

Abstract
AIM
To determine the effect of combined serelaxin and rosiglitazone treatment on established hepatic fibrosis.
METHODS
Hepatic fibrosis was induced in mice by carbon tetrachloride administration for 6 wk, or vehicle alone (nonfibrotic mice). For the final 2 wk, mice were treated with rosiglitazone, serelaxin, or both rosiglitazone and serelaxin. Serum liver enzymes and relaxin levels were determined by standard methods. The degree of liver collagen content was determined by histology and immunohistochemistry. Expression of type I collagen was determined by quantitative PCR. Activation of hepatic stellate cells was assessed by alpha-smooth muscle actin (SMA) levels. Liver peroxisome proliferator activated receptor-gamma coactivator 1 alpha (PGC1α) was determined by Western blotting.
RESULTS
Treatment of mice with CCl4 resulted in hepatic fibrosis as evidenced by increased liver enzyme levels (ALT and AST), and increased liver collagen and SMA. Monotherapy with either serelaxin or rosiglitazone for 2 wk was generally without effect. In contrast, the combination of serelaxin and rosiglitazone resulted in significantly improved ALT levels (P < 0.05). Total liver collagen content as determined by Sirius red staining revealed that only combination treatment was effective in reducing total liver collagen (P < 0.05). These results were supported by immunohistochemistry for type I collagen, in which only combination treatment reduced fibrillar collagen levels (P < 0.05). The level of hepatic stellate cell activation was modestly, but significantly, reduced by serelaxin treatment alone, but combination treatment resulted in significantly lower SMA levels. Finally, while hepatic fibrosis reduced liver PGC1α levels, the combination of serelaxin and rosiglitazone resulted in restoration of PGC1α protein levels.
CONCLUSION
The combination of serelaxin and rosiglitazone treatment for 2 wk was effective in significantly reducing established hepatic fibrosis, providing a potential new treatment strategy.
Keywords: Relaxin, Peroxisome proliferator-activated receptors, Liver cirrhosis, Liver diseases, Fibrosis
Core tip: Hepatic fibrosis is a chronic condition that can lead to cirrhosis, but treatment options are limited and ineffective. Agonists of peroxisome proliferator-activated receptor gamma (PPARγ), such as rosiglitazone have shown limited efficacy. The hormone relaxin has antifibrotic effects, and increases the activity of PPARγ, leading to the hypothesis that combination treatment may be more effective. Mice with established hepatic fibrosis were treated with relaxin and rosiglitazone alone or in combination. Combination treatment reduced liver fibrosis, and increased the level of a PPARγ coactivator. These results suggest that relaxin and PPARγ co-therapy could be a more effective treatment for hepatic fibrosis.
INTRODUCTION
Relaxin is a polypeptide hormone of the insulin/relaxin superfamily[1]. One important action of relaxin is the widespread remodeling of extracellular matrix, which involves altered secretion and degradation of matrix components[1,2]. The case for a role for relaxin as a general protective agent against fibrosis was dramatically strengthened by observations made using the relaxin-null mouse. These mice spontaneously developed age-related pulmonary, cardiac, dermal, and renal fibrosis[2-4]. This has led to the use of relaxin in the treatment of experimentally-induced pulmonary and renal fibrosis in rodents, which could be reversed by systemic relaxin treatment[5,6].
Relaxin also has effects in the liver. Relaxin treatment of rats caused acute changes in the hepatic microcirculation[7], and morphological changes were detected in nonparenchymal sinusoidal cells[8]. In addition, the relaxin-null mouse developed increased liver weight[9]. Work by our laboratory and others showed that relaxin had antifibrotic effects on activated hepatic stellate cells (HSC), which are the major collagen-producing cells in liver injury. Relaxin treatment of activated HSC had numerous effects, including decreased total collagen deposition, collagen synthesis, and collagen-I secretion, and decreased smooth muscle actin expression, but had no effect on HSC proliferation or apoptosis[10,11]. Relaxin promoted a matrix degrading phenotype in HSCs by increasing matrix metalloproteinase expression and activity, and inhibiting secretion of the tissue inhibitors of metalloproteinases[10,11]. The effects of relaxin were mediated by activation of the relaxin family peptide 1 (RXFP1) receptor, which is expressed predominantly in the HSC in liver[12,13]. Finally, using in vivo models of experimental hepatic fibrosis, relaxin prevented hepatic collagen content[10,14], and was effective in treating established hepatic fibrosis[13,15]. Therefore, there is considerable evidence to support a functional role for relaxin effects in the liver.
A second critical regulatory element in HSC activation is the PPARγ pathway. PPARγ is a transcription factor activated by the antidiabetic thiazolidinedione (TZD) drugs, such as rosiglitazone and pioglitazone, and some prostaglandins[16]. Expression of PPARγ is detectable in quiescent HSC, but is lacking in activated HSC and myofibroblasts[17]. Restoration of PPARγ expression, either by treatment of activated HSC with PPARγ ligands or by forced expression of PPARγ, induced a reversion of the HSC to a state that closely resembled the quiescent phenotype, as shown by decreased proliferation, reduced SMA, collagen and TIMP expression, increased MMP-13 expression, and restoration of lipid-storage[18]. Importantly, treatment of experimentally-induced fibrosis with PPARγ ligands prevented hepatic fibrosis in some in vivo models[19-21]. However, recent studies have suggested that TZD treatment may be ineffective for established fibrosis in rodents, casting some doubt on the utility of using TZDs alone for this purpose[22-24].
As discussed above, PPARγ has numerous antifibrotic effects, and relaxin reduced many of the same markers reported for PPARγ agonists in HSC in culture and in vivo. We reported that relaxin activates PPARγ transcriptional activity in cells expressing RFXP1 in a manner that did not require the addition of exogenous PPARγ ligands[25]. More recently, we identified the mechanism for this stimulation[26]. Relaxin increased the expression of a coactivator protein in activated HSC, known as PPARγ coactivator 1α (PGC1α) through cAMP and p38-MAPK dependent pathways, and that these pathways were intact in the human hepatic stellate cell line LX2. Therefore, relaxin treatment may enhance the response to TZDs in hepatic fibrosis. To test this hypothesis, we compared the effectiveness of the recombinant form of relaxin (serelaxin), rosiglitazone, or their combination, in the treatment of established models of hepatic fibrosis.
MATERIALS AND METHODS
Mouse model of hepatic fibrosis and treatment
Fibrosis was induced in male C57BL/6 mice (20-24 g, Charles River Laboratories, Wilmington, MA) as described[15]. Briefly, mice received twice-weekly intraperitoneal injections of CCl4 (diluted 1:7 in sunflower oil) at 1 mL/kg body weight, for a total of 6 wk to induce hepatic fibrosis. Control (nonfibrotic) mice received oil alone. For the final 2 wk of treatment, mice were randomly assigned to receive implantation of subcutaneous osmotic pumps (model 1002, Durect, Cupertino CA) to deliver serelaxin (generously provided by Dennis Stewart, Novartis) at 150 μg/g per day, or vehicle (citrate buffer). Rosiglitazone (4 mg/kg per day Enzo Life Sciences, Farmingdale, CA) or vehicle (5% DMSO in phosphate buffered saline) was also administered daily by oral gavage for the final 2 wk. Each group contained 5 mice. Mice were sacrificed 72 h after the final CCl4 injection, and liver and blood were collected. Mice were maintained at 22 °C under 12-h light/dark cycles, and had free access to food and water throughout the study. All procedures were conducted in accordance with The Guide for the Care and Use of Laboratory Animals[27], and were approved by the VA Nebraska Western-Iowa Institutional Animal Care and Use Committee.
Histology and immunohistochemistry
Liver tissue was fixed in 4% buffered formalin, embedded in paraffin, and sections were mounted onto slides. Sections were dewaxed and then stained with picrosirius red to visualize total collagen, as described[28]. For immunohistochemistry, tissues were subject to antigen unmasking by heating in citrate buffer (Vector Labs, Burlingame CA), then probed overnight at 4 °C with antibodies directed against type I collagen (ab21286, Abcam, Cambridge, MA) at 1:250 dilution, or α-smooth muscle actin (SMA, clone 1A4, Sigma Chemical, St. Louis, MO) at 1:400 dilution. Positive staining was detected using the DAB Envision System (Dako, Carpenteria, CA). Images were captured and analyzed using ImageJ software as described previously[15].
Gene expression analysis
Total liver RNA was extracted using the Purelink kit (Thermo Fisher, Carlsbad, CA), with on-column DNase treatment as per the manufacturer’s instructions. RNA integrity and lack of contaminating genomic DNA was confirmed by visualization on agarose gels, and RNA concentration was determined using the Ribogreen assay (Thermo Fisher, Carlsbad, CA). A total of 2 μg of RNA was converted to cDNA using the TaqMan High Capacity Reverse Transcription kit (Thermo Fisher, Carlsbad, CA) in a final volume of 20 μL. Quantitative PCR was conducted using TaqMan hydrolysis probe assays, using 2 μL of cDNA (diluted 1:15), 10 μL Taqman universal PCR master mix, 1.0 μL Taqman primer/probe mix in a final volume of 20 μL per reaction. The mouse gene expression assays used included procollagen type Iα2 (Col1a2; Mm00483888_m1), αSMA (Acta2, Mm01546133_m1), and TATA-box binding protein (Tbp; Mm01277045_m1). All expression levels were normalized to that of Tbp in the same sample, and the data expressed as the expression level relative to nonfibrotic controls, using the ΔΔCT method.
Serum measurements
Serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were determined by standard clinical chemistry assays. Human relaxin levels were determined using the Quantikine kit (R&D Systems, Minneapolis MN), which does not detect mouse relaxin or other insulin- and relaxin-related peptides. Mouse adiponectin levels were measured by immunoassay (Alpco, Salem NH).
Western blotting
Lysates were prepared from liver tissue and protein levels were determined by the bicinchoninic acid assay (Thermo Fisher, Carlsbad, CA). A total of 50 μg protein was applied to 10% SDS-PAGE gel, then transferred to PVDF membranes. The membranes were probed overnight at 4 °C with antibodies directed against PGC1α (#101707, Cayman Chemical, Ann Arbor, MI, 1:500) or GAPDH (MAB374, Millipore, Temecula, CA, 1:2000). After washing, membranes were probed with fluorescently-labeled secondary antibodies (Li-Cor, Lincoln, NE), and immunoreactive proteins detected using an Odyssey fluorescent scanner (Li-Cor, Lincoln, NE).
Statistical analysis
Statistical analysis was performed using Prism5 software (GraphPad, La Jolla, CA). Differences between groups were analyzed using one-way analysis of variance (ANOVA) with the Newman-Keuls post-test. Data are expressed as mean ± SE of means.
RESULTS
Serum levels of serelaxin were analyzed by a specific assay that does not detect mouse relaxin. Serelaxin was successfully delivered, as evidenced by detectable human relaxin in treated mice, but not control mice (Table 1). As expected, rosiglitazone treatment caused an increase in serum adiponectin levels, confirming successful treatment and bioactivity of rosiglitazone. Fibrotic mice (CCl4 group) had significantly elevated levels of ALT and AST. None of the treatments resulted in a significant change in ALT or AST levels compared with CCl4 treatment alone. A significant difference was detected between Rosi and Rln treatments alone, due to opposite but statistically insignificant differences caused by each treatment individually. There was no significant difference in body or liver weight under any treatment condition (Table 1).
Table 1.
Serum measurements in control and fibrotic mice
| Control | CCl4 only | Rosi | Rln | Rosi + Rln | |
| Body weight (g) | 26.7 ± 0.9 | 25.6 ± 0.8 | 24.8 ± 1.0 | 24.9 ± 1.0 | 24.7 ± 0.9 |
| Liver weight (g) | 1.7 ± 0.1 | 1.7 ± 0.1 | 1.5 ± 0.1 | 1.6 ± 0.1 | 1.4 ± 0.1 |
| Liver (% body wt) | 6.3 ± 0.1 | 6.7 ± 0.3 | 6.1 ± 0.3 | 6.4 ± 0.4 | 5.8 ± 0.2 |
| ALT | 91.8 ± 5.7 | 2656 ± 538a | 3892 ± 676a | 1755 ± 610ab | 3227 ± 313a |
| AST | 368 ± 33 | 1621 ± 282a | 2496 ± 339a | 1278 ± 277ab | 1863 ± 165a |
| Human relaxin (ng/mL) | ND | ND | ND | 28.6 ± 7.2 | 20.5 ± 7.4 |
| Adiponectin (ug/mL) | 35.0 ± 3.4 | 32.6 ± 3.9 | 86.0 ± 12.2c | 32.4 ± 2.2 | 147.5 ± 18.7c |
P < 0.05 vs control;
P < 0.05 vs rosiglitazone (Rosi);
P< 0.05 vs control, CCl4 only or relaxin (Rln). ND: Not detected.
The level of total collagen deposition determined by Sirius red staining was markedly increased with CCl4 treatment, confirming development of hepatic fibrosis (Figure 1). As demonstrated previously[14], 2 wk treatment with relaxin alone did not reduce Sirius red staining. Similarly, rosiglitazone alone had no significant effect on the total collagen deposition. In contrast, the combination of relaxin and rosiglitazone significantly reduced the degree of Sirius red staining (Figure 1). To more precisely assess the relative levels of fibrillar collagen, immunohistochemistry for type I collagen was performed (Figure 2). Consistent with the Sirius red staining, only the combination of relaxin and rosiglitazone reduced the overall level of type I collagen.
Figure 1.
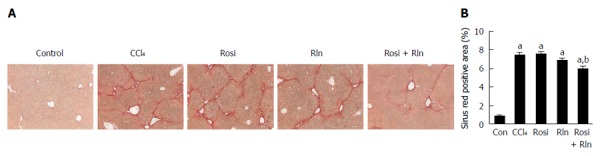
Total liver collagen content. A: Sirius red staining of liver tissue from control (Con), fibrotic (CCl4), rosiglitazone (Rosi), serelaxin (Rln) or combination-treated (Rosi + Rln) mice. Bar: 100 μm. B: Sirius red staining quantified. Data are expressed as mean ± SE, and analyzed by ANOVA (n = 5). aP < 0.001 vs Con; bP < 0.05 vs CCl4, Rosi, or Rln.
Figure 2.
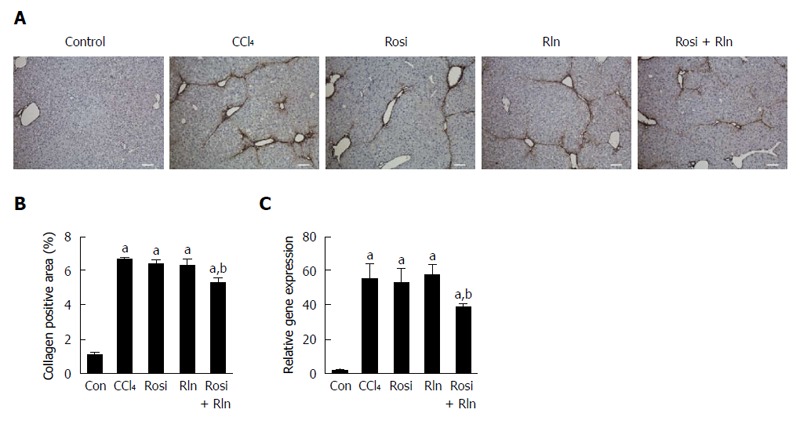
Liver type I collagen content. A: Immunohistochemical staining of liver tissue from control (Con), fibrotic (CCl4), rosiglitazone (Rosi), serelaxin (Rln) or combination-treated (Rosi + Rln) mice. Bar: 100 μm; B: Type I collagen staining quantified; C: Type I collagen gene expression determined by qPCR. Data are expressed as mean ± SE, and analyzed by ANOVA (n = 5). aP < 0.05 vs Con; bP < 0.05 vs CCl4, Rosi, or Rln.
The primary cell type responsible for the deposition of collagen in fibrosis are the activated hepatic stellate cells (HSC). Using immunohistochemistry for the activated HSC marker α-smooth muscle actin (αSMA), robust induction of HSC activation was induced by CCl4 treatment (Figure 3). Treatment with rosiglitazone was without effect, while relaxin caused a modest but significant, decrease in αSMA staining. The combination of relaxin and rosiglitazone induced a significant reduction in the level of HSC activation as exemplified by the reduction in αSMA level. These effects were confirmed at the transcriptional level, as similar effects were observed on the gene expression level of type I collagen as determined by qPCR.
Figure 3.
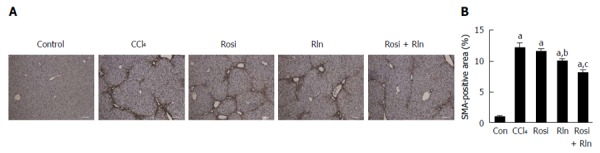
Liver smooth muscle actin content. A: Immunohistochemistry of liver tissue for SMA content from control (Con), fibrotic (CCl4), rosiglitazone (Rosi), serelaxin (Rln) or combination-treated (Rosi+Rln) mice. Bar: 100 μm; B: SMA staining quantified. Data are expressed as mean ± SE, and analyzed by ANOVA (n = 5). aP < 0.001 vs Con; bP < 0.05 vs CCl4; cP < 0.05 vs CCl4, Rosi, or Rln.
Relaxin was previously shown to increase the levels of PGC1α in cultured hepatic stellate cells[26]. To determine the effect of serelaxin and rosiglitazone treatment on PGC1α protein levels in hepatic fibrosis in vivo, Western blotting was performed on liver lysates. The level of PGC1α was decreased after CCl4 treatment (Figure 4). While single treatment with rosiglitazone was without effect, serelaxin alone or in combination with rosiglitazone restored PGC1α levels.
Figure 4.
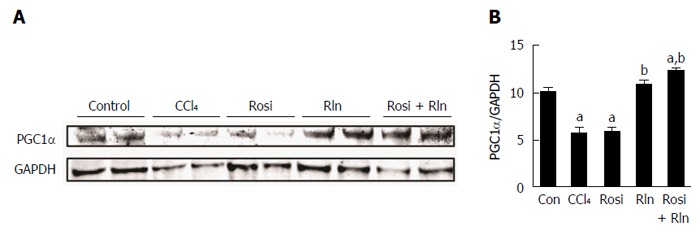
Western blotting of PGC1α content in livers from control (Con), fibrotic (CCl4), rosiglitazone (Rosi), serelaxin (Rln) or combination-treated (Rosi + Rln) mice. A: Liver tissue extracts were analyzed by Western blotting for PGC1α. The levels of GAPDH are shown as a loading control; B: The levels of PGC1α relative to GAPDH were determined by densitometry. Data are shown as mean Data are expressed as mean ± SE, and analyzed by ANOVA (n = 5). aP < 0.05 vs Con; bP < 0.01 vs CCl4 or Rosi.
DISCUSSION
While hepatic fibrosis is a major health concern worldwide, the options for treatment are limited. The effectiveness of TZDs in the treatment of human liver disease remains to be studied. Early studies of the antidiabetic PPARγ agonists of the thiazolidinedione class, including rosiglitazone and pioglitazone, reduced the activation of HSCs[29-31], and had preventive effects in rat models of hepatic fibrosis[20,21,32,33]. However, in more clinically relevant studies exploring the effectiveness of TZDs in the treatment of established hepatic fibrosis in rats, pioglitazone was only effective when introduced very early in the course of the disease[23]. Furthermore, pioglitazone was ineffective in reducing the fibrotic phenotype of mouse HSC, and did not prevent CCl4-induced hepatic fibrosis in mice[22]. These findings dampened enthusiasm for the utility of TZD treatment of hepatic fibrosis.
The reason for the failure of mice to respond to thiazolidinediones is unknown, but may be related to the lack of PPARγ expression in activated mouse HSC[23]. If this is the case, then strategies to increase PPARγ signaling might restore responsivity to TZDs. We previously identified PPARγ as a downstream target of relaxin signaling through its receptor, RXFP1[25]. Furthermore, we demonstrated that relaxin activated PPARγ through a ligand-dependent mechanism mediated by increased expression of the PPARγ coactivator PGC1α[26]. The co-treatment of cells with relaxin and the PPARγ agonist rosiglitazone resulted in greater PPARγ transcriptional activity than either relaxin or rosiglitazone alone, suggesting that relaxin was acting to enhance the activity of PPARγ[25]. The purpose of the present study was to test this relationship using an in vivo model of established hepatic fibrosis. In earlier studies, we found that short-term relaxin treatment of hepatic fibrosis (2 wk) was insufficient to significantly reduce collagen deposition, and that 4 wk of treatment was required for significant results[14,15]. We therefore chose to 2 wk of serelaxin treatment for this study. While serelaxin alone had no effect on total collagen or type I collagen, it did significantly reduce αSMA content and therefore, HSC activation. This suggests that the effects of serelaxin on HSC activation precede the degradation of excess collagen. We also confirmed, using rosiglitazone, the lack of effectiveness of TZD treatment alone on mouse hepatic fibrosis, reported previously using pioglitazone[23]. However, consistent with our earlier cell culture studies[25], the combination of relaxin and rosiglitazone significantly decreased collagen content and HSC activation, and reduced AST levels. The effects occurred with only 2 wk of treatment, and in the face of continued delivery of toxin (CCl4), suggesting that the combination treatment accelerated the rate of the antifibrotic effect.
Our previous findings showed that relaxin enhanced PPARγ signaling through increased expression of PGC1α[26]. In the present study, CCl4 treatment reduced the level of PGC1α, as shown previously in models of liver injury[34,35]. Treatment with serelaxin, or the combination of serelaxin and rosiglitazone, restored PGC1α levels. This finding supports the previous findings suggesting that relaxin acts to enhance PPARγ activity through increased expression of PGC1α. However, since relaxin treatment alone for 2 wk failed to reduce collagen levels, induction of PGC1α alone is not sufficient for resolution of hepatic fibrosis, and the presence of PPARγ agonists is necessary for maximum effectiveness.
Taken together, these data suggest that the combination of serelaxin and rosiglitazone may be a more effective treatment for hepatic fibrosis than either agent alone. This raises the possibility of new approaches to the treatment of hepatic fibrosis that can exploit the combined effects of both RXFP1 and PPARγ activation. Further studies are needed to determine if combination therapy can be effective in alternative models of hepatic fibrosis, or extended to extrahepatic fibrotic conditions.
COMMENTS
Background
Hepatic fibrosis is characterized by excess collagen deposition in response to a variety of causes of liver injury. There are currently no effective treatments for hepatic fibrosis and cirrhosis. The hormone relaxin has antifibrotic effects in a number of tissues, and was recently found to increase the activity of peroxisome proliferator-activated receptor γ (PPARγ).
Research frontiers
Relaxin is quickly emerging as an antifibrotic agent, and in preclinical studies has shown efficacy in the treatment of a variety of fibrosis models. The use of PPARγ agonists have also been explored for antifibrotic effects, but have had limited success in models of hepatic fibrosis.
Innovations and breakthroughs
This is the first study to explore the effect of combined relaxin and PPARγ agonist treatment of established hepatic fibrosis. The results suggested that the combination treatment was more effective that either treatment alone.
Applications
The findings provide evidence that combined use of relaxin and PPARγ agonists may represent a potential new approach for the treatment of hepatic fibrosis.
Terminology
Relaxin is a polypeptide hormone with important roles in pregnancy, cardiovascular function, and extracellular matrix regulation. PPARγ is a nuclear transcription factor that regulates the expression of target genes.
Peer-review
The authors have provided evidence that combined treatment with relaxin and rosiglitazone was effective in a model of hepatic fibrosis.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: United States
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): B
Grade C (Good): 0
Grade D (Fair): D
Grade E (Poor): 0
Institutional animal care and use committee statement: All procedures were conducted in accordance with The Guide for the Care and Use of Laboratory Animals, and were approved by the VA Nebraska Western-Iowa Institutional Animal Care and Use Committee.
Conflict-of-interest statement: The authors have no conflicts of interest.
Data sharing statement: No additional data are available.
Peer-review started: December 29, 2016
First decision: February 10, 2017
Article in press: May 9, 2017
P- Reviewer: Daliry A, Shieh KR, Elsiesy H S- Editor: Qi Y L- Editor: A E- Editor: Wang CH
References
- 1.Sherwood OD. Relaxin’s physiological roles and other diverse actions. Endocr Rev. 2004;25:205–234. doi: 10.1210/er.2003-0013. [DOI] [PubMed] [Google Scholar]
- 2.Samuel CS, Royce SG, Hewitson TD, Denton KM, Cooney TE, Bennett RG. Anti-fibrotic actions of relaxin. Br J Pharmacol. 2017;174:962–976. doi: 10.1111/bph.13529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Samuel CS, Zhao C, Bathgate RA, DU XJ, Summers RJ, Amento EP, Walker LL, McBurnie M, Zhao L, Tregear GW. The relaxin gene-knockout mouse: a model of progressive fibrosis. Ann N Y Acad Sci. 2005;1041:173–181. doi: 10.1196/annals.1282.025. [DOI] [PubMed] [Google Scholar]
- 4.Bennett RG. Relaxin and its role in the development and treatment of fibrosis. Transl Res. 2009;154:1–6. doi: 10.1016/j.trsl.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garber SL, Mirochnik Y, Brecklin CS, Unemori EN, Singh AK, Slobodskoy L, Grove BH, Arruda JA, Dunea G. Relaxin decreases renal interstitial fibrosis and slows progression of renal disease. Kidney Int. 2001;59:876–882. doi: 10.1046/j.1523-1755.2001.059003876.x. [DOI] [PubMed] [Google Scholar]
- 6.Unemori EN, Pickford LB, Salles AL, Piercy CE, Grove BH, Erikson ME, Amento EP. Relaxin induces an extracellular matrix-degrading phenotype in human lung fibroblasts in vitro and inhibits lung fibrosis in a murine model in vivo. J Clin Invest. 1996;98:2739–2745. doi: 10.1172/JCI119099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bani D, Nistri S, Quattrone S, Bigazzi M, Bani Sacchi T. The vasorelaxant hormone relaxin induces changes in liver sinusoid microcirculation: a morphologic study in the rat. J Endocrinol. 2001;171:541–549. doi: 10.1677/joe.0.1710541. [DOI] [PubMed] [Google Scholar]
- 8.Bani D, Nistri S, Quattrone S, Bigazzi M, Sacchi TB. Relaxin causes changes of the liver. In vivo studies in rats. Horm Metab Res. 2001;33:175–180. doi: 10.1055/s-2001-14935. [DOI] [PubMed] [Google Scholar]
- 9.Du XJ, Samuel CS, Gao XM, Zhao L, Parry LJ, Tregear GW. Increased myocardial collagen and ventricular diastolic dysfunction in relaxin deficient mice: a gender-specific phenotype. Cardiovasc Res. 2003;57:395–404. doi: 10.1016/s0008-6363(02)00663-6. [DOI] [PubMed] [Google Scholar]
- 10.Williams EJ, Benyon RC, Trim N, Hadwin R, Grove BH, Arthur MJ, Unemori EN, Iredale JP. Relaxin inhibits effective collagen deposition by cultured hepatic stellate cells and decreases rat liver fibrosis in vivo. Gut. 2001;49:577–583. doi: 10.1136/gut.49.4.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bennett RG, Kharbanda KK, Tuma DJ. Inhibition of markers of hepatic stellate cell activation by the hormone relaxin. Biochem Pharmacol. 2003;66:867–874. doi: 10.1016/s0006-2952(03)00403-9. [DOI] [PubMed] [Google Scholar]
- 12.Bennett RG, Dalton SR, Mahan KJ, Gentry-Nielsen MJ, Hamel FG, Tuma DJ. Relaxin receptors in hepatic stellate cells and cirrhotic liver. Biochem Pharmacol. 2007;73:1033–1040. doi: 10.1016/j.bcp.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 13.Fallowfield JA, Hayden AL, Snowdon VK, Aucott RL, Stutchfield BM, Mole DJ, Pellicoro A, Gordon-Walker TT, Henke A, Schrader J, et al. Relaxin modulates human and rat hepatic myofibroblast function and ameliorates portal hypertension in vivo. Hepatology. 2014;59:1492–1504. doi: 10.1002/hep.26627. [DOI] [PubMed] [Google Scholar]
- 14.Bennett RG, Heimann DG, Tuma DJ. Relaxin reduces fibrosis in models of progressive and established hepatic fibrosis. Ann N Y Acad Sci. 2009;1160:348–349. doi: 10.1111/j.1749-6632.2008.03783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennett RG, Heimann DG, Singh S, Simpson RL, Tuma DJ. Relaxin decreases the severity of established hepatic fibrosis in mice. Liver Int. 2014;34:416–426. doi: 10.1111/liv.12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Everett L, Galli A, Crabb D. The role of hepatic peroxisome proliferator-activated receptors (PPARs) in health and disease. Liver. 2000;20:191–199. doi: 10.1034/j.1600-0676.2000.020003191.x. [DOI] [PubMed] [Google Scholar]
- 17.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsukamoto H, She H, Hazra S, Cheng J, Miyahara T. Anti-adipogenic regulation underlies hepatic stellate cell transdifferentiation. J Gastroenterol Hepatol. 2006;21 Suppl 3:S102–S105. doi: 10.1111/j.1440-1746.2006.04573.x. [DOI] [PubMed] [Google Scholar]
- 19.Galli A, Crabb DW, Ceni E, Salzano R, Mello T, Svegliati-Baroni G, Ridolfi F, Trozzi L, Surrenti C, Casini A. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro. Gastroenterology. 2002;122:1924–1940. doi: 10.1053/gast.2002.33666. [DOI] [PubMed] [Google Scholar]
- 20.Kawaguchi K, Sakaida I, Tsuchiya M, Omori K, Takami T, Okita K. Pioglitazone prevents hepatic steatosis, fibrosis, and enzyme-altered lesions in rat liver cirrhosis induced by a choline-deficient L-amino acid-defined diet. Biochem Biophys Res Commun. 2004;315:187–195. doi: 10.1016/j.bbrc.2004.01.038. [DOI] [PubMed] [Google Scholar]
- 21.Kon K, Ikejima K, Hirose M, Yoshikawa M, Enomoto N, Kitamura T, Takei Y, Sato N. Pioglitazone prevents early-phase hepatic fibrogenesis caused by carbon tetrachloride. Biochem Biophys Res Commun. 2002;291:55–61. doi: 10.1006/bbrc.2002.6385. [DOI] [PubMed] [Google Scholar]
- 22.Leclercq IA, Sempoux C, Stärkel P, Horsmans Y. Limited therapeutic efficacy of pioglitazone on progression of hepatic fibrosis in rats. Gut. 2006;55:1020–1029. doi: 10.1136/gut.2005.079194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Da Silva Morais A, Abarca-Quinones J, Horsmans Y, Stärkel P, Leclercq IA. Peroxisome proliferated-activated receptor gamma ligand, Pioglitazone, does not prevent hepatic fibrosis in mice. Int J Mol Med. 2007;19:105–112. [PubMed] [Google Scholar]
- 24.Marra F. Thiazolidinediones and hepatic fibrosis: don’t wait too long. Gut. 2006;55:917–919. doi: 10.1136/gut.2005.085399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh S, Bennett RG. Relaxin signaling activates peroxisome proliferator-activated receptor gamma. Mol Cell Endocrinol. 2010;315:239–245. doi: 10.1016/j.mce.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh S, Simpson RL, Bennett RG. Relaxin activates peroxisome proliferator-activated receptor γ (PPARγ) through a pathway involving PPARγ coactivator 1α (PGC1α) J Biol Chem. 2015;290:950–959. doi: 10.1074/jbc.M114.589325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.National Research Council. Guide for the Care and Use of Laboratory Animals: Eighth Edition. Washington, DC: The National Academies Press; 2011. p. 246. [Google Scholar]
- 28.Junqueira LC, Bignolas G, Brentani RR. Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem J. 1979;11:447–455. doi: 10.1007/BF01002772. [DOI] [PubMed] [Google Scholar]
- 29.Galli A, Crabb D, Price D, Ceni E, Salzano R, Surrenti C, Casini A. Peroxisome proliferator-activated receptor gamma transcriptional regulation is involved in platelet-derived growth factor-induced proliferation of human hepatic stellate cells. Hepatology. 2000;31:101–108. doi: 10.1002/hep.510310117. [DOI] [PubMed] [Google Scholar]
- 30.Marra F, Efsen E, Romanelli RG, Caligiuri A, Pastacaldi S, Batignani G, Bonacchi A, Caporale R, Laffi G, Pinzani M, et al. Ligands of peroxisome proliferator-activated receptor gamma modulate profibrogenic and proinflammatory actions in hepatic stellate cells. Gastroenterology. 2000;119:466–478. doi: 10.1053/gast.2000.9365. [DOI] [PubMed] [Google Scholar]
- 31.Miyahara T, Schrum L, Rippe R, Xiong S, Yee HF, Motomura K, Anania FA, Willson TM, Tsukamoto H. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J Biol Chem. 2000;275:35715–35722. doi: 10.1074/jbc.M006577200. [DOI] [PubMed] [Google Scholar]
- 32.Marra F, DeFranco R, Robino G, Novo E, Efsen E, Pastacaldi S, Zamara E, Vercelli A, Lottini B, Spirli C, et al. Thiazolidinedione treatment inhibits bile duct proliferation and fibrosis in a rat model of chronic cholestasis. World J Gastroenterol. 2005;11:4931–4938. doi: 10.3748/wjg.v11.i32.4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bruck R, Weiss S, Aeed H, Pines M, Halpern Z, Zvibel I. Additive inhibitory effect of experimentally induced hepatic cirrhosis by agonists of peroxisome proliferator activator receptor gamma and retinoic acid receptor. Dig Dis Sci. 2009;54:292–299. doi: 10.1007/s10620-008-0336-5. [DOI] [PubMed] [Google Scholar]
- 34.Kang JW, Hong JM, Lee SM. Melatonin enhances mitophagy and mitochondrial biogenesis in rats with carbon tetrachloride-induced liver fibrosis. J Pineal Res. 2016;60:383–393. doi: 10.1111/jpi.12319. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y, He Y, Yu H, Ma F, Wu J, Zhang X. Liquiritigenin Protects Rats from Carbon Tetrachloride Induced Hepatic Injury through PGC-1α Pathway. Evid Based Complement Alternat Med. 2015;2015:649568. doi: 10.1155/2015/649568. [DOI] [PMC free article] [PubMed] [Google Scholar]