

Summary
The extent and nature of epigenomic changes associated with melanoma progression is poorly understood. Through systematic epigenomic profiling of 35 epigenetic modifications and transcriptomic analysis, we define chromatin state changes associated with melanomagenesis using a cell phenotypic model of non-tumorigenic and tumorigenic states. Computation of specific chromatin state transitions showed loss of histone acetylations and H3K4me2/3 on regulatory regions proximal to specific cancer-regulatory genes in important melanoma-driving cell signaling pathways. Importantly, such acetylation changes were also observed between benign nevi and malignant melanoma human tissues. Intriguingly, only a small fraction of chromatin state transitions correlated with expected changes in gene expression patterns. Restoration of acetylation levels on deacetylated loci by HDAC inhibitors selectively blocked excessive proliferation in tumorigenic cells and human melanoma cells suggesting functional roles of observed chromatin state transitions in driving hyper-proliferative phenotype. Taken together, we define functionally relevant chromatin states associated with melanoma progression.
Graphical abstract
Using comprehensive profiling of 35 epigenetic marks and determination of chromatin state transitions between non-tumorigenic and tumorigenic systems, Fiziev et al. find that in tumorigenic cells, loss of histone acetylation and H3K4 methylation occur on regulatory regions proximal to specific cancer-regulatory genes.
Introduction
Cancer cells acquire genetic and epigenetic alterations that increase fitness and drive progression through multiple steps of tumor evolution. However, the understanding of the roles of epigenetic alterations in cancer is lagging, in part due to challenges of generation of large-scale data for multiple epigenomes across tissues/time per individual and lack of “germline normal” equivalence.
The epigenome consists of an array of modifications, including DNA methylation and histone marks, which associate with dynamic changes in various cellular processes in response to stimuli. Although detailed profiles of specific epigenetic marks have been characterized in a number of normal tissues (Encode_Project_Consortium, 2012; Ernst et al., 2011; Roadmap Epigenomics et al., 2015) and some cancers including DNA-methylation in human tumors, genome-wide profiles of multiple histone marks and combinatorial chromatin states in cancer progression remain largely uncharacterized. Recently, enhancer aberrations were shown in diffuse large B-cell lymphoma, colorectal and gastric cancers by mapping H3K4me1/H3K27Ac (Akhtar-Zaidi et al., 2012; Chapuy et al., 2013; Muratani et al., 2014). Although these studies provide insight into the correlation of isolated epigenetic marks with cancer stage, more than 100 epigenetic modifications have been identified (Kouzarides, 2007; Tan et al., 2011) without clear understanding of their biological roles and interdependence. Furthermore, there are an even larger number of possible combinatorial patterns of these histone and DNA modifications, and it is these combinatorial patterns – not individual modifications - that dictate epigenetic states (Strahl and Allis, 2000). With the development of high-throughput ChIP-Sequencing methodology (Garber et al., 2012), it is now possible to systematically and comprehensively profile many epigenetic marks with relative ease. Here we profiled 35 epigenetic modifications in an isogenic cell system with distinct non-tumorigenic and tumorigenic phenotypes and defined chromatin state alterations associated with transition to tumorigenesis. Further, we determined chromatin changes correlation with stable RNA-expression patterns, assessed their role in tumorigenesis and established relevance premalignant to malignant transition in human melanoma.
Results
Systematic epigenomic profiling to define pro-tumorigenic changes in melanoma
To identify melanoma associated changes, we leveraged a melanocyte cell model system with two characterized biological phenotypes, namely non(or weakly)-tumorigenic (NTM) and tumorigenic (TM) phenotypes (Figure 1A). The NTM phenotype is defined here as one poised to switch to the TM state but require additional cooperative driver alterations. Specifically, we used the well-characterized system of TERT-immortalized human primary foreskin melanocytes engineered with dominant negative p53 and overexpression of CDK4R24C and BRAFV600E (Garraway et al., 2005). In two early passage (n <10) clonal variants (HMEL and PMEL), isogenic cells were created with knockdown of either GFP (non-tumorigenic) or PTEN (tumorigenic). Non-tumorigenic cells were confirmed to be inefficient in driving tumor formation (average tumor latency = 22 weeks) with low penetrance (10-20%) in nude mice (Figure 1A). In comparison, tumorigenic cells expressing shPTEN (∼75% knockdown; Figure S1A) were able to drive tumorigenesis within 10-12 weeks with high penetrance (∼80%) (Figure 1A). Similarly, tumorigenic cells showed aggressive behavior in proliferation, clonogenic and invasion assays (Figure 1B, S1B-E). Hereafter, these two duplicate biological pairs are referred as (1) NTMH (HMEL-BRAFV600E-shGFP, non-tumorigenic melanocytes) and TMH (HMEL-BRAFV600E-shPTEN, tumorigenic melanocytes); (2) NTMP (PMEL-BRAFV600E-shGFP, non-tumorigenic melanocytes) and TMP (PMEL-BRAFV600E-shPTEN, tumorigenic melanocytes). Unless specified otherwise, we have designated NTMH and TMH as the primary pair for discovery and the NTMP and TMP as the pair for additional validation (Methods). These two isogenic but phenotypically distinct melanocyte-derived cells provide a practical and relevant system for understanding epigenomic alterations that are associated with transition to tumorigenesis in melanoma.
Figure 1. Cell line based model of melanoma progression and epigenome profiling.
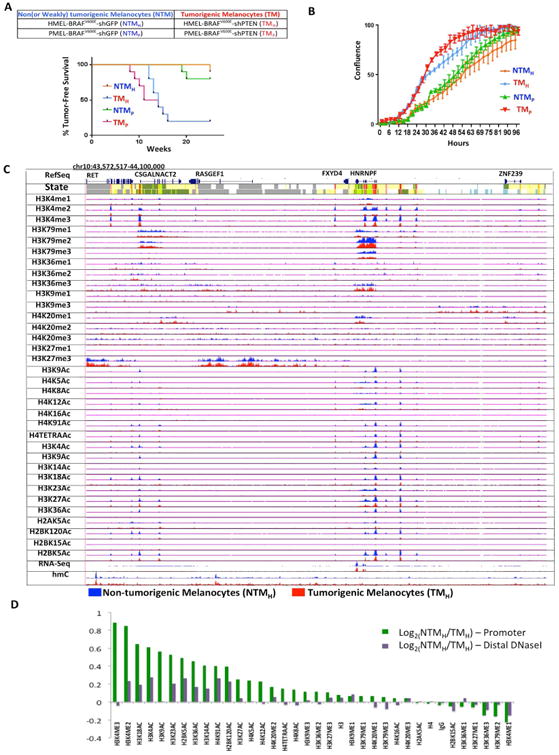
(A) Brief description of the primary melanocyte based model system that consists of two replicates of paired isogenic non (or weakly)-tumorigenic (NTMH, NTMP) and tumorigenic (TMH and TMP) cells. Kaplan-Meier curve showing tumor formation efficiency of NTMH, NTMP, TMH and TMP cells. NTMH and NTMP cells display long latency whereas TMH and TMP cells show shorter latency for tumor formation. Mantle-Cox p = .0007 for NTMH vs TMH and p = 0.0016 for NTMP vs TMP (B) Proliferation curve showing differences in cell confluence (Y-axis) in NTMH vs TMH and NTMP vs TMP as a function of time (X-axis). (C) Normalized signal of all profiled chromatin marks, IgG control and RNA-Seq in an example region (chr10:43,572,517-44,100,000) for NTMH (blue) and TMH (red) cells. Chromatin state tracks and gene annotations are also shown. (D) Log2 ratio between NTMH and TMH cells for the average signal strength of each chromatin mark in a window of 2kb around annotated transcription start sites from RefSeq (Green) and on DNaseI hypersensitive sites from ‘Melano’ (Purple) cell lines from ENCODE. See also Figure S1 and Table S1.
To define the epigenome, we determined the status of 33 histone modifications, histone H3, H4, and IgG marks using a high-throughput ChIP-sequencing method (chromatin immunoprecipitation followed by next-generation sequencing) (Garber et al., 2012) (Figure 1C). We confirmed that the pairwise relationship between the occupancy patterns of different histone marks was consistent with known associations among the marks (Figure S3A). We also predicted combinatorial patterns of marks presented as ‘chromatin states’ and annotated each cell type based on them (Ernst and Kellis 2010). In addition, we profiled 5-methylcytosine using a 450K Illumina array and 5-hydroxymethylcytosine using hMeDIP-Seq. In total we generated 3.08 billion uniquely aligned reads and produced 142 chromatin maps (Table S1). Furthermore, we performed RNA-Seq experiments to define the transcriptomes of these two biological states based on over 1 billion uniquely aligned reads (Table S1; Supplementary Methods).
Changes in histone marks during transition of non-tumorigenic phenotypic state to tumorigenic phenotypic state
We first compared the differences in occupancy of individual chromatin marks between cells in NTM and TM biological states. An analysis for relative enrichment of individual marks at all RefSeq annotated promoters revealed that multiple acetylation marks were consistently depleted in tumorigenic compared to non-tumorigenic cells (Figures 1D, S1F) (Methods). Similarly, a subset of acetylation marks was consistently depleted at a set of distal DNaseI hypersensitive sites (in ENCODE melanocytes; Supplementary Methods) in tumorigenic cells compared to non-tumorigenic cells in both replicates (Figures 1D, S1F). We also identified H3K4me2/3 marks in promoter regions as higher in NTM biological state relative to TM biological state (Figures 1D, S1F). Interestingly, we did not observe any difference in global levels of histone acetylations or H3K4 methylations by mass spectrometry based quantitation (of the measurable marks) or by western blotting (Figures S1G-I). Overall, these data suggest that transition from non-tumorigenic phenotype to tumorigenic phenotype is accompanied by switch to reduced acetylation and H3K4me2/3 methylation at nucleosomes in specific regions, but not at the global level.
To demonstrate human relevance of these observations from the cell model system, we assessed the status of representative marks in human benign nevi and melanoma samples representing pre-malignant (non-tumorigenic) and malignant (tumorigenic) biological states, respectively. We first developed a validation strategy using a ChIP-String assay (Ram et al., 2011) and designed a 96-test probe ‘codeset’ (Methods, Table S2) that could be used to evaluate recapitulation of key epigenetic features observed in the isogenic cell models. This ChIP-String codeset was designed to measure enrichment of 6 selected histone marks (H3K27Ac, H2BK5Ac, H4K5Ac, H3K4me1, H3K27me3 and H3K4me3) that showed consistent differences between the non-tumorigenic and tumorigenic cells and were part of three groups of epigenetic elements: promoters, enhancers and Polycomb-repressed regions. To verify that the codeset for this ChIP-String assay performs as expected, we assayed them in both NTMH and TMH and showed that there was good correlation between the signal intensity from ChIP-String with ChIP-Seq intensity (R = 0.62 to 0.81) for the tested probes (Figure S2A-F).
Chromatin immunoprecipitated DNA for two of the marks (H2BK5Ac and H4K5Ac) that provided sufficient yield after ChIP from 9 melanoma tumors and 4 nevi samples (Table S3), was tested for enrichment on the designed code set. As shown in Figures 2A-B, unsupervised hierarchical clustering analysis showed that nevi samples cluster with the non-tumorigenic cells, whereas melanoma samples were more similar to the tumorigenic cells (Figures 2A-B). Average mark levels across all designated probes showed that enrichment for these marks in nevi and tumor samples was similar to those seen in NTMH and TMH cells, respectively (Figures 2C-D, S2G-J). Further, principal component analysis (PCA) based on the enrichment of these chromatin marks at selected genomic regions showed that human nevi clustered tightly with NTMH cells, but not the melanoma samples. Interestingly, although away from the NTMH cells, the human melanoma samples do not cluster tightly around the TMH cells suggesting that the melanoma samples were more variable among themselves but collectively more different from the non-tumorigenic cells and the nevi samples (Figures 2E-F). This may foretell that, like the genome, the epigenome is more heterogeneous and complex in tumors than benign neoplasms or normal cells, with the caveat that the assay was limited to 96 probes. In summary, a subset of chromatin changes observed in the non-tumorigenic / tumorigenic melanocytic system displayed similar patterns as those observed in the benign / malignant human melanocytic lesions.
Figure 2. Chromatin changes are reflected in human tumors. (A-B).
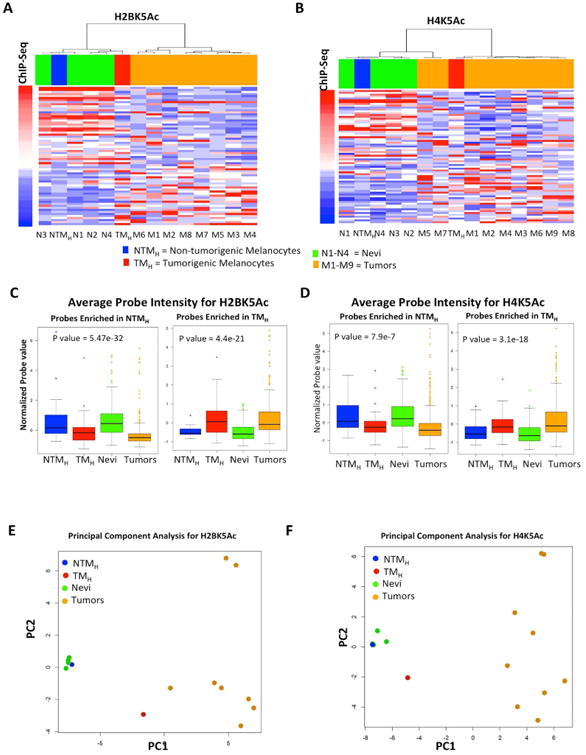
Heat map for H2BK5Ac (A) and H4K5Ac (B) showing enrichment in NTMH, TMH, 4 nevi samples (N1-N4) and in up to 9 melanoma tumor samples (M1-M9) as calculated by ChIP-String assay. Probes are ordered with increasing ChIP-Seq signal in TMH cells. Columns are ordered based on hierarchical clustering. (C-D) Boxplots showing average normalized intensity for ChIP-string probes across NTMH, TMH, nevi and tumors (Averaged over all enriched probes across all samples for Nevi and tumors). (E-F) PCA plot for H2BK5Ac (C) and H4K5Ac (D) showing relationship between NTMH, TMH, 4 nevi samples (N1-N4) and up to 9 melanoma tumor samples (M1-M9) as calculated by ChIP-String assay. Asterisk (*) represents p<0.05 and double asterisk (**) represents p<0.001. See also Figure S2 and Tables S2-S3.
Chromatin state changes between non-tumorigenic and tumorigenic states
To assess whether and how the combinatorial and spatial patterns of chromatin marks (Ernst and Kellis, 2010; Ernst et al., 2011) may change with transition from NTM to TM biological states, we employed ChromHMM (Ernst and Kellis, 2012) to discover and define a set of chromatin states based on the 33 histone modification profiles, in addition to H3, H4, and IgG controls in both NTM and TM cells. Briefly, by concatenating the chromatin maps for each mark, ChromHMM derived a common set of chromatin state definitions with cell type specific state assignments in both non-tumorigenic and tumorigenic cells. A final model with 18-states was adopted for further downstream analyses (Figures 3A, S3B), based on the observations that it effectively achieved a balance of (1) capturing important biological distinctions while (2) generating a manageable set of pairwise state combinations (Methods). We found for some states of the model the assignments to be substantially recoverable by multiple different individual marks, found other states that required a specific mark in order to be able to recover their assignments, and also some states that would need multiple marks to recover them (Supplementary Methods, Table S4). By triangulating the defined chromatin states with known genome organization features (Figures S3C-D), we then grouped the 18 chromatin states by the following putative annotations: promoter regions (States 1-3), enhancers (States 4-6), transcribed enhancers (States 7-9), transcribed (States 10-12), active proximal (State 13), low signal (State 14), polycomb repressed (State 15), H3K9me3 heterochromatin (State 16), quiescent (State 17) and artifact/repetitive elements (State 18).
Figure 3. Chromatin state predictions for non-tumorigenic and tumorigenic melanocytes.
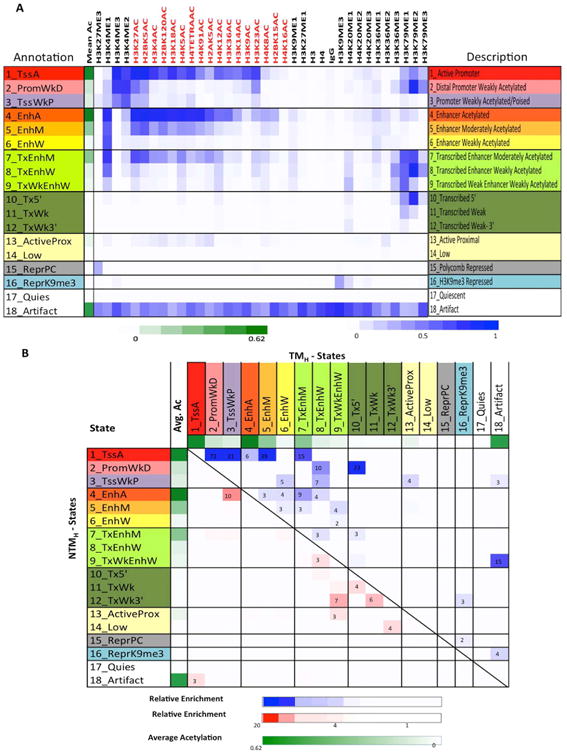
(A) Emission probabilities of the 18-state ChromHMM model (see Figure S3A for transition probabilities). Each row represents one chromatin state. First column gives state number and mnemonic and last column gives the candidate state description. Second column indicates the intensity of mean acetylation from zero (white) to 0.62 (green), which is the maximum mean acetylation across all states. Remaining columns each correspond to one chromatin mark with the intensity of the color in each cell reflecting the frequency of occurrence of that mark in the corresponding chromatin state on the scale from 0 (white) to 1 (blue). (B) Heat map showing fold enrichment of transitions of chromatin states in non-tumorigenic (NTMH) to tumorigenic (TMH) cells controlling for the overall state size and similarity (Supplementary Methods). The color intensities above (below) the main diagonal range from white (relative enrichment <1) to blue (red) (relative enrichment > 20), thus indicating chromatin state transitions that lose acetylation marks from NTMH to TMH within the same category are more enriched compared to the reverse chromatin state transition (i.e. from TMH to NTMH) and the lack of those that gain acetylations. See also Figure S3, S4 and Table S4.
Within each of these groupings, enrichment of specific genomic structures was as expected (Figures S3C-D). For example, regions within 2Kb of RefSeq annotated TSSs were enriched specifically in chromatin states 1-3, corresponding to promoter regions and CpG islands in the genome. Consistently, 5-methylcytosine (5-MeC) containing sites were weakly enriched in promoter-associated states, whereas 5-hydroxymethylcytosine (5-hMeC) showed complimentary patterns to 5-MeC. RefSeq gene annotations were enriched in regions associated with chromatin states containing transcription marks H3K79me2/H3K79me1/H3K36me3, primarily states 1, 2 and 7-12. LaminB1-associated domain association (Guelen et al., 2008) was specifically seen in H3K9me3 enriched state 16. These enrichments support the biological relevance of this 18-state model and the annotation assigned to each state.
Next we sought to define associations of chromatin states with non-tumorigenic and tumorigenic cell phenotypes. To this end, we identified regions that transition to a different chromatin state in non-tumorigenic and tumorigenic conditions. Calculation of coverage changes for each state in NTMH and TMH cells revealed that genome-wide occupancies of the most acetylated promoter state (State_1_TssA) and the most acetylated enhancer state (State_4_EnhA) were reduced from NTMH to TMH, by 4.5 and 2.9 fold, respectively (p<1e-15, Figure S4A). On the other hand, we noticed a 2.6 fold increase in the H3K9me3 repressive State_16_ReprK9me3 in TMH cells when compared to NTMH cells (Figure S4A; p<1e-15).
To understand the global state transitions, we analyzed the pairwise state transition enrichments between NTMH and TMH relative to the same pair in the opposite direction, which controls for overall state similarity (Supplementary Methods) (Figures 3B, S4B). This analysis revealed that, globally, there was a significant shift (transition) from strongly acetylated promoter and enhancer states to more weakly acetylated states accompanying the evolution from NTM to TM biological states (Figures 3B, S4B). For instance, the pairwise state transition from the strongly acetylated promoter state (State_1_TssA) in NTMH to a more weakly acetylated promoter State_2_PromWkD or State_3_TssWkP in TMH was 72 and 21 times, respectively, more enriched than observing a reverse transition from TMH to NTMH (p<1e-15). Similarly, the pairwise state transition from the strongly acetylated enhancer state (State_4_EnhA) in NTMH to a more weakly acetylated but transcribed enhancer state (State_7_TxEnhM) in TMH was 9 times more enriched - and to a weakly acetylated non-transcribed state 5_EnhM in TMH was 3 times more enriched - than the reverse transition between the same pair of states from TMH to NTMH (p<1e-15). The overall trends in chromatin state changes were similar after quantile normalization or downsampling to the same read depth (Figure S4C-D) as well as being replicated in NTMP and TMP cells (Figure S4E). Finally, we evaluated the correlation between mean histone acetylation and H3K4me2/3 changes on the same promoter and found them to be well correlated (Figures S5A-B). Together, these data suggest that during a non-tumorigenic to tumorigenic phenotype switch, certain promoter and enhancer regions with specific chromatin states harboring higher acetylations and H3K4me2/3 transition to those with lower acetylation and H3K4me2/3 levels.
Chromatin state changes enrich on genes regulating cancer-associated processes
To begin to explore the biological significance of prominent chromatin state transitions between NTM and TM biological states, we next performed pathway enrichment analysis [Gene Ontology (GO)] for genes associated with a specific pairwise transition in the promoter region (Supplementary Methods) (Figure 4A and Table S5). We found specific enrichments for cancer-associated processes and metabolic processes. For example, promoters harboring highly acetylated State_1_TssA in NTMH that transitioned to weakly acetylated State_2_PromWkD and State_3_TssWkP in TMH were found preferentially by genes regulating cell cycle and apoptosis as well as various cellular metabolic processes and protein modifications. These included important melanoma cell cycle inhibitors CDKN1B and CDKN2A (Bennett, 2016) as well as melanoma pro-apoptotic genes BAD and APAF1 (Campioni et al., 2005; Sheridan et al., 2008) (Figures 4B, S5C-D) suggesting increased proliferation and reduced apoptosis in tumorigenic cells. Interestingly, homophilic cell adhesion genes such as proto-cadherins were associated with the transition from a weakly poised promoter (State_3_TssWkP) or a quiescent state (State_17_Quies) to a more strongly H3K9me3 associated chromatin state (State_16_ReprK9me3) in tumorigenic cells (Figures 4A,C). This suggests that upon acquisition of a tumorigenic fate, genes promoting cell adhesion acquire a repressive chromatin signature, possibly contributing to loss of cell - cell adhesion in cancer.
Figure 4. Chromatin state changes during transition to tumorigenic state mark specific cancer pathways.
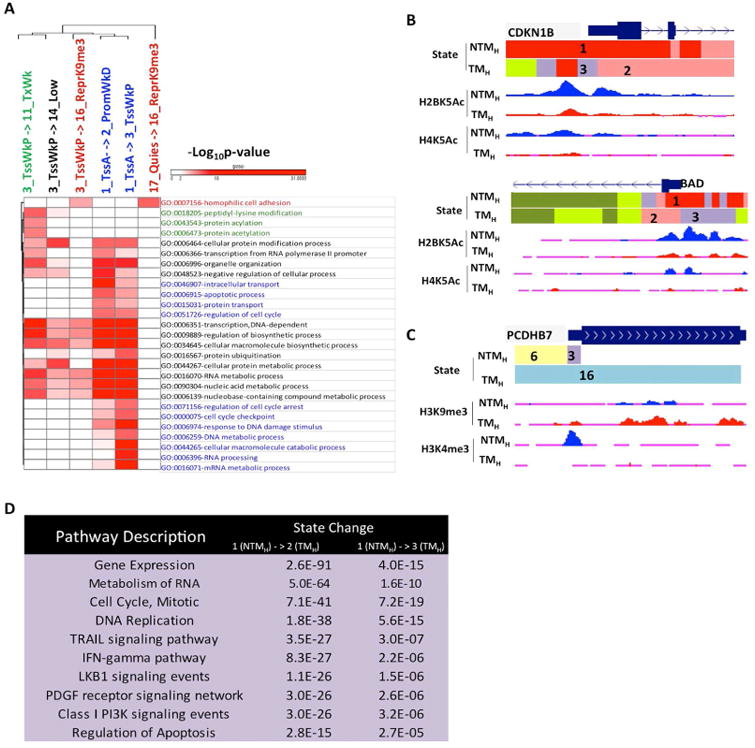
(A) Heat map showing -log10(p-value) for top GO terms enriched in specific promoter state transitions between non-tumorigenic (NTMH) to tumorigenic (TMH) cells. (B) UCSC genome browser view of chromatin states as well as selected histone acetylation profiles (H2BK5Ac and H4K5Ac) for loci encompassing cell cycle regulator CDKN1B and apoptotic genes BAD, which showed loss from NTMH to TMH cells. (C) UCSC genome browser view of chromatin states as well as selected histone mark H3K9me3 and H3K4me3 profiles for loci encompassing pro-adhesion PCDHB7 in NTMH and TMH. (D) Top 10 most significant pathways (pathway commons) associated with promoters displaying state transitions from State 1_TssA in non-tumorigenic cells (NTMH) to States 2_PromWkD and 3_TssWkP in tumorigenic (TMH) cells. See also Figure S5 and Table S5, S6.
Further, a pathway enrichment analysis of genes displaying chromatin state transition revealed additional association of cell signaling pathways with chromatin states (Figure 4D and Table S6; Supplementary Methods). We found significant enrichments of important melanoma cell signaling pathways such as PI3 kinase, IFNγ, LKB1, TRAIL and PDGF mediated signaling (Paluncic et al., 2016) in promoters transitioning from State_1_TssA to either State_2_PromWkD or State_3_TssWkP during non-tumorigenic (NTM) to tumorigenic (TM) phenotype switch (Figure 4D and Table S6).
Similar analysis of enhancer regions in two of the most significant chromatin state transitions, State_4_EnhA in NTMH to State_7_TxEnhM or State_5_EnhM in TMH cells, showed enrichment of nearest genes in important melanoma cell signaling events such as integrin, TGFβ and MAPK signaling (Busse and Keilholz, 2011; Pinon and Wehrle-Haller, 2011; Sullivan and Flaherty, 2013) (Figures S5E-F; Table S6 and Supplementary Methods). Overall, these data suggest that chromatin state changes during transition to tumorigenic phenotype occur on promoters and enhancers of a large number genes that are known to regulate relevant cancer processes such as proliferation, apoptosis and adhesion.
Complex relationship between gene expression and chromatin states
To understand relationships between chromatin state and gene expression, we integrated RNA-Seq profiles of non-tumorigenic and tumorigenic cells with the chromatin states individually in each cell type. As expected, promoters of highly expressed genes (FPKM > 5) displayed enrichment in chromatin states 1 and 2 that are marked by H3K4me3 and acetylations (Figure S3E). These promoters were depleted in repressed states 15-16 whereas their gene bodies were enriched in states 7-12 with transcription marks (H3K79me2/3 and H3K36me3) (Figure S3E). Furthermore, while comparing between different states within the same cell type, differences in acetylation content were associated with gene expression differences, particularly within the enhancer state group (Figure S3E).
Analysis of significant changes between NTM and TM states in the expression levels of known RefSeq transcripts revealed that changes in gene expression are bidirectional with similar numbers of genes upregulated or downregulated (Figure 5A). Next we sought to determine associations of chromatin state transitions with gene expression changed between non-tumorigenic and tumorigenic cells. To this end, we calculated the relative enrichment of all possible chromatin state transitions at the promoters +2kb and -2kb from TSS of genes that were up-regulated, down-regulated or unchanged (Supplementary Methods) (Figures 5B, S6A-F) with an expectation that genes downregulated in TMH cells in comparison to NTMH cells would show global switch from active chromatin states to repressed/low states on their promoters and vice versa. However, we observed overall similar patterns of chromatin state enrichments and with few exceptions did not see substantial chromatin state switches in up-regulated, down-regulated or unchanged genes between non-tumorigenic and tumorigenic cells (Figures 5B, S6A-F). This observation suggests that regulation of steady state levels of RNA transcripts in this system involves more than chromatin modification at the promoters.
Figure 5. Correspondence of chromatin state changes with RNA expression changes during transition to tumorigenesis.
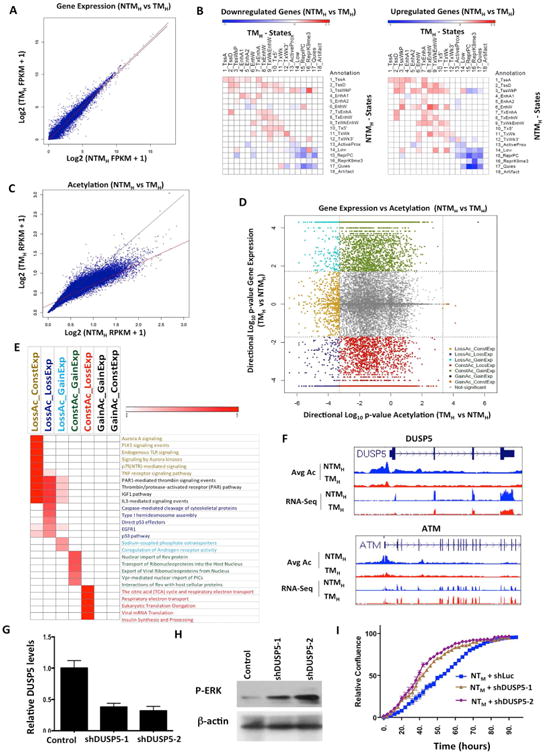
(A) Scatter plot comparing gene expression values [log2 (FPKM+1)] in NTMH and TMH for RefSeq genes. (B) Relative enrichment of chromatin state transitions at promoters of down-regulated genes compared to up-regulated genes (left panel) or up-regulated genes promoters compared to down-regulated (right panel) for all pairs of chromatin state transitions. Red shows enrichment whereas blue is depletion. (C) Scatter plot comparing promoter acetylations [log2(RPKM+1)] around +/-2kb of each RefSeq gene in NTMH and TMH. The line in red is a regression line, while in black is the y=x line. (D) Scatter plot displays directional log10(p-value) for acetylation and gene expression changes between TMH and NTMH. Negative values represent genes with decreased expression or acetylation levels in TMH compared to NTMH cells. Dashed lines show the significance cut-off for acetylation or expression changes. Genes with significant gene expression and/or acetylation changes are colored based on grouping indicated. (E) Heat map represents enriched pathways (pathway commons) for each group identified in Figure 5D. Color scale represents –log10(HyperFdrQ corrected). (F) UCSC genome browser view of average acetylation and RNA-Seq for an example from each of the LossAc_LossExp (DUSP5) (top) and LossAc_ConstExp (ATM) groups (bottom). (G) Graph showing relative levels of DUSP5 in NTMH cells harboring either control or DUSP5 shRNAs. (H) Western blot showing levels of p-ERK in NTMH cells harboring either control or DUSP5 shRNAs. (I) Growth curve showing proliferative capacity of NTMH cells harboring control or DUSP5 shRNAs (shDUSP5-1 and shDUSP5-2). See also Figure S6 and Table S7.
Since changes in acetylation marks were prominent between non-tumorigenic and tumorigenic cells, we quantitatively compared aggregate acetylation changes in promoter regions of all 17 acetylation marks profiled with gene expression changes. Here, we first identified a set of promoters for which the change in their average acetylation signal over all acetylation marks was statistically significant at a FDR of 1% (Supplementary Methods) when comparing between NTMH and TMH (Figure 5C). In stark contrast to gene expression patterns, changes in acetylation levels were unidirectional, with most changed regions having a lower average acetylation in tumorigenic cells compared to non-tumorigenic cells (Figures 5C-D, S6G).
Given these differences between patterns of acetylation changes and stable gene expression changes, we explored the possibility that different subsets of genes are responding differently to acetylation changes on their promoters. To test this directly, we systematically overlapped gene expression changes with changes in promoter acetylation to define nine possible subsets (Figure 5D, S6H; Supplementary Methods for definitions) and performed enrichment analysis of the genes to determine if different cellular processes were enriched in different subsets (Figure 5E and Table S7). Indeed, we found that prominent cancer-relevant pathways were enriched among genes that were downregulated and showed loss of acetylation (LossAc_LossExp), including EGFR pathway targets, p53 regulated genes and Caspase-mediated apoptotic signaling genes (Figure 5E and Table S7). These genes are epigenetically regulated by specific chromatin alterations and may regulate melanoma growth. For example, DUSP5 showed loss of acetylation on its promoters and concomitant downregulation in TM cells in comparison to NTM cells (Figure 5F). DUSP5 is a negative regulator of MAPK pathway (Caunt and Keyse, 2013) that functions by reducing nuclear phosphorylated ERK and therefore its loss can potentially provide positive feedback to MAPK signaling enhancing p-ERK levels. To test this hypothesis, we reduced DUSP5 levels in NTMH cells by generating stable cell lines bearing two specific shRNAs (Figure 5G) and tested p-ERK levels as well as proliferative capacity. Indeed, NTMH cells bearing DUSP5 shRNAs showed increased p-ERK levels (Figure 5H) and proliferated faster in comparison to NTMH cells harboring control shRNA (Figure 5I).
On the other hand, genes in certain signaling pathways such as aurora kinase and PLK signaling showed only acetylation loss in the promoters without expression change (LossAc_ConstExp group) (Figure 5E). For example, ATM, a critical mediator of the DNA damage checkpoint pathway (Shiloh and Ziv, 2013), was found to follow this pattern (Figure 5F). Possible deacetylation changes without accompanying alterations in steady-state RNA levels may reflect multi-level control of transcription requiring either upstream regulation such as promoter-enhancer interactions or downstream regulation by an additional event such as miRNA-mediated post-transcriptional regulation. Conversely, genes with differential expression but without acetylation changes (ConstAc_GainExp and ConstAc_LossExp) were enriched for various transport pathways, TCA cycle and translation, raising the possibility that these pathways are less likely to be regulated on the epigenomic level through promoter acetylation during tumorigenesis.
Taken together, these integrative analyses showed that some well-characterized cancer signaling pathways exhibit promoter acetylation-correlated expression regulation, suggesting that these pathways can be regulated by epigenomic modifications. At the same time, it is intriguing that changes in expression of some pathway genes such as those related to metabolism or transport do not appear to show correlation with changes in their promoter acetylation.
Loss of CBP (CREB-Binding Protein) creates pro-tumorigenic chromatin patterns and accelerates tumorigenic properties
Next, we asked whether decreased expression of a histone acetyltransferase or increased expression of a histone deacetylase in this system might be responsible for observed loss of acetylation peaks. We checked the expression differences of 32 known histone acetyltransferases and deacetylases between the NTM and TM models (Sammons et al., 2016) (Figure 6A). The expression of CBP acetyltransferase showed consistent patterns to observed acetylation loss in that its expression was downregulated > 2 fold in both variants of TM cells compared to their counterpart NTM cells (Figure 6A-B). We knocked down CBP mRNA in NTMH cells using two specific shRNAs (Figure 6C) and checked the levels of H2BK5Ac and H4K5Ac using the ChIP-string codeset that was utilized to validate acetylation changes in nevi/tumor samples. Indeed, stable cells harboring CBP shRNAs showed similar patterns of these two acetylations as seen in TM cells compared to NTM cells (Figure 6D-G). Consistently, the NTMH cells harboring CBP shRNAs showed significantly enhanced tumorigenesis compared to control shRNA-bearing cells (Figure 6H). These data argue that a tumorigenic phenotype might be associated with loss of acetylation irrespective of whether tumorigenic behavior was achieved by PTEN loss or by CBP loss in the same background (NTM background of TERT/p53DD/CDK4R24C). This hypothesis was further supported by our observations that NRASG12D overexpression in TERT/p53DD/CDK4R24C immortalized melanocytes created similar H4K5Ac and H2BK5Ac acetylation patterns to TMH cells (overexpression of BRAFV600E along with shPTEN) (Figures 6D-G). NRAS has been previously shown to activate MAPK pathway (result of BRAF activation) and PI3K pathway (result of PTEN loss) (Chudnovsky et al., 2005) thereby mimicking cellular phenotype of TM cells. Together, our data argue for a relatively uniform acetylation pattern of cells with tumorigenic behavior.
Figure 6. CBP loss in NTMH cells promotes tumorigenesis and mimics acetylation loss seen in TMH cells.
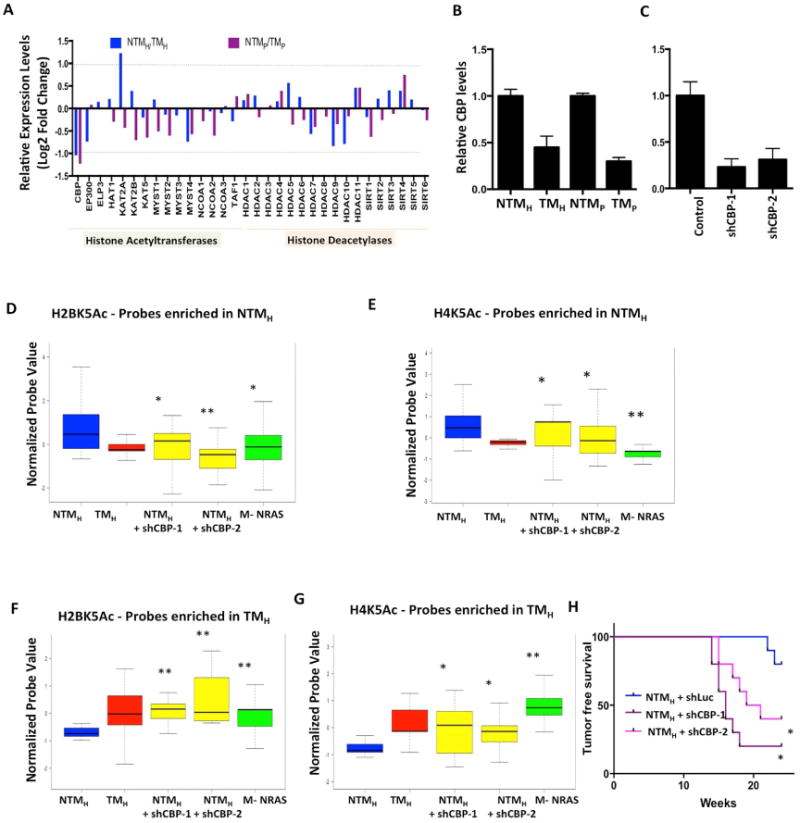
(A) Bar graph showing relative levels of 32 histone acetyltransferases and deacetylases between NTMH/TMH and NTMP/TMP cells. The Y–axis shows Log2 Fold Change values. The dotted line shows the cutoff of 2-fold change. (B-C) Graph showing relative levels of CBP histone acetyltransferase in (E) NTMH, TMH, NTMP and TMP cells and (F) NTMH cells harboring either control or CBP shRNAs. (D-G) Boxplots showing average normalized intensity for ChIP-string probes for (D, F) H2BK5Ac and (E, G) H4K5Ac in NTMH, TMH, NTMH cells harboring CBP shRNAs or NRASG12D expressing transformed melanocytes (M-NRAS). The plot is limited to those probes that were originally enriched in (D-E) NTMH cells or in (F-G) in TMH cells by ChIP-Seq experiments and validated by ChIP-String in Figure S2A-F. Asterisk (*) represents p<0.05 and double asterisk (**) represents p<0.001 (Wilcoxon Rank test) when comparisons are made to NTMH. (H) Kaplan-Meier curve showing tumor formation efficiency of NTMH cells harboring control or CBP shRNAs (shCBP-1 and shCBP-2).
HDAC inhibitors specifically reduce proliferative rate in tumorigenic cells
Next, we sought to determine if chromatin state changes seen during transition to tumorigenesis impart proliferative advantage to tumorigenic cells. Since loss of histone acetylation peaks was a consistent feature of all major chromatin state alterations, we tested the contribution of widespread acetylation loss to cell proliferation. Because steady state acetylation loss seen in tumorigenic cells could be an outcome of aberrations in histone acetylation-deacetylation cycle in favor of accelerated deacetylation or reduced acetylation, we sought to alter the former by inhibition of histone deacetylases, the primary driver enzymes of histone deacetylation in mammalian cells. We tested if treatment of tumorigenic cells with histone deacetylase (HDAC) inhibitors alters their acetylation levels toward those in non-tumorigenic cells. Indeed, measurement of H2BK5Ac, H4K5Ac and H3K27Ac levels in TMH cells treated with vehicle or two different HDAC inhibitors (vorinostat and entinotat) by ChIP-string revealed that the levels of the histone acetylations on loci highly acetylated in NTMH cells, but not in TMH cells, were partially restored to the levels seen in NTMH cells (Figures 7A-B, S7A). However, this treatment had minimal impact on acetylation levels on the loci seen to harbor higher levels of acetylation in TMH cells (Figures S7B-D). Next, we tested the impact of vorinostat and entinostat on the growth rate of non-tumorigenic and tumorigenic cells in a time-course experiment. Indeed, both of these inhibitors showed preferential effect on abrogation of proliferation in tumorigenic cells TMH and TMP compared to non-tumorigenic NTMH and NTMP (Figures 7C-D, S7E-F).
Figure 7. Acetylation status on deacetylated promoters in TH correlates with response to HDAC inhibitors. (A-B).
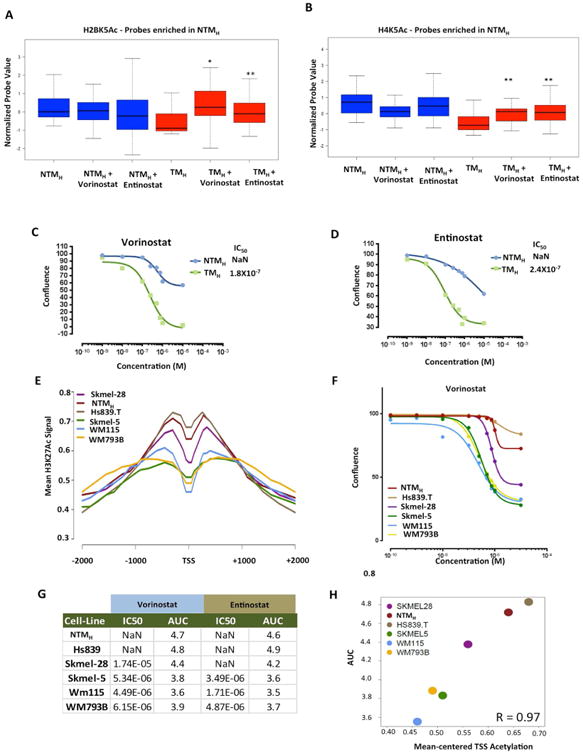
Boxplots showing average normalized intensity for (A) H2BK5Ac or (B) H4K5Ac on ChIP-string probes (that were enriched in NTMH cells by ChIP-Seq experiment) across NTMH and TMH cells that were either untreated or treated with vorinostat (200nM) or entinostat (300nM) for 72hrs. Asterisk (*) represents p<0.05 and double asterisk (**) represents p<0.001 (Wilcoxon Rank test) when comparisons are made to TMH. (C-D) Growth curves for NTMH and TMH cells grown under various concentrations of (C) vorinostat or (D) entinostat. (E) Aggregate plot showing H3K27Ac levels around +/-2Kb of deacetylated gene promoters (in TH cells) in various melanoma cell lines. (F) Growth curves for melanoma cell lines grown under various concentrations of vorinostat. (G) Table showing IC50 values (the concentration at which 50% response is achieved) and area under the curve (AUC) for two HDAC inhibitors, vorinostat and entinostat, in melanoma cells lines. Immmeasurable IC50 values are shown as NaN which refers to ‘not a number’. (H) Correlation plot between AUC and average H3K27Ac levels at TSS of gene promoters that showed loss of histone acetylation in TMH cells compared to NTMH cells. See also Figure S7.
Since these observations were made in an artificial model system that mimics melanoma progression, we next tested if levels of histone acetylation in melanoma-derived cell lines could indicate vulnerability to histone deacetylase inhibitors. To this end, we performed H3K27Ac ChIP-Seq in five melanoma cell lines and measured relative acetylation levels on promoters deacetylated in TMH cells. NTMH and Hs839.T contained relatively higher levels, Skmel-28 moderate levels and WM115, Skmel-5 and WM793B showed lower levels of H3K27Ac (Figure 7E). To determine whether the acetylation levels correlated with their response to HDAC inhibitors, we determined IC50 values and area under curve (AUC) for vorinostat and entinostat in each cell line (Figures 7F-H, S7G). Indeed, all three cell lines with lower acetylation levels (WM115, Skmel-5 and WM793B) showed substantially lower IC50/AUC values compared to those that had higher acetylation levels (NTMH and Hs839.T) to both treatments (Figures 7F-G, S7G). Skmel-28, which harbored intermediate levels of acetylation, displayed intermediate IC50/AUC values (Figures 7F-G, S7G). Correlation score between average acetylation at TSS and AUC values was calculated to be 0.97 (Figure 7H). These data confirm disease relevance of our observations that lower acetylation levels in tumorigenic cells functionally contribute to the proliferative phenotype and suggest that responsiveness to HDAC inhibitors may associate with histone acetylation levels on specific genomic loci.
Discussion
We have generated snapshots of the epigenome landscape at two phenotypically distinct biological states (e.g. non-tumorigenic and tumorigenic) as a way to delineate changes that are associated with tumorigenesis by leveraging an isogenic cell model system. Although artificial, this system is well-suited for this study for the following reasons: 1) these cells are derived from primary melanocytes, the appropriate cells of origin for melanoma; 2) the cell system recapitulates known genetic alterations observed in human melanoma tumors, in particular in p53, CDK4, BRAF and PTEN (Hodis et al., 2012; Krauthammer et al., 2012); 3) non-tumorigenic cells (shGFP) and tumorigenic cells (shPTEN) are otherwise isogenic. Finally, this melanocyte-based cell system has been previously used in other mechanistic studies related to regulation of melanomagenesis (Garraway et al., 2005; Rai et al., 2015).
We show that a predominant feature of chromatin state changes during progression to tumorigenic state in melanoma is lowering of frequency of detectable locations of acetylation modifications. Two independent observations suggest that these changes are relevant to human disease and could play a functional role: first, a selected subset of acetylation changes between non-tumorigenic and tumorigenic cells was reproduced between human benign nevi and malignant tumors. Second, the treatment with HDAC inhibition, which restored acetylation patterns on deacetylated loci, was able to abrogate high proliferation rate of tumorigenic cells and melanoma cells that contained lower acetylation than non-tumorigenic cells. Overall, our data suggests that a specific chromatin environment around certain loci in the genome can have pro-tumorigenic function.
One can hypothesize that such a state of chromatin can be established by one or multiple tumor-promoting genetic events such as PTEN deletion/mutation or other alterations in epigenetic machinery. This is supported by our observations that knockdown of CBP histone acetyltransferase in NTMH cells or overexpression of NRAS (which recapitulates BRAF activation + PTEN loss due to its ability to activate MAPK and PI3K pathways (Chudnovsky et al., 2005)) in Tert/p53/CDK4R24C immortalized melanocytes showed similar histone acetylation profiles as in TMH.
Functional characterization of the regions displaying altered chromatin states suggested that, consistent with observed phenotypes, promoters of genes with important roles in cancer progression show preferential deacetylation, such as cell cycle regulation and apoptosis, in tumorigenic cells. Further, we noted that a number of genes in important melanoma cell signaling pathways such as TRAIL, IFNγ, LKB1, PDGF, PI3K, ITGβ1, TGFβ and cytokine signaling were associated with chromatin state changes involving histone acetylations (Figure 4). For example, TGFβ and INTGβ1 are known to regulate cell invasion and adhesion properties (Jakowlew, 2006; Trikha et al., 1997), consistent with observed invasive properties of tumorigenic cells. Enrichment of multiple such signaling events linked to observed tumorigenic phenotypes suggests that, in this model system, chromatin associated changes are likely regulators of cancer progression, underscoring important roles of chromatin in tumorigenesis. This hypothesis is reinforced by abrogation of hyperproliferative phenotype by HDAC inhibitors, which restores the acetylation on deacetylated loci.
Interestingly, although acetylation intensity measurements based on ChIP-Seq profiles revealed a loss of peaks of acetylation marks in tumorigenic cells, we did not observe any changes in total levels of histone acetylation marks either by western blotting in whole cell lysate and chromatin fraction or by mass spectrometry analysis of acid extracted histones (Figure S1 and data not shown). Two independent observations in addition to ChIP-Seq reinforced the results of loss of acetylation peaks in tumorigenic cells and diminished the possibility of this being a technical or experimental bias. First, ChIP enrichment measurement by either nanostring at 96 probes or by qPCR at 5 loci revealed results consistent with the ChIP-Seq signal (Figures 2, S2 and not shown). Second, biological replicates for ChIP followed by either nanostring or qPCR measurement revealed similar enrichment profiles (data not shown). Based on these observations we speculate that, although both tumorigenic and non-tumorigenic cells harbor the same levels of acetylated histones in the cell and on chromatin, acetylated histones are more diffusely incorporated throughout the genome in tumorigenic cells, including at locations where it is not present in normal cells.
Overlap of epigenomic and transcriptomic data revealed that there was little correlation between chromatin changes and gene expression changes at the global level between the tumorigenic and non-tumorigenic cells in this system. Some other recent studies have also shown low- correlation between gene expression and acetylation changes in specific systems (Sen et al., 2016; Sun et al., 2016). A plausible explanation for such an observation is that the steady-state RNA-levels may not completely be reflective of all chromatin associated gene expression changes during biological state switches in tumorigenesis process and are influenced by other post-transcriptional regulatory molecular processes. Nonetheless, systematic analysis of gene sets in different groups clearly suggests that the set of genes that had both lower acetylations and lower gene expression enriched for pathways with known roles in tumor progression underscoring the importance of chromatin associated gene expression changes in cancer progression.
Taken together, our study provides a first systematic view of the epigenomic, as well as transcriptomic, landscape evolutions between two distinct biological states (e.g. non-tumorigenic and tumorigenic) associated with melanoma tumorigenesis.
Experimental Proceedures
Cell Culture, generation of stable cells and drug treatment
HMEL-BRAFV600E, PMEL-BRAFV600E cells were obtained from Dr. David Fisher's laboratory (Garraway et al., 2005) and maintained in standard tissue-culture conditions in DMEM media with 10% FBS. Stable knockdown of GFP (control) or PTEN (experimental) in early passage (n <10) was performed using pMKO-shGFP or pMKO-shPTEN vectors (Addgene) to create NTMH (HMEL-BRAFV600E-shGFP), TMH (HMEL-BRAFV600E-shPTEN), NTMP (PMEL-BRAFV600E-shGFP) and TMP (PMEL-BRAFV600E-shPTEN) cells. Control and experimental cells were passaged together for the same time before harvesting cells for ChIP-Seq experiments. Hs839.T, Skmel-28, Skmel-5, WM115 and WM793B cells were obtained from ATCC and grown according to manufacturer's recommendation. Cells were treated with Vorinostat (Sigma), entinostat (MS-275, SelleckChem) or vehicle (DMSO) by direct addition to media.
ChIP-Seq
Chromatin immunoprecipitation was performed as described earlier (Garber et al., 2012) with optimized shearing conditions and minor modifications for melanocytes. For more details, see supplementary methods.
ChIP-Seq and Chromatin State Analysis
ChIP-Seq reads were aligned using Bowtie (version 1.0.0) (Langmead et al., 2009) to human genome assembly NCBI Build 37 (UCSC hg19) and uniquely mapped reads with one mismatch were retained. ChromHMM (Ernst and Kellis, 2012) was used with default parameters to derive genome-wide chromatin state maps for all cell types. We binarized the input data with ChromHMM's BinarizeBed method using a p-value cutoff of 10-4. Chromatin state models were learnt jointly on all chromatin marks from NTMH and TMH ranging from 10 to 120 states. A model with 18 states was chosen for detailed analysis and is presented throughout the manuscript. Chromatin state annotations of NTMH, TMH, NTMP and TMP were produced subsequently by applying this model to the original binarized, quantile normalized or downsampled chromatin data from these cell types. For details, see supplementary methods.
RNA-Seq
Strand specific libraries were constructed using ScriptSeq Kit (Epicenter/Illumina). Reads were mapped to the human genome (hg19) using Mapsplice algorithm version 2.1.4 (Wang et al., 2010). Transcript expression was estimated using Cuffdiff 2.11. Further details are in supplementary methods.
ChIP-String
Nano-string experiments were run on a custom ChIP-string array according to manufacturer's recommendation using ChIP-DNA for shown marks (Figure 2, S2 from NTMH and TMH cells and nevi and tumor cells. A custom ChIP-string array containing probes for 96 genomic locations (Table S2) was used. Details of the design are in supplementary methods. The analysis was done as previously described in (Ram et al., 2011). The details are in extended supplementary methods.
Supplementary Material
Highlights.
Epigenomic alterations in non-tumorigenic to tumorigenic transition in melanoma.
Chromatin states harboring acetylation and H3K4me2/3 are lost in tumorigenic cells.
Chromatin state transitions preferentially occur on melanoma-regulatory pathways.
HDAC inhibition preferentially impacts proliferative ability of tumorigenic cells.
Acknowledgments
We thank Siavash Kurdistani, Sharmistha Sarkar, Yonathan Lissanu Deribe, Ian R. Watson and Lawrence Kwong for useful discussions or comments on the manuscript. We thank Marcus Coyle, Curtis Gumbs, SMF core at MDACC for sequencing support. The work described in this article was supported by grants from the NIH (5U01 CA141508 and U01 CA168394 to L. C.; R01ES024995 and U01 HG007912 to J. E.; CA016672 to SMF Core; GM110174 and CA196539 to B.G.), NCI (1K99CA160578 and R00CA160578 to K. R.), Cancer Prevention and Research Institute of Texas (R1204 to L.C.), National Science Foundation (1254200 to J. E.) and Center for Cancer Epigenetics at MDACC (K.R.). The following fellowship support is acknowledged: K. R. (Charles A. King postdoctoral fellowship), J.E. (Alfred P. Sloan fellowship), and P. F. (California Institute for Regenerative Medicine Training Grant TG2-01169, Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research at UCLA Training Program).
Footnotes
Conflict of Interest Statement: Authors declare no conflict of interest.
Accession: Please refer to GEO accession number GSE58953 for datasets in this manuscript.
Author Contributions: Conceptualization, L.C., J.E., K.R., K.C.A., P.F.; Methodology, A. P., S.S., C.S.C., I.A., P.F., K.C.A., K.R., J.E. ; Investigation P.F., K.C.A., K.R., J.E., J.P.M., E.Z.K., N.S.S., S.S., C.A.N., C.J.T., M.M., S.B.A., J.M.L., M.D., J.B.A., A.S., H.M., B.A.G.; Writing, J.E., L.C., K.R., K.C.A., and P.F.; Funding Acquisition, L.C., J.E., K.R.; Resources, K.Y.T., M.A.D., J.E., L.C., K.R.; Supervision, L.C., J.E., K.R., M.C.B.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akhtar-Zaidi B, Cowper-Sal-lari R, Corradin O, Saiakhova A, Bartels CF, Balasubramanian D, Myeroff L, Lutterbaugh J, Jarrar A, Kalady MF, et al. Epigenomic enhancer profiling defines a signature of colon cancer. Science. 2012;336:736–739. doi: 10.1126/science.1217277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DC. Genetics of melanoma progression: the rise and fall of cell senescence. Pigment Cell Melanoma Res. 2016;29:122–140. doi: 10.1111/pcmr.12422. [DOI] [PubMed] [Google Scholar]
- Busse A, Keilholz U. Role of TGF-beta in melanoma. Curr Pharm Biotechnol. 2011;12:2165–2175. doi: 10.2174/138920111798808437. [DOI] [PubMed] [Google Scholar]
- Campioni M, Santini D, Tonini G, Murace R, Dragonetti E, Spugnini EP, Baldi A. Role of Apaf-1, a key regulator of apoptosis, in melanoma progression and chemoresistance. Exp Dermatol. 2005;14:811–818. doi: 10.1111/j.1600-0625.2005.00360.x. [DOI] [PubMed] [Google Scholar]
- Caunt CJ, Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. The FEBS journal. 2013;280:489–504. doi: 10.1111/j.1742-4658.2012.08716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer cell. 2013;24:777–790. doi: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudnovsky Y, Adams AE, Robbins PB, Lin Q, Khavari PA. Use of human tissue to assess the oncogenic activity of melanoma-associated mutations. Nature genetics. 2005;37:745–749. doi: 10.1038/ng1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encode_Project_Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nature biotechnology. 2010;28:817–825. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nature methods. 2012;9:215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber M, Yosef N, Goren A, Raychowdhury R, Thielke A, Guttman M, Robinson J, Minie B, Chevrier N, Itzhaki Z, et al. A high-throughput chromatin immunoprecipitation approach reveals principles of dynamic gene regulation in mammals. Molecular cell. 2012;47:810–822. doi: 10.1016/j.molcel.2012.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–122. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakowlew SB. Transforming growth factor-beta in cancer and metastasis. Cancer metastasis reviews. 2006;25:435–457. doi: 10.1007/s10555-006-9006-2. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nature genetics. 2012;44:1006–1014. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratani M, Deng N, Ooi WF, Lin SJ, Xing M, Xu C, Qamra A, Tay ST, Malik S, Wu J, et al. Nanoscale chromatin profiling of gastric adenocarcinoma reveals cancer-associated cryptic promoters and somatically acquired regulatory elements. Nature communications. 2014;5:4361. doi: 10.1038/ncomms5361. [DOI] [PubMed] [Google Scholar]
- Paluncic J, Kovacevic Z, Jansson PJ, Kalinowski D, Merlot AM, Huang ML, Lok HC, Sahni S, Lane DJ, Richardson DR. Roads to melanoma: Key pathways and emerging players in melanoma progression and oncogenic signaling. Biochimica et biophysica acta. 2016;1863:770–784. doi: 10.1016/j.bbamcr.2016.01.025. [DOI] [PubMed] [Google Scholar]
- Pinon P, Wehrle-Haller B. Integrins: versatile receptors controlling melanocyte adhesion, migration and proliferation. Pigment Cell Melanoma Res. 2011;24:282–294. doi: 10.1111/j.1755-148X.2010.00806.x. [DOI] [PubMed] [Google Scholar]
- Rai K, Akdemir KC, Kwong LN, Fiziev P, Wu CJ, Keung EZ, Sharma S, Samant NS, Williams M, Axelrad JB, et al. Dual Roles of RNF2 in Melanoma Progression. Cancer discovery. 2015 doi: 10.1158/2159-8290.CD-15-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram O, Goren A, Amit I, Shoresh N, Yosef N, Ernst J, Kellis M, Gymrek M, Issner R, Coyne M, et al. Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell. 2011;147:1628–1639. doi: 10.1016/j.cell.2011.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammons MA, Zhu J, Berger SL. A Chromatin-Focused siRNA Screen for Regulators of p53-Dependent Transcription. G3 (Bethesda) 2016;6:2671–2678. doi: 10.1534/g3.116.031534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, Tsao HW, Godec J, LaFleur MW, Brown FD, et al. The epigenetic landscape of T cell exhaustion. Science. 2016;354:1165–1169. doi: 10.1126/science.aae0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan C, Brumatti G, Martin SJ. Oncogenic B-RafV600E inhibits apoptosis and promotes ERK-dependent inactivation of Bad and Bim. The Journal of biological chemistry. 2008;283:22128–22135. doi: 10.1074/jbc.M800271200. [DOI] [PubMed] [Google Scholar]
- Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nature reviews Molecular cell biology. 2013;14:197–210. [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Sullivan RJ, Flaherty K. MAP kinase signaling and inhibition in melanoma. Oncogene. 2013;32:2373–2379. doi: 10.1038/onc.2012.345. [DOI] [PubMed] [Google Scholar]
- Sun W, Poschmann J, Cruz-Herrera Del Rosario R, Parikshak NN, Hajan HS, Kumar V, Ramasamy R, Belgard TG, Elanggovan B, Wong CC, et al. Histone Acetylome-wide Association Study of Autism Spectrum Disorder. Cell. 2016;167:1385–1397 e1311. doi: 10.1016/j.cell.2016.10.031. [DOI] [PubMed] [Google Scholar]
- Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trikha M, Timar J, Lundy SK, Szekeres K, Cai Y, Porter AT, Honn KV. The high affinity alphaIIb beta3 integrin is involved in invasion of human melanoma cells. Cancer research. 1997;57:2522–2528. [PubMed] [Google Scholar]
- Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic acids research. 2010;38:e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.