

Abstract
The tobacco specific nitrosamine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) is a potent pulmonary carcinogen in laboratory animals. It is classified as a Group 1 human carcinogen by the International Agency for Cancer Research. NNK is bioactivated upon cytochrome P450 catalyzed hydroxylation of the carbon atoms adjacent to the nitrosamino group to both methylating and pyridyloxobutylating agents. Both pathways generate a spectrum of DNA damage that contributes to the overall mutagenic and toxic properties of this compound. NNK is also reduced to form 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) which is also carcinogenic. Like NNK, NNAL requires metabolic activation to DNA alkylating agents. Methyl hydroxylation of NNAL generates pyridylhydroxybutyl DNA adducts and methylene hydroxylation leads to DNA methyl adducts. The consequence of this complex metabolism is that NNK generates a vast spectrum of DNA damage, any of which can contribute to the overall carcinogenic properties of this potent pulmonary carcinogen. This article reviews the chemistry and the genotoxic properties of the collection of DNA adducts formed from NNK. In addition, it provides evidence that multiple adducts contribute to the overall carcinogenic properties of this chemical. Which adduct contributes to the genotoxic effects of NNK depends on the context, such as the relative amounts of each DNA alkylating pathway occurring in the model system, the levels and genetic variants of key repair enzymes and the gene targeted for mutation.
Keywords: NNK, nitrosamine, DNA adduct, tobacco smoke, mutagenicity, toxicity, DNA repair
Graphical abstract
1. Introduction
Lung cancer is the leading cause of cancer mortality in the United States as well as globally.1, 2 While the majority of all lung cancer cases are attributed to tobacco use, only a fraction of smokers get lung cancer, indicating that lung cancer risk is driven by gene-environment interactions.3–6 While smoking cessation is an effective method of lung cancer prevention, many smokers find it difficult to quit due to their nicotine addiction. Therefore, identification of risk variants may assist in the identification of high risk individuals who then can be targeted for screening and/or prevention.
Tobacco smoke is a toxic mixture of over 5000 chemicals, including over 70 established carcinogens that have “sufficient evidence for carcinogenicity in laboratory animals or humans” according to the International Agency for Research on Cancer.7, 8 Sixteen of these chemicals are classified as Group 1 human carcinogens.8 Many of these chemicals require metabolic activation to be converted to electrophiles that bind covalently to DNA. The resulting DNA adducts are critical for tumor formation. If the DNA adducts persist and escape DNA repair processes, they will cause miscoding during DNA replication, leading to permanent mutations in critical growth control genes such as ras and p53. This results in genomic instability, loss of normal cellular growth control mechanisms and ultimately lung cancer.
The focus of this perspective is the tobacco chemical, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK). Formed from nicotine during the curing process of tobacco, NNK is a potent lung carcinogen in laboratory animals; it also induces liver, nasal and pancreatic tumors.9–11 This compound induces lung adenocarcinomas in rodents at doses that are comparable to those experienced by smokers.12 This type of cancer is now the most common type of lung cancer observed in humans, having surpassed squamous cell carcinoma. This shift in histology has not been attributed to improvements in diagnoses but rather to the changes in cigarette design, which increases the exposure of humans to tobacco-specific nitrosamines.13 NNK metabolites have been detected in urine of smokers and individuals exposed to second hand smoke, indicating that humans are exposed to and metabolize this carcinogen.14–17 Elevated levels of NNK metabolites in human urine are associated with increased lung cancer risk. 18–20 All these data support the classification of NNK as a human carcinogen.21
2. DNA reactive metabolites of NNK
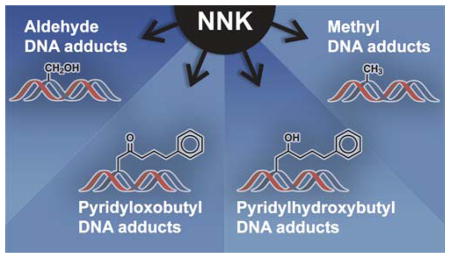
NNK requires metabolic activation to DNA reactive metabolites to exert its tumorigenic effects.22 Cytochrome P450 enzymes catalyze the oxidation of the α-carbon adjacent to the nitrosamine leading to the formation of unstable α-hydroxy metabolites that break down into DNA reactive metabolites (Figure 1). Since NNK is an unsymmetrical nitrosamine, it has two different α-hydroxylation pathways (Figure 1).23–27 Methyl hydroxylation of NNK generates a metabolite that decomposes to 4-(3-pyridyl)-4-oxobutanediazohydroxide and formaldehyde. The alkanediazohydroxide proceeds to 4-(3-pyridyl)-4-oxobutanediazonium ion which reacts with DNA bases to generate pyridyloxobutyl DNA adducts. Methylene hydroxylation of NNK leads to the formation of methanediazohydroxide and an aldehyde (4-oxo-1-(3-pyridyl)-1-butanone, OPB). The resulting methanediazonium ion reacts with DNA to generate methyl DNA adducts.
Figure 1.

Proposed bioactivation pathways for NNK and its metabolite, NNAL.
A major metabolic pathway of NNK is the reduction of the carbonyl group to form 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) as a mixture of (R)- and (S)-enantiomers.12 This is not a detoxification reaction because NNAL is carcinogenic.12, 28 Like NNK, NNAL requires metabolic activation to DNA alkylating agents (Figure 1). Methyl hydroxylation of NNAL generates pyridylhydroxybutyl DNA adducts and methylene hydroxylation leads to DNA methyl adducts.28–30
The consequence of this complex metabolism is that NNK generates a vast spectrum of DNA damage, any one of which can contribute to the overall carcinogenic properties of this potent pulmonary carcinogen. The next section reviews the chemistry and toxicological effects of the multitude of adducts derived from NNK.
3. Chemistry and toxicology of NNK-derived DNA damage
3.1. Methyl DNA adducts
Methylene hydroxylation of NNK and NNAL generates 7-methyl-2′-deoxyguanosine (7-mdGuo) and O6-methyl-2′-deoxyguanosine (O6-mdGuo) (Figure 2) as well as other methyl DNA damage.12 While not generally measured in DNA from NNK-treated animals, it can be assumed that the methanediazonium ion generated from NNK produces the same spectrum of methyl adducts as other methylating nitrosamines. The expected order of formation of adducts is 7-methylguanine > methyl phosphotriesters > O6-methylguanine > 3-methyladenine > 1-methyl adenine ≅ 7-methyladeninine ≅ 3-methylcytosine ≅ 3-methylguanine > 3-methylthymidine ≅ O2-methylthymine > O4-methylthymine ≅ O4-methylcytosine.31, 32 While formamidopyrimidine (fapy) methyl adducts have been observed in animals treated with methylating nitrosamines,31 this adduct has not been measured in DNA from NNK-treated animals.
Figure 2.

Structures of NNK-derived DNA adducts.
3.1.1. Genotoxic properties of O6-mdGuo
The major mutagenic and toxic methyl adduct is O6-mdGuo.33 It induces primarily GC to AT transition mutations.34, 35 This adduct is predominantly repaired by O6-alkylguanine DNA alkyltransferase (AGT). This protein facilitates the transfer of the methyl group from the O6-position of guanine to a cysteinyl residue at the active site.33 This transfer reaction renders AGT inactive and it is degraded.36, 37 Therefore, constitutive levels of AGT determine the initial repair capacity of a cell. Mismatch repair (MMR) plays a major role in the toxic effects of O6-mdGuo.38–42 During the replication of this adduct, polymerases preferentially place T opposite the modified G. The MMR machinery recognizes this as a mismatch and removes the T. If the adduct is not repaired, the mismatch process enters a futile cycle since DNA replication will result again in a mismatch. This futile cycle can trigger apoptosis.32, 43, 44
3.1.2. Genotoxic properties of 7-mdGuo
While 7-mdGuo is the dominant adduct, it neither miscodes nor blocks replication.44–46 The ring-opened methyl fapy adduct is a block to replication and can be mutagenic, depending on the sequence context.46–50 Studies with site-specifically incorporated methyl FAPY adducts in primate cells indicated that this adduct induced base substitutions (primarily T but also A and C) and single and dinucleotide deletions.50 Depurination of 7-mdGuo leaves behind an abasic site which is mutagenic and toxic if not repaired.44 It is rapidly repaired by base excision repair (BER).
3.1.3. Genotoxic properties of minor methyl DNA adducts
There is evidence that the minor methyl adducts could contribute to the genotoxic effects of the methylating pathway. For example, O4-methyl-2′-deoxythymidine (O4-mdThd) induces primarily TA to CG and AT to TA mutations.34, 35 While studies with site specifically incorporated adducts indicated that O4-mdThd is more mutagenic than O6-mdGuo, O6-mdGuo is a more abundant adduct.34, 35 3-Methyl-2′-deoxyadenosine blocks replication, leading to cytotoxicity.32, 44 There is also evidence that this adduct is weakly mutagenic.32 Finally, O2-methyl-2′-deoxythymidine (O2-mdThd) is mutagenic inducing TA to GC and TA to AT mutations in E. coli and TA to AT mutations in human cells.51, 52 This adduct also blocks DNA replication.51, 52
3.2. Pyridyloxobutyl DNA adducts
The chemical characterization of pyridyloxobutyl DNA damage was challenging since a majority of these adducts dealkylate, releasing 4-hydroxy-1-(3-pyridyl)-1-butanone (HPB) under strong acid or neutral thermal DNA hydrolysis conditions (Figure 1).12, 53 The first chemically characterized pyridyloxobutyl DNA adduct was O6-[4-3-(pyridyl)-4-oxobut-1-yl]-2′-deoxyguanosine (O6-pobdGuo) (Figure 2).54 Subsequent studies led to the characterization of three additional pyridyloxobutyl DNA adducts: 7-[4-3-(pyridyl)-4-oxobut-1-yl]-2′-deoxyguanosine (7-pobdGuo), O2-[4-3-(pyridyl)-4-oxobut-1-yl]-2′-deoxycytosine (O2-pobdCyd), and O2-[4-3-(pyridyl)-4-oxobut-1-yl]-2′-deoxythymidine (O2-pobdThd) (Figure 2).55,56 Standards for N2-[4-3-(pyridyl)-4-oxobut-1-yl]-2′-deoxyguanosine were prepared but this adduct was not observed in pyridyloxobutylated DNA.57 The presence of phosphate adducts in pyridyloxobutylated DNA has been recently reported.58 These adducts were detected as phosphotriesters (PTEs), with two nucleosides linked together by an alkylated phosphate group; 30 of the possible 32 PTEs were detected.
The pyridyloxobutyl DNA adducts have varying stability (Table 1). The most stable adducts are O6-pobdGuo, O2-pobdThd and the phosphate adducts.54, 56, 58 O6-pobdGuo and O2-pobdThd will depurinate/depyrimidate when heated in 0.1 N HCl.54, 56 The other pyridyloxobutyl DNA adducts, 7-pobdGuo and O2-pobdCyd, undergo one of two reactions under neutral thermal hydrolysis conditions: they lose the sugar moiety to generate the nucleobase adducts, 7-[4-3-(pyridyl)-4-oxobut-1-yl]guanine (7-pobGua) and O2-[4-3-(pyridyl)-4-oxobut-1-yl]cytosine (O2-pobCyt) or they dealkylate to release HPB.55, 56 The dealkylation reaction increases in strong acid hydrolysis conditions. Another possible source of HPB-releasing DNA adducts are 2-(3-pyridyl)-2,3,4,5-tetrahydrofuran adducts. Studies with model pyridyloxobutylating agents provide evidence for a reactive cyclic oxonium ion in the alkylation of nucleophiles by the pyridylxobutylating intermediate.59 This cyclic oxonium ion was trapped with nucleophiles such as thiols. While there is no direct evidence for these adducts in DNA, they are expected to be unstable and release HPB under all DNA hydrolysis conditions.12, 53
Table 1.
Stability of pyridyloxobutyl DNA adducts
| DNA adduct | Conditions | product | reference |
|---|---|---|---|
| O6-pobdGuo | pH 7, 30 min at 100° C | Stable | 54 |
| 0.1 N HCl, 30 min at 80° C | O6-pobGua | 54 | |
| 0.8 N HCl, 1 h at 80° C | HPB and Gua | 54 | |
| 7-pobdGuo | Enzyme hydrolysis of DNA: pH 7, 70 min at 37° C | Stable | 55 |
| pH 7, 1 h at 100° C | 7-pobGua and HPB | 55 | |
| 0.1 N HCl, 1 h at 80° C | 7-pobGua and HPB | 55 | |
| O2-pobdCyd | Enzyme hydrolysis of DNA: pH 7, 70 min at 37° C | Stable | 56 |
| pH 7, 1 h at 100° C | O2-pobCyt and HPB | 56 | |
| O2-pobdThd | Enzyme hydrolysis of DNA: pH 7, 70 min at 37° C | Stable | 56 |
| pH 7, 1 h at 100° C | Stable | 56 | |
| 0.1 N HCl, 1 h at 85° C | O2-pobThy | 56 | |
| Phosphate adducts | pH 7, 48 h at 37° C | Stable | 58 |
The relative levels of pyridyloxobutyl DNA adducts were determined in calf thymus DNA alkylated with the model pyridyloxobutylating agent, 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone (NNKOAc); in the presence of esterase, NNKOAc generates the same pyridyloxobutylating agent formed following the oxidation of NNK by cytochrome P450 (Figure 3).59 Quantitation of all these adducts in NNKOAc-treated DNA indicates that HPB-releasing adducts represent about 65% of the total pyridyloxobutyl DNA adducts.60 The four characterized adducts account for the remaining amount. The relative levels of the four adducts is 7-pobdGuo > O6-pobdGuo > O2-pobdThd ≥ O2-pobdCyd.61 The phosphate adducts represented about 5% of the total DNA adducts in NNKOAc-treated calf thymus DNA.58
Figure 3.

Structures and hydrolysis of model alkylating agents.
Indirect evidence for the formation of fapy adducts in DNA from NNKOAc-treated human cells was obtained using the comet assay in the presence and absence of formamidopyrimidine glycosylase.62 Since more breaks were observed in the presence of the glycolyase, it was concluded that pyridyloxobutyl fapy adducts were present in the alkylated DNA. It is not known if pyridyloxobutyl fapy adducts are repaired as efficiently as methyl fapy adducts.
3.2.1. Genotoxic properties of O6-pobdGuo
Several of the pyridyloxobutyl DNA adducts are mutagenic. O6-pobdGuo is mutagenic, generating exclusively GC to AT mutations in bacteria and predominantly GC to AT mutations with some GC to TA transversion mutations in human cells.63 It is repaired by AGT.54, 64, 65 The bulky pyridyloxobutyl group gets transferred to the protein’s active site, inactivating AGT.65, 66 AGT is able to modulate the mutagenic properties of DNA pyridyloxobutylating agents at GC base pairs in both mammalian and bacterial systems, supporting the mutagenic properties of O6-pobdGuo.67, 68 Unlike other bulky O6-alkyguanine adducts, O6-pobdGuo is not a substrate for nucleotide excision repair (NER).68 Studies with site-specifically incorporated O6-pobdGuo indicated that this adduct was also not repaired by bacterial AlkD glycosylase or human alkyladenine glycosylase.69
3.2.2. Genotoxic properties of O6-pobdThd
Another mutagenic pyridyloxobutyl DNA adduct is O2-pobdThd. The major mutation induced by this adduct is TA to AT mutations with small amounts of TA to GC and/or TA to CG mutations in human embryonic kidney 293T (HEK293T) cells.52 TA to GC mutations were the major mutation (37%) compared to TA to AT (12%) induced by this adduct in bacterial strains.51 The removal of O2-pobdThd from DNA was slower in Chinese hamster ovary (CHO) cells lacking the ERCC2 (XPD) gene (UV5 cells);68 this is an important protein in the NER pathway. Therefore, O2-pobdThd is likely a substrate for NER repair. Knockout of this repair pathway was also associated with increased NNKOAc-derived cytotoxicity in UV5 cells relative to the control cell line.68 Consistent with these observation, this adduct is a block to replication in E. coli and HEK293T cells.51, 52 The absence of NER in the UV5 CHO cell line also resulted in an increase in NNKOAc-induced hprt mutations, specifically TA to AT mutations, providing support for the importance of O2-pobdThd in the overall mutagenic activity of pyridyloxobutylating agents.68 Pol ζ and other Y-family polymerases are thought to play a major role in the error-prone and error-free bypass of this bulky lesion.52 This adduct is not a substrate for AGT, bacterial AlkD glycosylase or human Aag.68, 69
3.2.3. Genotoxic properties of 7-pobdGuo
The miscoding properties of 7-pobdGuo have not been investigated. A number of 7-alkylguanine adducts undergo imidazole ring-opening to mutagenic fapy adducts. Examples include fapy-7-methylguanine and aflatoxin B1-fapy adducts which both trigger GC to TA mutations.70–72 It is not known if 7-pobdGuo forms a fapy adduct, but it is worth noting that pyridyloxobutylated DNA contains fapy glycosylase-sensitive adducts.62 Since 7-pobdGuo spontaneously depurinates from DNA generating abasic sites,55, 60 it is possible that this adduct contributes to the genotoxic properties of the pyridyloxobutyl pathway by generating abasic sites. There does not appear to be active repair of this adduct in mammalian cells.68, 73
3.2.4. Genotoxic properties of O2-pobdCyd
The miscoding properties of O2-pobdCyd have also not been studied. This adduct spontaneously depyrimidates from DNA, generating abasic sites.56 While there is no evidence of active repair in mammalian cell lines,68 the levels of this adduct are, in general, 10 times lower relative to the other pyridyloxobutyl DNA adducts in vivo than what is reported in in vitro alkylated DNA.73 Therefore, active repair of this adduct may occur in vivo.
3.2.5. Genotoxic properties of abasic sites
It is possible that abasic sites could contribute to the overall genotoxic properties of NNK since there are multiple adducts that spontaneously decompose leaving behind abasic sites (see above). Abasic sites within GC base pairs cause GC to AT and GC to TA mutations.74 Consistent with this hypothesis, the cytotoxic and mutagenic effects of NNKOAc were roughly doubled in CHO cell lines lacking functional XRCC1 (EM9 cells).68 XRCC1 is an important scaffold protein that participates in numerous steps in the base excision repair (BER) pathway.75 Therefore, the pyridyloxobutyl pathway generates DNA damage that requires XRCC1 activity for the cell to escape the cytotoxic and mutagenic effects of this damage. There may be specific pyridyloxobutyl DNA adducts that are substrates for BER glycosylases that initiate the BER process. Alternatively, pyridyloxobutylation of DNA generates abasic sites since both 7-pobdGuo and O2-pobdCyd spontaneously depurinate/depyrimidate from DNA. These resultant abasic sites are expected to be repaired by the BER machinery. Since there was an increase in the mutation frequency of NNKOAc-induced AT to TA mutations in EM9 cells, abasic sites may be forming in AT base pairs. Studies with site-specific abasic sites indicate that they cause more AT to TA mutations than AT to GC mutations.74 Therefore, abasic sites generated either by glycosylase catalyzed removal of pyridyloxobutylated adducts or by spontaneous depurination/depyrimidation may contribute to the mutagenic activity of pyridyloxobuylating agents.
3.3. Aldehyde DNA adducts
The aldehydes formed in NNK or NNAL metabolism are also capable of reacting with DNA. Formaldehyde reacts with the exocyclic amines of dAdo, dCyd and dGuo to form reversible hydroxymethyl adducts, N6-hydroxymethyl-2′-deoxyadenosine, N4-hydroxy-2′-deoxycytotidine and N2-hydroxymethyl-2′-deoxyguanosine (Figure 2).76–81 These initial adducts lead to DNA-DNA crosslinks such as dAdo-CH2-dAdo, dGuo-CH2-dGuo and dAdo-CH2-dAdo.76, 77, 81 They also lead to the formation of DNA-protein crosslinks.82 Formaldehyde is mutagenic in CHO, V79 and mouse lymphoma cells as well as human cells.83–87 Formaldehyde induced AT to TA, AT to CG and GC to TA mutations in CHO cells.85 Recent studies in CHO cells provide evidence that that formaldehyde could contribute to the mutagenic properties of nitrosamines.88 A number of these formaldehyde DNA adducts have been detected in NNK-treated rodents and in NNKOAc-treated calf thymus DNA.81, 89 There are no studies investigating the formation of DNA-protein crosslinks in tissue DNA following NNK exposure.
While OPB-derived DNA adducts have not been directly detected, indirect evidence for DNA damage by this aldehyde exists. It induced sister chromatid exchanges in V79 cells (OPB concentrations from 0.01 to 0.5 mM) and caused DNA single strand breaks in V79 cells (OPB concentrations from 0.01 to 1 mM) and hepatocytes (OPB concentrations > 5 mM).90–92
3.4. Pyridylhydroxybutyl DNA adducts
Methyl hydroxylation of NNAL leads to the formation of at least four distinct DNA adducts: 7-[4-3-(pyridyl)-4-hydroxybut-1-yl]-2′-deoxyguanosine (7-phbdGuo), O6-[4-3-(pyridyl)-4-hydroxybut-1-yl]-2′-deoxyguanosine (O6-phbdGuo), O2-[4-3-(pyridyl)-4-hydroxybut-1-yl]-2′-deoxycytosine (O2-phbdCyd), and O2-[4-3-(pyridyl)-4-hydroxybut-1-yl]-2′-deoxythymidine (O2-phbdThd) (Figure 2).56, 93 The mutagenic activity of these adducts has not been investigated. However, (R)-NNAL is a pulmonary carcinogen and it generates significantly more pyridylhydroxybutyl DNA adducts than pyridyloxobutyl DNA adducts and O6-mdGuo.28 Therefore, the genotoxic potential of this DNA damaging pathway should be explored.
4. Contribution of specific DNA adducts to the genotoxic and carcinogenic activity of NNK in model systems
Which adduct contributes to the genotoxic effects of a nitrosamine will depend on the context. As outlined in the examples below, it depends on the relative amounts of each DNA alkylating pathway occurring in the model system, the levels and genetic variants of key repair enzymes and the gene which is targeted for mutation.
4.1. Bacterial models of genotoxicity
In most bacterial systems tested, the pyridyloxobutylating pathway was more genotoxic than the methylating pathway. Deuterium substitution on the α-methyl but not the α-methylene carbons of NNK reduced the mutagenic activity of NNK in S. typhimurium strains TA 1535 and TA100 in the presence of Aroclor induced rat liver S9, indicating that pyridyloxobutylation pathway as more active as a mutagen in the bacteria. 94 Consistently, model pyridyloxobutylating agents are more potent bacterial mutagens than methylating agents.94, 95 The pyridyloxobutylating agent, 4-(carbethoxynitrosamino)-1-(3-pyridyl)-butanone, was more mutagenic than the methylating agent carbethoxynitrosaminomethane in the same strains.94 In the Salmonella typhimurium mutagenesis test, NNKOAc produced 770 – 1400 revertants/μmol in strains TA98, TA100 and TA1535 whereas the model methylating agent, acetoxymethylmethylnitrosamine (AMMN, Figure 3) caused 130 – 170 revertants/μmol in these strains.95 These three strains are measurements of mutation frequency at GC base pairs; TA100 and TA1535 requires a point mutation whereas TA98 detects frameshift mutations.96
One reason that these bacterial strains are less susceptible to the mutagenic effects of methylating agents is that they have a robust repair defense against O6-mdGuo. The ogt gene codes for a constitutively active form of AGT whereas the expression of the ada protein is inducible during alkylation stress. Knockout of these two proteins causes bacteria to become very sensitive to the mutagenic and toxic properties of methylating agents.97 Studies with recombinant forms of AGT indicated that O6-pobdGuo is a poor substrate for the equivalent bacterial repair proteins Ogt and Ada.65 These proteins repair O6-pobdGuo more than ten times more slowly than O6-mdGuo, explaining, in part, the increased mutagenic activity of pyridyloxobutylating agents over methylating agents in bacterial systems. When mutagenic activity was compared to DNA adduct levels in the TA100 strain, the mutations per mutagenic adduct were comparable between NNKOAc and AMMN; all HPB-releasing adducts were assumed to be mutagenic whereas O6-mdGuo was considered as the only mutagenic adduct formed.95 Furthermore, the expression of human AGT in S. typhimurium strain YG7108 protects against the mutagenic activity of methylating and pyridyloxobutylating agents.67, 98 This strain is a derivative of TA1535 which lacks both the ada and ogt genes detects mutations at GC base pairs and is exquisitely sensitive to mutagenic properties of methylating agents.98 Collectively, these data support the hypothesis that the different repair rates for O6-mdGuo and O6-pobdGuo contributes to the different activities of methylating and pyridyloxobutylating agents in these bacterial strains.
The two O6-alkylguanine DNA adducts are also differentially mutagenic in bacterial systems as demonstrated in site-specific mutagenesis studies in E. coli DH10B cells.63, 99 O6-pobdGuo is very mutagenic when presented as a single stranded substrate, generating a mutation rate of 96%, with all of these mutations being GC to AT mutations.63 In contrast, O6-mdGuo in an identical construct had a mutation rate of 60%, with all of the mutants being GC to AT mutations.99
Another factor likely influencing the mutation frequency of methylating and pyridyloxobutylating agents in these bacterial strains is the differing cytotoxic properties of the O6-alkylguanine adducts. Unlike O6-mdGuo, O6-pobdGuo does not appear to be a cytotoxic adduct in bacteria. Modulation of adduct levels by the expression of human AGT in S. typhimurium strain YG7108 had no impact on the cytotoxicity of NNKOAc whereas it dramatically reduced the cytotoxic properties of a methylating agent.67 Cytotoxicity can limit the mutagenic properties of an alkylating agent since the cells are dying and therefore, are not able to undergo mutation.
Neither NNKOAc or AMMN were very active at inducing mutations in S. typhimurium strains TA102, which contains an AT base pair at the reversion site.95, 96 Recent studies demonstrate that O2-pobdThd blocks replication in E. coli which will negatively affect the mutation frequency at AT base pairs.51 Some mutagenic activity was observed, supporting the formation of methyl and pyridyloxobutyl DNA adducts at AT base pairs. The pyridyloxobutyl DNA adduct, O2-pobdThd, is modestly mutagenic in bacterial strains; TA to GC was the major mutation (37%) compared to TA to AT (12%) induced by this adduct.51 The pyridyloxobutyl analog was more miscoding than the corresponding methyl analog.51
A more complex picture emerges when testing the mutagenesis of NNK in genes that do not require mutations at specific base pairs. For example, mutations in the lacI gene in E. coli exposed to NNK in the presence of Arochlor-induced S9 indicate that multiple mutagenic NNK-derived DNA adducts are formed. While GC to AT mutations were dominant, representing 55% of all the mutations, GC to TA (10%), GC to CG (7%), AT to GC (5%), AT to CG (5%) and AT to TA (3%), frameshifts (5%), deletions (3%) and duplication (5%).100 Since these studies were performed in E. coli strains with active bacterial DNA repair and damage response genes, it is likely that O6-pobdGuo is responsible for the GC to AT mutations and O2-pobdThd is causing the AT to GC mutations in this gene. Similar studies with methylating agents indicate they generated roughly 90% GC to AT mutations.101, 102
4.2. Mammalian cell lines
In mammalian cell lines, the relative mutagenic activity of the pathways is reversed, with methylating agents often being more mutagenic than the pyridyloxobutylation pathway. In a transgenic Chinese hamster V79 cell line expressing the Escherichia coli xanthine-guanine phosphoribosyl transferase (gpt) gene (G12 cells),103 the methylating agent AMMN was more mutagenic than the pyridyloxobutylating chemical NNKOAc.95 Similarly, methylating agents are much more toxic on a molar basis than NNKOAc in CHO cells.68, 88 Much of these differences is explained by differences in the levels of DNA damage;68, 88, 95 the reactive methanediazonium ion is a more potent DNA alkylating species than an equal molar amount of a corresponding pyridyloxobutyl analog.
When looking at relative mutagenic activity of specific adducts, O6-mG in a gapped vector is more mutagenic than O6-pobdGuo when HEK293T cells have full repair capacity.34, 63 The bulky O6-pobdGuo is more mutagenic than O6-mdGuo when AGT is depleted in these cell lines.34, 63 The relative mutagenic properties of O2-pobdThd and O2-mdThd have also been compared in HEK293T cells.52 The smaller adduct was 20% more mutagenic than the larger one in part because O2-pobdThd was a greater block to translesional synthesis than O2-mdThd. Both adducts primarily induce TA to AT mutations in this model.
In CHO cells, O6-mdGuo was primarily responsible for the cytotoxic and mutagenic properties of methylating agents.39, 40, 88 The situation is more complicated for the pyridyloxobutylation pathway. This question was explored in CHO cells with different repair capacities.68 CHO cells do not express AGT. Expression of human AGT in these cells protected the cells against the toxic but not mutagenic effects of NNKOAc. Since the expression of AGT led to the complete removal of O6-pobdG, it appears that this adduct does not contribute significantly to the genotoxic effects of NNKOAc in the hprt gene in this model. Deficiencies in NER or BER lead to both increased sensitivity of CHO cells to the toxic and mutagenic activity of NNKOAc to a similar extent.68 The removal of O2-pobdThd was affected by knocking out the NER pathway; none of the other adduct levels were influenced by the absence of NER. Therefore, O2-pobdThd likely contributes to the cytotoxicity and mutagenicity of NNKOAc. This observation is consistent with the report that this adduct blocks translesional synthesis and gives rise to TA to AT mutations with small amounts of TA to GC and/or TA to CG mutations.52 Knockout of BER in these cells did not influence the repair of any of the pyridyloxobutyl DNA adducts since the missing protein is XRCC1 which is downstream from adduct removal in the BER pathway.
The activity of hypoxanthine-guanine phosphoribosyltransferase (hprt) can be knocked out by a wide variety of hprt gene mutations.104 Sequencing of the hprt mutants indicated that the majority of NNKOAc-induced mutations in the hprt gene were at AT base pairs (Table 2). The expression of AGT reduced the extent of GC to AT mutations consistent with the complete repair of O6-pobdGuo in this cell line (CHOAGT cells, Table 2). This reduction did not impact the overall mutagenesis rate in this gene since GC to AT mutations were a small percentage of the overall total. Furthermore, it is possible that 7-pobdGuo and O2-pobdCyd could contribute to the mutation frequency at GC basepairs. NNKOAc-induced TA to AT and TA to CG were significantly increased in the absence of NER (UV5 cells, Table 2). Since the repair of O2-pobdThd was reduced in these cells, it is hypothesized to be the source of these mutations. Finally, BER protected against NNKOAc-induced hprt mutations at AT base pairs as evidenced by the increase in mutations at AT base pairs when XRCC1 is knocked out (EM9 cells, Table 2). It is not known whether O2-pobdThd is a substrate for BER glycosylases. There may also be other uncharacterized adenine or thymidine DNA adducts. Another possibility is that formaldehyde which is generated from the decomposition of NNKOAc (Figure 3) is responsible for some of the NNKOAc-derived mutagenic DNA damage.81 Formaldehyde induced AT to TA (3/6), AT to CG (2/6) and GC to TA (1/6) mutations in CHO cells85 so it could be an important mutagenic adduct formed along with the pyridyloxobutyl DNA damage derived from NNKOAc.
Table 2.
Base substitution mutations observed in 6-thioguanine-resistant mutant colonies in CHO cell lines.68
| % of base substitution | |||||||
|---|---|---|---|---|---|---|---|
| Class of mutations | 24 μM NNKOAc
|
Spontaneous
|
|||||
| AA81 | UV5 | EM9 | CHOpcDNA3 | CHOAGT | AA82 | EM93 | |
|
|
|
||||||
| Transitions: | |||||||
| A:T→G:C | 4 | 13 | 13 | 13 | 12 | 2 | 0 |
| G:C→A:T | 25 | 17 | 17 | 17 | 5 | 16 | 16 |
| Transversions: | |||||||
| A:T→C:G | 63 | 35 | 52 | 70 | 74 | 12 | 0 |
| A:T→T:A | 4 | 22 | 17 | 0 | 5 | 6 | 8 |
| G:C→C:G | 0 | 4 | 0 | 0 | 0 | 12 | 8 |
| G:C→T:A | 4 | 9 | 0 | 0 | 5 | 35 | 69 |
|
|
|
||||||
The parental cell line, AA8, does not express the repair protein AGT 135. UV5 has a mutation in the ERCC-2 gene (XPD) and, as a result, is deficient in NER.136. EM9 lacks XRCC1 and is compromised in BER.137 CHOpcDNA3 cells are derived from AA8 cells and are stably transfected with the empty pCMV-neo-Bam vector.135 CHOAGT cells are stably transfected with the pCMV-hAGT expressing human AGT. 135
As previously reported.138
As previously reported.139
4.3. Animal models
Investigations in animal models support the hypothesis that the DNA adducts that contribute to the carcinogenic properties of NNK depends on the model which is being tested. The relative contribution of each α-hydroxylation pathway and its associated DNA damage to the overall carcinogenic properties of nitrosamines is species and tissue dependent.
4.3.1. Mouse models
DNA methylation is the critical pathway for NNK-induced lung tumorigenesis in the A/J mouse lung tumor model.27, 105 Compounds that only pyridyloxobutylate DNA are weak lung carcinogens in this model.27 Consistently, deuterium substitution of the methylene protons of NNK significantly reduced the levels of O6-mdGuo and, consequently, the pulmonary carcinogenic activity of NNK.105 Deuteration of the methyl group caused a significant increase in lung tumorigenic activity over the unlabeled compound. A comparison of methyl and pyridyloxobutyl DNA adduct levels indicates that there is substantially more O6-mdGuo adducts than pyridyloxobutyl DNA adducts formed in A/J mouse lungs (Figure 4), supporting the major role of the methylation pathway in the overall lung tumorigenic activity of NNK.
Figure 4.

Levels of NNK-derived DNA damage in lungs of A/J mice 8 h after treatment with 10 μmol NNK.27, 73 The error bars represent standard deviation of 3 – 5 replicates.
The formation and persistence of O6-mdGuo is critical for NNK-induced lung tumor formation in A/J mice.27, 106 The mutagenic adduct O6-mdGuo persists in lung DNA for at least two weeks; these levels are highly correlated to pulmonary tumorigenic activity in A/J mice 27, 106 The persistence of this adduct is, in part, driven by the relatively poor repair capacity of the lung. The levels of AGT are much lower in the lung than the liver (Table 3).106 Furthermore, the activity of this important repair protein is suppressed for much longer in the target tissue (Table 3).106 The overexpression of human AGT in transgenic mice reduced the potency of NNK as a lung carcinogen.107
Table 3.
Levels of AGT in lung and liver in A/J mice treated with NNK. 106
| fmol AGT/mg proteinb | |||
|---|---|---|---|
| Tissue | Treatmenta | 4 h | 96 h |
| Lung | 0 μmol NNK | 28 ± 5 | 33 ± 6 |
| 10 μmol NNK | 7 ± 2 | 8 ± 2 | |
| Liver | 0 μmol NNK | 110 ± 30 | 110 ± 20 |
| 10 μmol NNK | 0 ± 0 | 110 ± 30 | |
Groups of 30 mice were given i.p. injections NNK (0 or 10 umol) in saline (0.2 mL). Each group was subdivided into 2 groups for sacrifice at 4 or 96 h.
Mean of 4 - 13 samples ± SD.
Additional support for the role of O6-mdGuo in the tumorigenic activity of NNK in the A/J mouse comes from investigations into the K-ras mutation spectra induced by NNK and model alkylating compounds (Table 4). K-ras mutation and activation is closely associated with pulmonary tumor formation in NNK-treated A/J mice.108, 109 Ninety six percent of the K-ras mutations in tumors from NNK-treated mice were GC to AT mutations in the second base of the twelfth codon, consistent with the formation of O6-mdGuo. All of the K-ras mutations in tumors from AMMN-treated mice were GC to AT mutations at this loci. NNKOAc induced GC to TA as well as GC to AT mutations in the twelfth codon of the K-ras oncogene of tumors in A/J mice.110 Together, these data indicate that metabolic activation of NNK to a DNA methylating agent is critical for lung tumor formation in the A/J mouse.
Table 4.
K-ras mutations in tumors from carcinogen-treated A/J mice.110
| Compound | No. of tumors | GGT (wt) | GAT | TGT | GTT |
|---|---|---|---|---|---|
| NNK | 28 | 0 % | 96% | 4% | 0% |
| NNKOAc | 21 | 19% | 38% | 24% | 19% |
| AMMN | 18 | 0 | 100% | 0 | 0 |
Point mutations in codon 12, 13 or 61 of the ras gene leads to amino acid substitution in the protein that impairs the GTPase activity of the protein, resulting in constitutive activation of ras signaling pathways, leading to tumor formation.reviewed in 111 In the codon 12, these activating mutations primarily occur at GC base pairs.112 Therefore, it is not surprising that alkylating agents are particularly good at generated activated K-ras mutations.
As with the bacterial mutagenesis experiments, investigations into the mutagenic properties of NNK in mice using genes that are knocked out by a broad spectrum of mutations indicate that NNK is able to trigger a more complex spectrum of mutations. Studies in Muta™ transgenic mice demonstrated that NNK was a potent inducer of lacZ and cII mutations in both lungs and liver.113 Unlike the mutation spectrum in K-ras when the mutations were almost exclusively GC to AT mutations, the dominant mutation in the cII gene was AT to TA mutations, followed by AT to CG mutations. There was a more modest increase in GC to AT mutations at this gene loci. When the AGT activity was knocked out in this mouse strain, a modest increase in GC to AT mutations were observed in the lacI gene in the lungs of NNK-treated mice.114 These data indicate that NNK is capable of generating mutations at both GC and AT base pairs in mouse lungs, indicating that multiple mutagenic adducts are formed from NNK.
While the formation of O6-mdGuo is critical for lung tumor formation in the A/J mouse, there is strong evidence that the pyridyloxobutylation pathway also contributes to the overall tumorigenic activity of NNK in this model. When the methylating agent, AMMN, was co-administered with the pyridyloxobutylating agent, NNKOAc, the tumor yield was higher.27 Because this combination resulted in higher levels of O6-mdGuo, the ability of NNKOAc to increase the tumorigenic activity of AMMN was attributed to its ability to enhance the persistence of O6-mdGuo in lung DNA.27 Subsequent studies indicate that this increased persistence is in part due to competition between O6-pobdGuo and O6-mdGuo for repair by AGT.27, 66, 106
Both O6-pobdGuo and O6-mdGuo are substrates for AGT as discussed above. Studies with purified proteins indicate that these adducts compete efficiently with one another for repair by AGT variants. 65 Therefore, the presence of both O6-alkylguanine adducts will lead to their increased persistence in lung DNA. However, the in vivo levels of O6-pobdGuo are not sufficient to explain the persistence of O6-mdGuo in NNK-treated mouse lung. At 8 h after treatment, the levels of O6-pobdGuo are 1/20th of those of O6-mdGuo (Figure 4).27, 73 Even taking into account that some repair of O6-pobdGuo has occurred, the levels of O6-pobdGuo are not high enough for competition for AGT to explain the extended persistence of O6-mdGuo. Similarly, the adduct level measured for a 4.2 μmol dose of NNKOAc (3 pmol/μmol guanine) is not sufficient to explain the doubling of O6-mdGuo levels observed in lung DNA (from 20 pmol O6-mdGuo/μmol guanine to 41 pmol O6-mdGuo/μmol guanine at 4 h) when a 1.0 μmol dose of AMMN is combined with 4.2 μmol dose of NNKOAc.27, 73 Therefore, other mechanisms of interaction need to be considered. One possibility is that the pyridyloxobutylation pathway reduces the expression of AGT through an indirect route. Support for this hypothesis comes from the observation that levels of NER proteins are reduced in lungs of A/J mice following NNK exposure.115
Another way that the pyridyloxobutylation pathway can contribute to the tumorigenic properties of NNK is through increasing the mutagenic burden in lung DNA. Adduct studies indicate that the pyridyloxobutyl DNA persist for more than 96 h following treatment with NNK.73 The levels of 7-pobdGuo and O2-pobdThd did not change significantly over this period of time. Furthermore, pyridylhydroxylbutyl DNA adducts have been detected in lung DNA from NNK-treated mice.116 These adducts result from the in vivo reduction of NNK to NNAL followed by α-methyl hydroxylation generating a pyridylhydroxybutylating agent in the lungs. The levels of O6-hpbdGuo, O2-hpbdThd and 7-hpbdGuo were slightly higher than the levels of pyridyloxobutyl DNA adducts but still much lower than the levels of O6-mdGuo in the target tissue. All these bulky adducts could contribute to the tumorigenic activity of NNK by causing mutations in genes other than K-ras.
4.3.2. Rat models
In rats, the relative contribution of the DNA adducts to the pulmonary carcinogenic effects of NNK is more complicated. The available experimental evidence is consistent with the hypothesis that both DNA methylation and DNA pyridyloxobutylation are important for tumor formation. Compounds that generate only DNA methyl adducts (i.e dimethylnitrosamine) or only pyridyloxobutyl DNA adducts (i.e. N-nitrosonornicotine) are not potent lung carcinogens in this rodent model.12, 24 Furthermore, [4,4-2H2]NNK and [C2H3]NNK were comparable in potency as rat pulmonary carcinogens.117 DNA adduct studies indicate that both methyl and pyridyloxobutyl DNA adducts persist in rat lung DNA following NNK treatment. The mutagenic methyl adduct, O6-mdGuo, was selectively persistent in Clara cells for up to 8 days after NNK treatment (10 mg/kg/day for 4 days); it is efficiently removed from DNA in other lung cell types.118 A strong correlation between tumor formation and levels of O6-mdGuo in Clara cells was observed in NNK-treated rats.118 While there was not a similar correlation between O6-mdGuo in type II cells and tumor formation,118 HPB-releasing DNA adduct in Type II cells were strongly correlated to lung tumor formation in rats.119 In this study, no correlation was not observed between HPB-releasing adduct levels in Clara cells and tumor formation. Type II cells are the source of NNK-derived lung tumors in rats.22 These data suggest that there may be communication from initiated Clara cells that influence the transformation of NNK-induced tumorigenesis in Type II cells.25
More recently, specific pyridyloxobutyl or pyridylhydroxybutyl DNA damage have been quantified in lung DNA of NNK-treated rats.28, 29, 61, 89, 120 The most abundant pyridyloxobutyl adducts in lung DNA are 7-pobdGuo and O2-pobdThd. The latter adduct accumulates more readily in chronic dosing studies.28, 120 The levels of O6-pobdGuo are very low, indicating that it is rapidly repaired in this animal model.28, 29, 61, 89, 120 The levels of hydroxybutyl DNA adducts are lower than the levels of the pyridyloxobutyl DNA adducts.28, 121 As with O2-pobdThd, O2-phbdT accumulates in lung DNA with chronic treatment.121
Formaldehyde DNA adducts as measured by the conversion of N6-HOCH2-dA to N6-mdA were detected in lung DNA from rats treated with 4 daily s.c. injections of NNK.89 The levels of this adduct were comparable to the levels of O6-pobdG in these animals. Given that the extent of conversion of the adduct to the stable form analyzed for, the amounts measured are likely an underestimation of the levels of this adduct present in vivo.
A comparison of the levels of methyl, pyridyloxobutyl and pyridylhydroxylbutyl DNA adducts in lungs of rats chronically treated with 10 ppm NNK indicates that the levels of O6-mdGuo are comparable to levels of 7-pobdGuo and O2-pobdThd at early time points but O2-pobdThd is more likely to accumulate in lung DNA (Figure 5).121 A more recent study in animals receiving 5 ppm NNK indicated that O2-pobdThd was the major adduct in lung DNA with 7-pobdGuo and O6-mdGuo being the next most abundant adducts.28 As in the previous study, the levels of O6-pobdG were very low. Collectively, these experimental data support a hypothesis that O2-pobdThd and O6-mdGuo are the primary adducts that may contribute to the formation of lung tumors in NNK-treated rats.
Figure 5.

Levels of adducts in lungs of rats treated with 10 ppm NNK in the drinking water for 2, 10 and 20 weeks.29, 120, 121 The error bars represent standard deviation of 3 replicates
5. Summary and Future Directions
Together, these data provide strong evidence for why NNK is such a potent carcinogen. There are multiple pathways that lead to the formation of a variety of genotoxic DNA adducts. Each pathway by itself has carcinogenic potential, capable of generating significant damage at both GC and AT base pairs. Which adduct will contribute to the overall carcinogenic activity depends not only on the amount formed, but the activity of the various repair pathways present at the site of DNA damage and on what types of mutations that are required to activate oncogenes or turn off tumor suppressor genes.
Smokers are exposed to and metabolize NNK.122–124 α-Hydroxylation is the major metabolic pathway in humans125 Evidence for both methylation and pyridyloxobutylation of DNA in smokers is available.126–130 It is likely that these adducts contribute to the overall tumorigenic properties of tobacco smoke in humans. Which NNK adducts contribute to the carcinogenic properties of NNK in humans is not known. Unlike rodents, human AGT repairs O6-pobdGuo more slowly than O6-mdGuo indicating that this mutagenic pyridyloxobutyl DNA adduct may be more persistent in humans than it is in the rodent models.65 A detailed investigation into this question in human cell lines would provide valuable insights into this question.
Genetic variations in metabolism and DNA repair and response proteins may influence which adduct or spectrum of adducts is harmful to any one individual. Very little is known how humans vary in their sensitivity to the DNA damage generated from NNK. Given the wide variety of DNA damage coming from a single molecule, one can envision that different individuals will be more or less sensitive to specific types of NNK-derived DNA damage. For example, some epidemiological studies indicate that smoking related O6-alkylguanine adducts contribute to lung cancer risk from tobacco smoke exposure. Genetic variations in the mgmt gene which codes for AGT are associated with increased risk of lung cancer in smokers or those exposed to second hand smoke.131–133 My research group has shown that the genetic variants of AGT that impact the binding pocket size have differing abilities to repair O6-pobdGuo versus O6-mdGuo in vitro.65, 134 Understanding how these in vitro differences translate into susceptibility to mutagenesis requires further study.
A complete understanding of the role of specific adducts to the carcinogenic activity of NNK will also require a more complete understanding of the toxicological properties of specific DNA damage. The mutagenic characteristics of 7-pobdGuo should be defined. This adduct persists in vivo and may contribute to NNK-derived mutations at GC base pairs. Furthermore, it should be determined if this adduct can undergo imidazole ring opening to mutagenic fapy adducts; methyl-fapy and aflatoxin B1-fapy adducts caused GC to TA transversion mutations, providing support that a pyridyloxobutyl fapy adduct might be an important NNK-derived DNA damage.70–72 The role of aldehyde DNA damage in NNK-induced mutagenesis and carcinogenesis should also be defined. Our studies in CHO cells indicate that these adducts may play an important role in the toxicological properties of NNK. Finally, the genotoxic properties of the pyridyloxobutyl phosphate adducts as well as all of the pyridylhydroxybutyl DNA adducts are still to be explored.
Acknowledgments
Funding Sources.
The research on NNK conducted in Lisa Peterson’s lab is or has been funded by the following research grants from the National Institutes of Health: CA59887, CA115309, CA138338 and CA184987. The Analytical Biochemistry Shared Resource at the University of Minnesota is supported in part by Cancer Center Support Grant CA-77598.
I thank my collaborators and all the people in my research group who have contributed to this work over the years. I also thank Bob Carlson for his assistance with the figures.
Abbreviations
- 7-mdGuo
7-methyl-2′-deoxyguanosine
- 7-phbdGuo
7 [4-3-(pyridyl)-4-hydroxybut-1-yl]-2′-deoxyguanosine
- 7-pobGua
7-[4-3-(pyridyl)-4-oxobut-1-yl]guanine
- 7-pobdGuo
7-[4-3-(pyridyl)-4-oxobut-1-yl]-2′-deoxyguanosine
- AGT
O6-alkylguanine DNA alkyltransferase
- AMMN
N-acetoxymethyl-N-methylnitrosamine
- BER
base excision repair
- CHO
Chinese hamster ovary
- fapy
formamidopyrimidine
- HEK293T cells
human embryonic kidney 293T cells
- HPB
4-hydroxy-1-(3-pyridyl)-1-butanone
- hprt
hypoxanthine-guanine phosphoribosyl transferase
- MMR
mismatch repair
- NER
nucleotide excision repair
- NNAL
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol
- NNK
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- NNKOAc
4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone
- O2-phbdCyd
O2-[4-3-(pyridyl)-4-hydroxybut-1-yl]-2′-deoxycytosine
- O2-pobdCyd
O2-[4-3-(pyridyl)-4-oxobut-1-yl]-2′-deoxycytosine
- O2-pobCyt
O2-[4-3-(pyridyl)-4-oxobut-1-yl]cytosine
- O6-mdGuo
O6-methyl-2′-deoxyguanosine
- O6-phbdGuo
O6-[4-3-(pyridyl)-4-hydroxybut-1-yl]-2′-deoxyguanosine
- O6-pobGua
O6-[4-3-(pyridyl)-4-oxobut-1-yl]-guanine
- O6-pobdGuo
O6-[4-3-(pyridyl)-4-oxobut-1-yl]-2′-deoxyguanosine
- O2-mdThd
O2-methyl-2′-deoxythymidine
- O4-mdThd
O4-methyl-2′-deoxythymidine
- O2-phbdThd
O2-[4-3-(pyridyl)-4-hydroxybut-1-yl]-2′-deoxythymidine
- O2-pobdThd
O2-[4-3-(pyridyl)-4-oxobut-1-yl]-2′-deoxythymidine
- O2-pobdThy
O2-[4-3-(pyridyl)-4-oxobut-1-yl]-thymine
- OPB
4-oxo-1-(3-pyridyl)-1-butanone
Biography
Lisa Petersons is a professor in the Division of Environmental Health Sciences and co-Leader of the Carcinogenesis and Chemoprevention program in the Masonic Cancer Center, University of Minnesota. Her PhD is in Pharmaceutical Chemistry from University of California, San Francisco where she studied with Neal Castagnoli, Jr. After postdoctoral studies with Fred Guengerich at Vanderbilt University, she joined Stephen Hecht’s group at the American Health Foundation in Valhalla, NY. In 1997, Lisa moved to the University of Minnesota. Her research focuses on mechanisms by which chemicals initiate carcinogenesis. She is an Associate Editor for Chemical Research in Toxicology
Reference List
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 3.Islami F, Torre LA, Jemal A. Global trends of lung cancer mortality and smoking prevalence. Transl Lung Cancer Res. 2015;4:327–338. doi: 10.3978/j.issn.2218-6751.2015.08.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thun MJ, Henley SJ, Calle EE. Tobacco use and cancer: an epidemiologic perspective for geneticists. Oncogene. 2002;21:7307–7325. doi: 10.1038/sj.onc.1205807. [DOI] [PubMed] [Google Scholar]
- 5.Haiman CA, Stram DO, Wilkens LR, Pike MC, Kolonel LN, Henderson BE, Le Marchand L. Ethnic and racial differences in the smoking-related risk of lung cancer. N Engl J Med. 2006;354:333–342. doi: 10.1056/NEJMoa033250. [DOI] [PubMed] [Google Scholar]
- 6.Cote ML, Kardia SL, Wenzlaff AS, Ruckdeschel JC, Schwartz AG. Risk of lung cancer among white and black relatives of individuals with early-onset lung cancer. J Am Med Assoc. 2005;293:3036–3042. doi: 10.1001/jama.293.24.3036. [DOI] [PubMed] [Google Scholar]
- 7.International Agency for Research on Cancer. Tobacco Smoke and Involuntary Smoking. Vol. 83. IARC; Lyon, France: 2004. [Google Scholar]
- 8.International Agency for Research on Cancer. Personal Habits and Indoor Combustions vol. 100E. IARC; Lyon, France: 2012. [Google Scholar]
- 9.Hecht SS, Chen CB, Ohmori T, Hoffmann D. Comparative carcinogenicity in F344 rats of the tobacco specific nitrosamines, N′-nitrosonornicotine and 4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone. Cancer Res. 1980;40:298–302. [PubMed] [Google Scholar]
- 10.Rivenson A, Hoffmann D, Prokopczyk B, Amin S, Hecht SS. Induction of lung and exocrine pancreas tumors in F344 rats by tobacco-specific and areca-derived N-nitrosamines. Cancer Res. 1988;48:6912–6917. [PubMed] [Google Scholar]
- 11.Hecht SS, Morse MA, Amin SG, Stoner GD, Jordan KG, Choi CI, Chung FL. Rapid single dose model for lung tumor induction in A/J mice by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and the effect of diet. Carcinogenesis. 1989;10:1901–1904. doi: 10.1093/carcin/10.10.1901. [DOI] [PubMed] [Google Scholar]
- 12.Hecht SS. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol. 1998;11:559–603. doi: 10.1021/tx980005y. [DOI] [PubMed] [Google Scholar]
- 13.Burns DM. Changing rates of adenocarcinoma of the lung. Chem Res Toxicol. 2014;27:1330–1335. doi: 10.1021/tx500161m. [DOI] [PubMed] [Google Scholar]
- 14.Hecht SS, Carmella SG, Murphy SE, Akerkar S, Brunnemann KD, Hoffmann D. A tobacco-specific lung carcinogen in the urine of men exposed to cigarette smoke. N Engl J Med. 1993;329:1543–1546. doi: 10.1056/NEJM199311183292105. [DOI] [PubMed] [Google Scholar]
- 15.Parsons WD, Carmella SG, Akerkar S, Bonilla LE, Hecht SS. A metabolite of the tobacco-specific lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in the urine of hospital workers exposed to environmental tobacco smoke. Cancer Epidemiol Biomarkers Prev. 1998;7:257–260. [PubMed] [Google Scholar]
- 16.Lackmann GM, Salzberger U, Tollner U, Chen M, Carmella SG, Hecht SS. Metabolites of a tobacco-specific carcinogen in urine from newborns. JNCI. 1999;91:459–465. doi: 10.1093/jnci/91.5.459. [DOI] [PubMed] [Google Scholar]
- 17.Hecht SS. Carcinogen biomarkers for lung or oral cancer chemoprevention trials. IARC Sci Publ. 2001;154:245–255. [PubMed] [Google Scholar]
- 18.Hecht SS, Murphy SE, Stepanov I, Nelson HH, Yuan JM. Tobacco smoke biomarkers and cancer risk among male smokers in the Shanghai cohort study. Cancer Lett. 2013;334:34–38. doi: 10.1016/j.canlet.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stepanov I, Sebero E, Wang R, Gao YT, Hecht SS, Yuan JM. Tobacco-specific N-nitrosamine exposures and cancer risk in the Shanghai cohort study: Remarkable coherence with rat tumor sites. Int J Cancer. 2013 doi: 10.1002/ijc.28575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yuan JM, Butler LM, Stepanov I, Hecht SS. Urinary tobacco smoke-constituent biomarkers for assessing risk of lung cancer. Cancer Res. 2014;74:401–411. doi: 10.1158/0008-5472.CAN-13-3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.International Agency for Research on Cancer. Smokeless tobacco and tobacco-specific nitrosamines. Vol. 89. IARC; Lyon, France: 2007. [Google Scholar]
- 22.Weng Y, Fang C, Turesky RJ, Behr M, Kaminsky LS, Ding X. Determination of the role of target tissue metabolism in lung carcinogenesis using conditional cytochrome P450 reductase-null mice. Cancer Res. 2007;67:7825–7832. doi: 10.1158/0008-5472.CAN-07-1006. [DOI] [PubMed] [Google Scholar]
- 23.Belinsky SA, White CM, Boucheron JA, Richardson FC, Swenberg JA, Anderson M. Accumulation and persistence of DNA adducts in respiratory tissue of rats following multiple administrations of the the tobacco specific carcinogen 4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone. Cancer Res. 1986;46:1280–1284. [PubMed] [Google Scholar]
- 24.Hecht SS, Trushin N, Castonguay A, Rivenson A. Comparative tumorigenicity and DNA methylation in F344 rats by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and N-nitrosodimethylamine. Cancer Res. 1986;46:498–502. [PubMed] [Google Scholar]
- 25.Belinsky SA, Foley JA, White CM, Anderson MW, Maronpot RR. Dose-response relationship between O6-methylguanine formation in Clara cells and induction of pulmonary neoplasia in the rat by NNK. Cancer Res. 1990;50:3772–3780. [PubMed] [Google Scholar]
- 26.Murphy SE, Palomino A, Hecht SS, Hoffmann D. Dose-response study of DNA and hemoglobin adduct formation by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in F344 rats. Cancer Res. 1990;50:5446–5452. [PubMed] [Google Scholar]
- 27.Peterson LA, Hecht SS. O6-Methylguanine is a critical determinant of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone tumorigenesis in A/J mouse lung. Cancer Res. 1991;51:5557–5564. [PubMed] [Google Scholar]
- 28.Balbo S, Johnson CS, Kovi RC, James-Yi SA, O’Sullivan MG, Wang M, Le CT, Khariwala SS, Upadhyaya P, Hecht SS. Carcinogenicity and DNA adduct formation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in F-344 rats. Carcinogenesis. 2014;35:2798–2806. doi: 10.1093/carcin/bgu204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Upadhyaya P, Kalscheuer S, Hochalter JB, Villalta PW, Hecht SS. Quantitation of pyridylhydroxybutyl-DNA adducts in liver and lung of F-344 rats treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem Res Toxicol. 2008;21:1468–1476. doi: 10.1021/tx8001109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang S, Wang M, Villalta PW, Lindgren BR, Upadhyaya P, Lao Y, Hecht SS. Analysis of pyridyloxobutyl and pyridylhydroxybutyl DNA adducts in extrahepatic tissues of F344 rats treated chronically with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem Res Toxicol. 2009;22:926–936. doi: 10.1021/tx900015d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beranek DT. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat Res. 1990;231:11–30. doi: 10.1016/0027-5107(90)90173-2. [DOI] [PubMed] [Google Scholar]
- 32.Wyatt MD, Pittman DL. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem Res Toxicol. 2006;19:1580–1594. doi: 10.1021/tx060164e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pegg AE. Repair of O6-alkylguanine by alkyltransferase. Mutat Res. 2000;462:83–100. doi: 10.1016/s1383-5742(00)00017-x. [DOI] [PubMed] [Google Scholar]
- 34.Pauly GT, Moschel RC. Mutagenesis by O6-methyl-, O6-ethyl- and O6-benzylguanine and O4-methylthymine in human cells: effects of O6-alkylguanine DNA alkyltransferase and mismatch repair. Chem Res Toxicol. 2001;14:894–900. doi: 10.1021/tx010032f. [DOI] [PubMed] [Google Scholar]
- 35.Altshuler KB, Hodes CS, Essigmann JM. Intrachromosomal probes for mutagenesis by alkylated DNA bases replicated in mammalian cells: a comparison of the mutagenicities of O4-methylthymine and O6-methylguanine in cells with different DNA repair backgrounds. Chem Res Toxicol. 1996;9:980–987. doi: 10.1021/tx960062w. [DOI] [PubMed] [Google Scholar]
- 36.Daniels DS, Mol CD, Arvai AS, Kanugula S, Pegg AE, Tainer JA. Active and alkylated human AGT structures: a novel zinc site, inhibitor and extrahelical base binding. EMBO J. 2000;19:1719–1730. doi: 10.1093/emboj/19.7.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu-Welliver M, Pegg AE. Degradation of the alkylated form of the DNA repair protein, O6-alkylguanine-DNA alkyltransferase. Carcinogenesis. 2002;23:823–830. doi: 10.1093/carcin/23.5.823. [DOI] [PubMed] [Google Scholar]
- 38.Dolan ME, Norbeck L, Clyde C, Hora NK, Erickson LC, Pegg AE. Expression of mammalian O6-alkylguanine-DNA alkyltransferase in a cell line sensitive to alkylating agents. Carcinogenesis. 1989;10:1613–1619. doi: 10.1093/carcin/10.9.1613. [DOI] [PubMed] [Google Scholar]
- 39.Kaina B, Fritz G, Mitra S, Coquerelle T. Transfection and expression of human O6-methylguanine-DNA methyltransferase (MGMT) cDNA in Chinese hamster cells: the role of MGMT in protection against the genotoxic effects of alkylating agents. Carcinogenesis. 1991;12:1857–1867. doi: 10.1093/carcin/12.10.1857. [DOI] [PubMed] [Google Scholar]
- 40.Cai Y, Wu MH, Xu-Welliver M, Pegg AE, Ludeman SM, Dolan ME. Effect of O6-benzylguanine on alkylating agent-induced toxicity and mutagenicity. In Chinese hamster ovary cells expressing wild-type and mutant O6-alkylguanine-DNA alkyltransferases. Cancer Res. 2000;60:5464–5469. [PubMed] [Google Scholar]
- 41.Hickman MJ, Samson LD. Role of DNA mismatch repair and p53 in signaling induction of apoptosis by alkylating agents. PNAS. 1999;96:10764–10769. doi: 10.1073/pnas.96.19.10764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaina B, Christmann M, Naumann S, Roos WP. MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair (Amst) 2007;6:1079–1099. doi: 10.1016/j.dnarep.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 43.Christmann M, Tomicic MT, Roos WP, Kaina B. Mechanisms of human DNA repair: an update. Toxicology. 2003;193:3–34. doi: 10.1016/s0300-483x(03)00287-7. [DOI] [PubMed] [Google Scholar]
- 44.Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer. 2012;12:104–120. doi: 10.1038/nrc3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boiteux S, Laval J. Imidazole open ring 7-methylguanine: an inhibitor of DNA synthesis. Biochem Biophys Res Commun. 1983;110:552–558. doi: 10.1016/0006-291x(83)91185-3. [DOI] [PubMed] [Google Scholar]
- 46.Larson K, Sahm J, Shenkar R, Strauss B. Methylation-induced blocks to in vitro DNA replication. Mutat Res. 1985;150:77–84. doi: 10.1016/0027-5107(85)90103-4. [DOI] [PubMed] [Google Scholar]
- 47.Asagoshi K, Terato H, Ohyama Y, Ide H. Effects of a guanine-derived formamidopyrimidine lesion on DNA replication: translesion DNA synthesis, nucleotide insertion, and extension kinetics. J Biol Chem. 2002;277:14589–14597. doi: 10.1074/jbc.M200316200. [DOI] [PubMed] [Google Scholar]
- 48.Christov PP, Angel KC, Guengerich FP, Rizzo CJ. Replication past the N5-methyl-formamidopyrimidine lesion of deoxyguanosine by DNA polymerases and an improved procedure for sequence analysis of in vitro bypass products by mass spectrometry. Chem Res Toxicol. 2009;22:1086–1095. doi: 10.1021/tx900047c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Christov PP, Yamanaka K, Choi JY, Takata K, Wood RD, Guengerich FP, Lloyd RS, Rizzo CJ. Replication of the 2,6-diamino-4-hydroxy-N(5)-(methyl)-formamidopyrimidine (MeFapy-dGuo) adduct by eukaryotic DNA polymerases. Chem Res Toxicol. 2012;25:1652–1661. doi: 10.1021/tx300113e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Earley LF, Minko IG, Christov PP, Rizzo CJ, Lloyd RS. Mutagenic spectra arising from replication bypass of the 2,6-diamino-4-hydroxy-N(5)-methyl formamidopyrimidine adduct in primate cells. Chem Res Toxicol. 2013;26:1108–1114. doi: 10.1021/tx4001495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jasti VP, Spratt TE, Basu AK. Tobacco-specific nitrosamine-derived O2-alkylthymidines are potent mutagenic lesions in SOS-induced Escherichia coli. Chem Res Toxicol. 2011;24:1833–1835. doi: 10.1021/tx200435d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weerasooriya S, Jasti VP, Bose A, Spratt TE, Basu AK. Roles of translesion synthesis DNA polymerases in the potent mutagenicity of tobacco-specific nitrosamine-derived O2-alkylthymidines in human cells. DNA Repair (Amst) 2015;35:63–70. doi: 10.1016/j.dnarep.2015.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hecht SS, Spratt TE, Trushin N. Evidence for 4-(3-pyridyl)-4-oxobutylation of DNA in F344 rats treated with the tobacco specific nitrosamines 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and N′-nitrosonornicotine. Carcinogenesis. 1988;9:161–165. doi: 10.1093/carcin/9.1.161. [DOI] [PubMed] [Google Scholar]
- 54.Wang L, Spratt TE, Liu XK, Hecht SS, Pegg AE, Peterson LA. Pyridyloxobutyl adduct O6-[4-oxo-4-(3-pyridyl)butyl]guanine is present in 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone-treated DNA and is a substrate for O6-alkylguanine-DNA alkyltransferase. Chem Res Toxicol. 1997;10:562–567. doi: 10.1021/tx9602067. [DOI] [PubMed] [Google Scholar]
- 55.Wang M, Cheng G, Sturla SJ, McIntee EJ, Villalta PW, Upadhyaya P, Hecht SS. Identification of adducts formed by pyridyloxobutylation of deoxyguanosine and DNA by 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone, a chemically activated form of tobacco specific carcinogens. Chem Res Toxicol. 2003;16:616–626. doi: 10.1021/tx034003b. [DOI] [PubMed] [Google Scholar]
- 56.Hecht SS, Villalta PW, Sturla SJ, Cheng G, Yu N, Upadhyaya P, Wang M. Identification of O2-substituted pyrimidine adducts formed in reactions of 4-(acetoxymethylnitrosamino)- 1-(3-pyridyl)-1-butanone and 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanol with DNA. Chem Res Toxicol. 2004;17:588–597. doi: 10.1021/tx034263t. [DOI] [PubMed] [Google Scholar]
- 57.Spratt TE, Trushin N, Lin D, Hecht SS. Analysis for N2-(pyridyloxobutyl)deoxyguanosine adducts in DNA of tissues exposed to tritium labelled 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and N′-nitrosonornicotine. Chem Res Toxicol. 1989;2:169–173. doi: 10.1021/tx00009a008. [DOI] [PubMed] [Google Scholar]
- 58.Ma B, Villalta PW, Zarth AT, Kotandeniya D, Upadhyaya P, Stepanov I, Hecht SS. Comprehensive high-resolution mass spectrometric analysis of DNA phosphate adducts formed by the tobacco-specific lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Chem Res Toxicol. 2015 doi: 10.1021/acs.chemrestox.5b00318. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spratt TE, Peterson LA, Confer WL, Hecht SS. Solvolysis of model compounds for α-hydroxylation of N′-nitrosonornicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone: Evidence for a cyclic oxonium ion intermediate in the alkylation of nucleophiles. Chem Res Toxicol. 1990;3:350–356. doi: 10.1021/tx00016a013. [DOI] [PubMed] [Google Scholar]
- 60.Sturla SJ, Scott J, Lao Y, Hecht SS, Villalta PW. Mass spectrometric analysis of relative levels of pyridyloxobutylation adducts formed in the reaction of DNA with a chemically activated form of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Chem Res Toxicol. 2005;18:1048–1055. doi: 10.1021/tx050028u. [DOI] [PubMed] [Google Scholar]
- 61.Lao Y, Villalta PW, Sturla SJ, Wang M, Hecht SS. Quantitation of pyridyloxobutyl DNA adducts of tobacco-specific nitrosamines in rat tissue DNA by high-performance liquid chromatography-electrospray ionization-tandem mass spectrometry. Chem Res Toxicol. 2006;19:674–682. doi: 10.1021/tx050351x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lacoste S, Castonguay A, Drouin R. Formamidopyrimidine adducts are detected using the comet assay in human cells treated with reactive metabolites of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) Mutat Res. 2006;600:138–149. doi: 10.1016/j.mrfmmm.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 63.Pauly GT, Peterson LA, Moschel RC. Mutagenesis by O6-[4-oxo-4-(3-pyridyl)butyl]guanine in Escherichia coli and human cells. Chem Res Toxicol. 2002;15:165–169. doi: 10.1021/tx0101245. [DOI] [PubMed] [Google Scholar]
- 64.Wang L, Spratt TE, Pegg AE, Peterson LA. Synthesis of DNA oligonucleotides containing site-specifically incorporated O6-[4-oxo-4-(3-pyridyl)butyl]guanine and their reaction with O6-alkylguanine-DNA alkyltransferase. Chem Res Toxicol. 1999;12:127–131. doi: 10.1021/tx980251+. [DOI] [PubMed] [Google Scholar]
- 65.Mijal RS, Thomson NM, Fleischer NL, Pauly GT, Moschel RC, Kanugula S, Fang Q, Pegg AE, Peterson LA. The repair of the tobacco-specific nitrosamine derived adduct O6-[4-oxo-4-(3-pyridyl)butyl]guanine by O6-alkylguanine-DNA alkyltransferase variants. Chem Res Toxicol. 2004;17:424–434. doi: 10.1021/tx0342417. [DOI] [PubMed] [Google Scholar]
- 66.Peterson LA, Liu XK, Hecht SS. Pyridyloxobutyl DNA adducts inhibit the repair of O6-methylguanine. Cancer Res. 1993;53:2780–2785. [PubMed] [Google Scholar]
- 67.Mijal RS, Loktionova NA, Vu CC, Pegg AE, Peterson LA. O6-Pyridyloxobutylguanine adducts contribute to the mutagenic properties of pyridyloxobutylating agents. Chem Res Toxicol. 2005;18:1619–1625. doi: 10.1021/tx050139t. [DOI] [PubMed] [Google Scholar]
- 68.Li L, Perdigao J, Pegg AE, Lao Y, Hecht SS, Lindgren BR, Reardon JT, Sancar A, Wattenberg EV, Peterson LA. The influence of repair pathways on the cytotoxicity and mutagenicity induced by the pyridyloxobutylation pathway of tobacco-specific nitrosamines. Chem Res Toxicol. 2009;22:1464–1472. doi: 10.1021/tx9001572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rubinson EH, Gowda AS, Spratt TE, Gold B, Eichman BF. An unprecedented nucleic acid capture mechanism for excision of DNA damage. Nature. 2010;468:406–411. doi: 10.1038/nature09428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tudek B, Graziewicz M, Kazanova O, Zastawny TH, Obtulowicz T, Laval J. Mutagenic specificity of imidazole ring-opened 7-methylpurines in M13mp18 phage DNA. Acta Biochim Pol. 1999;46:785–799. [PubMed] [Google Scholar]
- 71.Smela ME, Hamm ML, Henderson PT, Harris CM, Harris TM, Essigmann JM. The aflatoxin B(1) formamidopyrimidine adduct plays a major role in causing the types of mutations observed in human hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2002;99:6655–6660. doi: 10.1073/pnas.102167699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Woo LL, Egner PA, Belanger CL, Wattanawaraporn R, Trudel LJ, Croy RG, Groopman JD, Essigmann JM, Wogan GN, Bouhenguel JT. Aflatoxin B1-DNA adduct formation and mutagenicity in livers of neonatal male and female B6C3F1 mice. Toxicol Sci. 2011;122:38–44. doi: 10.1093/toxsci/kfr087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Urban AM, Upadhyaya P, Cao Q, Peterson LA. Formation and repair of pyridyloxobutyl DNA adducts and their relationship to tumor yield in A/J mice. Chem Res Toxicol. 2012;25:2167–2178. doi: 10.1021/tx300245w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Simonelli V, Narciso L, Dogliotti E, Fortini P. Base excision repair intermediates are mutagenic in mammalian cells. NAR. 2005;33:4404–4411. doi: 10.1093/nar/gki749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fan J, Wilson DM., III Protein-protein interactions and posttranslational modifications in mammalian base excision repair. Free Radic Biol Med. 2005;38:1121–1138. doi: 10.1016/j.freeradbiomed.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 76.Chaw YFM, Crane LE, Lange P, Shapiro R. Isolation and identification of cross-links from formaldehyde-treated nucleic acids. Biochemistry. 1980;19:5525–5531. doi: 10.1021/bi00565a010. [DOI] [PubMed] [Google Scholar]
- 77.Beland FA, Fullerton NF, Heflich RH. Rapid isolation, hydrolysis and chromatography of formaldehyde-modified DNA. J Chromatogr. 1984;308:121–131. doi: 10.1016/s0021-9673(01)87539-7. [DOI] [PubMed] [Google Scholar]
- 78.Huang H, Solomon MS, Hopkins PB. Formaldehyde preferentially interstrand cross-links duplex DNA through deoxyadenosine residues at the sequence 5′-d(AT) JACS. 1992;114:9240–9241. [Google Scholar]
- 79.Huang H, Hopkins PB. DNA interstrand cross-linking by formaldehyde: Nucleotide sequence preference and covalent structure of the predominant cross-link formed in synthetic oligonucleotides. JACS. 1993;115:9402–9408. [Google Scholar]
- 80.Cheng G, Shi Y, Sturla SJ, Jalas JR, McIntee EJ, Villalta PW, Wang M, Hecht SS. Reactions of formaldehyde plus acetaldehyde with deoxyguanosine and DNA: formation of cyclic deoxyguanosine adducts and formaldehyde cross-links. Chem Res Toxicol. 2003;16:145–152. doi: 10.1021/tx025614r. [DOI] [PubMed] [Google Scholar]
- 81.Cheng G, Wang M, Upadhyaya P, Villalta PW, Hecht SS. Formation of formaldehyde adducts in the reactions of DNA and deoxyribonucleosides with alpha-acetates of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL), and N-nitrosodimethylamine (NDMA) Chem Res Toxicol. 2008;21:746–751. doi: 10.1021/tx7003823. [DOI] [PubMed] [Google Scholar]
- 82.International Agency for Research on Cancer. A Review of Human Carcinogens. F. Chemical Agents and Related Occupations. IARC; Lyon, France: 2012. Formaldehyde; pp. 401–436. [Google Scholar]
- 83.Grafström RC, Curren RD, Yang LL, Harris CC. Genotoxicity of formaldehyde in cultured human bronchial fibroblasts. Science. 1985;228:89–91. doi: 10.1126/science.3975633. [DOI] [PubMed] [Google Scholar]
- 84.Liber HL, Benforado K, Crosby RM, Simpson D, Skopek TR. Formaldehyde-induced and spontaneous alterations in human hprt DNA sequence and mRNA expression. Mutat Res. 1989;226:31–37. doi: 10.1016/0165-7992(89)90089-4. [DOI] [PubMed] [Google Scholar]
- 85.Graves RJ, Trueman P, Jones S, Green T. DNA sequence analysis of methylene chloride-induced HPRT mutations in Chinese hamster ovary cells: comparison with the mutation spectrum obtained for 1,2-dibromoethane and formaldehyde. Mutagenesis. 1996;11:229–233. doi: 10.1093/mutage/11.3.229. [DOI] [PubMed] [Google Scholar]
- 86.Merk O, Speit G. Significance of formaldehyde-induced DNA-protein crosslinks for mutagenesis. Environ Mol Mutagen. 1998;32:260–268. doi: 10.1002/(sici)1098-2280(1998)32:3<260::aid-em9>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 87.Speit G, Merk O. Evaluation of mutagenic effects of formaldehyde in vitro: detection of crosslinks and mutations in mouse lymphoma cells. Mutagenesis. 2002;17:183–187. doi: 10.1093/mutage/17.3.183. [DOI] [PubMed] [Google Scholar]
- 88.Peterson LA, Urban AM, Vu CC, Cummings ME, Brown LC, Warmka JK, Li L, Wattenberg EV, Patel Y, Stram DO, Pegg AE. Role of aldehydes in the toxic and mutagenic effects of nitrosamines. Chem Res Toxicol. 2013;26:1464–1473. doi: 10.1021/tx400196j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang M, Cheng G, Villalta PW, Hecht SS. Development of liquid chromatography electrospray ionization tandem mass spectrometry methods for analysis of DNA adducts of formaldehyde and their application to rats treated with N-nitrosodimethylamine or 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Chem Res Toxicol. 2007;20:1141–1148. doi: 10.1021/tx700189c. [DOI] [PubMed] [Google Scholar]
- 90.Alaoui-Jamali MA, Gagnon R, El Alami N, Castonguay A. Cytotoxicity, sister-chromatid exchanges and DNA single-strand breaks induced by 4-oxo-4-(3-pyridyl)butanal, a metabolite of a tobacco-specific N-nitrosamine. Mutat Res. 1990;240:25–33. doi: 10.1016/0165-1218(90)90005-m. [DOI] [PubMed] [Google Scholar]
- 91.Liu L, Alaoui-Jamali MA, Alami NE, Castonguay A. Metabolism and DNA single strand breaks induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and its analogues in primary culture of rat hepatocytes. Cancer Res. 1990;50:1810–1816. [PubMed] [Google Scholar]
- 92.Demkowicz-Dobrazanski K, Castonguay A. Comparison of DNA alkali-labile sites induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and 4-oxo-4-(3-pyridyl)butanal in rat hepatocytes. Carcinogenesis. 1991;12:2135–2140. doi: 10.1093/carcin/12.11.2135. [DOI] [PubMed] [Google Scholar]
- 93.Upadhyaya P, Sturla SJ, Tretyakova N, Ziegel R, Villalta PW, Wang M, Hecht SS. Identification of adducts produced by the reaction of 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanol with deoxyguanosine and DNA. Chem Res Toxicol. 2003;16:180–190. doi: 10.1021/tx0256376. [DOI] [PubMed] [Google Scholar]
- 94.Hecht SS, Lin D, Castonguay A. Effects of α-deuterium substitution on the mutagenicity of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) Carcinogenesis. 1983;4:305–310. doi: 10.1093/carcin/4.3.305. [DOI] [PubMed] [Google Scholar]
- 95.Foiles PG, Peterson LA, Miglietta LM, Ronai Z. Analysis of mutagenic activity and ability to induce replication of polyoma DNA sequences by different model compounds of the carcinogenic tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Mutat Res. 1992;279:91–101. doi: 10.1016/0165-1218(92)90250-4. [DOI] [PubMed] [Google Scholar]
- 96.Maron DM, Ames BN. Revised methods for the salmonella mutagenicity test. Mutat Res. 1983;113:173–215. doi: 10.1016/0165-1161(83)90010-9. [DOI] [PubMed] [Google Scholar]
- 97.Yamada M, Matsui K, Sofuni T, Nohmi T. New tester strains of Salmonella typhimurium lacking O6-methylguanine DNA methyltransferases and highly sensitive to mutagenic alkylating agents. Mutat Res. 1997;381:15–24. doi: 10.1016/s0027-5107(97)00139-5. [DOI] [PubMed] [Google Scholar]
- 98.Yamada M, Sedgwick B, Sofuni T, Nohmi T. Construction and characterization of mutants of Salmonella typhimurium deficient in DNA repair of O6-methylguanine. J Bacteriol. 1995;177:1511–1519. doi: 10.1128/jb.177.6.1511-1519.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pauly GT, Hughes SH, Moschel RC. Mutagenesis in Escherichia coli by three O6-substituted guanines in double-stranded or gapped plasmids. Biochemistry. 1995;34:8924–8930. doi: 10.1021/bi00027a045. [DOI] [PubMed] [Google Scholar]
- 100.Jiao J, Zielenska M, Anderson MW, Glickman BW. Mutational specificities of environmental carcinogens in the lacI gene of Escherichia coli: IV. The tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis. 1991;12:221–224. doi: 10.1093/carcin/12.2.221. [DOI] [PubMed] [Google Scholar]
- 101.Horsfall MJ, Glickman BW. Mutational specificities of environmental carcinogens in the lacI gene of Escherichia coli. I The direct-acting analogue N-nitroso-N-methyl-N-α-acetoxymethylamine. Carcinogenesis. 1989;10:817–822. doi: 10.1093/carcin/10.5.817. [DOI] [PubMed] [Google Scholar]
- 102.Horsfall MJ, Zeilmaker MJ, Mohn GR, Glickman BW. Mutational specificities of environmental carcinogens in the lacl gene of Escherichia coli. II: A host-mediated approach to N-nitroso-N,N-dimethylamine and endogenous mutagenesis in vivo. Mol Carcinog. 1989;2:107–115. doi: 10.1002/mc.2940020210. [DOI] [PubMed] [Google Scholar]
- 103.Klein CB, Rossman TG. Transgenic chinese hamster V79 cell lines which exhibit varialble levels of gpt mutagenesis. Environ Mol Mutagen. 1990;16:1–12. doi: 10.1002/em.2850160102. [DOI] [PubMed] [Google Scholar]
- 104.Albertini RJ. Somatic gene mutations in vivo as indicated by the 6-thioguanine-resistant T-lymphocytes in human blood. Mutat Res. 1985;150:411–422. doi: 10.1016/0027-5107(85)90138-1. [DOI] [PubMed] [Google Scholar]
- 105.Hecht SS, Jordan KG, Choi CI, Trushin N. Effects of deuterium substitution on the tumorigenicity of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in A/J mice. Carcinogenesis. 1990;11:1017–1020. doi: 10.1093/carcin/11.6.1017. [DOI] [PubMed] [Google Scholar]
- 106.Peterson LA, Thomson NM, Crankshaw DL, Donaldson EE, Kenney PJ. Interactions between methylating and pyridyloxobutylating agents in A/J mouse lungs: implications for 4-(methylnitrosamino)-1-(3-pyridyl)-1- butanone-induced lung tumorigenesis. Cancer Res. 2001;61:5757–5763. [PubMed] [Google Scholar]
- 107.Liu L, Qin X, Gerson SL. Reduced lung tumorigenesis in human methylguanine DNA-methyltransferase transgenic mice achieved by expression of transgene within the target cell. Carcinogenesis. 1999;20:279–284. doi: 10.1093/carcin/20.2.279. [DOI] [PubMed] [Google Scholar]
- 108.Belinsky SA, Devereux TR, Maronpot RR, Stoner GD, Anderson MW. Relationship between the formation of promutagenic adducts and the activation of the K-ras protooncogene in lung tumors from A/J mice treated with nitrosamines. Cancer Res. 1989;49:5305–5311. [PubMed] [Google Scholar]
- 109.Kawano R, Takeshima Y, Inai K. Effects of K-ras gene mutations in the development of lung lesions induced by 4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone in A/J mice. Jpn J Cancer Res. 1996;87:44–50. doi: 10.1111/j.1349-7006.1996.tb00198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ronai ZA, Gradia S, Peterson LA, Hecht SS. G to A transitions and G to T transversions in codon 12 of the Ki-ras oncogene isolated from mouse lung tumors induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and related DNA methylating and pyridyloxobutylating agents. Carcinogenesis. 1993;14:2419–2422. doi: 10.1093/carcin/14.11.2419. [DOI] [PubMed] [Google Scholar]
- 111.Riely GJ, Marks J, Pao W. KRAS mutations in non-small cell lung cancer. Proc Am Thorac Soc. 2009;6:201–205. doi: 10.1513/pats.200809-107LC. [DOI] [PubMed] [Google Scholar]
- 112.Rodenhuis S, Slebos RJ. Clinical significance of ras oncogene activation in human lung cancer. Cancer Res. 1992;52:2665s–2669s. [PubMed] [Google Scholar]
- 113.Hashimoto K, Ohsawa K, Kimura M. Mutations induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) in the lacZ and cII genes of Muta Mouse. Mutat Res. 2004;560:119–131. doi: 10.1016/j.mrgentox.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 114.Sandercock LE, Hahn JN, Li L, Luchman HA, Giesbrecht JL, Peterson LA, Jirik FR. Mgmt deficiency alters the in vivo mutational spectrum of tissues exposed to the tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) Carcinogenesis. 2008;29:866–874. doi: 10.1093/carcin/bgn030. [DOI] [PubMed] [Google Scholar]
- 115.Brown PJ, Massey TE. In vivo treatment with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) induces organ-specific alterations in in vitro repair of DNA pyridyloxobutylation. Mutat Res. 2009;663:15–21. doi: 10.1016/j.mrfmmm.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 116.Narayanapillai SC, Balbo S, Leitzman P, Grill AE, Upadhyaya P, Shaik AA, Zhou B, O’Sullivan GO, Peterson LA, Lu J, Hecht SS, Xing C. Dihydromethysticin (DHM) from kava blocks tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-induced lung tumorigenesis and differentially reduces DNA damage in A/J mice. Carcinogenesis. 2014;35:2365–2372. doi: 10.1093/carcin/bgu149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hecht SS, Lin D, Castonguay A, Rivenson A. Effects of α-deuterium substitution on the tumorigenicity of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in F344 rats. Carcinogenesis. 1987;8:291–294. doi: 10.1093/carcin/8.2.291. [DOI] [PubMed] [Google Scholar]
- 118.Belinsky SA, Dolan ME, White CM, Maronpot RR, Pegg AE, Anderson MW. Cell specific differences in O6-methylguanine-DNA methyltransferase activity and removal of O6-methylguanine in rat pulmonary cells. Carcinogenesis. 1988;9:2053–2058. doi: 10.1093/carcin/9.11.2053. [DOI] [PubMed] [Google Scholar]
- 119.Staretz ME, Foiles PG, Miglietta LM, Hecht SS. Evidence for an important role of DNA pyridyloxobutylation in rat lung carcinogenesis by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone: effects of dose and phenethyl isothiocyanate. Cancer Res. 1997;57:259–266. [PubMed] [Google Scholar]
- 120.Lao Y, Yu N, Kassie F, Villalta PW, Hecht SS. Formation and accumulation of pyridyloxobutyl DNA adducts in F344 rats chronically treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem Res Toxicol. 2007;20:235–245. doi: 10.1021/tx060207r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Upadhyaya P, Lindgren BR, Hecht SS. Comparative levels of O6-methylguanine, pyridyloxobutyl-, and pyridylhydroxybutyl-DNA adducts in lung and liver of rats treated chronically with the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Drug Metab Dispos. 2009;37:1147–1151. doi: 10.1124/dmd.109.027078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yuan JM, Gao YT, Murphy SE, Carmella SG, Wang R, Zhong Y, Moy KA, Davis AB, Tao L, Chen M, Han S, Nelson HH, Yu MC, Hecht SS. Urinary levels of cigarette smoke constituent metabolites are prospectively associated with lung cancer development in smokers. Cancer Res. 2011;71:6749–6757. doi: 10.1158/0008-5472.CAN-11-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yuan JM, Koh WP, Murphy SE, Fan Y, Wang R, Carmella SG, Han S, Wickham K, Gao YT, Yu MC, Hecht SS. Urinary levels of tobacco-specific nitrosamine metabolites in relation to lung cancer development in two prospective cohorts of cigarette smokers. Cancer Res. 2009;69:2990–2995. doi: 10.1158/0008-5472.CAN-08-4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Church TR, Anderson KE, Caporaso NE, Geisser MS, Le CT, Zhang Y, Benoit AR, Carmella SG, Hecht SS. A prospectively measured serum biomarker for a tobacco-specific carcinogen and lung cancer in smokers. Cancer Epidemiol Biomarkers Prev. 2009;18:260–266. doi: 10.1158/1055-9965.EPI-08-0718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hecht SS, Carmella SG, Stepanov I, Jensen J, Anderson A, Hatsukami DK. Metabolism of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone to its biomarker total NNAL in smokeless tobacco users. Cancer Epidemiol Biomarkers Prev. 2008;17:732–735. doi: 10.1158/1055-9965.EPI-07-2843. [DOI] [PubMed] [Google Scholar]
- 126.Lewis SJ, Cherry NM, Niven RM, Barber PV, Povey AC. Associations between smoking, GST genotypes and N7-methylguanine levels in DNA extracted from bronchial lavage cells. Mutat Res. 2004;559:11–18. doi: 10.1016/j.mrgentox.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 127.Foiles PG, Akerkar SA, Carmella SG, Kagan M, Stoner GD, Resau JH, Hecht SS. Mass spectrometric analysis of tobacco-specific nitrosamine DNA adducts in smokers and non-smokers. Chem Res Toxicol. 1991;4:364–368. doi: 10.1021/tx00021a017. [DOI] [PubMed] [Google Scholar]
- 128.Holzle D, Schlobe D, Tricker AR, Richter E. Mass spectrometric analysis of 4-hydroxy-1-(3-pyridyl)-1-butanone-releasing DNA adducts in human lung. Toxicology. 2007;232:277–285. doi: 10.1016/j.tox.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 129.Schöbe D, Hölze D, Richter E, Ostermeier-Hatz D, Von Meyer L, Lindner M, Tricker AR. Determination of tobacco-specific nitrosamine DNA adducts in human tissues. Proc Am Assoc Cancer Res. 2002;43:346. [Google Scholar]
- 130.Ma B, Ruszczak C, Jain V, Khariwala SS, Lindgren B, Hatsukami DK, Stepanov I. Optimized Liquid Chromatography Nanoelectrospray-High-Resolution Tandem Mass Spectrometry Method for the Analysis of 4-Hydroxy-1-(3-pyridyl)-1-butanone-Releasing DNA Adducts in Human Oral Cells. Chem Res Toxicol. 2016 doi: 10.1021/acs.chemrestox.6b00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kaur TB, Travaline JM, Gaughan JP, Richie JP, Jr, Stellman SD, Lazarus P. Role of polymorphisms in codons 143 and 160 of the O6-alkylguanine DNA alkyltransferase gene in lung cancer risk. Cancer Epidemiol Biomarkers Prev. 2000;9:339–342. [PubMed] [Google Scholar]
- 132.Cohet C, Borel S, Nyberg F, Mukeria A, Bruske-Hohlfeld I, Constantinescu V, Benhamou S, Brennan P, Hall J, Boffetta P. Exon 5 polymorphisms in the O6-alkylguanine DNA alkyltransferase gene and lung cancer risk in non-smokers exposed to second-hand smoke. Cancer Epidemiol Biomarkers Prev. 2004;13:320–323. doi: 10.1158/1055-9965.epi-03-0120. [DOI] [PubMed] [Google Scholar]
- 133.Chae MH, Jang JS, Kang HG, Park JH, Park JM, Lee WK, Kam S, Lee EB, Son JW, Park JY. O6-alkylguanine-DNA alkyltransferase gene polymorphisms and the risk of primary lung cancer. Mol Carcinog. 2006;45:239–249. doi: 10.1002/mc.20171. [DOI] [PubMed] [Google Scholar]
- 134.Mijal RS, Kanugula S, Vu CC, Fang Q, Pegg AE, Peterson LA. DNA sequence context affects repair of the tobacco-specific adduct O6-[4-oxo-4-(3-pyridyl)butyl]guanine by human O6-alkylguanine-DNA alkyltransferases. Cancer Res. 2006;66:4968–4974. doi: 10.1158/0008-5472.CAN-05-3803. [DOI] [PubMed] [Google Scholar]
- 135.Loktionova NA, Pegg AE. Point mutations in human O6-alkylguanine DNA alkyltransferase prevent the sensitization of O6-benzylguanine to killing by N,N′-bis(2-chloroethyl)-N-nitrosourea. Cancer Res. 1996;56:1578–1583. [PubMed] [Google Scholar]
- 136.Thompson LH, Brookman KW, Dillehay LE, Mooney CL, Carrano AV. Hypersensitivity to mutation and sister-chromatid-exchange induction in CHO cell mutants defective in incising DNA containing UV lesions. Somatic Cell Genet. 1982;8:759–773. doi: 10.1007/BF01543017. [DOI] [PubMed] [Google Scholar]
- 137.Thompson LH, Brookman KW, Dillehay LE, Carrano AV, Mazrimas JA, Mooney CL, Minkler JL. A CHO-cell strain having hypersensitivity to mutagens, a defect in DNA strand-break repair, and an extraordinary baseline frequency of sister-chromatid exchange. Mutat Res. 1982;95:427–440. doi: 10.1016/0027-5107(82)90276-7. [DOI] [PubMed] [Google Scholar]
- 138.Xu Z, Yu Y, Schwartz JL, Meltz ML, Hsie AW. Molecular nature of spontaneous mutations at the hypoxanthine-guanine phosphoribosyltransferase (hprt) locus in Chinese hamster ovary cells. Environ Mol Mutagen. 1995;26:127–138. doi: 10.1002/em.2850260206. [DOI] [PubMed] [Google Scholar]
- 139.Coryell VH, Stearns DM. Molecular analysis of hprt mutations generated in Chinese hamster ovary EM9 cells by uranyl acetate, by hydrogen peroxide, and spontaneously. Mol Carcinog. 2006;45:60–72. doi: 10.1002/mc.20155. [DOI] [PubMed] [Google Scholar]