

Abstract
Thrombomodulin (TBM) is an important vascular anticoagulant that has species specific effects. When expressed as a transgene in pigs, human (h)TBM might abrogate thrombotic manifestations of acute vascular rejection (AVR) that occur when GalT-KO and/or complement regulator transgenic pig organs are transplanted to primates. hTBM transgenic mice were generated and characterized to determine whether this approach might show benefit without the development of deleterious hemorrhagic phenotypes. hTBM mice are viable and are not subject to spontaneous hemorrhage, although they have a prolonged bleeding time. They are resistant to intravenous collagen-induced pulmonary thromboembolism, stasis-induced venous thrombosis and pulmonary embolism. Cardiac grafts from hTBM mice to rats treated with cyclosporine in a model of AVR have prolonged survival compared to controls. hTBM reduced the inflammatory reaction in the vein wall in the stasis-induced thrombosis and mouse-to-rat xenograft models and reduced HMGB1 levels in LPS-treated mice. These results indicate that transgenic expression of hTBM has anticoagulant and antiinflammatory effects that are graft-protective in murine models.
Keywords: Acute, clotting factors, coagulation, coagulation factors, rejection, thrombosis, transgenic, vascular, xenotransplantation
Introduction
The contribution of thrombosis to the pathogenesis of rejection of vascularized organ xenografts is well established. It varies from being florid, immediate and pan-vascular in hyperacute rejection (HAR) occurring as a consequence of high titer antibodies against endothelial antigens in nonmanipulated recipients of unmodified grafts, to subtle, delayed and microvascular in well immunosuppressed recipients of genetically modified grafts lacking αGal or expressing supraphysiological levels of complement regulators. Although these genetic modifications of the pig donor have largely prevented HAR, there has been little progress in preventing graft loss due to acute vascular rejection (AVR) (also known as delayed xenograft rejection and acute humoral xenograft rejection) and failure of these grafts is inevitable within 6 months. Our group has demonstrated marked attenuation of consumptive coagulopathy and thrombosis and a near doubling of survival of nonimmunosuppressed pig to baboon kidney grafts with antithrombin III treatment (1), indicating the importance of thrombosis in this model, however others have been unsuccessful in obtaining lasting benefit with a variety of anticoagulants in immunosuppressed xenograft recipients (2,3).
These studies have improved our understanding of the pathogenesis of AVR including the antecedents and consequences of thrombosis, and discussion generally involves two main themes. First, as in severe AVR in allografts, thrombosis is predominantly a consequence of rejection of the endothelium (4), which appears to be largely due to antiendothelial antibody and possibly to various cellular immune mechanisms. Shimizu (5–8) analyzed the pathology of renal and cardiac xenografts performed by the Boston group and showed that microvascular thrombosis correlated strongly with markers of humoral rejection and Byrne et al. (3) found that the level of immunosuppression was a major determinant of graft loss due to microthrombotic delayed xenograft rejection. Previous studies had indicated that endothelial damage results in loss of its natural anticoagulant properties, including loss of coagulation regulators, and expression of a procoagulant phenotype (see Robson et al. [9], for review). These observations strongly suggest that prevention and/or control of rejection is essential to prevent the development of thrombosis. However, as a prescription this alone may be insufficient and is yet to be achieved. Yamada et al. (10) used a highly effective immunosuppressive/protolerogenic protocol that prevented graft loss due to rejection but nevertheless was unable to completely prevent microvascular thrombosis.
The second theme concerns the contribution of molecular incompatibilities between various receptors or ligands on the porcine endothelium and their partner molecules in the circulating primate blood. Two such incompatibilities which have been demonstrated by in vitro studies may be relevant: porcine von Willebrand factor (vWF) can bind human GP1b and activate human platelets without the need for sheer stress (11), and pig thrombomodulin is able to bind human thrombin but is a poor cofactor for the activation of human protein C (12,13). The importance of the latter incompatibility and the consequent inability to efficiently generate activated Protein C (aPC) at sites of endothelial injury can be surmised from the tendency to spontaneous venous thrombosis of people with Protein C deficiency (14).
A further factor which may also contribute is the relative lack of expression of endothelial protein C receptor (EPCR) on small vessels (15), which are the focus of microvascular thrombosis. EPCR is an amplifier of aPC generation by the thrombomodulin–thrombin complex (16) and the relatively small amount of EPCR in these vessels may explain why they are the focus of rejection-induced thrombosis. Indeed, these small vessels may be particularly vulnerable. Even low-level perturbation of xenograft microvascular endothelium made thrombophilic by porcine vWF, and probably lacking any ability to generate aPC because of the dual effect of porcine TBM incompatibility and lack of EPCR, is likely to precipitate thrombosis. These considerations have led us to suggest that an array of corrective and therapeutic transgenes will be needed to overcome this barrier to clinical application of xenotransplantation (17,18).
This study was undertaken to assess whether transgenic expression of human thrombomodulin (hTBM) might prove sufficiently effective in preventing thrombosis in a small animal model to support the inclusion of hTBM in multi-transgenic pigs as potential organ xenograft donors. It was first necessary to validate the mouse as a model by demonstrating that hTBM was functional in mice, that is, able to bind mouse thrombin, bind mouse EPCR and activate mouse Protein C. The data presented indicate that the model is valid and that transgenically expressed hTBM is capable of preventing thrombosis in two clinically relevant models.
Materials and Methods
Construction of the transgene and generation of hTBM-Tg mice
A 3.7-kb hTBM cDNA was cloned as an EcoRI fragment into a plasmid containing the murine H-2Kb promoter. A 9.0-kb fragment containing the promoter, intron, hTBM cDNA and polyadenylation signal was excised from the vector backbone using XhoI/NotI and microinjected at 10 ng/L into fertilized C57BL/6 mouse oocytes. Seventeen transfers, yielded 12 pregnancies and 54 live born pups. Tail genomic DNA was prepared from the offspring with a DNA extraction kit (Invitrogen, CA) and genotyped by PCR using hTBM-specific primers 5′-CCCTCGGCTTACAGCTAATG-3′ and 5′-CCGCACAGGTGCCAGATGTT-3′, which generated a 551 base pair (bp) product (19). PCR screening identified seven transgenics (13%). Three of these expressed hTBM strongly on PBL by flow cytometry and were used to establish independent lines, with the transgene transmitted at the expected Mendelian frequency in each case. The line with the highest expression of hTBM by flow cytometry was used for all experiments described in this study.
The mice were maintained in individually ventilated cages in a specific pathogen-free environment and bred on a C57BL/6 background. Male hTBM-Tg and wild-type (WT) littermates 10–12 weeks old and weighing 25.5 ± 5 g were used for experiments. All experiments were performed with the observer blinded to the genotype.
Flow cytometry
Mouse peripheral blood leukocytes were incubated with mouse anti-hTBM mAb-FITC (1:100) and analyzed on a FACScalibur flow cytometer (Becton Dickinson, NJ).
Tail bleeding times were performed under anesthesia as previously described (20).
APC generation
106 WT or hTBM-Tg thymocytes were incubated with 0.125 U/mL of human thrombin and 0–16 μg/mL human PC (Calbiochem/Merck, Sydney, Australia) at 37°C for 30 min. The reaction was terminated by addition of 2 U/mL hirudin and the cells pelleted. Spectrozyme APC (100 μL of 1 mM) (American Diagnostica, CT) was added to 100 μL of the supernate in a 96-well flat-bottom plate, and the change in absorbance at 405 nm was read at 37°C using a FLUOStar Galaxy kinetic plate reader (BMG Labtech, Germany). The concentration of aPC generated was calculated using a standard curve of 0–60 nM aPC. In vitro assays for hTBM cofactor activity for murine thrombin activation of PC were also performed. Triplicate samples of COS-7 cells stably expressing hTBM or parental COS-7 cells were cultured with either mouse thrombin and mouse PC or human thrombin and human PC.
Model of collagen-induced thrombosis
Acute thrombosis was induced by injection of collagen (0.075 μg/g) (DKSH, Hallam, Australia) into the external jugular vein of anesthetized mice. Survival was determined at 1 h when survivors were sacrificed. Lungs were recovered, perfused with ice-cold normal saline and histological sections were examined after staining with H&E. Six random nonoverlapping fields from six mice per group were captured and digitized using a BX50 microscope attached to a Fujix HC5000 digital camera. Vessel size was determined using an Analytical Imaging Station Version 6.0 (AIS, Ontario, Canada). Vessel thrombosis is expressed as a percentage of total large vessels (>1000 μm2). We have previously demonstrated that this model is characterized by pulmonary artery thrombosis and pulmonary embolism (21).
Model of inferior vena caval thrombosis
Venous thrombosis was generated using the method of Myers et al. (22). The IVC was ligated immediately inferior to the entry of the renal veins and groups of mice were euthanased at 12, 24 and 48 h. The IVC was dissected and excised between the renal pedicle and the iliac branches and was weighed (g) and its length (cm) was measured, and the weight/length ratio calculated. Blood vessels were identified using anti-vWF antibody in paraffin-embedded sections (23), and sequential sections were used to determine the extent of thrombosis using martius/scarlet/blue (MSB) stain (magnification 200×) to identify fibrin (24). All blood vessels were counted in 10 independent fields from each group of mice (n = 6) and the clotted blood vessels were presented as percentages.
Vein wall morphometrics
Five contiguous representative high-power fields (HPFs; oil immersion, 1000×) around the vein wall of H&E stained paraffin sections were examined and the vein wall infiltrating cell count was determined. Neutrophils and monocytes were identified by standard histological criteria. Total cell counts from the five HPFs were summed for each section studied and mean ± SEM were calculated for each group.
HMGB1 assays
Mice were injected with a 4 mg/kg dose of LPS (Escherichia coli 026:B6; Sigma-Aldrich Chemical, St. Louis, MO) via the tail vein. Blood for plasma was collected 2.5 h following the injection and analyzed via ELISA for HMGB1 concentration (Shino-TEST, Kanzawa, Japan).
Cardiac transplants
Hearts from hTBM-Tg, CD31-TFPI-Tg (a kind gift of A. Dorling and D. Chen, Imperial College London, UK) (25) and WT littermate controls at 8–12 weeks of age were transplanted heterotopically into Lewis rat recipients (150–200 g). Heart function was monitored daily by abdominal palpation. Rejected hearts were excised, embedded in OCT and sections were stained with TCR αβ clone R73 (Serotec) and developed by the peroxidise anti-peroxidase technique as previously described (26). Infiltrating T cells were counted in six random HPFs per cardiac section. Some rats received cyclosporine A (CsA) (kind gift of Novartis Pharmaceuticals, Basel, Switzerland) by gavage at 20 mg/kg from day 1 to day 4 followed by 10 mg/kg daily until the end of the experiment. In all groups, n = 6 transplants.
For further information about antibodies used, histopathology, statistical methods and ethical approvals, see Supporting Information.
Results
Phenotype of hTBM transgenic mice
hTBM transgenic mice were indistinguishable from WT littermates in appearance, size, level of activity and fertility. There was no spontaneous bleeding diathesis and no excess bleeding was observed with parturition or during any experimental procedures. The mice had normal full blood counts, renal function, prothrombin and activated partial thromboplastin times, collagen-induced platelet aggregation and thrombin-induced platelet expression of P-selectin (CD62P) (see Supporting Information).
Consistent with previous experience using the H-2Kb promoter (27), expression of the transgene was directed to multiple tissue types including leucocytes, permitting screening of the mice by flow cytometry (Figure 1A). Expression on platelets was minimal (data not shown). hTBM was strongly expressed on the vascular endothelium of lung and heart and poorly expressed on cardiomyocytes and bronchial cells (Figures 1C and E).
Figure 1. (A) FACS profile of hTBM expression on leucocytes of WT and hTBM-Tg mice.

(B–E). Expression of hTBM on lung (B and C) and heart (D and E) of WT (B and D) and hTBM transgenic mice (C and E). Arrows point to the endothelium of small arteries.
Transgenically expressed hTBM is functional
Thymocytes were isolated from transgenic mice and littermate controls and incubated with human thrombin and protein C. The hTBM-Tg thymocytes generated higher levels of aPC than WT thymocytes at all concentrations of protein C tested (Figure 2A). In vitro assays using stable transfectants expressing hTBM showed that cofactor activity for aPC generation was similar using either mouse or human thrombin and protein C (Figure 2B), indicating the absence of any major molecular incompatibility across the species. This was supported in vivo by the significant prolongation of tail-tip bleeding time in hTBM-Tg mice compared to WT mice (Figure 2C). In contrast to WT mice, primary hemostasis in the hTBM-Tg mice was followed rapidly by reactionary or secondary hemorrhage.
Figure 2. (A) APC generation from PC by WT and hTBM-Tg thymocytes (n = 2 in each group, p.

< 0.05).
Representative of three similar experiments. (B) APC generation by hTBM from mouse and human PC and with mouse and human thrombin as a cofactor. (C) Tail bleeding times of WT and hTBM-Tg mice.
Transgenically expressed hTBM protects against pulmonary arterial thromboembolism
The prothrombotic agent collagen was administered via the jugular vein to determine whether the expression of hTBM would confer protection against systemically induced acute thrombosis. Immediately following injection both WT and transgenic mice developed cardiorespiratory compromise, however the hTBM-Tg mice recovered promptly and demonstrated significantly greater 1 h survival (82%) than WT mice (47%) (p = 0.04; Figure 3A). Histological examination of the lungs from hTBM-Tg mice revealed less thrombosis of small pulmonary arteries compared to lungs from WT mice (p = 0.04; Figures 3B and C).
Figure 3. IV collagen-induced thrombosis model.

(A) Survival analysis. (B) Quantification of pulmonary emboli in large vessels (surface area >1000 μm2). (C) Lung histopathology showing pulmonary emboli (H&E stain, 200× magnification).
Transgenically expressed hTBM protects against venous thrombosis
The effect of hTBM-Tg expression was assessed in a stasis-induced model of venous thrombosis/thromboembolism. The inferior vena cava (IVC) was ligated immediately below the renal veins and the extent of thrombosis was determined in a defined distal IVC segment, from the renal pedicle to the iliac bifurcation, after 12, 24 and 48 h. Thrombosis was significantly reduced in hTBM-Tg mice compared to WT mice at 12 h (0.98 ± 0.09 g/cm vs. 1.72 ± 0.07 g/cm, respectively, p < 0.001; Figure 4A). Differences at later time points were not significant.
Figure 4. Stasis-induced venous thrombosis model.

(A) Quantification of thrombosis at 12, 24 and 48 h in WT and hTBM-Tg mice. (B) Representative lung histopathology (MSB stain 200× magnification). (C) Quantification of pulmonary emboli in large vessels (surface area >1000 μm2).
There was also a significant difference (p = 0.015) in the 48 h mortality between WT (7/27; 26%) and hTBM-Tg (0/20).
Histopathology of lungs recovered at 48 h showed significantly more thromboembolism of large pulmonary vessels (>1000 μm2) in WT mice (80.6 ± 6.5%) compared to hTBM-Tg mice (43.3 ± 9.3%) (p = 0.017; Figures 4B and C). Sham-operated mice exhibited a 35.6 ± 8.0% occlusion rate despite gently flushing all lungs at the time of recovery, indicating a contribution of postmortem coagulation. These results suggest that pulmonary emboli, originating from thrombus developing proximal to the IVC ligation, were the cause of death, and that transgenic hTBM expression protects against acute thrombosis and thromboembolism in this stasis-induced model of venous thrombosis.
Transgenically expressed hTBM reduces the inflammatory infiltrate after IVC ligation
Thrombosis incites an inflammatory response in the vessel wall, which in turn exacerbates clot formation. Since TBM regulates both coagulation and inflammation (28), we investigated whether there were differences in thrombus-induced vessel wall inflammation in the IVC ligation model. The hTBM-Tg cohort showed a significant reduction in neutrophil and monocyte cell infiltrates compared to WT mice at 48 h, while monocyte numbers were also reduced at 24 h (Figure 5). These data are consistent with hTBM-Tg mediating an antiinflammatory action in this model.
Figure 5. Stasis-induced venous thrombosis model: vena cava histopathology.

(A) Representative histopathology (H&E stain, magnification 200×) of WT and hTBM-Tg vena cava showing thrombosis-associated inflammation. (B and C) Quantitation of the inflammatory infiltrate (B and C, neutrophils and monocytes, respectively) at 24 and 48 h (Sham: striped bars; WT: white bars; hTBM-Tg: black bars).
Transgenically expressed hTBM reduces HMGB1 levels
To further examine the antiinflammatory properties of TBM, levels of HMGB1 were examined following the induction of sepsis using LPS (29). HMGB1 is secreted by activated monocytes or macrophages (29) and released by necrotic or damaged cells, elevated levels of HMGB1 have been shown to correlate with the level of severity of sepsis in animal models (29). The plasma level of HMGB1 measured 2.5 h postinjection of LPS was significantly reduced in HTBM mice (8.6 ng/mL ± 0.4) versus WT mice (19.9 ng/ml ± 4.1) (Figure 6; p = 0.029).
Figure 6. Sepsis model. Mice were treated with 4 mg/kg of LPS Escherichia coli 026:B6 and plasma collected at 2.5 h for HMGB1 ELISA.
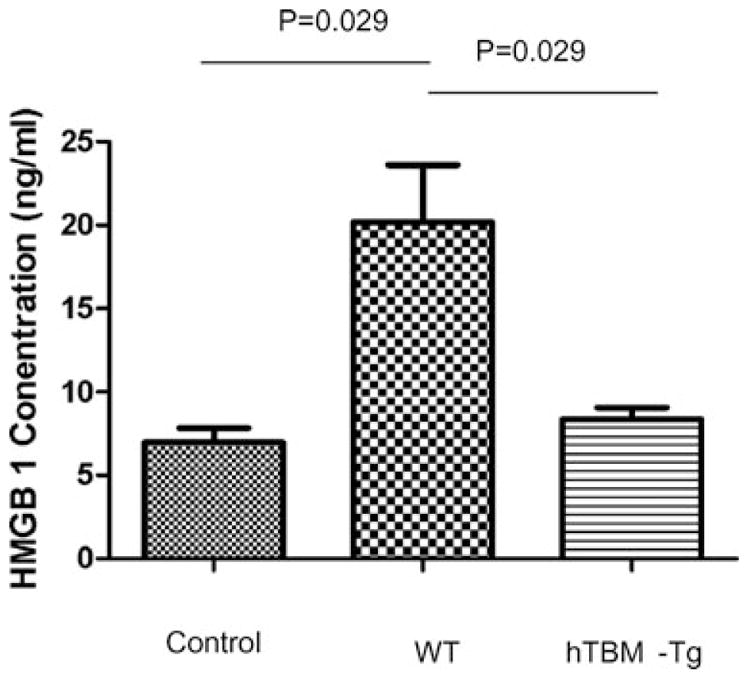
Saline-treated wild type control group n = 3, wild type and transgenic LPS-treated groups, each n = 4.
Transgenically expressed hTBM prolongs cardiac graft survival and reduces infiltrating T cells
The effect of hTBM transgene expression was examined in the concordant mouse-to-rat cardiac xenograft model, which does not model HAR (30) but shows the inflammatory (31) and thrombotic features (26,32,33) characteristic of AVR. Untreated rats rejected WT mouse heart xenografts within 3 days (median 2.5 days) (Figure 7A) and these showed extensive small vessel thrombosis and tissue disruption (data not shown). Survival of untreated hTBM-Tg hearts was similar (up to 4 days; median 3.3 days) and CD31-TFPI-Tg hearts, which express human TFPI, survived up to 5 days (median 4.7 days) (Figure 7A). Rejected hTBM-Tg donor hearts contained fewer infiltrating cells (p = 0.026) and rejected CD31-TFPI-Tg hearts contained more infiltrating cells (p = 0.003) than rejected WT hearts (Figure 7C).
Figure 7. Mouse-to-rat cardiac xenograft model.

(A) Graft survival of untreated WT hearts (dotted line), hTBM-Tg hearts (solid line) and CD31-TFPI-Tg hearts (heavy dotted line); N = 6 in all groups. (B) Graft survival of cyclosporin-treated WT hearts (dotted line), hTBM-Tg hearts (solid line) and CD31-TFPI-Tg hearts (heavy dotted line); N = 6 in all groups. (C) T-cell infiltrate in rejected hearts with and without cyclosporine therapy (WT: white bars; hTBM-Tg: black bars and CD31-TFPI-Tg: striped bars).
CsA treatment of rats receiving WT hearts did not improve graft survival (median = 2.4 days) (Figure 7B). However, CsA treatment of hTBM-Tg hearts recipients resulted in significant prolongation of the time to rejection (median = 4.3 days) (p = 0.016; Figure 7B). In keeping with the action of CsA as a modulator of effector T-cell function, fewer infiltrating T cells were evident in both WT and hTBM-Tg grafts (Figure 7C) in the treatment arm. CsA treatment did not significantly prolong the survival of CD31-TFPI-Tg cardiac xenografts (median = 6.5 days) (Figure 7B).
Discussion
AVR is an inflammatory process consequent upon endothelial damage and characterized by thrombosis. Its pathogenesis involves rejection as the prime driver of endothelial damage, initiating a vicious cycle of loss of the natural endothelial antithrombotic mechanisms and consequent thrombosis which itself is highly pro-inflammatory. The process may be exacerbated by molecular incompatibilities in the thrombomodulin–Protein C–endothelial Protein C receptor (TBM–PC–EPCR) system (12,13), which with other natural antithrombotic/antiinflammatory systems such as the anti-purinergic CD39–CD73 (21,34) system and TFPI (35) normally opposes and limits thrombosis.
Clearly prevention of endothelial damage, that is, prevention of rejection, will be the mainstay of preventing graft thrombosis. This rationale is supported by the finding that more efficient immunosuppression is beneficially associated with less evidence of micro-thrombosis and improved graft survival (3). However, even when the antirejection regimen is vigorous to the point of largely suppressing other manifestations of rejection, micro-thrombosis still occurs (10). The basic postulate of this study is that the transgenic expression of therapeutic levels of regulators of thrombosis and inflammation such as hTBM may be required to prevent induced thrombosis, particularly in the absence of effective natural systems. A parallel exists with the markedly beneficial effect of expression of complement regulators at therapeutic levels, that is, levels beyond those required to correct inter-species molecular incompatibilities, where HAR can be prevented even in the presence of undiminished anti-αGal–αGal interactions (36–38). Indeed, the parallel extends to the associated antiinflammatory effects of the regulators of complement and coagulation/thrombosis (39).
The TBM–PC–EPCR system is a prime candidate for manipulation by therapeutic level transgenic expression of either or both of the cell bound components. Expression of hTBM is particularly appealing as it should correct the molecular incompatibility as well as providing therapeutic anticoagulant and antiinflammatory effects. This study sought to characterize the hTBM transgenic mouse and to subject it to various models, which might indicate whether this genetic modification was worth transferring to the pig.
It demonstrated that the hTBM transgenic mouse was viable, reproduced normally and did not have a spontaneous bleeding diathesis, suggesting that it should be possible to generate a viable hTBM pig without a deleterious phenotype. Consistent with our previous experience with a variety of other transgenes expressed under the control of the H2-Kb promoter (27), hTBM was expressed widely but most importantly it was expressed strongly on vascular endothelium including that of the microvasculature (Figure 1). It was also important to demonstrate that hTBM was capable of working effectively in the mouse in concert with mouse thrombin and Protein C, otherwise the functional mouse models would be invalidated. Fortunately, the molecular incompatibility which is known to exist in this system between pig and human (12,13) is not evident between mouse and human. We confirmed that hTBM binds mouse thrombin, and is an equivalent cofactor for aPC generation in this setting (Figures 2A and B), allowing the effect of hTBM transgene expression to be assessed in in vivo models in the mouse.
hTBM mice show minor hemostatic issues, indicated by prolonged bleeding times, which imparted a survival advantage in models of thrombosis and transplantation while not effecting viability. The expression of hTBM did not result in excessive systemic anticoagulation as evidenced by normal hematological coagulation parameters. Interestingly, in the tail bleeding assays, primary hemostasis was obtained in all mice, but unlike the WT mice, hTBM-Tg mice were prone to reactionary or secondary hemorrhage. This may be explained by the fact that TBM acts at a stage beyond formation of the platelet plug via the generation of aPC, which regulates the speed and strength of clot formation.
The impact of thrombomodulin on the tail bleeding assay is in keeping with the results of several groups (40,41). Aoki demonstrated a prolongation of bleeding times using rhs-TBM, although the doses required were in excess of that needed to prevent tissue factor-induced disseminated intravascular coagulation in rats (41). Moreover, rhs-TBM has been shown to decrease the speed of clot growth after the start of coagulation (42).
hTBM-Tg mice demonstrated an increased survival rate after intrajugular collagen compared to WT mice, in keeping with reports that thrombomodulin from mouse lung inhibits thrombin-induced thromboembolism (43), and that intravenous rhs-TBM is highly effective for inhibiting thrombin-induced pulmonary thromboembolism in mice (44). Similarly, in the IVC ligation model, there was less thrombus formation in the IVC below the ligature as evidenced by lower weight:length ratio in the hTBM-Tg mice at 12 h, suggesting a direct anticoagulant effect of enhanced hTBM expression. The antiinflammatory effect of TBM was apparent as a significant reduction in IVC wall leukocyte infiltration in the hTBM-Tg mice at 24 and 48 h. Given that the actual clot burden was not different at these times, a direct antiinflammatory effect of hTBM is implicated. This is consistent with the effect obtained using rhs-TBM in models of DIC (45). The antiinflammatory effect inherent to TBM was further substantiated in a model of LPS sepsis. HMGB1 is a prognostic indicator for survival in endotoxemia (46). Recent studies have demonstrated that the N-terminal lectin domain of TBM binds to HMGB1, preventing in vitro leukocyte activation and in vivo LPS-induced lethality (47). Hence we sought to assess whether TG expression of TBM in a sepsis model of inflammation could reduce the plasma level of HMGB1. Our finding of reduced HMGB1 levels in hTBM-Tg mice following LPS sepsis implies an antiinflammatory effect of transgene expression.
The increased resistance to intravascular thrombosis exhibited by the hTBM-Tg mice in these in vivo studies in the absence of any demonstrable effect on in vitro platelet activity, hematological and clotting parameters suggests an effect of hTBM expression within the target tissue, a hypothesis supported by the transplantation studies. Transgenic expression of hTBM on donor hearts in conjunction with systemic immunosuppression conferred a survival advantage in the transplantation model. In the absence of immunosuppression, hTBM-Tg graft survival was not significantly prolonged but was associated with a reduction in the myocardial T-cell infiltrate. CsA treatment resulted in a significant prolongation of hTBM-Tg graft survival and reduced the T-cell infiltrate in both trans-genic and nontransgenic hearts. The effect on graft survival was not as marked as that observed in a previous study (48) using the CD31-TFPI-Tg transgenic mice, which express TFPI on the endothelium once these cells are activated (48). We used CD31-TFPI-Tg mice as a positive control in this model but were unable to replicate the reported CD31-TFPI-Tg graft survival of >100 days with CsA immunosuppression.
In this concordant xenograft model, rejection is humorally mediated by both T-cell-dependent (CsA-sensitive) IgG and T-cell-independent (CsA-insensitive) IgM xenoantibodies. One possible explanation is a higher level of IgM xenoantibodies in our rats, which were conventionally housed in contrast to the SPF housing used for rats in the earlier study. The observation that CD31-TFPI-Tg graft survival in untreated recipients was 5 days in our hands but 7 days in that study (25) is consistent, at least in part, with this explanation.
Our laboratory and others have generated pigs with one or more genetic modifications to prevent HAR, such as deletion of the α1,3 galactosyltransferase gene and/or expression of human complement regulators, however organs from these are still susceptible to rejection associated with micro-thrombosis. The data presented here support the addition of an hTBM transgene to this array in an attempt to overcome this barrier to clinical xenotransplantation of vascularized organs.
Supplementary Material
Acknowledgments
Funding sources: This work was supported by the NHMRC (Australia) and NIH (SCR). S.C. was the recipient of an NHMRC postgraduate research scholarship.
Footnotes
The following supporting information is available for this article online:
Response baseline hematological and biochemical profile of the hTBM Tg mouse.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Cowan PJ, Aminian A, Barlow H, et al. Protective effects of recombinant human antithrombin III in pig-to-primate renal xenotransplantation. Am J Transplant. 2002;2:520–525. doi: 10.1034/j.1600-6143.2002.20605.x. [DOI] [PubMed] [Google Scholar]
- 2.Cozzi E, Simioni P, Boldrin M, et al. Effects of long-term administration of high-dose recombinant human antithrombin in immunosuppressed primate recipients of porcine xenografts. Transplantation. 2005;80:1501–1510. doi: 10.1097/01.tp.0000178377.55615.8b. [DOI] [PubMed] [Google Scholar]
- 3.Byrne GW, Davies WR, Oi K, et al. Increased immunosuppression, not anticoagulation, extends cardiac xenograft survival. Transplantation. 2006;82:1787–1791. doi: 10.1097/01.tp.0000251387.40499.0f. [DOI] [PubMed] [Google Scholar]
- 4.Shimizu A, Meehan SM, Kozlowski T, et al. Acute humoral xenograft rejection: Destruction of the microvascular capillary endothelium in pig-to-nonhuman primate renal grafts. Lab Invest. 2000;80:815–830. doi: 10.1038/labinvest.3780086. [DOI] [PubMed] [Google Scholar]
- 5.Shimizu A, Yamada K, Yamamoto S, et al. Thrombotic microangiopathic glomerulopathy in human decay accelerating factor-transgenic swine-to-baboon kidney xenografts. J Am Soc Nephrol. 2005;16:2732–2745. doi: 10.1681/ASN.2004121148. [DOI] [PubMed] [Google Scholar]
- 6.Shimizu A, Yamada K. Pathology of renal xenograft rejection in pig to non-human primate transplantation. Clin Transplant. 2006;20(Suppl 15):46–52. doi: 10.1111/j.1399-0012.2006.00550.x. [DOI] [PubMed] [Google Scholar]
- 7.Shimizu A, Colvin RB. Pathological features of antibody-mediated rejection. Curr Drug Targets Cardiovasc Haematol Disord. 2005;5:199–214. doi: 10.2174/1568006054064744. [DOI] [PubMed] [Google Scholar]
- 8.Hisashi Y, Yamada K, Kuwaki K, et al. Rejection of cardiac xenografts transplanted from alpha1,3-galactosyltransferase gene-knockout (GalT-KO) pigs to baboons. Am J Transplant. 2008;8:2516–2526. doi: 10.1111/j.1600-6143.2008.02444.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robson S, Cooper D, d’Apice A. Disordered regulation of coagulation and platelet activation in xenotransplantation. Xenotransplantation. 2001;7:166–176. doi: 10.1034/j.1399-3089.2000.00067.x. [DOI] [PubMed] [Google Scholar]
- 10.Yamada K, Yazawa K, Shimizu A, et al. Marked prolongation of porcine renal xenograft survival in baboons through the use of a1,3-galactosyltransferase gene-knockout donors and the cotransplantation of vascularized thymic tissue. Nature Med. 2005;11:32–34. doi: 10.1038/nm1172. [DOI] [PubMed] [Google Scholar]
- 11.Schulte am EJ, Cruz MA, Siegel JB, Anrather J, Robson SC. Activation of human platelets by the membrane-expressed A1 domain of von Willebrand factor. Blood. 1997;90:4425–4437. [PubMed] [Google Scholar]
- 12.Siegel JB, Grey ST, Lesnikoski B, et al. Xenogeneic endothelial cells activate human prothrombin. Transplantation. 1997;64:888–896. doi: 10.1097/00007890-199709270-00017. [DOI] [PubMed] [Google Scholar]
- 13.Roussel JC, Moran CJ, Salvaris EJ, Nandurkar HH, d’Apice AJF, Cowan PJ. Pig thrombomodulin binds human thrombin but is a poor cofactor for activation of human protein C and TAFI. Am J Transplant. 2008;8:1101–1112. doi: 10.1111/j.1600-6143.2008.02210.x. [DOI] [PubMed] [Google Scholar]
- 14.Broekmans AW, Veltkamp JJ, Bertina RM. Congenital protein C deficiency and venous thromboembolism. A study of three Dutch families. New Engl J Med. 1983;309:340–344. doi: 10.1056/NEJM198308113090604. [DOI] [PubMed] [Google Scholar]
- 15.Laszik Z, Mitro A, Taylor FB, Jr, Ferrell G, Esmon CT. Human protein C receptor is present primarily on endothelium of large blood vessels: Implications for the control of the protein C pathway. Circulation. 1997;96:3633–3640. doi: 10.1161/01.cir.96.10.3633. [DOI] [PubMed] [Google Scholar]
- 16.Taylor FB, Jr, Peer GT, Lockhart MS, Ferrell G, Esmon CT. Endothelial cell protein C receptor plays an important role in protein C activation in vivo. Blood. 2001;97:1685–1688. doi: 10.1182/blood.v97.6.1685. [DOI] [PubMed] [Google Scholar]
- 17.Cowan PJ. Coagulation and the xenograft endothelium. Xenotransplantation. 2007;14:7–12. doi: 10.1111/j.1399-3089.2006.00368.x. [DOI] [PubMed] [Google Scholar]
- 18.Cowan PJ, d’Apice AJF. The coagulation barrier in xenotransplantation: incompatibilities and strategies to overcome them. Curr Opin Organ Transplant. 2008;13:178–183. doi: 10.1097/MOT.0b013e3282f63c74. [DOI] [PubMed] [Google Scholar]
- 19.Dwyer K. Intravascular Coagulation in Discordant Xenotransplantation. University Of Melbourne; Australia: 2003. [Google Scholar]
- 20.Dejana E, Callioni A, Quintana A, de Gaetano G. Bleeding time in laboratory animals. II–A comparison of different assay conditions in rats. Thromb Res. 1979;15:191–197. doi: 10.1016/0049-3848(79)90064-1. [DOI] [PubMed] [Google Scholar]
- 21.Dwyer KM, Robson SC, Nandurkar HH, et al. Thromboregulatory manifestations in human CD39 transgenic mice and the implications for thrombotic disease and transplantation. J Clin Invest. 2004;113:1440–1446. doi: 10.1172/JCI19560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Myers DD, Hawley AE, Farris DM, et al. P-selectin and leukocyte microparticles are associated with venous thrombogenesis. J Vasc Surg. 2003;38:1075–1089. doi: 10.1016/s0741-5214(03)01033-4. [DOI] [PubMed] [Google Scholar]
- 23.Galbusera M, Zoja C, Donadelli R, et al. Fluid shear stress modulates von Willebrand factor release from human vascular endothelium. Blood. 1997;90:1558–1564. [PubMed] [Google Scholar]
- 24.Lendrum AC, Fraser DS, Slidders DS, Henderson R. Studies on the character and staining of fibrin. J Clin Pathol. 1962;15:401–413. doi: 10.1136/jcp.15.5.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen D, Weber M, McVey JH, et al. Complete inhibition of acute humoral rejection using regulated expression of membrane-tethered anticoagulants on xenograft endothelium. Am J Transplant. 2004;4:1958–1963. doi: 10.1111/j.1600-6143.2004.00625.x. [DOI] [PubMed] [Google Scholar]
- 26.Bersztel A, Tufveson G, Gannedahl G, Johnsson C. A morphological sequential study of mouse-to-rat cardiac xenografts. Scand J Immunol. 1998;48:485–490. doi: 10.1046/j.1365-3083.1998.00306.x. [DOI] [PubMed] [Google Scholar]
- 27.Cowan PJ, Shinkel TA, Aminian A, et al. High-level co-expression of complement regulators on vascular endothelium in transgenic mice: CD55 and CD59 provide greater protection from human complement-mediated injury than CD59 alone. Xenotransplantation. 1998;5:184–190. doi: 10.1111/j.1399-3089.1998.tb00026.x. [DOI] [PubMed] [Google Scholar]
- 28.Van de WM, Collen D, Conway EM. Thrombomodulin-protein C-EPCR system: Integrated to regulate coagulation and inflammation. Arterioscler Thromb Vasc Biol. 2004;24:1374–1383. doi: 10.1161/01.ATV.0000134298.25489.92. [DOI] [PubMed] [Google Scholar]
- 29.Wang H, Yang H, Czura CJ, Sama AE, Tracey KJ. HMGB1 as a late mediator of lethal systemic inflammation. Am J Respir Crit Care Med. 2001;164(10 Pt 1):1768–1773. doi: 10.1164/ajrccm.164.10.2106117. [DOI] [PubMed] [Google Scholar]
- 30.Soares MP, Lin Y, Anrather J, et al. Expression of heme oxygenase-1 can determine cardiac xenograft survival. Nat Med. 1998;4:1073–1077. doi: 10.1038/2063. [DOI] [PubMed] [Google Scholar]
- 31.Lorant T, Engstrand M, Tufveson G, Johnsson C. Isolation of mouse-to-rat cardiac xenograft-infiltrating cells by ex vivo propagation. Xenotransplantation. 2002;9:209–219. doi: 10.1034/j.1399-3089.2002.01060.x. [DOI] [PubMed] [Google Scholar]
- 32.Gannedahl G, Fellstrom B, Larsson E, Roos-Engstrand E, Tufveson G. Characteristics of mouse to rat xenograft heart transplantation. Eur Surg Res. 1990;22:206–212. doi: 10.1159/000129102. [DOI] [PubMed] [Google Scholar]
- 33.Murase N, Starzl TE, Demetris AJ, et al. Hamster-to-rat heart and liver xenotransplantation with FK506 plus antiproliferative drugs. Transplantation. 1993;55:701–707. doi: 10.1097/00007890-199304000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dwyer KM, Deaglio S, Crikis S, et al. Salutary roles of CD39 in transplantation. Transplantation Reviews. 2007;21:54–63. [Google Scholar]
- 35.Lwaleed BA, Bass PS. Tissue factor pathway inhibitor: Structure, biology and involvement in disease. J Pathol. 2006;208:327–339. doi: 10.1002/path.1871. [DOI] [PubMed] [Google Scholar]
- 36.Cowan P, Aminian A, Barlow H, et al. Renal xenografts from triple-transgenic pigs are not hyperacutely rejected but cause coagulopathy in non-immunosuppressed baboons. Transplantation. 2000;69:2504–2515. doi: 10.1097/00007890-200006270-00008. [DOI] [PubMed] [Google Scholar]
- 37.Byrne G, McCurry KR, Martin MJ, McClellan SM, Platt JL, Logan JS. Transgenic pigs expressing human CD59 and decay-accelerating factor produce an intrinsic barrier to complement-mediated damage. Transplantation. 1997;63:149–155. doi: 10.1097/00007890-199701150-00027. [DOI] [PubMed] [Google Scholar]
- 38.McGregor CGA, Teotia SS, Byrne GW, et al. Cardiac transplantation: Progress toward the clinic. Transplantation. 2005;78:1569–1575. doi: 10.1097/01.tp.0000147302.64947.43. [DOI] [PubMed] [Google Scholar]
- 39.Cowan PJ, d’Apice AJ. Complement activation and coagulation in xenotransplantation. Immunol Cell Biol. 2009 doi: 10.1038/icb.2008.107. advance online publication, 20 January 2009. [DOI] [PubMed] [Google Scholar]
- 40.Nawa K, Itani T, Ono M, Sakano K, Marumoto Y, Iwamoto M. The glycosaminoglycan of recombinant human soluble thrombomodulin affects antithrombotic activity in a rat model of tissue factor-induced disseminated intravascular coagulation. Thromb Haemost. 1992;67:366–370. [PubMed] [Google Scholar]
- 41.Aoki Y, Takei R, Mohri M, et al. Antithrombotic effects of recombinant human soluble thrombomodulin (rhs-TM) on arteriovenous shunt thrombosis in rats. Am J Hematol. 1994;47:162–166. doi: 10.1002/ajh.2830470303. [DOI] [PubMed] [Google Scholar]
- 42.Mohri M, Suzuki M, Sugimoto E, Sata M, Yamamoto S, Maruyama I. Effects of recombinant human soluble thrombomodulin (rhs-TM) on clot-induced coagulation in human plasma. Thromb Haemost. 1998;80:925–929. [PubMed] [Google Scholar]
- 43.Kumada T, Dittman WA, Majerus PW. A role for thrombomodulin in the pathogenesis of thrombin-induced thromboembolism in mice. Blood. 1988;71:728–733. [PubMed] [Google Scholar]
- 44.Gomi K, Zushi M, Honda G, et al. Antithrombotic effect of recombinant human thrombomodulin on thrombin-induced thromboembolism in mice. Blood. 1990;75:1396–1399. [PubMed] [Google Scholar]
- 45.Mohri M. ART-123: Recombinant human soluble thrombomodulin. Cardiovasc Drug Rev. 2000;18:312–325. [Google Scholar]
- 46.Nagato M, Okamoto K, Abe Y, Higure A, Yamaguchi K. Recombinant human soluble thrombomodulin decreases the plasma high-mobility group box-1 protein levels, whereas improving the acute liver injury and survival rates in experimental endotoxemia. Crit Care Med. 2009;37:2181–2186. doi: 10.1097/CCM.0b013e3181a55184. [DOI] [PubMed] [Google Scholar]
- 47.Abeyama K, Stern DM, Ito Y, et al. The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J Clin Invest. 2005;115:1267–1274. doi: 10.1172/JCI22782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dorling A, Chen D, Riesbeck K, et al. Regulated endothelial cell expression of novel anticoagulants: A strategy for the prevention and therapy of intravascular thrombosis. Transplant Proc. 2000;32:971. doi: 10.1016/s0041-1345(00)01067-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.