

Abstract
The PKR-like ER kinase (PERK), a transmembrane protein, resides in the endoplasmic reticulum (ER). Its activation serves as a key sensor of ER stress, which has been implicated in traumatic brain injury (TBI). The loss of memory is one of the most common symptoms after TBI, but the precise role of PERK activation in memory impairment after TBI has not been well elucidated. Here, we have shown that blocking the activation of PERK using GSK2656157 prevents the loss of dendritic spines and rescues memory deficits after TBI. To elucidate the molecular mechanism, we found that activated PERK phosphorylates CAMP response element binding protein (CREB) and PSD95 directly at the S129 and T19 residues, respectively. Phosphorylation of CREB protein prevents its interaction with a coactivator, CREB-binding protein, and subsequently reduces the BDNF level after TBI. Conversely, phosphorylation of PSD95 leads to its downregulation in pericontusional cortex after TBI in male mice. Treatment with either GSK2656157 or overexpression of a kinase-dead mutant of PERK (PERK-K618A) rescues BDNF and PSD95 levels in the pericontusional cortex by reducing phosphorylation of CREB and PSD95 proteins after TBI. Similarly, administration of either GSK2656157 or overexpression of PERK-K618A in primary neurons rescues the loss of dendritic outgrowth and number of synapses after treatment with a PERK activator, tunicamycin. Therefore, our study suggests that inhibition of PERK phosphorylation could be a potential therapeutic target to restore memory deficits after TBI.
SIGNIFICANCE STATEMENT Traumatic brain injury (TBI) is the leading cause of death and disability around the world and affects 1.7 million Americans each year. Here, we have shown that TBI-activated PKR-like ER kinase (PERK) is responsible for memory deficiency, which is the most common problem in TBI patients. A majority of PERK's biological activities have been attributed to its function as an eIF2α kinase. However, our study suggests that activated PERK mediates its function via increasing phosphorylation of CAMP response element binding protein (CREB) and PSD95 after TBI. Blocking PERK phosphorylation rescues spine loss and memory deficits independently of phosphorylation of eIF2α. Therefore, our study suggests that CREB and PSD95 are novel substrates of PERK, so inhibition of PERK phosphorylation using GSK2656157 would be beneficial against memory impairment after TBI.
Keywords: CREB, GSK3B, PERK, PSD95, TBI
Introduction
Traumatic brain injury (TBI) is a major cause of morbidity and mortality and affects >1.7 million people throughout the United States annually (Nortje and Menon, 2004; Rondina et al., 2005). TBI is a contributing factor to a ∼1/3 (30.5%) of all injury-related deaths in the United States. Endoplasmic reticulum (ER) stress has recently received increased attention in the pathogenesis of various neurodegenerative disorders including TBI (Larner et al., 2006; Truettner et al., 2007; Begum et al., 2013; Begum et al., 2014; Logsdon et al., 2014).
Protein kinase-like endoplasmic reticulum kinase (PERK) is an eIF2α kinase and transmembrane protein resident in the ER membrane that couples ER stress signals to translation inhibition (Marciniak et al., 2006; Bánhegyi et al., 2007; Saito et al., 2011; Pereira et al., 2013; Liu et al., 2015). ER stress increases the activity of PERK, which then phosphorylates eIF2α to promote reduced translation (Vattem and Wek, 2004; Wek and Cavener, 2007; Saito et al., 2011; Liu et al., 2015). PERK is one of at least four eIF2α protein kinases that include the heme-regulated kinase (HRI), interferon-inducible RNA-dependent protein kinase (PKR), and GCN2. Among these, only PERK function is required for the cellular response to ER stress (Saito et al., 2011; Liu et al., 2015). Phosphorylation of PERK at Thr980 within the kinase activation loop is essential for the autocatalytic activity of the kinase (Harding et al., 1999). Treatment with GSK2656157 has been shown to inhibit phosphorylation of PERK protein both in vitro and in vivo (Moreno et al., 2013; Krishnamoorthy et al., 2014).
CAMP response element binding protein (CREB) is a transcription factor that binds the cAMP response element and activates the transcription of several memory-related genes, including BDNF. Activation of CREB is dependent upon the phosphorylation of serine 133 and interaction with a transcriptional coactivator CREB-binding protein (CBP) (Ginty et al., 1994; Bonni et al., 1995; Finkbeiner et al., 1997). However, phosphorylation of CREB at S129 leads to an attenuation of its transcriptional activity, although the mechanism has not been well elucidated (Wang et al., 2010). PSD-95 is an abundant scaffold protein in the postsynaptic density (PSD) of excitatory synapses (Kim and Sheng, 2004; Cheng et al., 2006). Activation of GSK3β phosphorylates PSD95 at threonine 19, which leads to destabilization of PSD-95 in spines (Nelson et al., 2013).
In the present study, we have shown that an increase in phosphorylation of PERK inactivates CREB and causes the loss of protein level of PSD95 that collectively contributes to the memory impairment after TBI. Manipulation of PERK phosphorylation mitigates synapse loss and improves memory functions by rescuing phosphorylation levels of CREB and PSD95 both in vivo and in vitro.
Materials and Methods
Drugs.
GSK2656157 was purchased from Calbiochem and was dissolved in DMSO. DMSO was delivered by intraperitoneal injection to sham and TBI groups of mice. GSK2656157 (50 mg/kg) was delivered by intraperitoneal injection to sham and TBI groups at 30 min after TBI, followed by a daily dose for 24 d. Because GSK2656157 was dissolved in DMSO, only sham and TBI mice received DMSO as controls. For the experiment on dose dependence, we administered GSK2656157 at various doses such as 10, 20, and 50 mg/kg at 30 min after TBI, followed by a daily dose for 24 d. Mice were treated with lithium (5 mg/kg) for 24 d with or without administration of GSK2656157 (50 mg/kg) after either sham or TBI procedures (Fig. 1). Male mice were used throughout all of the experiments. Biochemical analysis was done on the day 25 (Fig. 1A). In other experiments, mice were trained on the Morris water maze (MWM) test for 6 d after completion of GSK2656157 treatment and the probe trial was done on the day 31 of TBI (Fig. 1B). The same sets of mice were used to monitor the synaptic density of TBI or sham mice on day 31 after TBI. In Figure 1C, either PERK or PERK-K618A was overexpressed in mice brain 7 d before TBI. Then biochemical analysis was performed on day 25 after TBI. The procedure for TBI and overexpression of PERK/PERK-K618A are described in the following section.
Figure 1.

Experimental design for TBI mice after with or without treatment with GSK2656157. A, B, mice were treated with GSK2656157 for 24 d after TBI and then biochemical analysis was done on day 25. In other experiments, the mice were trained for the MWM test for 6 d after completion of GSK2656157 treatment and the probe trial was done on day 31 after TBI. The same sets of mice were used to monitor the synaptic density of TBI or sham mice on day 31 after TBI. C, Either PERK or PERK-K618A was overexpressed in the brain 7 d before TBI. Biochemical analysis was performed on day 25 after TBI. The procedure for TBI and overexpression of PERK/PERK-K618A were described in the Materials and Methods.
TBI procedures.
The Committee on Animal Use for Research and Education at the Augusta University approved all animal studies, in compliance with National Institutes of Health guidelines. The procedure was done based on our previously published protocol (Farook et al., 2013; Kapoor et al., 2013; Sen and Sen, 2016). Briefly, adult male C57BL/6 (Jackson Laboratory) mice were anesthetized with xylazine (8 mg/kg)/ketamine (60 mg/kg) and subjected to a sham injury or controlled cortical impact. Briefly, mice were placed in a stereotaxic frame (Ambient Instruments) and a 3.5 mm craniotomy was made in the right parietal bone midway between bregma and lambda with the medial edge lateral to the midline, leaving the dura intact. Mice were impacted at 4.5 m/s with a 20 ms dwell time and 1.2 mm depression using a 3-mm-diameter convex tip, mimicking a moderate TBI. Sham-operated mice underwent the identical surgical procedures but were not impacted. The incision was closed with VetBond and mice were allowed to recover. Body temperature was maintained at 37°C using a small animal temperature controller throughout all procedures (Kopf Instruments).
MWM test.
In the MWM test, the hidden platform procedure was performed in a circular tank filled with opaque water as described previously with minor modifications (Vorhees and Williams, 2006; Mir et al., 2014; Sen and Sen, 2016). Both sham and TBI-mice received GSK2656157 for 24 d after TBI. After completion of treatments, mice underwent training for 6 d. For training, mice were placed in the tank at four random points and allowed to search and find the platform. In the event the mouse did not find the platform within 60 s, it was placed manually on the platform for an extra 30 s. During the training, the mouse was allowed to search for the platform for the 60 s. Two training sessions were given every day and the latency after 2 training sessions was recorded for each day for 6 d. Altogether, 30 d (24 + 6) were used for treatments and training. Twenty-four hours after the last training session on day 30, a probe trial was performed on day 31 after TBI or sham. The mice were allowed to swim in the tank for the 60 s without the platform and performance was assessed on the basis of the time spent in the quadrant in which the hidden platform was originally located (the right quadrant).The mouse speed, time for thigmotaxis, and time in each quadrant were analyzed using the data generated by Any-maze software.
Neurite outgrowth.
The dendritic length assay was performed as described previously with modifications (Sen and Snyder, 2011). Briefly, primary neurons were transfected with GFP and images of neuronal morphologies were captured based on immunoreactivities against GFP using the 510 META confocal laser-scanning microscope (LSM) system (Zeiss) after treatment with either GSK2656157 (5 mg/ml) or GSK2656157 (200 μg/ml) + tunicamycin (3 μg/ml). Dendrites and axons were identified by standard morphological criteria. Because the majority of neurons in our cortical culture preparation possessed only one clearly classifiable axon and one or more dendrites, neurons with nonpyramidal morphological features (such as multiple axons or no classifiable processes) were excluded from analyses. The total length was determined manually using the Neuron J1.0.0 plug-in software for ImageJ. Representative images were acquired using the 510 META confocal LSM with a 40× objective. All analyses were performed by an observer blinded to the identity of the transfected constructs.
Cortical primary neuronal culture and treatment.
The preparation of the primary cortical neurons from wild-type mice was performed as described previously (Sen et al., 2008; Sen et al., 2009; Sen et al., 2012; Mir et al., 2014). Pregnant mice at embryonic day 18.5 gestational age based on the vaginal plug determination method were anesthetized with xylazine (8 mg/kg)/ketamine (60 mg/kg) and cleaned using 70% ethanol. The uterus was opened and pups were placed in sterile prechilled HBSS. Each pup was mounted on the stereomicroscope and cortices were dissected out. After enzyme-based digestion, cortical neurons were counted and plated on the sterilized cover glass or six-well plates in serum-free Neurobasal medium supplemented with appropriate growth supplements (B27 and N2; Life Technologies). The medium was changed very 3–4 d. For biochemical purposes, neurons were washed and scraped off and collected in lysis buffer with protease inhibitors. The protein content was estimated using the Bradford method. Primary neurons were treated with either tunicamycin (3 μg/ml) and/or GSK2656157 (200 μg/ml) for 24 h in subsequent experiments. All experiments were performed with 10- to 12-d-old cultures, at which time the cultures contained ∼95–98% neurons and 2–5% astrocytes.
Transfection experiments were performed using the NeuroPORTER transfection kit (Sigma-Aldrich) following a previously published protocol with modifications (Di Giovanni et al., 2005; Uchida et al., 2006; Chi and Nicol, 2007; Andres et al., 2011). The cells were rinsed once with Opti-MEM medium and incubated at 37°C for ∼30 min. The Neuroporter–DNA complex (100 nm) was added on day 3 in culture and then the neurons were exposed to either Myc-PERK or Myc-PERK-K618A construct or Neuroporter alone for 48 h at 37°C. After 2 d (day 5 in culture), the Neuroporter ± DNA constructs were washed out and the normal medium containing antibiotics and NGF was then added to the neurons and allowed to incubate for another 2 d before Western blots were performed.
Overexpression of constructs in mouse brain.
Plasmid/polyethyleneimine (PEI) complexes including either PERK or PERK-K618A were injected into the cortex as described previously (Jouvert et al., 2004; Wu et al., 2004; Schaffer et al., 2010; Uchida et al., 2010). A vector containing either Myc-PERK or Myc-PERK-K618A (7.5 μg; Addgene) were complexed with 15 nmol of linear 22 kDa PEI in 5% glucose solution. Mixtures were allowed to equilibrate for 15 min at room temperature and then injected (0.5 μg per injection) into the cortex before TBI. Next, 3.5 μl of PEI-plasmid complexes was injected using a 10 μl Hamilton syringe. After a wait of 5 min, the needle was slowly withdrawn.
Western blot and coimmunoprecipitation.
Whole tissue lysates were prepared from 3 mm coronal sections centered upon the site of impact. A 1 mm micropunch was collected from the pericontusional cortex or from the corresponding pericontusional hemisphere as described previously (Farook et al., 2013; Kapoor et al., 2013; Mir et al., 2014; Sen and Sen, 2016). Tissue was placed in complete RIPA buffer, sonicated, and centrifuged for 120 min at 124,000 × g at 4°C. Thirty micrograms of protein were resolved on a 4–20% SDS-polyacrylamide gel and transferred onto a polyvinylidene difluoride membrane. Blots were incubated overnight at 4°C in primary antibody phosphorylated-PERK (Thr980; Cell Signaling Technology), phosphorylated (phospho-) PSD95 (T19; Abcam), phospho-CREB (S129; Abcam), phospho-eIF2α (S51; Abcam), PSD95 (Abcam), CREB (Cell Signaling Technology), CBP (Santa Cruz Biotechnology), Actin (cell Signaling) and BDNF (Cell Signaling Technology), followed by a 2 h incubation with an Alexa Fluor-tagged secondary antibody at room temperature. Blots were visualized using a Li-Cor Odyssey near-infrared imaging system and densitometry analysis was performed using Quantity One software (Bio-Rad) (Farook et al., 2013; Kapoor et al., 2013; Mir et al., 2014). The intensity of each band was determined by ImageJ software and the changes in the experimental band was represented as the fold change as described previously (Sen et al., 2012) (Sen et al., 2008).
Protein–protein interactions were measured by coimmunoprecipitation assay per our method (Sen et al., 2008; Farook et al., 2013; Sen and Sen, 2016). Briefly, treated or untreated cells were lysed in lysis buffer containing 50 mm Tris, pH 7.4, 150 mm NaCl, 0.1% CHAPS, 100 μm deferoxamine, and 1 mm EDTA and homogenized by passage through a 26-gauge needle. Crude lysates were cleared of insoluble debris by centrifugation at 14,000 × g. Immunoprecipitation buffer containing 50 mm Tris, pH 7.4, 150 mm NaCl, 0.1% CHAPS, 100 μm deferoxamine, 1 mm EDTA, and 0.1 mg/ml BSA was added to 100 μg of cell lysates to bring samples to a total volume of 1000 μl. Cell lysates were incubated with either anti-CREB/CREB (S129) or anti-IgG antibody and 30 μl of protein G agarose was added and incubated on a rotator at 4°C. The protein G agarose was washed four times with lysis buffer and quenched with 30 μl of SDS sample buffer. Coimmunoprecipitates were resolved by SDS-PAGE and analyzed by Western blotting with anti-CBP antibodies.
Immunohistochemistry.
Deeply anesthetized mice were perfused with saline, followed by fixation with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. Brains were postfixed overnight in paraformaldehyde, followed by cryoprotection with 30% sucrose, pH 7.4, until brains permeated. Serial coronal sections were prepared using a cryostat microtome (Leica) and mounted directly onto glass slides. Sections were incubated at room temperature with 100% normal donkey serum in PBS containing 0.4% Triton X-100 for 12 h, followed by incubation with the primary antibody against phospho-PERK (Thr980), phospho-CREB (S129), phospho-PSD95 (T19), Myc-PERK, and Myc-PERK-K618A overnight at 4°C. Sections were then washed and incubated with the appropriate Alexa Fluor-tagged secondary antibody. The omission of primary antibody served as a negative control (Sen and Snyder, 2011; Farook et al., 2013; Kapoor et al., 2013; Mir et al., 2014; Sen and Sen, 2016).
Confocal microscopy.
Immunofluorescence was determined using an LSM 710 Meta confocal laser microscope (Zeiss), as described previously. Cellular colocalization was determined in a z-stack mode using a 63× oil-immersion Neofluor objective (numerical aperture 12.3) with the image size set at 512 × 512 pixels. The following excitation lasers/emission filters settings were used for various chromophores: an argon2 laser was used for Alexa Fluor 488, with excitation maxima at 490 nm and emission in the range of 505–530 nm. An HeNe1 laser was used for Alexa Fluor 594 with excitation maxima at 543 nm and emission in the range 568–615 nm. z-stacks (20 optical slices) were collected at optimal pinhole diameter at 12-bit pixel depth and converted into 3D projection images using LSM 510 Meta imaging software (Zeiss) (Sen et al., 2012; Kapoor et al., 2013; Sen and Sen, 2016).
DiI stain and spine evaluation.
Mice were deeply anesthetized with isoflurane and perfused transcardially with 0.9% saline solution. The brains were removed and stained using the Dil method as described previously (Stranahan et al., 2007; Erion et al., 2014). Coronal sections of 200 μm thickness from the cortex were obtained using a vibratome. These sections were collected on positive fixed microscope slides and dehydrated spontaneously. Spines were counted after image acquisition on secondary and tertiary branches of dendrites of neurons with or without treatment with GSK2656157 after either sham injury or TBI. Spine number was measured by ImageJ and analyzed by investigators who were blinded to protocol. For analysis of dendritic spine morphology, image stacks were filtered and analyzed using Zeiss LSM 710 software. First, spines were identified based on the presence of a clearly resolved head and neck. For analysis of spine length, the full extent of the tracing was determined manually from the point of contact with the dendritic shaft to the tip of the spine head.
In vitro kinase assay.
Recombinant PERK protein (2 μg; Thermo Fisher Scientific) was incubated with either recombinant CREB (1.2 μg; Novus Biologicals) or recombinant PSD95 protein (1.2 μg; Novus Biologicals) in a kinase buffer (CST) containing 25 mm Tris-HCl, pH 7.5, 5 mm b-glycerophosphate, 2 mm dithiothreitol (DTT), 0.1 mm Na3VO4, and 10 mm MgCl2 supplemented with ATP (200 μm) at 37°C for 1 h. Then, 40 μl of the total reaction mixture was run on an SDS-PAGE gel. The phosphorylation level of CREB (S129) and PSD95 (S9) was monitored by Western blot hybridization using the specific antibodies mentioned above.
Experimental design and statistical analysis.
All data are presented as the mean ± SEM. The effects of treatments were analyzed using a one-way ANOVA followed by Dunnett's post hoc test. Results are expressed as mean ± SEM (n = 5–7). A p-value <0.05 was considered to be statistically significant. Two-way repeated-measures ANOVAs with LSD post hoc tests were used to determine statistical significance between the TBI and TBI + GSK2656157 groups for all training days during MWM tests (n = 7). One-way ANOVAs with the appropriate LSD post hoc tests were used to compare experimental groups. For all analyses, p < 0.05 was considered significant. Male C57BL/6 mice were used for all experiments.
Results
Treatment with GSK2656157 prevents PERK phosphorylation but not phosphorylation of eIF2α after TBI
PERK is an ER kinase and, upon induction of either the unfolded protein response (UPR) or ER stress, PERK is activated by increasing its phosphorylation status (Harding et al., 2000; Zhang et al., 2002; Wek and Cavener, 2007; Saito et al., 2011). Activated PERK phosphorylates eIF2α, which is known to inhibit protein synthesis. To determine whether TBI has any influence on phosphorylation of PERK and/or eIF2α, we measured their levels in the pericontusional cortex. We found that TBI leads to an increase in the phosphorylation level of both PERK (Thr980) and eIF2α (S51) to ∼3.5- and 2.6-fold, respectively (Fig. 2A). We also tested the activation of ATF6 and IRE1α after TBI. We found that the cleaved level of ATF6 was not increased after TBI, which suggests that that ATF6 pathway was not activated after TBI (Fig. 2B). IRE1α is a Ser/Thr protein kinase that possesses endonuclease activity. ER stress can activate IRE1α by inducing its phosphorylation level. We found that the phosphorylation level of IRE1α was not altered after TBI (Fig. 2C). These data suggest that TBI does not induce the activation of IRE1α in our TBI experimental conditions.
Figure 2.

Treatment with GSK2656157 reduces PERK phosphorylation. A, Western blot analysis of the expression of PERK and eIF2α phosphorylation in pericontusional cortex after TBI. *p < 0.05, n = 9, one-way ANOVA, mean ± SEM. B, Western blot analysis to measure uncleaved and cleaved ATF6 after sham or TBI. C, Western blot analysis to measure phosphorylation level of IRE1α after TBI. D, Western blot analysis of phosphorylation of both PERK and eIF2α after treatment with GSK2656157 at 10, 20, and 50 mg/kg after TBI. *p < 0.05, n = 5, one-way ANOVA, mean ± SEM. E, Western blot analysis of eIF2α phosphorylation after overexpression of either PERK or PERK-K618A in the brain before TBI. The overexpression of PERK was monitored by Western blot hybridization. F, Confocal microscopic analysis to measure PERK phosphorylation with or without GSK2656157 treatment after TBI. G, Primary neurons were treated with tunicamycin (3 μg/ml) with or without GSK2656157 (15 μg/ml) treatment for 6 h and phosphorylation of PERK and eIF2α were monitored by Western blot hybridization. *p < 0.05, n = 9, one-way ANOVA, mean ± SEM.
We were interested to determine whether treatment with an inhibitor of PERK phosphorylation, GSK2656157, has any influence on the phosphorylation of both PERK and eIF2α. We treated wild-type mice with GSK2656157 in a dose-dependent manner (10–50 mg/kg) after TBI and phosphorylation of PERK and eIF2α was measured through Western blot analysis in the pericontusional cortex. Interestingly, we found that treatment with GSK2656157 caused a decrease in PERK phosphorylation in a dose-dependent manner; however, the phosphorylation level of eIF2α remains unaltered throughout the treatment (Fig. 2F). We also found that treatment with 50 mg/kg GK2656157 was enough to fully block the induction of phosphorylation of PERK after TBI (Fig. 2D); however, the phosphorylation level of eIF2α remained unaltered. Consistent with treatment with GSK2656157, we found that overexpression of the kinase-dead construct of PERK did not reduce the phosphorylation level of eIF2α (Fig. 2E). To further confirm the alteration of PERK phosphorylation after GSK2656157 treatment, we performed confocal microscopy analysis. Consistent with our Western blot data, we found that treatment with GSK2656157 (50 mg/kg) reduced PERK phosphorylation significantly (Fig. 2F).
In another set of experiments, primary neurons were treated with an ER stress inducer, tunicamycin, in the presence or absence of GSK2656157. We found that treatment with tunicamycin caused an increase in both the PERK and eIF2α phosphorylation level; however, the induction of PERK phosphorylation was abolished in the presence of GSK2656157 and the phosphorylation level of eIF2α remained unaltered (Fig. 2G).
Treatment with GSK2656157 restores spine density and memory deficits after TBI
Because treatment with GSK2656157 was shown to prevent PERK phosphorylation independently of eIF2α phosphorylation in pericontusional cortex after TBI, we were interested in determining whether it has any effect on spine density and performance in memory functions. Dendritic spines were visualized in slices from GSK2656157- and vehicle-treated wild-type mice using the lipophilic membrane tracer DiI. After Dil staining, TBI mice had fewer dendritic spines than wild-type mice. Moreover, GSK2656157 treatment normalized dendritic spine density (Fig. 3A,B) along the spine length (Fig. 3A,C) and spine surface area (Fig. 3A,D) after TBI.
Figure 3.
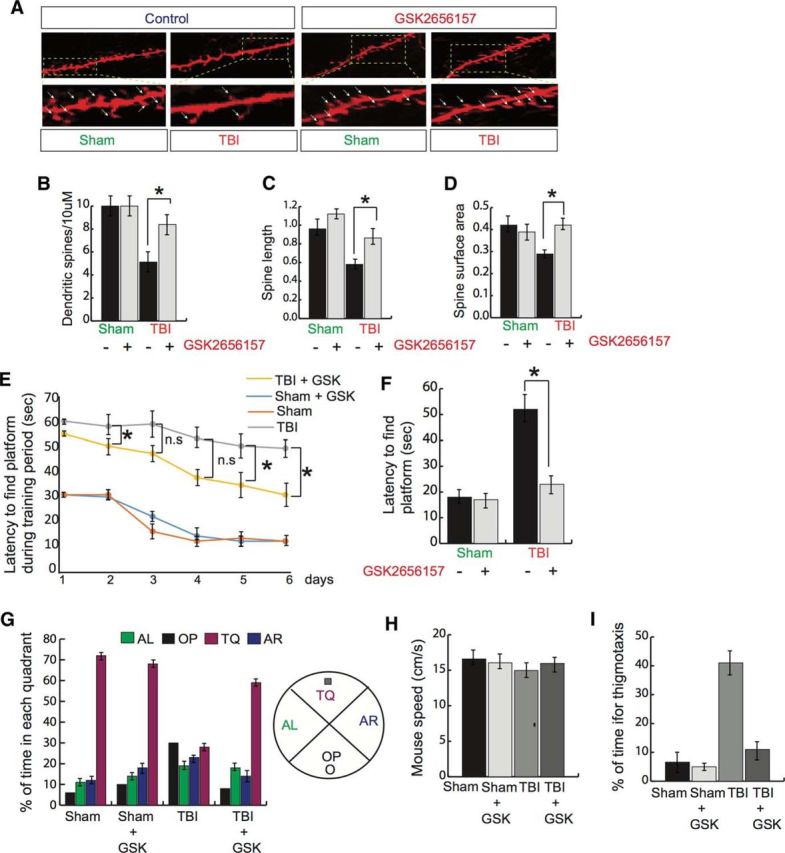
Effect of GSK2656157 on spine density and memory functions after TBI. A–D, Dendritic spines/10 μm (A, B), spine length (A, C), and spine surface area (A, D) were measured after treatment with GSK2656157 (50 mg/kg) after TBI. *p < 0.05, n = 5, one-way ANOVA, mean ± SEM. E, F, Latency to find the platform during training period of 6 d at the end of 2 trials in a day (E). *p < 0.05, n = 7, two-way ANOVA between TBI and TBI + GSK2656157 groups for all days during training (mean ± SEM) and the day of the probe trial (F). G, Percentage of time in each quadrant was measured for the sham-, sham + GSK2656157-, TBI-, and TBI + GSK2656157-treated groups. The quadrant with the platform was designated as TQ and the quadrant from which the mice started their swimming was designated as OP for “opposite.” The quadrant on the left side of OP was designated as AL for “adjacent left” and the quadrant on the right side of OP was designated as AR for “adjacent right.” *p < 0.05, n = 5–6, two-way ANOVA, mean ± SEM. H, Mouse speed was monitored in the sham-, sham + GSK2656157-, TBI-, and TBI + GSK2656157-treated groups. I, Percentage of time for thigmotaxis was monitored in the sham-, sham + GSK2656157-, TBI-, and TBI + GSK2656157-treated groups. *p < 0.05, n = 10–12, one-way ANOVA, mean ± SEM.
To monitor whether treatment with GSK2656157 has any influence on memory impairment after TBI, both the TBI and TBI + GSK2656157 groups, along with sham mice, were subjected to MWM tests after TBI. We found that, on the first day of training, the latency to find the platform for either sham- or sham + GSK2656157-treated mice started at ∼30 s; however, after 6 d of training, they could reach the platform in ∼15 s (Fig. 3E). Compared with sham mice, TBI mice did not improve their memory functions even after 6 d of training. In contrast, after treatment with GSK2656157, TBI mice gradually performed better than TBI mice. The latency for TBI + GSK2656157 on the first day was close to 58 s (vs TBI 60 s). After 6 d of training, the latency to find the platform for TBI + GSK2656157-treated mice was ∼45 s (vs TBI 58 s; Fig. 3E). Interestingly, two-way ANOVA analysis between the TBI and TBI + GSK2656157-treated groups for different time points shows that data are significant only at 2, 5, and 6 d after TBI. On the day of the probe trial, the trend for the latency to find the platform remained unaltered (Fig. 3F). To further confirm how the treatment with GSK2656157 affected memory function, we monitored the percentage of time spent in each quadrant.
We found that either sham or sham + GSK2656157-treated mice spent ∼70% of the time in the quadrant where the platform was located (TQ; target quadrant) compared with TBI mice, which spent ∼30% time in the TQ. Interestingly, treatment with GSK2656157 after TBI rescued the percentage of time in the TQ (Fig. 3G). We also measured mouse speed for sham-, sham + GSK2656157-, TBI-, and TBI + GSK2656157-treated mice. We did not observe any significant difference in mouse speed regardless of group (Fig. 3H). These data suggest that MWM learning impairments are independent of locomotor effects because land-based locomotor reductions did not affect swimming speed. Another important aspect of MWM tests is thigmotaxis, or the tendency to cling or follow the wall around the outer perimeter of the tank, which serves as one sign that the animal is not problem solving. On the day of probe trial, the percentage of time for thigmotaxis in either the sham or sham + GSK2656157 groups was not more than 5–7%; however, the percentage of the time for thigmotaxis of TBI mice was ∼20%. Interestingly, treatment with GSK2656157 reduced the percentage of time for thigmotaxis in TBI mice (Fig. 3I). Together, our data suggest that treatment with GSK2656157 improves learning and memory functions after TBI.
Active PERK inactivates CREB via phosphorylation at S129 after TBI
CREB is a transcription factor known to transcribe several genes, including BDNF, that regulate formation and maintenance of memory (Finkbeiner et al., 1997; Tao et al., 1998; Herold et al., 2011). To determine whether activation of PERK has any influence on CREB phosphorylation, we monitored its level via Western blot hybridization. We found that the phosphorylation level of CREB at serine 129 (S129) was increased significantly, but the phosphorylation level at serine 133 (S133) remained unaltered after TBI (Fig. 4A). To further confirm the alteration of CREB phosphorylation, we performed confocal microscopy analysis in which the phosphorylation CREB at S129 was monitored by green fluorescence intensity. We found that TBI led to an increase in CREB phosphorylation at the S129 residue, as indicated by an increase in green fluorescent intensity (Fig. 4B). Because activation of CREB depends on its interaction with a coactivator, CBP protein, we monitored the interaction between CREB and CBP by coimmunoprecipitation assay. We found that the interaction between CREB and CBP and phospho-CREB at the S129 residue and CBP was decreased significantly (Fig. 4C), along with a decrease in BDNF protein level (Fig. 4D), in pericontusional cortex after TBI.
Figure 4.

Effect of GSK2656157 on CREB phosphorylation and dendritic outgrowth. A, Western blot analysis of CREB phosphorylation at S129 and S133 in pericontusional cortex after TBI. *p < 0.05, n = 5, one-way ANOVA, mean ± SEM. B, Confocal microscopic analysis of CREB phosphorylation (S129). C, Coimmunoprecipitation assay to monitor the interaction between either CREB or phospho-CREB (S129) and CBP after TBI. D, Western blot analysis of BDNF protein level. E, Western blot analysis of the protein level of CREB phosphorylation (S129) and BDNF after GSK2656157 treatment after TBI. F, Confocal microscopic analysis of Myc in cortex after overexpression of either Myc-PERK or Myc-PERK-K618A. Western blot analysis to monitor CREB phosphorylation at S129, BDNF, Actin, and Myc protein levels after TBI. *p < 0.05, n = 8–10, one-way ANOVA, mean ± SEM. G, Western blot analysis to monitor CREB phosphorylation at S129 after treatment with lithium (5 mg/kg) with or without the addition of GSK2656157. *p < 0.05, n = 5, one-way ANOVA, mean ± SEM. H, In vitro kinase assay for CREB phosphorylation by PERK. Western blot analysis to monitor CREB phosphorylation at S129 residue. I, J, Primary neurons were overexpressed with GFP and treated with isoflurane. Neurons were imaged by confocal microscopy to monitor dendritic morphology (I). Total dendritic length was measured after GSK2656157 treatment and cells overexpressed with either PERK or PERK-K618A before treatment with tunicamycin (J). *p < 0.05, n = 8–10, one-way ANOVA, mean ± SEM.
To determine whether treatment with GSK2656157 can prevent CREB phosphorylation at the S129 and BDNF level, mice were treated with GSK2656157 after TBI and the phosphorylation level of CREB was monitored by Western blot hybridization assay. We found that the increase CREB phosphorylation at S129 was decreased significantly in mice treated with GSK2656157 after TBI (Fig. 3E). Similarly, the protein level of BDNF was restored in TBI + GSK2656157-treated mice compared with TBI mice (Fig. 4E). To further confirm whether the alteration of CREB phosphorylation is regulated directly by activation of PERK after TBI, we overexpressed a kinase-dead mutant of PERK (PERK-K618A) through injection in the cortex before TBI. Consistent with the data after treatment with GSK2656157, we found that overexpression of PERK-K618A prevented the induction of CREB phosphorylation at the S129 residue and rescued the protein level of BDNF compared with mice overexpressed with lentiviral particles of control vector construct (Fig. 4F). These data suggest that an increase in phosphorylation of CREB at S129 compromises its interaction with CBP and subsequent transcriptional activity and BDNF level after TBI. The loss of BDNF can be restored by preventing activation of PERK via preventing CREB's phosphorylation at S129 residue.
Previously, it was reported that activation of GSK3β can phosphorylate CREB directly at the S129 residue (Bullock and Habener, 1998; Grimes and Jope, 2001) and TBI can activate GSK3β by reducing the activation of a protein kinase, Akt (Farook et al., 2013). To determine whether induction of CREB phosphorylation at S129 can be blocked by a GSK3β inhibitor, lithium, we treated TBI mice with lithium in the presence or absence of GSK2656157. We found that treatment with lithium had no effect on CREB phosphorylation at S129; however, GSK2656157 could block CREB phosphorylation at S129 regardless of lithium treatment (Fig. 4G). Administration of lithium can block the activation of GSK3β by increasing the level of phosphorylation of GSK3β after TBI. Together, these data suggest that activated PERK can phosphorylate CREB at S129.
To further confirm whether PERK can phosphorylate CREB directly, we incubated recombinant PERK protein along with recombinant CREB protein in the kinase buffer in the presence or absence of ATP. The reaction mixtures ran into the SDS-PAGE gel and phosphorylation of CREB was detected by Western blot hybridization. We found that CREB phosphorylation at S129 was detected only in the sample containing ATP (Fig. 4H).
Because CREB's transcriptional activity and BDNF were decreased due to an increase in PERK phosphorylation, we were interested in determining whether the inhibition of PERK had any effect on dendritic length. Primary neurons were treated with GSK2656157 before exposure of cells to an ER stress inducer, tunicamycin. We found that the total dendritic length was decreased significantly after exposure to tunicamycin; however, treatment with GSK2656157 could rescue the loss of total dendritic length in primary neurons exposed to tunicamycin (Fig. 4I). Our data were further confirmed by overexpression of a kinase-dead mutant of PERK in primary neurons before treatment with tunicamycin. We found that tunicamycin results in the loss of total dendritic length, but cells overexpressed with PERK-K618A rescued the loss of total dendritic length after treatment with tunicamycin (Fig. 4J).
Phosphorylation of PSD95 at T19 by activated PERK causes its downregulation after TBI
PSD95 is a scaffold protein and is known to promote synapse maturation and to exert a major influence on synaptic stability, memory formation in the brain (El-Husseini et al., 2000; Béïque and Andrade, 2003; Kim and Sheng, 2004; Ehrlich et al., 2007). To determine whether TBI has any influence on phosphorylation of PSD95, we monitored the phosphorylation level of PSD95 by Western blot hybridization. We found that TBI leads to an increase in phosphorylation of PSD95 at the T19 residue (Fig. 5A). It was further confirmed by confocal microscopy in which phosphorylation of PSD95 was monitored by green fluorescence intensity (Fig. 5B).
Figure 5.

Effect of GSK2656157 on PSD95 phosphorylation and puncta numbers. A, Western blot analysis of PSD95 phosphorylation at T19 after TBI. *p < 0.05, n = 9, one-way ANOVA, mean ± SEM. B, Confocal microscopic analysis of PSD95 phosphorylation at T19 after TBI. C, Western blot analysis of PSD95 phosphorylation (T19) after treatment with GSK2656157. *p < 0.05, n = 5, one-way ANOVA, mean ± SEM. D, Western blot analysis and quantitative measurement to monitor PSD95 phosphorylation and protein level of PSD95 after overexpression of either PERK wild-type or PERK-K618A in the cortex. *p < 0.05, n = 5, one-way ANOVA, mean ± SEM. E, Western blot analysis to monitor PSD95 phosphorylation at T19 after treatment with lithium (1 mm) with or without the addition of GSK2656157. F, In vitro kinase assay for PSD95 phosphorylation by PERK. Western blot analysis to monitor PSD95 phosphorylation at the T19 residue. G–I, Confocal microscopic analysis of the number (G, H) and size (G, I) of PSD95-positive puncta after tunicamycin treatment with or without GSK2656157. *p < 0.05, n = 9, one-way ANOVA, mean ± SEM. J–L, Confocal microscopic analysis of the number (J, K) and size (J, L) of PSD95-positive puncta in cells overexpressed with either PERK or PERK-K618A mutant after TBI. *p < 0.05, n = 8–10, one-way ANOVA, mean ± SEM. M, Schematic representation showing that TBI leads to an increase in PERK phosphorylation that will subsequently phosphorylate CREB and PSD95 at the S129 and T19 residues, respectively. The increase in CREB phosphorylation causes a downregulation of BDNF level and PSD95 phosphorylation causes its downregulation after TBI. These events lead to spine loss and memory impairment after TBI.
To monitor whether treatment with GSK2656157 has any influence on phosphorylation of PSD95 at the T19 residue, we treated mice with GSK2656157 after TBI. Interestingly, we found that treatment with GSK2656157 markedly reduced the increase in TBI-induced augmentation of PSD95 phosphorylation at the T19 residue (Fig. 5C). To determine whether inhibition of PERK phosphorylation affects the phosphorylation of PSD95 directly, we overexpressed PERK-K618A into the cortex before TBI. we found that mice overexpressed with PERK-K618A prevent the increase in PSD95 phosphorylation at the T19 residue compared with mice overexpressed with the control vector (Fig. 5D). It was shown previously that phosphorylation of PSD95 at the T19 residue affects its stability (Nelson et al., 2013). We were interested in determining whether inhibition of PERK phosphorylation influences the protein level of PSD95 after TBI. We found that TBI leads to a significant decrease in the PSD95 protein level (Fig. 5D). However, overexpression of PERK-K618A rescues the loss of the PSD95 protein level, along with a decrease in phosphorylation of PSD95 at the T19 residue (Fig. 5D). These data suggest that the TBI-induced decrease in PSD95 is dependent on its phosphorylation status at the T19 residue, but this can be rescued by preventing PERK phosphorylation in mice after TBI.
Activation of GSK3β is known to phosphorylate PSD95 at T19. Because TBI leads to an activation of GSK3β, we were interested in determining whether blocking GSK3β activation could rescue PSD95 phosphorylation at the T19 residue. TBI mice were treated with a GSK3β inhibitor, lithium, along with a PERK-kinase inhibitor, GSK2656157. We found that treatment with lithium had no effect on PSD95 phosphorylation; however, preventing PERK phosphorylation by GSK2656157 significantly reduced PSD95 phosphorylation at the T19 residue independently of treatment with lithium (Fig. 5E). Administration of lithium can block the activation of GSK3β by increasing the level of phosphorylation of GSK3β after TBI. Together, these data suggest that activation of PERK is directly responsible for PSD95 phosphorylation at the T19 residue.
To further confirm whether PERK can phosphorylate PSD95 directly, we incubated a recombinant PERK protein along with recombinant PSD95 protein in the kinase buffer in the presence or absence of ATP. The reaction mixtures ran into the SDS-PAGE gel and phosphorylation of PSD95 was monitored by Western blot hybridization using an antibody specific to phospho-PSD95 at the T19 residue. We found that PSD95 phosphorylation at T19 was detected only in the sample containing ATP (Fig. 5F). The phosphorylated and unphosphorylated level of PSD95 was also detected by PSD95 antibody. These data validate the reliability of the antibody specific for phosphorylation of PSD95.
To determine whether activation of PERK has any influence on the number of PSD95-positive puncta, we treated primary neurons with GSK2656157 before exposure to the ER stress activator tunicamycin. We found that exposure to tunicamycin caused a significant decrease in both the number (Fig. 4G,H) and size (Fig. 5G,I) of PSD95-positive puncta; however, treatment with GSK2656157 before tunicamycin prevented the decrease in number (Fig. 5G,H) and size (Fig. 5G,I) of PSD95-positive puncta, along with a decrease in phosphorylation of PSD95 at the T19 residue in cells treated with tunicamycin. Consistent with these data, overexpression of PERK-K618A in primary neurons rescued the tunicamycin-induced decrease n number (Fig. 5J,K) and sixe (Fig. 5J,L) of PSD95-positive puncta. Together, these results suggest that activation of PERK affects the PSD95 level directly by increasing its phosphorylation level.
Discussion
In the present study, we have demonstrated a novel mechanism whereby activation of PERK causes an impairment of memory through inactivation of CREB protein and downregulation of PSD95. An increased level of phosphorylation of CREB at S129 prevents its transcriptional activity and phosphorylation of PSD95 at T19 leads to its downregulation after TBI (Fig. 5M). We have also shown that inhibiting phosphorylation of PERK by using GSK2656157 restores memory deficiency and neurite outgrowth by reducing phosphorylation of CREB and PSD95 without affecting the elevated level of phosphorylation of the initiation factor eIF2α.
PERK is a ubiquitous ER-localized protein kinase and global inactivation of PERK in mice results in multiple developmental defects, including early-onset diabetes, growth retardation, skeletal abnormalities, and pancreatic atrophy (Harding et al., 2001; Zhang et al., 2002). In the brain, PERK functions in the cortex as a physiological constraint of memory consolidation and its downregulation serves as a cognitive enhancement by phosphorylating eIF2α at the S51 residue. In wild-type mice, brain-specific deletion of the PERK protein has been shown to impair cognitive functions and behavioral flexibility (Trinh et al., 2012) and these data are consistent with studies in which either GCN2 KO mice (Costa-Mattioli et al., 2005) or eIF2α-S51A mutant mice (Costa-Mattioli et al., 2007) have shown facilitation of memory. In TBI, treatment with either salubrinal (Rubovitch et al., 2015) or docosahexaenoic acid (Begum et al., 2014; Desai et al., 2014) has been shown to prevent phosphorylation of eIF2α and to act as a neuroprotective agent. Consistent with TBI data, elevated phosphorylation of eIF2α has been observed in the brains of Alzheimer's disease (AD) patients and AD model mice (Chang et al., 2002; O'Connor et al., 2008; Ma et al., 2013). Genetic deletion of PERK prevented enhanced phosphorylation of eIF2α and deficits in protein synthesis, synaptic plasticity, and spatial memory in mice that express familial AD-related mutations in APP and PSEN1.
Here, we have shown that inhibition of PERK phosphorylation does not affect phosphorylation of eIF2a, but does rescue the deficiency of synaptic plasticity significantly. These data suggest that PERK phosphorylation may function independently of the phosphorylation of eIF2a, but it is not exclusively independent of eIF2a. Other studies have suggested that aberrant eIF2α phosphorylation was associated with synaptic pathophysiology and memory dysfunction after AD (Ma et al., 2013). Moreover, UPR-mediated translational failure was shown as a generic pathogenic mechanism in protein-misfolding disorders, including tauopathies (Radford et al., 2015). This suggests that phosphorylation of PERK and eIF2α both contribute to the deterioration of synaptic plasticity, but their individual contribution varies from context to context. Recent studies suggest that treatment with the small-molecule ISRIB, which restores translation downstream of eIF2α, conferred neuroprotection in prion-diseased mice without adverse effects on the pancreas (Halliday et al., 2015). We have used GSK2656157 as a PERK phosphorylation inhibitor to study its role in deficiencies in synaptic plasticity after TBI. We anticipate that treatment with ISRIB will lead to PSD95 reduction and CREB-related dysfunction.
In addition, our data are consistent with the previous finding that treatment with GSK2656157 inhibits PERK activity in the mouse pancreas (Atkins et al., 2013), but does not inhibit downstream PERK signaling such as phospho-eIF2α, ATF, or CHOP protein levels. These data suggest that inhibition of PERK activity by GSK2656157 does not always correlate with inhibition of eIF2α phosphorylation or mimic the biological effects of genetic inactivation of PERK. Moreover, the initiation factor eIF2α can be phosphorylated by several kinases other than PERK, such as GCN2, PKR, and HRI (Ohno, 2014). Therefore, it is possible that preventing phosphorylation of PERK does not necessarily block phosphorylation of eIF2α. At the same time, our data indicate that the phosphorylation of eIF2α could be dispensable to restoring memory deficits after TBI.
Multiple studies have shown that significant loss of synapses in the days after brain injury (Gao and Chen, 2011; Gao et al., 2011; Park and Biederer, 2013) are responsible for memory deficits after TBI. Dendritic spines are the postsynaptic protrusions through which neurons receive most of their excitatory input. Gao et al. (2011) showed in a mouse model that, after TBI, there are fewer dendritic synaptic spines in the cortex and hippocampus (Gao and Chen, 2011; Gao et al., 2011). The formation of synapses and spine density depends on the level of the postsynaptic scaffold protein PSD95 and the BDNF level, which can be transcribed by CREB.
The activation of CREB depends on its phosphorylation level. Compelling evidence has suggested that phosphorylation of CREB at S133 is essential for its transcription; however, studies of phosphorylation of CREB at S129 have reached two opposite conclusions. Fiol et al. (1994) reported that GSK3β facilitated activation of CREB by increasing its phosphorylation status. In contrast, Bullock and Habener (1998) found that phosphorylation of CREB by GSK3β attenuated the DNA-binding activity of CREB. This study was further supported by Grimes and Jope (2001), who showed that GSK3β regulates CREB DNA-binding activity negatively and that lithium and sodium valproate, inhibitors of GSK3β, facilitate CREB activation. Consistent with these studies, we found that PERK phosphorylates CREB directly at the S129 residue without altering its phosphorylation status at S133. To further elucidate the underlying mechanism, we have shown that phosphorylation of CREB at S129 prevents its interaction with a coactivator, CBP, that leads to its transcriptional inactivation and downregulation of BDNF. Conversely, PSD95, a major scaffold protein of PSD that promotes synaptic strength, can be phosphorylated by GSk3β at the T19 residue (Nelson et al., 2013). Phosphorylation of PSD95 affects stability in dendritic spines, which can be reversed by overexpression of the T19A mutant of PSD95.
The question arises whether activation of GSK3β can also contribute to the phosphorylation of both CREB and PSD95 proteins after TBI, particularly considering the fact that TBI leads to an activation of GSK3β by reducing its phosphorylation at the S9 residue (Farook et al., 2013). Here, we have shown that inhibition of GSK3β with lithium cannot prevent the phosphorylation of either CREB or PSD95 in the pericontusional cortex; however, inhibition of PERK phosphorylation abolish the phosphorylation level of CREB and PSD95 after TBI. These data suggest that activation of PERK is directly responsible for the phosphorylation of CREB and PSD95 after TBI, which was further supported by the in vitro phosphorylation assay. Moreover, treatment with a PERK inhibitor blocks CREB and PSD95 phosphorylation without having any effect on eIF2α phosphorylation. These data suggest that eIF2α is not responsible for phosphorylation of CREB and PSD95 after TBI. We have also shown that inhibition of PERK activation restores dendritic spines, spine length, and spine surface area and decreases PSD95 and CREB phosphorylation at the T19 and S129 residues, respectively, after TBI. To our knowledge, our study may provide the first evidence that CREB and PSD95 can be phosphorylated directly by PERK after TBI.
Collectively, these results provide a novel mechanism in which PERK phosphorylation impairs memory by inactivating CREB and downregulation of PSD95 after TBI. Inhibiting PERK phosphorylation rescues spine density and dendritic outgrowth after TBI. Therefore, our study provides a framework on which a PERK phosphorylation inhibitor such as GSK2656157 can be tested as a therapeutic approach to prevent memory impairment in TBI patients.
Footnotes
This work was supported by the National Institutes of Health (Grants RO1NS094516 and RO1EY025622 to N.S. and R.G.). We thank Dr. Alexis Stranahan from Augusta University for DiI staining to study synaptic morphology.
The authors declare no competing financial interests.
References
- Andres RH, Choi R, Pendharkar AV, Gaeta X, Wang N, Nathan JK, Chua JY, Lee SW, Palmer TD, Steinberg GK, Guzman R (2011) The CCR2/CCL2 interaction mediates the transendothelial recruitment of intravascularly delivered neural stem cells to the ischemic brain. Stroke 42:2923–2931. 10.1161/STROKEAHA.110.606368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins C, Liu Q, Minthorn E, Zhang SY, Figueroa DJ, Moss K, Stanley TB, Sanders B, Goetz A, Gaul N, Choudhry AE, Alsaid H, Jucker BM, Axten JM, Kumar R (2013) Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res 73:1993–2002. 10.1158/0008-5472.CAN-12-3109 [DOI] [PubMed] [Google Scholar]
- Bánhegyi G, Baumeister P, Benedetti A, Dong D, Fu Y, Lee AS, Li J, Mao C, Margittai E, Ni M, Paschen W, Piccirella S, Senesi S, Sitia R, Wang M, Yang W (2007) Endoplasmic reticulum stress. Ann N Y Acad Sci 1113:58–71. 10.1196/annals.1391.007 [DOI] [PubMed] [Google Scholar]
- Begum G, Harvey L, Dixon CE, Sun D (2013) ER stress and effects of DHA as an ER stress inhibitor. Transl Stroke Res 4:635–642. 10.1007/s12975-013-0282-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum G, Yan HQ, Li L, Singh A, Dixon CE, Sun D (2014) Docosahexaenoic acid reduces ER stress and abnormal protein accumulation and improves neuronal function following traumatic brain injury. J Neurosci 34:3743–3755. 10.1523/JNEUROSCI.2872-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béïque JC, Andrade R (2003) PSD-95 regulates synaptic transmission and plasticity in rat cerebral cortex. J Physiol 546:859–867. 10.1113/jphysiol.2002.031369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonni A, Ginty DD, Dudek H, Greenberg ME (1995) Serine 133-phosphorylated CREB induces transcription via a cooperative mechanism that may confer specificity to neurotrophin signals. Mol Cell Neurosci 6:168–183. 10.1006/mcne.1995.1015 [DOI] [PubMed] [Google Scholar]
- Bullock BP, Habener JF (1998) Phosphorylation of the cAMP response element binding protein CREB by cAMP-dependent protein kinase A and glycogen synthase kinase-3 alters DNA-binding affinity, conformation, and increases net charge. Biochemistry 37:3795–3809. 10.1021/bi970982t [DOI] [PubMed] [Google Scholar]
- Chang RC, Wong AK, Ng HK, Hugon J (2002) Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer's disease. Neuroreport 13:2429–2432. 10.1097/00001756-200212200-00011 [DOI] [PubMed] [Google Scholar]
- Cheng D, Hoogenraad CC, Rush J, Ramm E, Schlager MA, Duong DM, Xu P, Wijayawardana SR, Hanfelt J, Nakagawa T, Sheng M, Peng J (2006) Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol Cell Proteomics 5:1158–1170. 10.1074/mcp.D500009-MCP200 [DOI] [PubMed] [Google Scholar]
- Chi XX, Nicol GD (2007) Manipulation of the potassium channel Kv1.1 and its effect on neuronal excitability in rat sensory neurons. J Neurophysiol 98:2683–2692. 10.1152/jn.00437.2007 [DOI] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Harding H, Herdy B, Azzi M, Bruno M, Bidinosti M, Ben Mamou C, Marcinkiewicz E, Yoshida M, et al. (2005) Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature 436:1166–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Stern E, Gamache K, Cuello C, Sossin W, Kaufman R, Pelletier J, Rosenblum K, Krnjevic K, Lacaille J-C, Nader K, Sonenberg N (2007) eIF2a phosphorylation regulates the switch from short to long-term synaptic plasticity and memory. Cell 129:195–206. 10.1016/j.cell.2007.01.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai A, Kevala K, Kim HY (2014) Depletion of brain docosahexaenoic acid impairs recovery from traumatic brain injury. PLoS One 9:e86472. 10.1371/journal.pone.0086472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giovanni S, De Biase A, Yakovlev A, Finn T, Beers J, Hoffman EP, Faden AI (2005) In vivo and in vitro characterization of novel neuronal plasticity factors identified following spinal cord injury. J Biol Chem 280:2084–2091. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Klein M, Rumpel S, Malinow R (2007) PSD-95 is required for activity-driven synapse stabilization. Proc Natl Acad Sci U S A 104:4176–4181. 10.1073/pnas.0609307104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS (2000) PSD-95 involvement in maturation of excitatory synapses. Science 290:1364–1368. [PubMed] [Google Scholar]
- Erion JR, Wosiski-Kuhn M, Dey A, Hao S, Davis CL, Pollock NK, Stranahan AM (2014) Obesity elicits interleukin 1-mediated deficits in hippocampal synaptic plasticity. J Neurosci 34:2618–2631. 10.1523/JNEUROSCI.4200-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farook JM, Shields J, Tawfik A, Markand S, Sen T, Smith SB, Brann D, Dhandapani KM, Sen N (2013) GADD34 induces cell death through inactivation of Akt following traumatic brain injury. Cell Death Dis 4:e754. 10.1038/cddis.2013.280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME (1997) CREB: a major mediator of neuronal neurotrophin responses. Neuron 19:1031–1047. 10.1016/S0896-6273(00)80395-5 [DOI] [PubMed] [Google Scholar]
- Fiol CJ, Williams JS, Chou CH, Wang QM, Roach PJ, Andrisani OM (1994) A secondary phosphorylation of CREB341 at Ser129 is required for the cAMP-mediated control of gene expression. A role for glycogen synthase kinase-3 in the control of gene expression. J Biol Chem 269:32187–32193. [PubMed] [Google Scholar]
- Gao X, Chen J (2011) Mild traumatic brain injury results in extensive neuronal degeneration in the cerebral cortex. J Neuropathol Exp Neurol 70:183–191. 10.1097/NEN.0b013e31820c6878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Deng P, Xu ZC, Chen J (2011) Moderate traumatic brain injury causes acute dendritic and synaptic degeneration in the hippocampal dentate gyrus. PLoS One 6:e24566. 10.1371/journal.pone.0024566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginty DD, Bonni A, Greenberg ME (1994) Nerve growth factor activates a Ras-dependent protein kinase that stimulates c-fos transcription via phosphorylation of CREB. Cell 77:713–725. 10.1016/0092-8674(94)90055-8 [DOI] [PubMed] [Google Scholar]
- Grimes CA, Jope RS (2001) CREB DNA binding activity is inhibited by glycogen synthase kinase-3 beta and facilitated by lithium. J Neurochem 78:1219–1232. 10.1046/j.1471-4159.2001.00495.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday M, Radford H, Sekine Y, Moreno J, Verity N, le Quesne J, Ortori CA, Barrett DA, Fromont C, Fischer PM, Harding HP, Ron D, Mallucci GR (2015) Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis 6:e1672. 10.1038/cddis.2015.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397:271–274. 10.1038/16729 [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D (2000) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 5:897–904. 10.1016/S1097-2765(00)80330-5 [DOI] [PubMed] [Google Scholar]
- Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D (2001) Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell 7:1153–1163. 10.1016/S1097-2765(01)00264-7 [DOI] [PubMed] [Google Scholar]
- Herold S, Jagasia R, Merz K, Wassmer K, Lie DC (2011) CREB signalling regulates early survival, neuronal gene expression and morphological development in adult subventricular zone neurogenesis. Mol Cell Neurosci 46:79–88. 10.1016/j.mcn.2010.08.008 [DOI] [PubMed] [Google Scholar]
- Jouvert P, Revel MO, Lazaris A, Aunis D, Langley K, Zwiller J (2004) Activation of the cGMP pathway in dopaminergic structures reduces cocaine-induced EGR-1 expression and locomotor activity. J Neurosci 24:10716–10725. 10.1523/JNEUROSCI.1398-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor S, Kim SM, Farook JM, Mir S, Saha R, Sen N (2013) Foxo3a transcriptionally upregulates AQP4 and induces cerebral edema following traumatic brain injury. J Neurosci 33:17398–17403. 10.1523/JNEUROSCI.2756-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Sheng M (2004) PDZ domain proteins of synapses. Nat Rev Neurosci 5:771–781. 10.1038/nrn1517 [DOI] [PubMed] [Google Scholar]
- Krishnamoorthy J, Rajesh K, Mirzajani F, Kesoglidou P, Papadakis AI, Koromilas AE (2014) Evidence for eIF2alpha phosphorylation-independent effects of GSK2656157, a novel catalytic inhibitor of PERK with clinical implications. Cell Cycle 13:801–806. 10.4161/cc.27726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larner SF, Hayes RL, Wang KK (2006) Unfolded protein response after neurotrauma. J Neurotrauma 23:807–829. 10.1089/neu.2006.23.807 [DOI] [PubMed] [Google Scholar]
- Liu Z, Lv Y, Zhao N, Guan G, Wang J (2015) Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death Dis 6:e1822. 10.1038/cddis.2015.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon AF, Turner RC, Lucke-Wold BP, Robson MJ, Naser ZJ, Smith KE, Matsumoto RR, Huber JD, Rosen CL (2014) Altering endoplasmic reticulum stress in a model of blast-induced traumatic brain injury controls cellular fate and ameliorates neuropsychiatric symptoms. Front Cell Neurosci 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P, Cavener DR, Klann E (2013) Suppression of eIF2alpha kinases alleviates Alzheimer's disease-related plasticity and memory deficits. Nat Neurosci 16:1299–1305. 10.1038/nn.3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak SJ, Garcia-Bonilla L, Hu J, Harding HP, Ron D (2006) Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J Cell Biol 172:201–209. 10.1083/jcb.200508099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir S, Sen T, Sen N (2014) Cytokine-induced GAPDH sulfhydration affects PSD95 degradation and memory. Mol Cell 56:786–795. 10.1016/j.molcel.2014.10.019 [DOI] [PubMed] [Google Scholar]
- Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, Mallucci GR (2013) Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med 5:206ra138. 10.1126/scitranslmed.3006767 [DOI] [PubMed] [Google Scholar]
- Nelson CD, Kim MJ, Hsin H, Chen Y, Sheng M (2013) Phosphorylation of threonine-19 of PSD-95 by GSK-3beta is required for PSD-95 mobilization and long-term depression. J Neurosci 33:12122–12135. 10.1523/JNEUROSCI.0131-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nortje J, Menon DK (2004) Traumatic brain injury: physiology, mechanisms, and outcome. Curr Opin Neurol 17:711–718. 10.1097/00019052-200412000-00011 [DOI] [PubMed] [Google Scholar]
- O'Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA, Lammich S, Lichtenthaler SF, Hébert SS, De Strooper B, Haass C, Bennett DA, Vassar R (2008) Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 60:988–1009. 10.1016/j.neuron.2008.10.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M. (2014) Roles of eIF2α kinases in the pathogenesis of Alzheimer's disease. Front Mol Neurosci 7:22. 10.3389/fnmol.2014.00022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K, Biederer T (2013) Neuronal adhesion and synapse organization in recovery after brain injury. Future neurology 8:555–567. 10.2217/fnl.13.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira CF. (2013) Crosstalk between endoplasmic reticulum stress and protein misfolding in neurodegenerative diseases. ISRN Cell Biology 2013:256404. [Google Scholar]
- Radford H, Moreno JA, Verity N, Halliday M, Mallucci GR (2015) PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol 130:633–642. 10.1007/s00401-015-1487-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondina C, Videtta W, Petroni G, Lujan S, Schoon P, Mori LB, Matkovich J, Carney N, Chesnut R (2005) Mortality and morbidity from moderate to severe traumatic brain injury in Argentina. J Head Trauma Rehabil 20:368–376. 10.1097/00001199-200507000-00008 [DOI] [PubMed] [Google Scholar]
- Rubovitch V, Barak S, Rachmany L, Goldstein RB, Zilberstein Y, Pick CG (2015) The neuroprotective effect of salubrinal in a mouse model of traumatic brain injury. Neuromolecular Med 17:58–70. 10.1007/s12017-015-8340-3 [DOI] [PubMed] [Google Scholar]
- Saito A, Ochiai K, Kondo S, Tsumagari K, Murakami T, Cavener DR, Imaizumi K (2011) Endoplasmic reticulum stress response mediated by the PERK-eIF2(alpha)-ATF4 pathway is involved in osteoblast differentiation induced by BMP2. J Biol Chem 286:4809–4818. 10.1074/jbc.M110.152900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer DJ, Tunc-Ozcan E, Shukla PK, Volenec A, Redei EE (2010) Nuclear orphan receptor Nor-1 contributes to depressive behavior in the Wistar-Kyoto rat model of depression. Brain Res 1362:32–39. 10.1016/j.brainres.2010.09.041 [DOI] [PubMed] [Google Scholar]
- Sen N, Snyder SH (2011) Neurotrophin-mediated degradation of histone methyltransferase by S-nitrosylation cascade regulates neuronal differentiation. Proc Natl Acad Sci U S A 108:20178–20183. 10.1073/pnas.1117820108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Hara MR, Kornberg MD, Cascio MB, Bae BI, Shahani N, Thomas B, Dawson TM, Dawson VL, Snyder SH, Sawa A (2008) Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat Cell Biol 10:866–873. 10.1038/ncb1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Hara MR, Ahmad AS, Cascio MB, Kamiya A, Ehmsen JT, Aggrawal N, Hester L, Doré S, Snyder SH, Sawa A (2009) GOSPEL: a neuroprotective protein that binds to GAPDH upon S-nitrosylation. Neuron 63:81–91. 10.1016/j.neuron.2009.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Paul BD, Gadalla MM, Mustafa AK, Sen T, Xu R, Kim S, Snyder SH (2012) Hydrogen sulfide-linked sulfhydration of NF-kappaB mediates its antiapoptotic actions. Mol Cell 45:13–24. 10.1016/j.molcel.2011.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen T, Sen N (2016) Treatment with an activator of hypoxia-inducible factor 1, DMOG provides neuroprotection after traumatic brain injury. Neuropharmacology 107:79–88. 10.1016/j.neuropharm.2016.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Khalil D, Gould E (2007) Running induces widespread structural alterations in the hippocampus and entorhinal cortex. Hippocampus 17:1017–1022. 10.1002/hipo.20348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME (1998) Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20:709–726. 10.1016/S0896-6273(00)81010-7 [DOI] [PubMed] [Google Scholar]
- Trinh MA, Kaphzan H, Wek RC, Pierre P, Cavener DR, Klann E (2012) Brain-specific disruption of the eIF2alpha kinase PERK decreases ATF4 expression and impairs behavioral flexibility. Cell Rep 1:676–688. 10.1016/j.celrep.2012.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truettner JS, Hu B, Alonso OF, Bramlett HM, Kokame K, Dietrich WD (2007) Subcellular stress response after traumatic brain injury. J Neurotrauma 24:599–612. 10.1089/neu.2006.0186 [DOI] [PubMed] [Google Scholar]
- Uchida M, Enomoto A, Fukuda T, Kurokawa K, Maeda K, Kodama Y, Asai N, Hasegawa T, Shimono Y, Jijiwa M, Ichihara M, Murakumo Y, Takahashi M (2006) Dok-4 regulates GDNF-dependent neurite outgrowth through downstream activation of Rap1 and mitogen-activated protein kinase. J Cell Sci 119:3067–3077. 10.1242/jcs.03043 [DOI] [PubMed] [Google Scholar]
- Uchida S, Hara K, Kobayashi A, Funato H, Hobara T, Otsuki K, Yamagata H, McEwen BS, Watanabe Y (2010) Early life stress enhances behavioral vulnerability to stress through the activation of REST4-mediated gene transcription in the medial prefrontal cortex of rodents. J Neurosci 30:15007–15018. 10.1523/JNEUROSCI.1436-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vattem KM, Wek RC (2004) Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A 101:11269–11274. 10.1073/pnas.0400541101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorhees CV, Williams MT (2006) Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1:848–858. 10.1038/nprot.2006.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Iwasaki M, Ficara F, Lin C, Matheny C, Wong SH, Smith KS, Cleary ML (2010) GSK-3 promotes conditional association of CREB and its coactivators with MEIS1 to facilitate HOX-mediated transcription and oncogenesis. Cancer Cell 17:597–608. 10.1016/j.ccr.2010.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wek RC, Cavener DR (2007) Translational control and the unfolded protein response. Antioxid Redox Signal 9:2357–2371. 10.1089/ars.2007.1764 [DOI] [PubMed] [Google Scholar]
- Wu K, Meyers CA, Bennett JA, King MA, Meyer EM, Hughes JA (2004) Polyethylenimine-mediated NGF gene delivery protects transected septal cholinergic neurons. Brain Res 1008:284–287. 10.1016/j.brainres.2004.02.051 [DOI] [PubMed] [Google Scholar]
- Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, Cavener DR (2002) The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol 22:3864–3874. 10.1128/MCB.22.11.3864-3874.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]