

Abstract
Background:
Neuroendocrine tumors (NET) originate from the diffuse neuroendocrine system. These can arise in almost every organ of the body, although they are most commonly found in the gastrointestinal tract and respiratory system. The skull base and sellar region are extremely rare sites for neuroendocrine carcinoma. Consequently, in this case, both diagnosis and definition of surgical goals, as well as further treatment strategies were challenging.
Case Description:
A 65-year-old woman was admitted to our Neurosurgery Department with a rapidly progressive visus reduction, drowsiness, polyuria, and polydipsia. Neuroimaging showed a sellar/suprasellar mass (diameter of 2 cm) with a heterogeneous signal compressing the optic chiasm and extending laterally toward the cavernous sinus. Differential diagnosis based on imaging included pituitary macroadenoma or metastasis. The patient underwent endoscopic endonasal transsphenoidal surgery. A total resection of the mass was impossible because of the infiltration of the optic chiasm and the intraoperative histological diagnosis of malignant epithelial neoplasm. Further histological evaluation revealed that the lesion was a NET with no other primary or metastatic sites detectable. Subsequently, the patient was successfully treated with fractioned stereotactic radiotherapy and polychemotherapy. Four years after the surgery, follow-up magnetic resonance imaging showed stability of the residual disease. Neurologic examination revealed a complete visual recovery.
Conclusions:
Primary pituitary NET, though rare, should be included in the differential diagnosis of sellar lesions. A multimodality treatment approach is needed. Finally, the present case highlights, that in the case of a pituitary lesion infiltrating the optic chiasm, including NET, the endoscopic endonasal transsphenoidal subtotal resection followed by fractioned stereotactic radiotherapy and chemotherapy may represent an effective and safe choice of treatment.
Keywords: Endoscopic endonasal transsphenoidal approach, neuroendocrine tumor, pituitary, sellar region tumors
INTRODUCTION
This case report describes a neuroendocrine tumor (NET) arising from enterochromaffin cells that underwent neoplastic transformation.[2] These tumors can arise in almost every organ of the body although they are most commonly found in the gastrointestinal tract and respiratory system.[8] The skull base and sellar region are extremely rare sites for NET.[3] To date, only 6 cases of primary intracranial NET have been reported in the literature.[1,2,3,4,5,6,7,8] In this scenario, both diagnosis and definition of surgical goals, as well as further treatment strategies are challenging.
We present here a case of a pathologically diagnosed isolated pituitary NET, which underwent endoscopic endonasal transsphenoidal subtotal resection. Serial prospective and extensive metastatic workup revealed no other primary lesions during a 4-year follow-up period, indicating this to be the primary site of involvement. Finally, clinical and pathological aspects, imaging findings, surgical strategies, and the outcome of NET are discussed in the light of the pertinent literature.
CASE DESCRIPTION
A 65-year-old woman was admitted to our department following a 2-week history of rapidly progressive visual reduction, drowsiness, polyuria, and polydipsia. Her medical history included a temporal arachnoid cyst, symptomatic with seizures, and VIII cranial nerve schwannoma treated with gamma-knife radiosurgery 2 years before. Her clinical examination revealed no neurological deficit other than bitemporal hemianopsia; there was no diarrhea or flushing. Magnetic resonance imaging (MRI) showed a sellar/suprasellar mass (diameter of 2 cm) with a heterogeneous signal compressing the optic chiasm and extending laterally toward the left cavernous sinus. The lesion was predominantly isointense to the gray matter on T1-weighted images (WI) and hyperintense on T2-WI [Figure 1a]. The images showed diffuse homogeneous enhancement following contrast administration [Figure 1b and c]. Interestingly, an MRI examination performed 2 years before, for follow-up after radiosurgery for vestibular schwannoma and frontal arachnoid cyst, demonstrated a normal pituitary gland [Figure 1d–f]. Differential diagnosis based on imaging included pituitary macroadenoma or metastasis. Endocrinological evaluation showed the following levels – thyroid-stimulating hormone 1.31 mU/ml, free thyroxine 3.9 pg/ml, free triiodothyronine 2.4 pg/ml, and prolactin (PRL) 77 ng/ml. The patient underwent endoscopic endonasal transsphenoidal surgery with the aid of neuronavigation. On initial inspection, the tumor resembled a pituitary adenoma. However, on entering the lesion, it had a firm and solid consistency and was not removable with suction. Therefore, the main part was dissected with curettes and removed with punch forceps. Moreover, the upper part of the lesion presented clear infiltration of the optic chiasm without a safe plane of cleavage [Figure 2a]. Because the intraoperative histological diagnosis was consistent with malignant epithelial neoplasm, and the lesion was firmly adherent to the optic chiasm, total removal was not possible, and after decompression of the optic chiasm, a small residual mass of tumor was left in place.
Figure 1.

(a) Preoperative magnetic resonance (MRI) coronal T2-weighted imaging demonstrating sellar/suprasellar mass (diameter of 2 cm) with a heterogeneous signal, mainly hyperintense, compressing the optic chiasm and extended laterally toward the left cavernous sinus. (b and c) Preoperative magnetic resonance coronal and sagittal contrast-enhanced T1-weighted imaging showing an intense and diffuse enhancement. (d-f) MRI imaging performed 2 years before showing normal pituitary/sellar signal (white arrow), the frontal arachnoid cyst and left vestibular schwannoma
Figure 2.
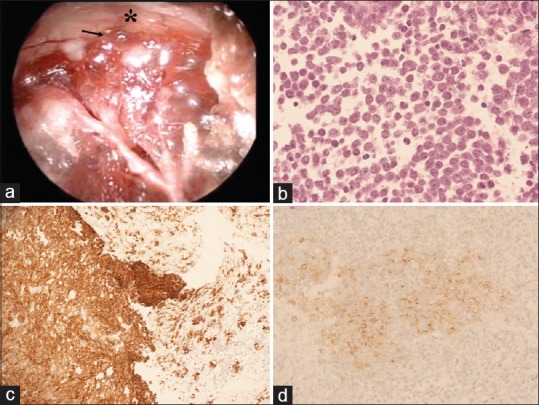
(a) Intraoperative endoscopic picture depicting that the upper part of the lesion (black arrow) presented clear infiltration of the optic chiasm (asterisk) without a safe plane of cleavage. (b) Hematoxylin-eosin (H and E) staining demonstrates cells with scant cytoplasm and nuclei with speckled chromatin. Mitotic activity was present and vigorous (magnification ×20). (c and d) Cells are positive for (c) cytokeratin Cam 5.2. (magnification, ×20) and (d) synaptophysin (magnification, ×20)
Histological and immunohistochemical analysis of the pituitary biopsy showed a neuroendocrine neoplasm of at least intermediate grade. The pituitary mass was characterized by cells with scant cytoplasm and nuclei with speckled chromatin. Mitotic activity was present and vigorous (up to 9–10 mitoses per high power field). There was no evidence of necrosis [Figure 2b]. In the background, multiple fragments of anterior pituitary tissue were noted. Immunohistochemical stains were performed on paraffin-embedded tissue using antibodies to synaptophysin, cytokeratin Cam 5.2, chromogranin, thyroid transcription factor (TTF-1), CDX2, Ki-67, p53 protein, and pituitary hormones. Tumor cells stained with antibodies to cytokeratin Cam 5.2. [Figure 2c], synaptophysin [Figure 2d], and chromogranin. The results for staining with TTF-1 and CDX2 were negative, and p53 protein was overexpressed in over 5% of tumor cells. The Ki-67 labeling index was approximately 25%. These findings support the diagnosis of neuroendocrine neoplasm.
The postoperative course was uneventful, and neurological examination revealed a complete visual recovery. Before the patient was discharged, an extensive search for a primary carcinoid tumor or other metastases was undertaken. Images from CT of the thorax and abdomen were normal. Both 11C-5- hydroxytryptophan PET and Somatostatin Receptor scintigraphy using Indium 111Pentetreotide [111In-DTPA-D-Phe-] (Octreoscan) showed no additional area of abnormal uptake; therefore, primary pituitary NET was confirmed.
Subsequent postoperative CT and MRI [Figure 3a] demonstrated the presence of an abnormal signal toward the cavernous sinus and suprasellar area, which was consistent with residual disease. Then, after consultation with the neuro-oncology group, the patient was successfully treated with fractioned stereotactic radiotherapy and polychemotherapy, (using a combination of cisplatinum, ifosfamide, and etoposide) with good tolerance.
Figure 3.
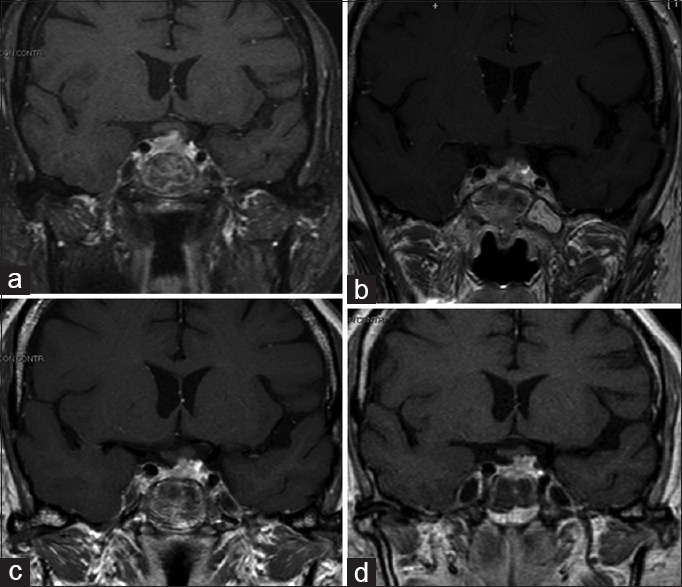
(a-d) Postoperative and follow-up at 12, 24 and 48 months, coronal MRI pictures depicting the stability of residual disease
Fractioned stereotactic radiotherapy (SRT), employing image integration techniques and a frame that could be relocated to facilitate a fractioned dosing scheme, was carried out under a plan for reducing the treatment risk of the optic apparatus. Postoperative CT and MRI imaging were employed for greater precision in identifying the target. The relevant anatomy and tumor residual volume were then outlined. Because the tumor was close to critical normal tissue such as the optic chiasm, optic nerves, and cavernous sinus, the rotational angles were planned to reduce the doses to these critical regions. The treatment consisted of 8 daily fractions of 3.5 Gy to the 65% isodose line and a total dose of 43.1 Gy. Thus, the optic chiasm was exposed to a mean total dose of 208.6 cGy. No deterioration of visual acuity or pituitary function occurred after SRT.
Follow-up MRI studies at 12, 24, and 48 months were performed [Figure 3b–d] showing the stability of the residual mass in the sellar region. Results from the neurological examination were negative.
DISCUSSION
We describe the treatment of a NET arising from enterochromaffin cells that had undergone neoplastic transformation.[2] Enterochromaffin cells are located in the crypts of Lieberkuhn of the intestinal lumen and in the bronchopulmonary tract. However, these epithelial cells or their progenitors are present in nearly all organs.[2,8] Therefore, NET can arise in almost every organ of the body although these tumors are most commonly found in the gastrointestinal tract and respiratory system.[1,2,3,5,8]
Metastatic carcinoid tumors in the central nervous system are an unusual but well-documented finding in several case reports.[4,6,8] In the series by Patchell and Posner, intracranial metastases were the second most common neurological complication arising from systemic disease.[6] In addition, a case of a carcinoid tumor metastasizing to the dura mater and simulating a meningioma has been previously reported, however, this patient had a known gastric primary tumor.[1] The skull base and sellar region are extremely rare sites for NET,[3] and to date, only 6 cases of primary intracranial NET are reported in the literature.[1,2,3,4,5,6,7,8]
Patients with NET commonly present with no specific clinical features, including focal neurological deficit and intracranial hypertension. Functional tumors secreting one or more hormones would result in the presence of some endocrinal symptoms, and nonfunctional tumors may affect pituitary function and lead to hypopituitarism.[4] In our reported case, lesions invaded the suprasellar region with optic chiasm compression and subsequent bitemporal hemianopsia.
Producing images of NET is usually challenging, requiring a combination of functional and anatomical techniques.[1,5,7] Scans by CT and MRI examination are nonspecific, and in the skull base and sellar region, as in our case, NET presented MRI findings similar to more common pathologies such as pituitary adenoma, meningioma, and metastasis.[7] Because most cases of NET express somatostatin receptor subtype 2 and 5, somatostatin receptor (SR) scintigraphy represents the primary imaging method for diagnosis, staging, and monitoring of this tumor.[4,5] Somatostatin receptor scintigraphy study using Indium 111 Pentetreotide (which has a high affinity for SR), can achieve a sensitivity that has been reported to be between 80% and 100%.[3] An MIBG (131I-metaiodobenzylguanidine) scan uses an analogue of a biogenic amine precursor, 131I-metaiodobenzylguanidine. This analogue is taken up by chromaffin cells and stored in the neurosecretory granules. The MIBG scan presented a sensitivity a little lower than that of the 111In-pentetreotide scintigraphy.[4,5] Positron emission tomography (PET) scanning using 18F-labeled fluorodeoxyglucose (18FDG) is widely used as a powerful imaging technique in clinical oncology. Unfortunately, monitoring increased FDG uptake in NET is limited because of the low cellular proliferative activity and high differentiation rate.[3] Therefore, several tracers directed toward the specific characteristics of carcinoid tumors were developed for PET imaging in these tumors, for example, 6-[18F] fluorodopamine (18F-dopa) and 11C-5-hydroxytryptophan.[4] A PET scan using 11C-5-hydroxytryptophan has been shown to be a sensitive method for the imaging of small NETs, and in most cases yields a significantly higher detection rate than somatostatin receptor scintigraphy.[5,6,7,8]
Histopathological diagnosis is based on morphological and immunohistochemical examination. The neoplasm comprises cells of different sizes with uniform nuclei and scant cytoplasm; in addition, nidulant, basophil granulocytes are found. Irregular mitosis is present based on the degree of differentiation. The diagnosis of carcinoid tumor was favored based on its immunohistochemical profile; CAM 5.2, a marker for low molecular weight cytokeratins, synaptophysin, and chromogranin are markers for cells of neuroendocrine origin, and supported the diagnosis of NET.[4,5,6,7,8] Glial fibrillary acidic protein (GFAP), carcinoembryonic antigen (CEA), S100, and calcitonin are negative in NET, contrary to other cerebral tumors. For NET in the sellar region, as in the present case, it is necessary to differentiate it from esthesioneuroblastoma.[4] Esthesioneuroblastoma comprises neuroblasts with uniform circular nuclei and longitudinal cerebromedullary tubes in the dendrites. However, NET originates from the epithelium, lacking neural features.[1,2,3,4,5,6]
To date, only 6 cases of primary intracranial NET have been reported in the literature [Table 1]. Therefore, the diagnosis, prognosis, and definition of treatment strategies are challenging. With the exception of the case presented by Deshaies et al.,[1] describing a frontal convexity mass with a prominent dural-based tail, the other five cases together with our case are located at the skull base, which appears to be the most frequent site of primary intracranial NET.[3,4,5,7] Porter et al.[7] reported a 62-year-old male patient who showed signs of elevated intracranial pressure, secondary to a tumor in the right cerebellopontine angle. The patient was treated with surgical subtotal resection of an extra-axial tumor by a retromastoid, retrosigmoid approach leaving a small remnant of tumor on the tentorial edge. The operative impression was of a tentorial edge meningioma. No primary tumor was found and no adjuvant therapy was administered. The follow-up for 5 years did not show any relapse.
Table 1.
Summary of clinical, radiological, and surgical findings of patients with neuroendocrine tumor reported in the literature; adjuvant therapy and outcome were also reported

Ibrahim et al.[4] described another NET located at the skull base in a 29-year-old woman who presented with multiple cranial nerve palsies. The tumor was located at the foramen jugulare extending into the cerebellomedullary angle cistern. A biopsy was performed that led to the diagnosis of NET. The patient was treated with monthly somatostatin injections only and did not receive any postoperative radiation therapy or complete resection; she was in stable condition 1 year after surgical removal. More recently, Hood et al.[3] illustrated the case of a 61-year-old woman with multimorbidity who presented with transient memory loss and a left cavernous sinus mass extending into the infratemporal fossa. A biopsy was performed, and the patient then underwent subtotal tumor resection, and finally received postoperative radiotherapy. Because the tumor was firm and not easily removable with suction, the final portion off the cavernous carotid artery was left in place. The pathology report indicated a low-grade NET.
Finally, Liu et al.[5] reported two cases of primary NET: the first located in the sellar/suprasellar area and the second in the anterior cranial fossa with extension into the paranasal sinus. The first patient underwent a gross removal of tumor during surgery with a single nostril transsphenoidal approach. Pathological diagnosis was high-grade small-cell NET, and the patient died of extensive metastases after approximately 3 months. For the second patient, surgery achieved a gross removal through a right frontal craniotomy. The pathological diagnosis was low-grade NET. Follow-up and other treatments were not reported.
Our case was the only one successfully treated with a multimodality approach. After a subtotal resection, the patient underwent to fractioned stereotactic radiotherapy (total irradiation dose 43.1 Gy, fractioned in 8 daily sessions), followed by polychemotherapy protocol (using a combination of cisplatinum, ifosfamide, and etoposide), which was well tolerated. Four years after the treatment, a follow-up MRI showed a complete stability of the residual disease and results from neurological examination were negative. Other particular features of our case were the location of NET and the use of an endoscopic technique. In fact, to the best of our knowledge, we present the second case of primary sellar NET. In contrast to the sellar NET presented by Liu et al.,[5] our case was preoperatively indistinguishable from non-functioning pituitary macroadenoma, both clinically and radiologically. However, because of the better visualization granted by the endoscopic technique (moving the lens and light source closer to the pathology), we were able to intraoperatively distinguish the lesion from normal macroadenoma. In appearance, it had a firm and solid consistency, and it was not removable with suction; in addition, the upper part of the lesion presented clear infiltration of the optic chiasm without a safe plane of cleavage. Therefore, the surgical strategy was immediately changed, and we proceeded with decompression of locoregional neurovascular structures, leaving a small residue of tumor infiltrating the optic chiasm.
CONCLUSIONS
Primary pituitary NET, though rare, should be included in the differential diagnosis of sellar lesions. A multimodality treatment approach is needed. Finally, the present case highlights that, in the case of a pituitary lesion infiltrating the optic chiasm, including NET, the endoscopic endonasal transsphenoidal subtotal resection followed by fractioned stereotactic radiotherapy and chemotherapy may represent an effective and safe choice of treatment.
Compliance with ethical standards.
Funding
No funding was received for this research.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Footnotes
Contributor Information
D. Nasi, Email: davidenasi83@gmail.com.
D. Perano, Email: perano.daniele@asmn.re.it.
R. Ghadirpour, Email: ghadirpour.reza@asmn.re.it.
C. Iaccarino, Email: ciaccarino@ao.pr.it.
F. Servadei, Email: fservadei@ao.pr.it.
A. Romano, Email: aromano@ao.pr.it.
REFERENCES
- 1.Deshaies EM, Adamo MA, Qian J, DiRisio DA. A carcinoid tumor mimicking an isolated intracranial meningioma. Case report. J Neurosurg. 2004;101:858–60. doi: 10.3171/jns.2004.101.5.0858. [DOI] [PubMed] [Google Scholar]
- 2.Faggiano A, Mansueto G, Ferolla P, Milone F, del Basso de Caro ML, Lombardi G, et al. Diagnosis and prognostic implication of the World Health Organization classification of neuroendocrine tumors. Endocrinol Invest. 2008;31:216–23. doi: 10.1007/BF03345593. [DOI] [PubMed] [Google Scholar]
- 3.Hood B, Bray E, Bregy A, Norenberg M, Weed D, Morcos JJ. Primary carcinoid tumor of the cavernous sinus. World Neurosurg. 2014;81:202.e9–13. doi: 10.1016/j.wneu.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 4.Ibrahim M, Yousef M, Bohnen N, Eisbruch A, Parmar H. Primary carcinoid tumor of the skull base: Case report and review of the literature. J Neuroimaging. 2010;20:390–2. doi: 10.1111/j.1552-6569.2008.00317.x. [DOI] [PubMed] [Google Scholar]
- 5.Liu H, Wang H, Qi X, Yu C. Primary intracranial neuroendocrine tumor: Two case reports. World J Surg Oncol. 2016;14:138. doi: 10.1186/s12957-016-0887-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patchell R, Posner J. Neurologic complications of carcinoid. Neurology. 1986;36:745–9. doi: 10.1212/wnl.36.6.745. [DOI] [PubMed] [Google Scholar]
- 7.Porter DG, Chakrabarty A, McEvoy A, Bradford R. Intracranial carcinoid without evidence of extracranial disease. Neuropathol Appl Neurobiol. 2000;26:298–300. doi: 10.1046/j.1365-2990.2000.00257.x. [DOI] [PubMed] [Google Scholar]
- 8.Sirsath NT, Babu KG, Das U, Premlatha CS. Paranasal sinus neuroendocrine carcinoma: A case report and review of the literature. Case Rep Oncol Med 2013. 2013 doi: 10.1155/2013/728479. 728479. [DOI] [PMC free article] [PubMed] [Google Scholar]