so-called xenobiotic response elements (XREs), in the promoter regions of target genes such as the cytochrome P4501A1 (CYP1A1) and cytochrome P4501B1 (CYP1B1) genes encoding xenobiotic-metabolizing enzymes (for a review, see reference 10). The activity of the ligand-activated AhR is controlled by several attenuating mechanisms. The half-life of the activated receptor is restricted by ubiquitinylation and, subsequently, proteasomal degradation (21). The availability of the activating ligand is controlled through metabolic clearance by the AhR-induced expression of metabolizing enzymes (12). Moreover, the AhR induces the expression of another bHLH-PAS protein, the repressor of AhR function (AhRR) (13). Previous studies by Oshima et al. showed that the AhRR competes with the AhR for heterodimerization with ARNT at XRE sites (14), thereby negatively regulating AhR signaling.
Besides functioning as a ligand-activated transcription factor, in 2007, the AhR was identified by Ohtake et al. as a ligand-dependent E3 ubiquitin ligase targeting substrate proteins for proteasomal degradation (15). Unlike typical CUL4BDCFA E3 ubiquitin ligases, which contain damaged DNA binding protein 1 (DDB1)- and CUL4-associated factors (DCFA), the CUL4AhR complex uses the activated AhR as an adaptor protein for substrate recognition. Consisting of the scaffold protein CUL4B, the ring finger protein RBX1, the β-transducin-like protein transducin-β-like 3 (TBL3), the proteasomal 19S particle, and DDB1, this atypical CUL4B ligase assembles and functions only in the presence of the activated AhR. Among substrate proteins identified for the CUL4AhR complex are steroid receptors such as estrogen receptor alpha (ERα), ERβ, and androgen receptor (15).
An antitumorigenic role of the AhR was observed in steroid receptor signaling-dependent tumors such as prostate and breast cancers (15, 16). The development and progression of breast cancer are highly linked to excessive ERα signaling due to the increased production of 17β-estradiol (E2) as well as high ERα levels. The activated AhR has been shown to interfere with ERα signaling through (i) the activation of ERα in the absence of E2 on ERα target genes (17), (ii) inhibitory XREs in the promoter region of ERα target genes (17), (iii) the modulation of E2-synthesizing enzymes such as CYP19 (18), (iv) competition for coactivators such as ARNT (19), and (v) targeting of ERα for proteasomal degradation (15).
To date, it is not clear how the dual function of the AhR is regulated. In this study, we investigate whether ARNT is the main factor governing the ability of the AhR to switch between the two different functions. Since AhR-mediated ERα degradation is not affected by ARNT knockdown, we hypothesize that the AhR functions as a transcription factor when ARNT is available but that in the absence of ARNT, the E3 ligase function is favored.
RESULTS AND DISCUSSION
ERα degradation by the CUL4BAhR complex is not dependent on ARNT.
The AhR is the only bHLH-PAS protein identified so far that possesses both an intrinsic transcription factor function as well as an E3 ubiquitin ligase function. Equipped with these two functions, the AhR is able to regulate cellular protein levels by two distinct processes, transcription and proteasomal degradation.
However, the molecular mechanisms regulating the transcription factor and E3 ubiquitin ligase functions of the AhR have not yet been identified. We used the human breast cancer cell line MCF7 expressing a functional ERα signaling pathway to study the molecular switch that regulates the dual functions of the AhR. As expected, we show that ligand activation of AhR function by treatment with 10 nM TCDD for 0 to 6 h markedly reduced total ERα levels (Fig. 1A, left) in MCF7 cells. The AhR itself was also degraded upon ligand treatment but only after 6 h, as previously shown (20, 21). Nevertheless, in cells in which the AhR was transiently downregulated by RNA interference, ERα degradation was almost completely abolished (Fig. 1A, right). Furthermore, immunoprecipitation studies with anti-ERα revealed that ERα was polyubiquitinated in MCF7 cells treated with 10 nM TCDD for 6 h in the presence of the proteasomal inhibitor MG132 (10 μM) but not in cells that received only vehicle or MG132 treatment (Fig. 1B). Taken together, these results confirm that the AhR functions as a ligand-activated E3 ubiquitin ligase targeting ERα for proteasomal degradation.
FIG 1.

The E3 ubiquitin ligase function of the AhR is favored in cells in which AhR transcriptional activity is impaired by ARNT knockdown. (A) Western blotting (top) and relative AhR protein levels (bottom). MCF7 cells were transfected with negative-control siRNA (siCtrl) or siRNA targeting the AhR (siAhR) and incubated for 48 h. The cells were then treated with TCDD (5 nM) for the indicated times. Cycloheximide at 10 μM was included in all treatment regimes. Whole-cell extracts were prepared, separated on SDS-PAGE gels, and analyzed by immunoblotting using ERα, AhR, and β-actin antibodies. The band intensities of ERα immunoblots were quantified by using ImageJ software, and the relative ERα protein levels were normalized to β-actin levels and are presented as mean values from three independent experiments. (B) Cells were treated with 10 nM TCDD in the presence of 10 μM the proteasomal inhibitor MG132 for up to 6 h, and whole-cell extracts were prepared. Extracts were analyzed following immunoprecipitation (IP) with an ERα antibody by immunoblotting using antiubiquitin (anti-Ub). (C) Following transfection with siRNA, MCF7 cells were treated with 10 nM TCDD for 0 to 6 h. Total RNA was prepared, and the expression of the AhR target gene CYP1A1 was analyzed by RT-qPCR. HPRT was used as an endogenous control for ΔΔCT analysis. Data are presented as means ± SD of results from three independent experiments performed in duplicate. (D) Western blotting (left) and relative AhR protein levels (right). Cells were treated with 10 nM TCDD for 0 to 6 h, and whole-cell extracts were collected. Cell extracts were separated on an SDS-PAGE gel and transferred onto a membrane for immunoblotting. The blots were analyzed by using ERα, ARNT, and β-actin antibodies. The band intensities of ERα immunoblots were quantified by using ImageJ software, and the relative ERα protein levels were normalized to β-actin levels and are presented as mean values from three independent experiments. (E) MCF7 cells were transiently transfected with negative-control siRNA or siRNA against ARNT. Forty-eight hours after transfection, cells were treated with 10 μM the proteasomal inhibitor MG132 and 10 nM TCDD. Cells were harvested, an immunoprecipitation assay was preformed with an ERα antibody, and cells were immunoblotted as indicated. (F) MCF7 cells were treated with the proteasomal inhibitor MG132 (10 μM) and TCDD (10 nM) for 6 h. (Left) Cell extracts were coimmunoprecipitated with anticullin, and immunoblotting was performed as indicated. (Right) MCF7 cells were transfected with Flag-tagged Arnt, and a Flag antibody was used in the coimmunoprecipitation assay. The immunoprecipitation experiments were repeated several times, and the data show the results of a representative experiment.
To function as a transcription factor, the AhR needs to heterodimerize with its partner protein ARNT. We therefore examined the role of ARNT in the regulation of the transcription factor and E3 ubiquitin ligase functions of the AhR. To examine the transcription capacity of the AhR, we used CYP1A1, which is one of the best-studied AhR target genes. MCF7 cells transiently transfected with scrambled small interfering RNA (siRNA) showed a tremendous increase in the CYP1A1 mRNA level with treatment with 10 nM TCDD over a time course of 0 to 6 h (Fig. 1C). However, MCF7 cells in which ARNT expression was downregulated by RNA interference responded to TCDD treatment with a significantly lower level of induction of CYP1A1 mRNA expression, suggesting that ARNT is essential as a partner protein for the AhR to function properly as a transcription factor. Surprisingly, when we investigated ERα degradation in TCDD-treated MCF7 cells in which ARNT was downregulated by siRNA, we observed an even better reduction in total ERα levels compared to those in cells receiving control siRNA (Fig. 1D). We further investigated the levels of endogenous ubiquitinated ERα in cells in which ARNT levels were greatly reduced due to RNA interference. In accordance with the ERα degradation data, immunoprecipitation experiments revealed that these cells showed very similar or even higher levels of ubiquitinated ERα than cells that received scrambled siRNA (Fig. 1E). ARNT was originally identified by Ohtake and colleagues (15) as part of the Cul4BAhR complex together with the activated AhR, cullin 4B, Rbx1, DDB1, TBL3, and the 19S regulatory particle. However, we could not identify Arnt as a part of the Cul4BAhR complex in MCF7 cells treated with TCDD when coimmunoprecipitated with a Cul4B antibody (Fig. 1F, left). In addition, MCF7 cells were transfected with Flag-tagged Arnt, and a Flag antibody was used in the coimmunoprecipitation assay. As expected, the AhR was a part of the coimmunoprecipitated complex, but neither Cul4B nor ERα was detected (Fig. 1F, right), in accordance with the Cul4B coimmunoprecipitation results. In contrast to our data, Ohtake et al. (15) used Flag-tagged AhR overexpressed in HeLa cell nuclear extracts to coimmunoprecipitate the Cul4BAhR complex and thus probably obtained a mix of AhR-ARNT transcription factor and Cul4BAhR E3 ubiquitin ligase complexes for detection. Their observation that ARNT is part of the E3 ligase complex may therefore be mistakenly based on the occurrence of ARNT within AhR-ARNT transcription factor complexes but not within the E3 ubiquitin ligase complexes (see Fig. 1 and 2D in reference 15).
FIG 2.
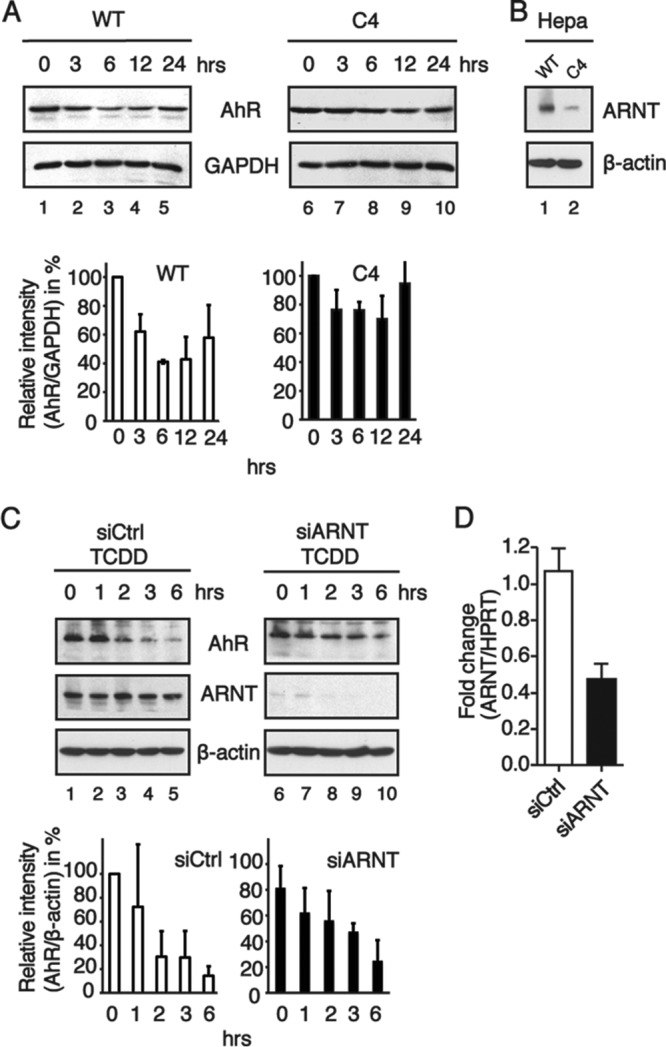
Degradation of the AhR is impaired in cells where ARNT protein levels are reduced. (A) Western blotting (top) and relative AhR protein levels (bottom). Whole-cell extracts were prepared from TCDD-treated Hepa-1c1c7 (wild-type [WT]) and Hepa C4 (C4) cells at the indicated time points and separated by SDS-PAGE. Immunoblots were analyzed by using AhR and GAPDH antibodies. The band intensities of AhR immunoblots were quantified by using ImageJ software, and the relative AhR protein levels were normalized to GAPDH levels and are presented as mean values from three independent experiments. (B) Western blot analysis of ARNT protein levels in Hepa-1c1c7 cells (WT) and Hepa C4 cells (C4). (C) Western blotting (top) and relative AhR protein levels (bottom). MCF7 cells were transiently transfected with negative-control siRNA and siRNA targeted to ARNT and treated with TCDD. At the indicated time points, cells were harvested, and whole-cell extracts were subjected to immunoblotting. Blots were analyzed for anti-AhR, anti-ARNT, and anti-β-actin. The data show results from a representative experiment. The band intensities of AhR immunoblots were quantified by using ImageJ software, and the relative AhR protein levels were normalized to β-actin levels and are presented as mean values from three independent experiments. (D) ARNT mRNA expression in MCF7 cells transfected with control siRNA or specific siRNA against ARNT. The data represent the means ± SD of results from three independent experiments performed in duplicate.
The degradation of the ligand-activated AhR is dependent on the ARNT.
An important mechanism to restrict the function and activity of transcription factors and E3 ubiquitin ligases is to highly control their expression levels. Also, AhR levels are tightly regulated in a time- and dose-dependent way when activated by ligands such as TCDD (21–24). Observations reported previously by Ma and Baldwin (21) suggest that functional ARNT is essential for the ligand-induced ubiquitinylation of the AhR. Indeed, AhR protein levels were downregulated in wild-type mouse hepatoma (Hepa) cells (Fig. 2A, left), whereas Hepa C4 cells harboring a point mutation in the PAS domain of ARNT (25, 26) showed very little ligand-induced AhR degradation (Fig. 2A, right). In these cells, mutant ARNT has highly increased protein turnover, leading to a large reduction of total ARNT protein levels (Fig. 2B). Similar to Hepa C4 cells, ligand-induced AhR degradation was delayed in MCF7 cells in which ARNT levels were reduced by RNA interference (Fig. 2C). Figure 2D shows that ARNT mRNA levels in MCF7 cells transfected with siRNA targeting ARNT are largely decreased. Taken together, these data show that ARNT has a pivotal role for ligand-induced AhR degradation but not for the E3 ubiquitin ligase function of the Cul4BAhR complex.
Overexpression of the AhRR disrupts DNA binding of the AhR and thereby negatively regulates AhR transcriptional function.
The availability of ARNT itself is modulated by another bHLH-PAS protein, the AhRR, as identified by Mimura and colleagues (13). The structural organization of this protein resembles that of the AhR, with the exception of the ligand binding domain and the transactivation domain. Thus, the AhRR cannot be activated by ligands. Oshima and colleagues (14) showed that mutual sumoylation enhanced the interaction between the AhRR and ARNT, thereby competing with the AhR/ARNT heterodimer for XRE binding and suppressing AhR signaling, consistent with both the AhR and AhRR utilizing ARNT as a dimerization partner. We generated MCF7 cells with stable overexpression of the Myc-tagged AhRR (Fig. 3A) and characterized these clones with regard to TCDD-induced AhR signaling. Clones overexpressing the AhRR showed both lower basal mRNA expression levels of the AhR target gene CYP1A1 as well as lower ligand-induced expression levels of the CYP1A1 gene (Fig. 3B). In accordance with the mRNA data, CYP1A1 protein levels and CYP1A1 enzyme activity were upregulated in response to TCDD only in the clone expressing an empty control vector, whereas clones overexpressing the AhRR showed no upregulation of protein levels and enzyme activity (data not shown). Furthermore, the recruitment of the AhR to its DNA binding site (XRE) on the target gene CYP1A1 was impaired when the AhRR was overexpressed (Fig. 3C, right), and the sequestering of the AhR from the XRE was due to the binding of the AhRR to the same sequence (Fig. 3C, left). These data clearly indicate that both the DNA binding and the transactivation properties of the ligand-activated AhR are disrupted by the overexpression of the AhRR.
FIG 3.
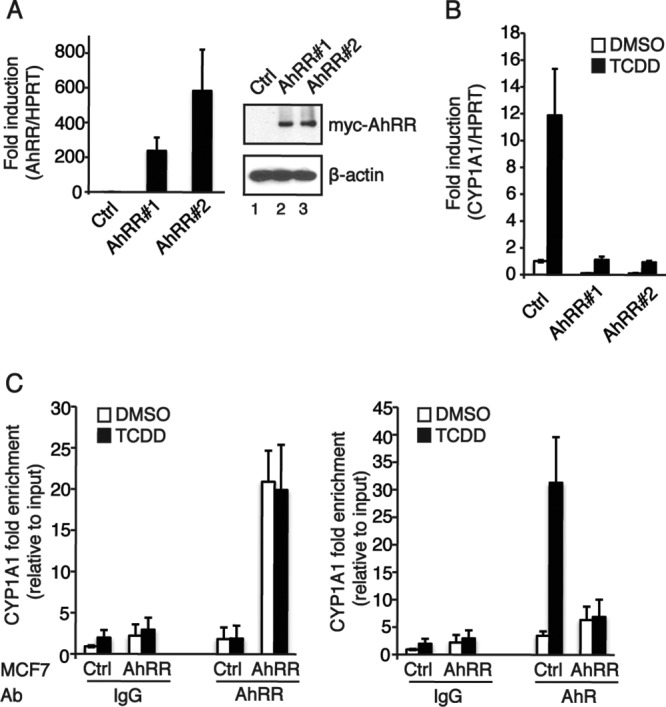
Overexpression of the AhRR affects AhR signaling by diminishing the transcription factor function of the AhR. (A) AhRR expression levels in MCF7 cells stably overexpressing the AhRR. Clones stably overexpressing the Myc-tagged AhRR were generated by transfection with pEFIRESpuro or pEFIRESpuromycAhRR constructs and subsequent selection with puromycin. Shown are data from analyses of AhRR mRNA expression levels (left) and protein levels (right) in MCF7 control cells and two selected clones. (B) Effect of AhRR overexpression on AhR-regulated gene expression. MCF7 cells were treated with 10 nM TCDD for 6 h. Total RNA was prepared, and the expression of CYP1A1 was analyzed by RT-qPCR. HPRT was used as an endogenous control for ΔΔCT analysis. Data are presented as means ± SD of results from three independent experiments performed in duplicate. (C) Recruitment of the AhR to the CYP1A1 promoter is impaired by overexpressed AhRR. ChIP was performed on MCF7 cells treated with DMSO or 10 nM TCDD for 2 h, using antibodies (Ab) against AhRR, AhR, and IgG (as a negative control). The immunoprecipitated DNAs were analyzed by quantitative PCR using primers spanning an XRE-containing region of CYP1A1. Data are presented as means ± SD of results from three independent experiments performed in duplicate.
Estrogen responsiveness and ERα protein levels are reduced in MCF7 cells stably overexpressing the AhRR.
We further investigated whether the E3 ubiquitin ligase function of the AhR is affected by the overexpression of the repressor of AhR function. Since the ubiquitin ligase function of the AhR targets ERα for proteasomal degradation, we compared the responses to estrogen treatment (E2) in MCF7 cells with or without overexpression of AhRR. MCF7 cells stably transfected with an empty control vector responded to 24 h of E2 treatment with an upregulation of the ERα target genes cathepsin D (CTSD) (Fig. 4A, left) and progesterone receptor (PR) (Fig. 4A, right). In contrast to the control cells, AhRR-overexpressing clones (AhRR#1 and AhRR#2) showed a reduced responsiveness to E2 treatment, suggesting that their unresponsiveness may be due to lower ERα protein levels. We therefore investigated ERα protein and ERα ubiquitinylation levels in MCF7 cells overexpressing the AhRR. Clones that overexpress the AhRR had in general lower ERα protein levels than did control cells. TCDD-induced ERα degradation was observed in control cells as well as AhRR-overexpressing clones, with the exception of clone 2, which showed the lowest ERα protein levels and seemed not to be further degradable (Fig. 4B). A similar pattern was also observed for ubiquitinylated ERα. While control cells and clone 1 showed TCDD-induced ERα ubiquitinylation, high levels of ubiquitinylated ERα were detected in clone 2 irrespective of TCDD treatment (Fig. 4C). By sequestering ARNT, the AhRR impairs the transcription factor function of the AhR, thereby switching the receptor toward its E3 ubiquitin ligase function. It was previously shown that the ARNT could function as a coactivator for both ERα and ERβ regulating estrogen-dependent transcription (19). It is therefore possible that competition for the recruitment of coactivators plays a role in the responsiveness of ERα target genes to estrogen treatment. It is plausible that the overexpression of the AhRR can affect other signaling pathways that regulate ERα stability and function; however, our results obtained in experiments on ERα protein levels and ubiquitinylation using siRNA targeting ARNT rather point toward a role of ARNT in the regulation of the transcription factor and E3 ligase function of the AhR.
FIG 4.
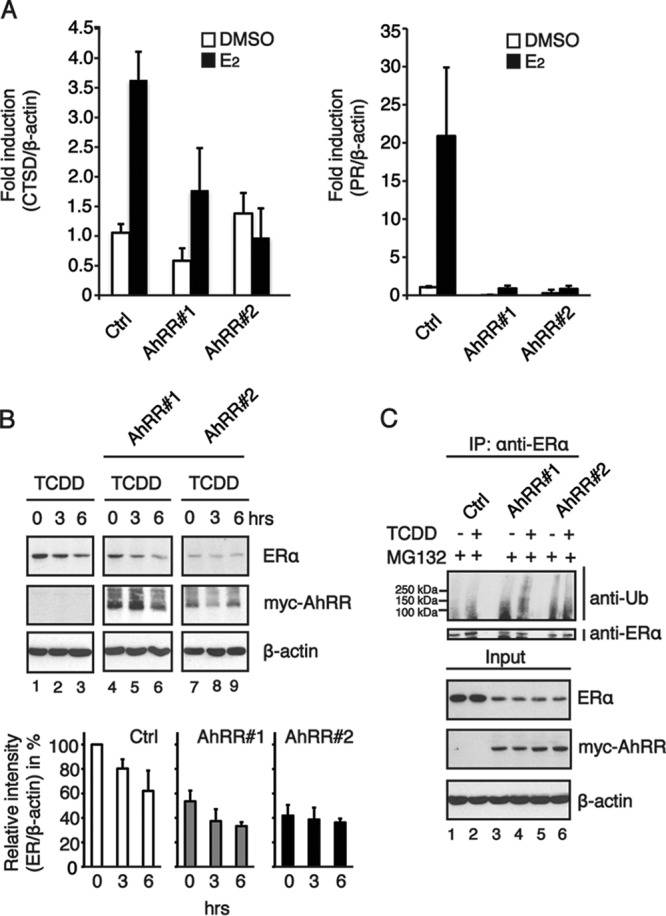
Estrogen receptor alpha signaling is impaired in MCF7 breast cancer cells overexpressing the repressor of AhR function. (A) MCF7 cells were treated with 10 nM E2 for 24 h, total RNA was prepared, and the expression levels of the estrogen receptor target genes CTSD (left) and PR (right) were analyzed by RT-qPCR. Beta-actin was used as an endogenous control for ΔΔCT analysis. Data are presented as means ± SD of results from three independent experiments performed in duplicate. (B) TCDD-induced ERα degradation is increased in MCF7 cells stably overexpressing the Myc-tagged AhRR. Shown are data for Western blotting (top) and relative AhR protein levels (bottom). Cells were treated with 10 nM TCDD for the indicated times, and whole-cell extracts were prepared and subjected to immunoblotting with anti-ERα, anti-Myc tag, and anti-β-actin. The band intensities of ERα immunoblots were quantified by using ImageJ software, and the relative ERα protein levels were normalized to β-actin levels and are presented as mean values from three independent experiments. (C) TCDD-induced ERα ubiquitinylation in MCF7 cells stably overexpressing the AhRR. Cells were treated with 10 μM the proteasomal inhibitor MG132 and 10 nM TCDD. Whole-cell extracts were immunoprecipitated with anti-ERα and immunoblotted with antiubiquitin.
Reduced tumor volume and ERα expression in MCF7 xenograft tumors stably overexpressing the AhRR.
By targeting the ERα for proteasomal degradation through its E3 ubiquitin ligase function, the AhR may serve as an important new therapeutic target in ER-dependent tumors. In order to investigate the functional consequences of reduced ERα protein levels for tumor cell behavior, we investigated tumor growth of MCF7 breast cancer cells in which AhR function was modulated by AhRR overexpression in vitro and in vivo. MCF7 cells with overexpressed AhRR (AhRR#2 cells, which also have low levels of ERα without ligand treatment) showed a lower growth rate than that of control cells in vitro, while there was no statistically significant difference between control cells and AhRR#1 (Fig. 5A). This is in line with previous work done by Zhang and coworkers that showed an enhanced proliferation of MCF7 and BT474 cells, both estrogen-dependent cell lines, when the AhR was silenced with siRNA. In contrast, the knockdown of the AhR in MDA-MB-468 cells, an estrogen-independent cell line, did not affect cell proliferation (27). Hypoxia-inducible transcription is important for tumor progression (28). Since hypoxia-inducible factors (HIFs) also use ARNT as a partner factor, the AhRR may affect HIF signaling. In order to investigate whether AhRR overexpression influences hypoxia-inducible gene transcription, we cultured MCF7 cells with and without the overexpressed AhRR in 21% or 1% oxygen for 18 h and investigated the expression of three well-characterized hypoxia-inducible genes, phosphoglycerate kinase 1 (PGK1), Dec1, and prolyl hydroxylase 3 (PHD3) (29). As shown in Fig. 5B, the induction of these genes was not affected by the overexpression of the AhRR, suggesting that the AhRR does not alter the HIF signaling pathway. It was previously shown that HIF-1α exhibited a very high affinity for Arnt, resulting in competition with the ligand-activated AhR for the recruitment of Arnt (30), and we believe that HIF1α has a higher affinity for Arnt than for the AhRR. Since AhRR#2 cells showed reduced growth without a ligand, we injected these cells and control cells into SCID mice. Interestingly, xenograft tumor volumes were reduced in animals that received MCF7 cells with high levels of the AhRR in comparison to xenograft tumors that contained control cells (Fig. 5C). Figure 5D shows that AhRR mRNA expression levels are indeed higher in these tumors. We further investigated ERα protein levels in these tumors. In accordance with the data from the cell studies, ERα protein levels were lower in tumors from mice that received MCF7 cells overexpressing the AhRR (Fig. 5E). Furthermore, paraffin sections were prepared from the tumors and immunostained for ERα (Fig. 5F). Reduced ERα staining was detected in sections of tumors of mice that received MCF7 cells overexpressing the repressor of AhR function (Fig. 5F, right). Taken together, our data clearly point toward a tumor-suppressive role of the AhR in MCF7 xenograft tumors. In contrast to our study, Moennikes et al. and Andersson et al. (31, 32) described that the AhR functions in a protumorigenic manner, promoting the development of liver and stomach tumors in transgenic mice expressing a constitutively active AhR (CA-AhR). In these mice, the constitutively active AhR, which was constructed by deleting the ligand binding domain, mimics the situation of constant activation by persistent ligands, implying that prolonged receptor activation can be deleterious or that the mutated AhR may not have ubiquitinylation activity. However, in accordance with our data, Kawajiri and colleagues (7) observed a higher incidence of gastrointestinal tumors in AhR−/− mice due to abnormally high levels of β-catenin, suggesting an important antitumorigenic role of the AhR. As described here for ERα, even β-catenin serves as a substrate protein for the E3 ubiquitin ligase function of the AhR. Thus, we propose a tumor-suppressive function for the E3 ubiquitin ligase function of the AhR.
FIG 5.

Estrogen-dependent tumor growth and ERα levels are reduced in SCID mice injected with MCF7 breast cancer cells stably overexpressing the AhRR. (A) MCF7 control cells and MCF7 cells stably overexpressing the AhRR (AhRR#1 and AhRR#2) were cultured for up to 96 h. At the indicated time points, cells were harvested, and the optical density at 600 nm (OD 600) was measured. Data are presented as means ± SD of results from three independent experiments in quadruplicate (P value of 0.0126). (B) AhRR overexpression does not affect hypoxia-inducible gene factors in MCF7 cells. MCF7 control and AhRR-overexpressing (AhRR#1 and AhRR#2) cells were cultured for 18 h in 21% or 1% oxygen. Cells were subsequently harvested, and expression levels of the hypoxia target genes PGK1, Dec1, and PHD3 were analyzed by RT-qPCR. HPRT was used as an endogenous control for ΔΔCT analysis. Data are presented as means ± SD from three independent experiments. (C) SCID mice (n = 10) were injected with MCF7 cancer cells with or without overexpressed AhRR (AhRR#2) in the presence of estrogen-releasing pellets. Tumors were measured by palpation. After 22 days, the mice were sacrificed, tumors were removed, and the tumor volume was determined. Data are presented as means ± standard errors of the means (n = 10) (*, P ≤ 0.05; **, P ≤ 0.01 [by two-way analysis of variance]). (D) AhRR mRNA levels in xenograft tumors from SCID mice injected with control cells (Ctrl) or AhRR-overexpressing cells (AhRR#2). Expression levels were analyzed by RT-qPCR. TBP was used as an endogenous control for ΔΔCT analysis. Data are presented as means ± SD of results from four different tumors. (E) ERα protein levels in tumors from SCID mice injected with control cells (Ctrl) and cells overexpressing the AhRR (AhRR#2). The data show results from a representative experiment with 6 different tumors. (F) Immunohistochemical analysis of ERα levels in tumors from mice injected with (right) or without (left) AhRR-overexpressing cells. The tumors were stained with an antibody against ERα and counterstained with hematoxylin. Data show results from a representative experiment.
Hormone-dependent tumors, e.g., breast cancer, are widely treated with aromatase inhibitors, inhibiting the production of E2, or with so-called selective estrogen receptor modulators such as tamoxifen. Unfortunately, resistance to the above-mentioned treatment is frequently observed, with progression to more aggressive phenotypes in which ERα phosphorylation renders the receptor in an activated conformation independent of ligand activation (33). Therapeutic treatment that can affect the cellular levels of activated ERα therefore needs to be developed.
As schematically summarized in Fig. 6, our data suggest an important role of ARNT in the modulation of the dual functions of the AhR. We show that ARNT may serve as the molecular switch that determines whether the AhR will serve as a ligand-activated, sequence-specific transcription factor or be part of the CUL4BAhR ubiquitin ligase complex targeting, e.g., ERα for proteasomal degradation. The knockdown of ARNT or overexpression of the AhRR, another dimerization partner for ARNT, impaired dramatically the transcriptional activation properties of the AhR but mostly did not affect the recognition of substrate proteins by the CUL4BAhR complex. Thus, the availability of ARNT seems to determine whether the AhR is directed toward an E3 ligase function or acts as a transcription factor. Furthermore, we show that MCF7 cells overexpressing the AhRR have lower growth rates, lower levels of ERα protein, and a reduced estrogen-dependent xenograft tumor growth capacity, clearly indicating that AhRR protein levels can modulate AhR function via ARNT. More work utilizing both in vitro and in vivo models is needed to elucidate the full function of the AhR as an E3 ubiquitin ligase. Nevertheless, our work provides evidence for a tumor-suppressive role of the E3 ubiquitin ligase function of the AhR. It will therefore be interesting to assess if the AhR can be used as a therapeutic target for the treatment of estrogen-dependent tumors.
FIG 6.
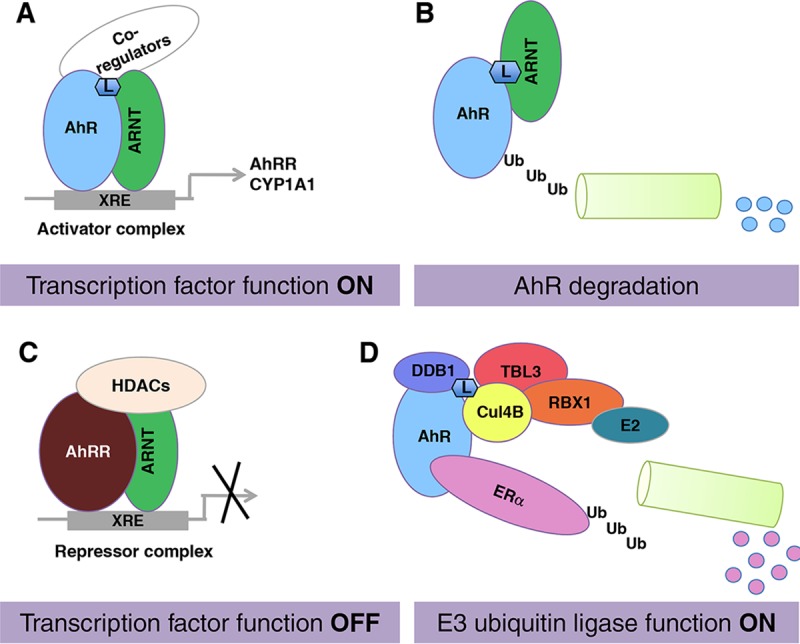
Arnt availability regulates the AhR switch from a transcription factor to an E3 ubiquitin ligase. (A and B) If ARNT is available, the AhR functions as a ligand-induced transcription factor (A) and is subsequently degraded via the proteasome complex (B). (C and D) If ARNT is occupied by other partner factors, e.g., the AhRR (C), the AhR functions as an E3 ubiquitin ligase targeting substrate proteins for proteasomal degradation, for instance, ERα. L, AhR ligand; HDACs, histone deacetylases.
MATERIALS AND METHODS
Reagents.
TCDD (Wellington Laboratories, Guelph, Ontario, Canada) and the proteasomal inhibitor MG132 (Calbiochem, Merck, Darmstadt, Germany) were dissolved in dimethyl sulfoxide (DMSO) (Sigma-Aldrich, St. Louis, MO). E2 and cycloheximide (CHX) (both from Sigma-Aldrich) were dissolved in ethanol. The final concentration of the vehicle did not exceed 0.1% (vol/vol). Puromycin and N-ethylmaleimide were purchased from Sigma-Aldrich and dissolved in H2O.
Cell culture conditions and treatments.
The ERα+ human breast cancer cell line MCF7 was cultured in Dulbecco's minimum Eagle medium (DMEM) (4.5 g/liter glucose) supplemented with 10% fetal bovine serum (FBS), streptomycin (100 μg/ml), and penicillin (100 U/ml) at 37°C in a 5% CO2 atmosphere. For analysis of ERα protein levels, cells were grown in phenol red-free DMEM supplemented with 5% charcoal-treated FBS at least 48 h before treatment regimes. To generate MCF7 cells stably overexpressing the Myc-tagged AhRR, MCF7 cells were transfected with either pEFIRESpuro (for control cells) or pEFIRESpuromycAhRR using Lipofectamine 2000 according to the manufacturer's recommendations. Cells were then selected based on puromycin resistance and routinely propagated in DMEM additionally supplemented with 0.5 μg/ml puromycin. The murine liver hepatoma cell lines Hepa-1c1c7 and Hepa-1c1c7c4 were routinely grown in minimal essential medium (MEM) supplemented with 10% FBS, streptomycin (100 μg/ml), and penicillin (100 U/ml) at 37°C in a 5% CO2 atmosphere. The Hepa-1c1c7c4 cell line contains a point mutation in the PAS domain of ARNT, which markedly contributes to the increased turnover of the protein. All cell culture growth media and supplements were purchased from Life Technologies (Carlsbad, CA).
Plasmids.
The pEFIRESpuromycAhRR construct was generated by subcloning an XbaI-NotI fragment containing the Myc-tagged human AhRR (hAhRR) from pCMV-myc-AhRR into the XbaI-NotI-digested pEFIRESpuro vector. The cytomegalovirus (CMV)-Myc-hAhRR construct was generated by the insertion of a SalI-BamHI fragment from pBS-hAhRR (a kind gift from Yoshiaki Fujii-Kuriyama, Tohoku University, Japan) into SalI-BglII-digested pCMV-myc (Clontech). To generate the hAhRR in frame with the Myc tag, a PCR fragment containing the hAhRR (400 bp) with EcoI-NsiI ends was subcloned into the former construct. All constructs were confirmed by sequencing.
RNA interference experiments.
For RNA interference experiments, MCF7 cells were seeded into 6-well plates and grown for 12 h before transfection. Negative-control siRNA and siRNA targeted against the AhR or ARNT were purchased from Qiagen (Hilden, Germany). The levels of the AhR and ARNT were downregulated by transient transfection with siRNA interference (20 nM) using Lipofectamine 2000 according to the manufacturer's recommendations. Upon transfection, cells were incubated for 48 h before the start of treatment.
Total RNA isolation and RT-qPCR.
Total RNA was prepared from MCF7 cells by using the NucleoSpin RNA II kit from Macherey-Nagel (Düren, Germany). To obtain total RNA from tumors, the tumors were disrupted in TRIzol (Invitrogen) by using a Qiagen TissueLyser II instrument (frequency of 22.0 Hz, three times for 2 min), and total RNA was isolated according to the manufacturer's protocols. RNA (0.5 μg) was reverse transcribed by using the Maxima first strand cDNA synthesis kit for reverse transcription-quantitative PCR (RT-qPCR) (Thermo Scientific) according to the manufacturer's recommendations. RT-qPCR was performed by using the ABI7300 instrument with PowerSYBR green or TaqMan PCR master mix (Applied Biosystems). The primers used are listed in Table 1.
TABLE 1.
Primersa
| Target | Forward primer sequence | Reverse primer sequence |
|---|---|---|
| AhRR | 5′-TCT TAA TGG CTT TGC TCT GGT CG-3′ | 5′-GTT CTG GTG CAT TAC ATC CGT CTG-3′ |
| ARNT | 5′-GAA CCA GCT CCT TTA CTT TCC-3′ | 5′-GGC TAG AGT TCT TCA CAT TGG-3′ |
| CTSD | 5′-ACA GGC ACT TCC CTC ATG GT-3′ | 5′-GGG ATC ATG TAC TCG CCC TGA-3′ |
| CYP1A1 | 5′-CCT TCA TCC TGG AGA CCT TCC-3′ | 5′-ATG GTT GAT CTG CCA CTG GTT T-3′ |
| PR | 5′-CGC GCT CTA CCC TGC ACT C-3′ | 5′-TGA ATC CGG CCT CAG GTA GTT-3′ |
| HPRT | 5′-GCA GCC CTG GCG TCG TGA TTA G-3′ | 5′-TGA TGG CCT CCC ATC TCC TTC ATC-3′ |
| TBP | 5′-CCA CTC ACA GAC TCT CAC AAC-3′ | 5′-CTG CGG TAC AAT CCC AGA AC-3′ |
| PGK1 | 5′-GGA ACA CGG AGG ATA AAG TCA G-3′ | 5′-CAG GAA CTA AAA GGC AGG AAA G-3′ |
| PHD3 | 5′-ATC GAC AGG CTG GTC CTC TA-3′ | 5′-CTT GGC ATC CCA ATT CTT GT-3′ |
For Dec1, TaqMan assay Hs01041212_m1 was used, and for β-actin, TaqMan assay 4326315E was used.
The comparative threshold cycle (ΔΔCT) method was used for the relative quantification of target DNA amplification. Hypoxanthine phosphoribosyltransferase (HPRT), β-actin, and TATA binding protein (TBP) served as endogenous controls. All measurements were performed three times in duplicate.
Preparation of cell extracts and immunoblotting.
For the analysis of total protein levels, whole-cell extracts were prepared in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 50 mM Tris-HCl [pH 7.5], 0.1% SDS, 0.5% sodium deoxycholate, 1% NP-40) supplemented with 1 mM dithiothreitol (DTT) and a complete protease inhibitor cocktail (Roche, Basel, Switzerland). The protein concentration was then determined by using a colorimetric assay (Bio-Rad). Proteins were separated by SDS-PAGE, transferred onto a nitrocellulose membrane, and blotted according to standard procedures with the following antibodies: anti-hAhR (1:500) (catalog number sc-133088), anti-ERα (1:1,000) (catalog number sc-543; both from Santa Cruz Biotechnology, Dallas, TX), antiubiquitin (catalog number Z0458; Dako), anti-β-actin (1:20,000) (catalog number AC-15; Abcam, Cambridge, UK), anti-mouse AhR (mAhR) (1:500) (catalog number BML-SA210; Enzo, New York, NY), anti-ARNT (1:1,000) (catalog number 611079; BD, Franklin Lakes, NJ), anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) (1:10,000) (Fitzgerald Industries, Acton, MA), and anti-Myc (1:2,000) (catalog number 631206; Clontech, Mountain View, CA). Beta-actin and GAPDH served as loading controls. Bands were visualized by using enhanced chemiluminescence reagents (GE Healthcare, Pittsburgh, PA).
The tumors were disrupted in lysis buffer (500 mM NaCl, 20% glycerol, 50 mM Tris-HCl [pH 7.4], and 1% NP-40), including a complete protease inhibitor cocktail (Roche), 1 mM DTT, and 5 mM β-mercaptoethanol, using the Qiagen TissueLyser II instrument (frequency of 22.0 Hz, three times for 2 min). Lysates were centrifuged at 14,000 rpm for 30 min, and the protein concentrations in the supernatants (whole-cell extracts) were determined by using the Bradford method (34). The proteins were then separated on an 8% SDS-PAGE gel and transferred onto a nitrocellulose membrane by using a semiwet method (Pierce G2 Fast Blotter; Thermo Scientific). Subsequently, the membranes were incubated with antibodies against human ERα (1:5,000) (catalog number sc-543; Santa Cruz) or actin (1:20,000) (catalog number AC-15; Abcam) at 4°C overnight, followed by incubation with horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse secondary antibodies (Dako). Proteins were visualized by using enhanced chemiluminescence (Pierce ECL Western blotting substrate) according to the manufacturer's instructions. Band intensities were compared on the same blots and quantified by using ImageJ.
Immunoprecipitation.
For immunoprecipitation experiments, cell lysates were collected in BC buffer (20% glycerol, 0.2 mM EDTA, 0.05% NP-40, 180 mM KCl, 20 mM HEPES [pH 7.9]) supplemented with 1 mM DTT, a complete protease inhibitor cocktail (Roche), and 10 mM N-ethylmaleimide. In Fig. 1F (right), the cells were transfected with the pCMV-Flag-Arnt plasmid before cell lysates were prepared. A total of 1 to 2 mg of the precleared lysate was immunoprecipitated with anti-ERα (catalog number sc-543; Santa Cruz), anti-Flag (M2; Sigma-Aldrich), or anti-cullin 4B (catalog number ab67035; Abcam) overnight at 4°C with rotation before absorption on a 50% protein G-Sepharose slurry at 4°C for 90 min. After five washes with BC buffer supplemented with 0.1% NP-40, precipitated proteins were separated by SDS-PAGE and transferred onto a nitrocellulose membrane. Blots were analyzed by using antiubiquitin (catalog number Z0458; Dako, Glostrup, Denmark), anti-ERα (catalog number sc-543; Santa Cruz), anti-Arnt (BD Biosciences), anti-AhR (Santa Cruz), and anti-cullin 4B (Abcam). Beta-actin (Abcam) served as a loading control. Proteins were visualized by using enhanced chemiluminescence (Pierce ECL Western blotting substrate) according to the manufacturer's instructions.
AhRR antibody.
A peptide corresponding to amino acids 370 to 385 of the human AhRR was synthesized, and antisera were produced in rabbits by standard techniques (MedProbe). The sera were further purified with an AhRR-specific antigen (Innovagen), and the antibody was found to have a strong binding affinity for the human AhRR in a chromatin immunoprecipitation (ChIP) assay.
ChIP assays.
Briefly, MCF7 cells with or without the overexpressed AhRR were treated with 10 nM TCDD or the vehicle only (0.1% DMSO) for 2 h. ChIP analyses combined with quantitative PCR were performed as previously described (35), with minor changes, using antibodies against AhRR, AhR (36), or IgG (as a negative control) (Jackson ImmunoResearch). PCR primers flanking the XRE of the CYP1A1 gene (position −1020/−889) were forward primer 5′-GAA TGG GTC GGC TGG GTG GC-3′ and reverse primer 5′-GCG CCG GCG ACA TCC CTC TA-3′, giving a product of 131 bp. The ΔΔCT method was used for the relative quantification of target DNA amplification using SYBR green. All of the ChIP assays were performed three times in duplicate.
Crystal violet staining.
Crystal violet staining was used to measure cell growth, and stained cells are proportional to the number of cells per unit area. MCF7 cells with or without the overexpressed AhRR were seeded onto 96-well plates at a density of 1,200 cells/well, and the cells were grown for 24, 48, 72, or 96 h before harvest. The cells were then fixed with 2% formaldehyde for 20 min, stained with a 0.04% crystal violet solution for 30 min, and allowed to dry. The crystal violet stain was dissolved in 1% SDS, and the intensity was measured at 600 nm in a plate reader. Data are presented as means ± standard deviations (SD) of results from three independent experiments in quadruplicate.
Mice and xenograft tumor study.
All animals were housed and maintained at the MTC breeding unit according to Karolinska Institute (KI) guidelines. Mice were kept on a 12-h-light/dark cycle with ad libitum access to water and chow. Six days prior to tumor cell injections, an estrogen pellet was placed subcutaneously in 20 SCID (CM17sc) female mice (approximately 9 weeks of age). Mice were kept under anesthesia (2.4% isoflurane) during the entire procedure. Hair was removed, a small incision was made between the shoulders and carefully expanded with closed scissors, the pellet (0.72 mg estradiol, 60-day release) (catalog number SE 121; Innovative Research of America, Sarasota, FL) was placed subcutaneously with forceps, and the wound was closed with Vetbond tissue adhesive (3M9). MCF7 breast cancer cells with or without the overexpressed AhRR (2 × 106 cells) in Matrigel (BD Matrigel Matrix, growth factor reduced, high concentration; BD Bioscience, Bedford, MA) were injected into the right flank of female SCID mice. Mice were sacrificed 22 days after injection, and tumors were dissected. The tumor volume was calculated by using the formula X × Y × Y × π/6, where X and Y are the long and short tumor dimensions, respectively, and π equals 3.14. All experiments were performed according to rules stipulated and approved by an animal ethics permit of the Swedish board of agriculture.
Immunohistochemistry.
Tumors were fixed in 4% formaldehyde and paraffin embedded. Sections (7 μm thick) were dewaxed by using Histolab-Clear (Histolab Products, Spånga, Sweden) and hydrated with decreasing concentrations of ethanol. Microwave antigen retrieval was performed in target retrieval solution (pH 6.1) (Dako, Glostrup, Denmark). The endogenous peroxidase activity was blocked with 3% H2O2 in methanol. All sections were incubated with mouse-on-mouse (MOM) blocking (Vector Laboratories, Burlingame, CA) for 60 min and CAS blocking (Invitrogen) for 30 min, before incubation with the anti-ERα primary antibody (catalog number M7047; Dako) overnight at 4°C. Subsequently, the sections were incubated with an HRP-conjugated secondary antibody for 1 h at room temperature, and the immunoreactive signal was visualized with a 3,3′-diaminobenzidine (DAB) substrate kit (Vector Laboratories). Finally, the sections were counterstained with hematoxylin, dehydrated with increasing concentrations of ethanol, and mounted with Pertex mounting medium (Histolab Products). Chromogenic signals were detected by using a Leica DM5500 B microscope. To determine tissue and tumor morphology, tissue sections were stained with hematoxylin and eosin (Histolab Products).
ACKNOWLEDGMENTS
We thank Yoshiaki Fujii-Kuriyama (Tokyo Medical and Dental University) and Murray Whitelaw (University of Adelaide) for critical reading of the manuscript, Pernilla Roswall (Karolinska Institutet) for technical help with ERα immunohistochemistry, and Jorge L. Ruas (Karolinska Institutet) for appreciated help during the revision stage.
REFERENCES
- 1.Bersten DC, Sullivan AE, Peet DJ, Whitelaw ML. 2013. bHLH-PAS proteins in cancer. Nat Rev Cancer 13:827–841. doi: 10.1038/nrc3621. [DOI] [PubMed] [Google Scholar]
- 2.Poland A, Knutson JC. 1982. 2,3,7,8-Tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: examination of the mechanism of toxicity. Annu Rev Pharmacol Toxicol 22:517–554. doi: 10.1146/annurev.pa.22.040182.002505. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt JV, Su GH, Reddy JK, Simon MC, Bradfield CA. 1996. Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc Natl Acad Sci U S A 93:6731–6736. doi: 10.1073/pnas.93.13.6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B. 2008. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453:106–109. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- 5.Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL. 2008. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453:65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- 6.Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. 2008. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci U S A 105:9721–9726. doi: 10.1073/pnas.0804231105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawajiri K, Kobayashi Y, Ohtake F, Ikuta T, Matsushima Y, Mimura J, Pettersson S, Pollenz RS, Sakaki T, Hirokawa T, Akiyama T, Kurosumi M, Poellinger L, Kato S, Fujii-Kuriyama Y. 2009. Aryl hydrocarbon receptor suppresses intestinal carcinogenesis in ApcMin/+ mice with natural ligands. Proc Natl Acad Sci U S A 106:13481–13486. doi: 10.1073/pnas.0902132106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Korecka A, Dona A, Lahiri S, Tett AJ, Al-Asmakh M, Braniste V, D'Arienz R, Abbaspour A, Reichardt N, Fujii-Kuriyama Y, Rafter J, Narbad A, Holmes E, Nicholson J, Arulampalam V, Pettersson S. 2016. Bidirectional communication between the aryl hydrocarbon receptor (AhR) and the microbiome tunes host metabolism. NPJ Biofilms Microbiomes 2:16014. doi: 10.1038/npjbiofilms.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hahn ME. 2002. Aryl hydrocarbon receptors: diversity and evolution. Chem Biol Interact 141:131–160. doi: 10.1016/S0009-2797(02)00070-4. [DOI] [PubMed] [Google Scholar]
- 10.Denison MS, Soshilov AA, He G, DeGroot DE, Zhao B. 2011. Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol Sci 124:1–22. doi: 10.1093/toxsci/kfr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reference deleted.
- 12.Chiaro CR, Patel RD, Marcus CB, Perdew GH. 2007. Evidence for an aryl hydrocarbon receptor-mediated cytochrome p450 autoregulatory pathway. Mol Pharmacol 72:1369–1379. doi: 10.1124/mol.107.038968. [DOI] [PubMed] [Google Scholar]
- 13.Mimura J, Ema J, Sogawa K, Fujii-Kuriyama Y. 1999. Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev 13:20–25. doi: 10.1101/gad.13.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oshima M, Mimura J, Sekine H, Okawa H, Fujii-Kuriyama Y. 2009. SUMO modification regulates the transcriptional repressor function of aryl hydrocarbon receptor repressor. J Biol Chem 284:11017–11026. doi: 10.1074/jbc.M808694200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohtake F, Baba A, Takada I, Okada M, Iwasaki K, Miki H, Takahashi S, Kouzmenko A, Nohara K, Chiba T, Fujii-Kuriyama Y, Kato S. 2007. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature 446:562–566. doi: 10.1038/nature05683. [DOI] [PubMed] [Google Scholar]
- 16.Fritz WA, Lin TM, Peterson RE. 2008. The aryl hydrocarbon receptor (AhR) inhibits vanadate-induced vascular endothelial growth factor (VEGF) production in TRAMP prostates. Carcinogenesis 29:1077–1082. doi: 10.1093/carcin/bgn069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohtake F, Takeyama K, Matsumoto T, Kitagawa H, Yamamoto Y, Nohara K, Tohyama C, Krust A, Mimura J, Chambon P, Yanagisawa J, Fujii-Kuriyama Y, Kato S. 2003. Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature 423:545–550. doi: 10.1038/nature01606. [DOI] [PubMed] [Google Scholar]
- 18.Chan MY, Huang H, Leung LK. 2010. 2,3,7,8-Tetrachlorodibenzo-para-dioxin increases aromatase (CYP19) mRNA stability in MCF-7 cells. Mol Cell Endocrinol 317:8–13. doi: 10.1016/j.mce.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 19.Brunnberg S, Pettersson K, Rydin E, Matthews J, Hanberg A, Pongratz I. 2003. The basic helix-loop-helix-PAS protein ARNT functions as a potent coactivator of estrogen receptor-dependent transcription. Proc Natl Acad Sci U S A 100:6517–6522. doi: 10.1073/pnas.1136688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davarinos NA, Pollenz RS. 1999. Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytosplasmic [sic] proteasome following nuclear export. J Biol Chem 274:28708–28715. doi: 10.1074/jbc.274.40.28708. [DOI] [PubMed] [Google Scholar]
- 21.Ma Q, Baldwin KT. 2000. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced degradation of aryl hydrocarbon receptor (AhR) by the ubiquitin-proteasome pathway. Role of the transcription activaton [sic] and DNA binding of AhR. J Biol Chem 275:8432–8438. doi: 10.1074/jbc.275.12.8432. [DOI] [PubMed] [Google Scholar]
- 22.Pollenz RS, Sullivan HR, Holmes J, Necela B, Peterson RE. 1996. Isolation and expression of cDNAs from rainbow trout (Oncorhynchus mykiss) that encode two novel basic helix-loop-helix/PER-ARNT-SIM (bHLH/PAS) proteins with distinct functions in the presence of the aryl hydrocarbon receptor. Evidence for alternative mRNA splicing and dominant negative activity in the bHLH/PAS family. J Biol Chem 271:30886–30896. doi: 10.1074/jbc.271.48.30886. [DOI] [PubMed] [Google Scholar]
- 23.Pollenz RS, Santostefano MJ, Klett E, Richardson VM, Necela B, Birnbaum LS. 1998. Female Sprague-Dawley rats exposed to a single oral dose of 2,3,7,8-tetrachlorodibenzo-p-dioxin exhibit sustained depletion of aryl hydrocarbon receptor protein in liver, spleen, thymus, and lung. Toxicol Sci 42:117–128. doi: 10.1006/toxs.1998.2439. [DOI] [PubMed] [Google Scholar]
- 24.Roman BL, Pollenz RS, Peterson RE. 1998. Responsiveness of the adult male rat reproductive tract to 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure: Ah receptor and ARNT expression, CYP1A1 induction, and Ah receptor down-regulation. Toxicol Appl Pharmacol 150:228–239. doi: 10.1006/taap.1998.8388. [DOI] [PubMed] [Google Scholar]
- 25.Numayama-Tsuruta K, Kobayashi A, Sogawa K, Fujii-Kuriyama Y. 1997. A point mutation responsible for defective function of the aryl-hydrocarbon-receptor nuclear translocator in mutant Hepa-1c1c7 cells. Eur J Biochem 246:486–495. doi: 10.1111/j.1432-1033.1997.00486.x. [DOI] [PubMed] [Google Scholar]
- 26.Reyes H, Reisz-Porszasz S, Hankinson O. 1992. Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science 256:1193–1195. doi: 10.1126/science.256.5060.1193. [DOI] [PubMed] [Google Scholar]
- 27.Zhang S, Lei P, Liu X, Li X, Walker K, Kotha L, Rowlands C, Safe S. 2009. The aryl hydrocarbon receptor as a target for estrogen receptor-negative breast cancer chemotherapy. Endocr Relat Cancer 16:835–844. doi: 10.1677/ERC-09-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wigerup C, Pahlman S, Bexell D. 2016. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol Ther 164:152–169. doi: 10.1016/j.pharmthera.2016.04.009. [DOI] [PubMed] [Google Scholar]
- 29.Holmquist-Mengelbier L, Fredlund E, Lofstedt T, Noguera R, Navarro S, Nilsson H, Pietras A, Vallon-Christersson J, Borg A, Gradin K, Poellinger L, Pahlman S. 2006. Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell 10:413–423. doi: 10.1016/j.ccr.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 30.Gradin K, McGuire J, Wenger RH, Kvietikova I, Whitelaw ML, Toftgard R, Tora L, Gassmann M, Poellinger L. 1996. Functional interference between hypoxia and dioxin signal transduction pathways: competition for recruitment of the Arnt transcription factor. Mol Cell Biol 16:5221–5231. doi: 10.1128/MCB.16.10.5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moennikes O, Loeppen S, Buchmann A, Andersson P, Ittrich C, Poellinger L, Schwarz M. 2004. A constitutively active dioxin/aryl hydrocarbon receptor promotes hepatocarcinogenesis in mice. Cancer Res 64:4707–4710. doi: 10.1158/0008-5472.CAN-03-0875. [DOI] [PubMed] [Google Scholar]
- 32.Andersson P, McGuire J, Rubio C, Gradin K, Whitelaw ML, Pettersson S, Hanberg A, Poellinger L. 2002. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proc Natl Acad Sci U S A 99:9990–9995. doi: 10.1073/pnas.152706299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anbalagan M, Rowan BG. 2015. Estrogen receptor alpha phosphorylation and its functional impact in human breast cancer. Mol Cell Endocrinol 418(Part 3):264–272. doi: 10.1016/j.mce.2015.01.016. [DOI] [PubMed] [Google Scholar]
- 34.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 35.Zheng X, Linke S, Dias JM, Zheng X, Gradin K, Wallis TP, Hamilton BR, Gustafsson M, Ruas JL, Wilkins S, Bilton RL, Brismar K, Whitelaw ML, Pereira T, Gorman JJ, Ericson J, Peet DJ, Lendahl U, Poellinger L. 2008. Interaction with factor inhibiting HIF-1 defines an additional mode of cross-coupling between the Notch and hypoxia signaling pathways. Proc Natl Acad Sci U S A 105:3368–3373. doi: 10.1073/pnas.0711591105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gradin K, Toftgard R, Poellinger L, Berghard A. 1999. Repression of dioxin signal transduction in fibroblasts. Identification of a putative repressor associated with Arnt. J Biol Chem 274:13511–13518. doi: 10.1074/jbc.274.19.13511. [DOI] [PubMed] [Google Scholar]