

Abstract
Nonribosomal peptide synthesis involves the interplay between covalent protein modifications, conformational fluctuations, catalysis, and transient protein-protein interactions. Delineating the mechanisms involved in orchestrating these various processes will deepen our understanding of domain-domain communication in nonribosomal peptide synthetases (NRPSs) and lay the groundwork for the rational reengineering of NRPSs by swapping domains handling different substrates to generate novel natural products. Although many structural and biochemical studies of NRPSs exist, few studies have focused on the energetics and dynamics governing the interactions in these systems. Here, we present detailed binding studies of an adenylation domain and its partner carrier protein in apo-, holo-, and substrate-loaded forms. Results from fluorescence anisotropy, isothermal titration calorimetry, and NMR titrations indicated that covalent modifications to a carrier protein modulate domain communication, suggesting that chemical modifications to carrier proteins during NRPS synthesis may impart directionality to sequential NRPS domain interactions. Comparison of the structure and dynamics of an apo-aryl carrier protein with those of its modified forms revealed structural fluctuations induced by post-translational modifications and mediated by modulations of protein dynamics. The results provide a comprehensive molecular description of a carrier protein throughout its life cycle and demonstrate how a network of dynamic residues can propagate the molecular impact of chemical modifications throughout a protein and influence its affinity toward partner domains.
Keywords: antibiotics, nuclear magnetic resonance (NMR), protein dynamic, protein-protein interaction, structural biology, adenylation domain, allostery, aryl carrier protein, nonribosomal peptide synthetase
Introduction
Nonribosomal peptide synthetases (NRPSs)4 are microbial enzymatic systems that produce secondary metabolites with important biological functions and pharmaceutical properties. Examples include antibiotics (vancomycin), immunosupressants (cyclosporin), or anti-cancer agents (epothilone) (1). Other NRPS products promote virulence (enterobactin, yersiniabactin (2)) and represent targets for designing new inhibitors.
NRPSs employ a modular, multidomain architecture and a series of enzymatic reactions to covalently tether simple substrates to so-called carrier proteins (CPs) and assemble them into natural products via peptide bonds (3, 4). During synthesis, CPs are modified from inactive apo-forms (Fig. 1A, shown with an aryl carrier protein (ArCP)) to active holo-forms (Fig. 1B) via attachment of phosphopantetheine (PP) arms. PP then harbors monomeric substrates (Fig. 1C, shown with a salicylate substrate) or metabolite intermediates through a thioester bond. First, a phosphopantetheinyl transferase (PPTase) introduces PP onto a conserved serine. An adenylation (A) domain then catalyzes both substrate adenylation and subsequent thioesterification of the substrate onto PP, with a reorientation of N- and C-terminal subdomains (A(N) and A(C)) between both reactions. Thus, an adenylation conformation is competent for substrate activation (Fig. 1E), and a thioester conformation catalyzes substrate loading (Fig. 1F); an open conformation (Fig. 1D) is probably adopted for initial substrate and adenosine triphosphate (ATP) binding (5–9). Next, a condensation or cyclization domain catalyzes peptide bond formation between the substrate of an upstream CP and that of a downstream CP. Peptide bond formation restores the upstream carrier protein to its holo-form, whereas the downstream CP now harbors an elongated intermediate. A thioesterase domain typically releases the product from the terminal carrier protein, either by hydrolysis or macrocyclization.
Figure 1.
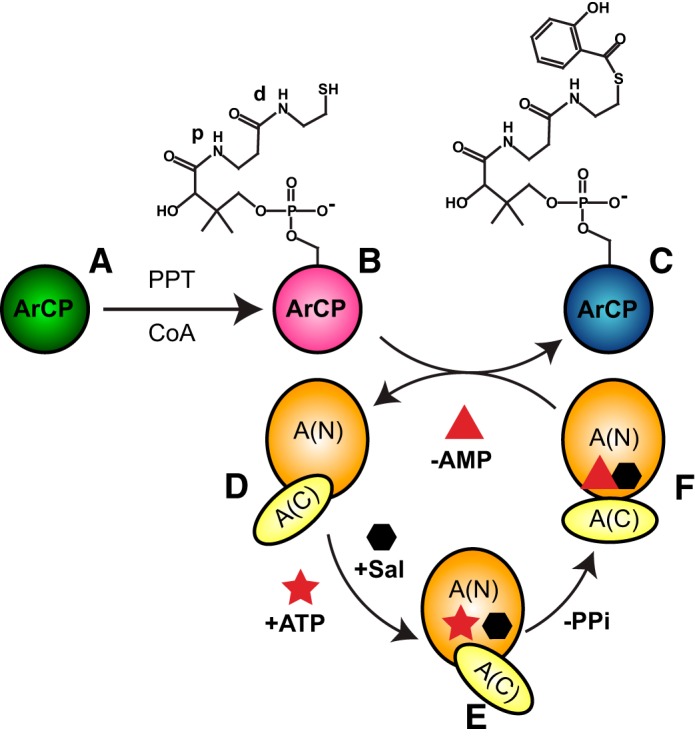
Covalent modifications of the yersiniabactin synthetase ArCP during NRPS synthesis. A PP transferase (PPT) converts apo-ArCP (A) to holo-ArCP (B) upon attachment of PP to Ser52. An adenylation domain loads salicylate (hexagon) on holo-ArCP (C). This A-domain first adopts an adenylation conformation (E) to activate the substrate through ATP (star) into a high-energy adenylate, SalAMP, where AMP is represented as a triangle. Next, a thioester conformation (F) allows for tethering the substrate to holo-CP with release of AMP. A third conformation may exist for the free domain (D). The proximal and distal amide groups in PP, discussed in the legend to Fig. 5, are labeled with p and d, respectively.
The modular organization of NRPSs and the conservation of their domains suggest that novel pharmaceuticals may be produced by swapping domains to modify substrate incorporation. In recent years, it has emerged that NRPSs are not rigid assemblies of domains but dynamic systems subject to inter- and intradomain structural fluctuations (5, 8–16). In particular, carrier proteins are dynamic domains (11, 17) that successively visit catalytic domains in a series of transient domain/domain interactions during synthesis (8, 9, 12, 13). If one is to successfully reprogram NRPSs, it will be critical to ensure proper translocation of carrier proteins in their various forms. This task necessitates the characterization of both the energetics governing the domain interactions and the molecular determinants that affect these affinities. Here, we demonstrate that covalent modifications alter the affinity of an ArCP of the yersiniabactin synthetase protein HMWP2 toward its partner adenylation domain, YbtE, suggesting that the modifications play an active role in driving CPs toward their partners. Further, we show that the same modifications induce structural fluctuations through an allosteric effect mediated by protein dynamics and propagated throughout the protein and up to 20 Å away from the site, including all regions shown to be involved in domain communication.
Results
Structure and dynamics of apo-ArCP
The solution structure of apo-ArCP was determined through 1128 unambiguous distance restraints and 141 angle constraints (see “Experimental Procedures”). Apo-ArCP shows the four-helix bundle typical of carrier proteins (18–21) (Fig. 2, A and B), with three longer helices, α1 (Ala19–Glu31), α2 (Ser52–Arg63), and α4 (Leu80–Leu88), mostly parallel to one another and running in an up-down-down fashion with a fourth, shorter helix, α3 (Leu71–Leu76), lying at a large angle to the others. Ser52, at the N terminus of α2, is the phosphopantetheinylation site. α1 and α2 are connected by a long loop (loop 1), which can be viewed as a flap covering the helical bundle. Loop 1 can be separated into a C-terminal region that appears to interact closely with the core (Glu42–Asp51) and a less compact N-terminal end (Leu32–Glu41), in which residues Pro36–Gln38 display some helical characteristics and are denoted by “α0”. A similar conformation can be observed in a subset of acyl carrier proteins (18, 22–25) and is somewhat apparent in holo-ArCP and loaded ArCP (17). Within the C-terminal end of loop 1 lies an additional single-turn helix, αI, which is also found in the holo-structures and salicylate-loaded structures (17). Other helices are connected by shorter loops (loops 2 and 3).
Figure 2.
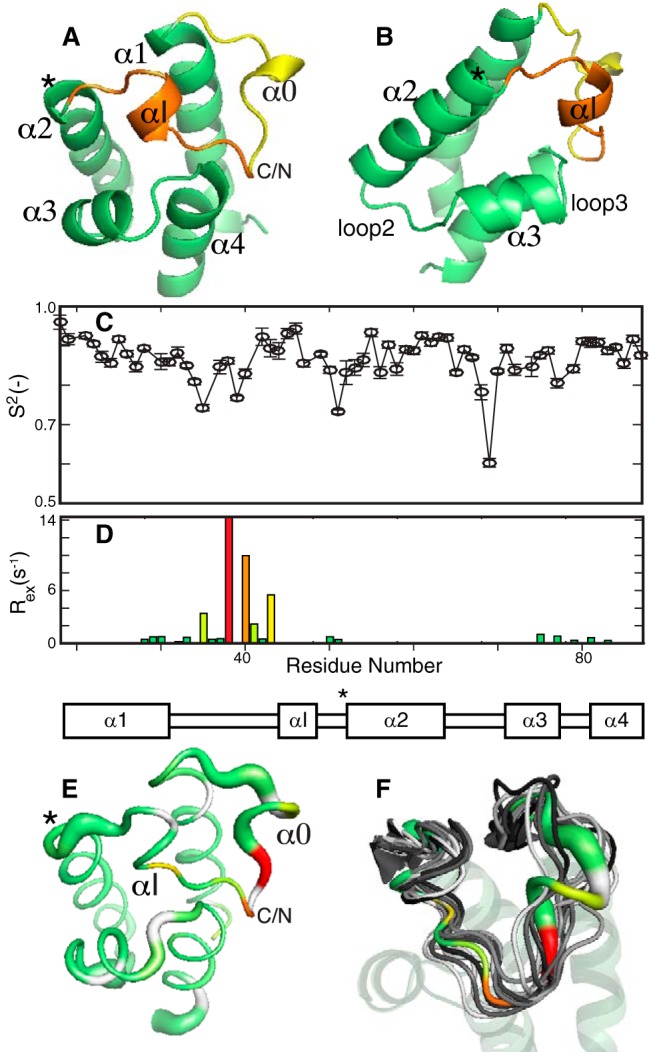
Structure and dynamics of apo-ArCP. A, the mean from the 20 lowest-energy conformers (see supplemental data) is shown. The N-terminal end of loop 1 is colored yellow, and its C-terminal end is orange. The phosphopantetheinylation site is highlighted by an asterisk. B, alternative view obtained by rotation of 60° along a vertical axis. Shown are the order parameter (C) and exchange rates (D) obtained by model-free analysis of 15N relaxation for the apo-ArCP core, excluding disordered N and C termini. Full profiles can be seen in supplemental Figs. S2 and S3. E, 3D representation of ArCP dynamics. A thicker sausage indicates a lower-order parameter and picosecond-to-nanosecond dynamics, whereas microsecond-to-millisecond fluctuations are color-coded according to the magnitude of Rex, from yellow to red. F, correlation of ArCP dynamics with structural variations in crystal structures of carrier proteins in NRPS multidomains. Thirteen carrier proteins were aligned according to their α-helical core (transparent) and compared with the 3D representation of ArCP dynamics. The ribbon in αI has been removed to highlight the fluctuation of its orientation.
Apo-ArCP displays a patch of surface-exposed hydrophobic moieties (Ile46 in αI, as well as Ile53, Met56, and Leu59 in α2, and Leu71 and Tyr75 in α3) that can also be observed in the four structures of (wild-type) apo-carrier proteins available for NRPSs (11, 19, 26, 27). This area encompasses residues found to interact with a phosphopantetheinyl transferase and its bound cofactor, CoA (28), with a subset of these residues also binding with prosthetic groups in holo- and substrate-loaded carrier proteins (17, 29). This observation suggests that the transient interactions between ArCP and its prosthetic groups (17) are constraints imposed by surface-exposed hydrophobic residues needed for efficient phosphopantetheinylation and, thus, should be well-conserved.
The dynamics of apo-ArCP were characterized through model-free analysis (30–33) of 15N NMR relaxation parameters (Fig. 2C). Here, so-called order parameters, S2, depict flexibility at picosecond-to-nanosecond time scales, with S2 = 0 interpreted as complete disorder and S2 = 1 indicating rigidity. However, S2 values are sensitive to anisotropic motions in an orientation-dependent manner, and variations in S2 reflect any alteration in the nature of the picosecond-to-nanosecond motion and not necessarily a rigidification or an increase in flexibility. The parameter Rex (relaxation induced by exchange; Fig. 2D) identifies slow conformational fluctuations (microsecond-to-millisecond) and is affected by the number of conformations involved, their populations, their change in environment, and the rate of the fluctuation. In our measurement conditions, Rex reports on time scales faster than those studied by relaxation dispersion or chemical exchange saturation transfer (see “Experimental Procedures”). Because measurements were taken at only a single field, variations in Rex will be interpreted in a semi-quantitative manner.
Loop 1 features three hinges that are flexible at picosecond-to-nanosecond time scales, two of which book-end α0 (Leu34, Leu39) and one in the C-terminal end of loop 1 immediately preceding α2 (Gly49, Asp51), with connecting regions subject to slower conformational fluctuations (Fig. 2E). This profile closely resembles that reported for the holo-forms and loaded forms and is reminiscent of those observed in fatty acid synthase and polyketide synthase acyl carrier proteins (17). We reasoned that such a conserved profile should leave a signature in crystallographic studies involving NRPS carrier proteins communicating with various domains. We aligned structures of 13 NRPS carrier proteins (3RG2 (34), 4MRT (28), 4ZXI (8), 4ZXJ (8), 5ES8 (9), 5ES9 (9), 4PWV (35), 4PXH (35), 4DG9 (36), 4IZ6 (37), 2VSQ (13), 2JGP (38), 2FQ1 (26)) and found that regions showing conformational variability coincided with residues that were dynamic (Fig. 2F). The ensemble suggests that Rex at Leu45 may reflect fluctuations of αI, whereas Rex at the center of loop 1 (His40, Glu42) may depict a hinge for microsecond-to-millisecond fluctuations in which the N-terminal end swivels while the C-terminal end remains docked against the core. The fact that α0 is not always observed and the presence of Rex at Gln37 probably reflect that α0 is a transient feature. Although many other factors can contribute to conformational changes in the ensemble (e.g. different sequences of amino acids), Fig. 2F suggests that dynamics should be taken into account when analyzing loop 1 in carrier proteins. In particular, whereas 239 distances constrain the conformation of ArCP loop 1 and lead to a low root mean square S.D. of 0.27 Å (Cα), Fig. 2 (E and F) indicates that loop 1 undergoes conformational fluctuations. In such a case, distance constraints reflect weighted averages of those of individual conformers and lead to an apparent, observed conformation (39). Consequently, changes in conformations discussed when comparing apo-, holo-, and loaded ArCP structures (see “Discussion”) correspond to a redistribution of (transient) distance constraints. Here, the distinction between changes in conformations and variations in structural fluctuations becomes ill-defined, and we will refer to apparent changes in conformation when discussing loop 1 as defined by distances, whereas variations of fluctuations refer to changes in Rex.
Modification of apo-ArCP with a nonhydrolyzable analog of salicylate-loaded pantetheine
To characterize the binding of the adenylation domain YbtE with its product, loaded ArCP, we synthesized a nonhydrolyzable analog of salicylate-loaded pantetheine (SalNH-pant) in which the labile thioester bond has been replaced with a stable amide linkage (see supplemental material). We then used a one-pot reaction (40) to both convert SalNH-pant into a CoA analog and attach it onto apo-ArCP, thus generating SalNH-ArCP. This strategy was used to solve the structure of a pyrrole-loaded CP (PltL) (29), and the interaction between the tethered substrate and PltL was similar to that observed in our structure of loaded ArCP, which contained the native thioester (17, 29). This suggests that amide-containing analogs faithfully recapitulate the structure of native substrate-loaded CPs and are suitable for our binding studies.
Characterization of binding through fluorescence anisotropy
Fluorescent labeling of a single-amino acid variant of ArCP, ArCP_R16C, permitted us to monitor binding with YbtE via fluorescence anisotropy (FA) (Fig. 3, A–C). YbtE shows a weak interaction with apo-ArCP, with an apparent dissociation constant KD of 69 ± 10 μm, in accordance with the fact that the apo-form is not a substrate for YbtE. Phosphopantetheinylation induces a marked increase in affinity, with a KD of 7 ± 1 μm for holo-ArCP. Surprisingly, YbtE binds to its product mimic, SalNH-ArCP, with an affinity comparable with that of holo-ArCP, with KD = 5 ± 1 μm. Thus, YbtE discriminates against apo-ArCP but binds to holo-ArCP and loaded ArCP with comparable affinities.
Figure 3.
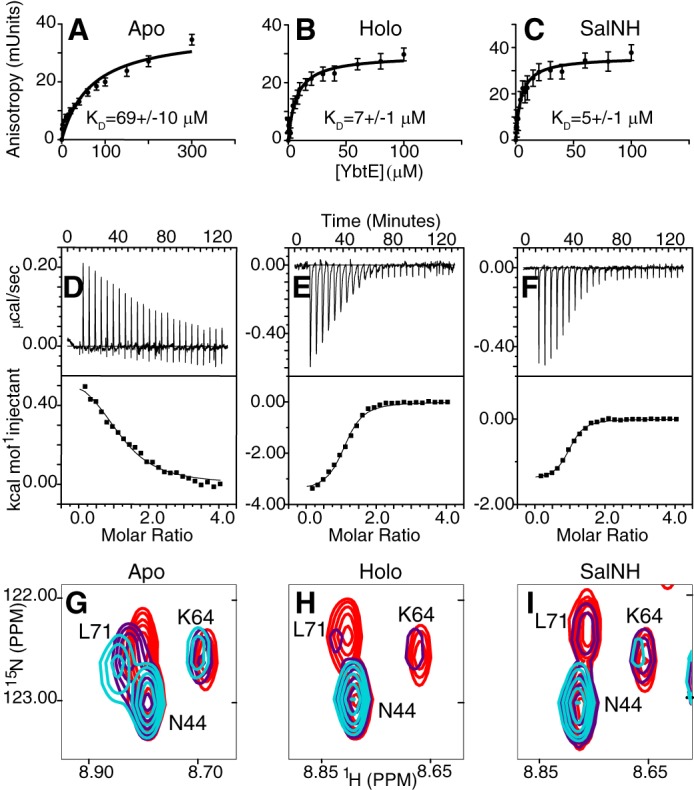
Binding of YbtE and ArCP in apo-form (A, D, and G), holo-form (B, E, and H), and SalNH-loaded form (C, F, and I). A–C, fluorescence anisotropy titrations show that YbtE binds to holo- and SalNH-ArCP with dissociation constants 10-fold lower than that of apo-ArCP. Error bars, S.D. of six (apo-ArCP) or five (holo- and SalNH-ArCP) titrations. The concentration of ArCP was 100 nm. D–F, isothermal titration calorimetry reveals that the interaction with apo-ArCP is driven entirely by entropy and slightly endothermic (D), whereas those of holo- and SalNH-ArCP are exothermic and supplemented with favorable entropy. ArCP was in the cell and at a concentration of 40 μm. The top panels show baseline-corrected raw data, and the bottom panels show the integrated heats. G–I, different forms of ArCP show different spectroscopic responses to binding by YbtE. Select regions of HN-HSQCs of 0.1 mm 15N-ArCP in the presence of YbtE at stoichiometries of 1:0 (red), 1:0.125 (purple), and 1:0.25 (cyan) are shown for apo-ArCP (G), holo-ArCP (H), and SalNH-ArCP (I). All spectra were scaled to identical contour levels.
Isothermal titration calorimetry
The dissociation constants obtained by ITC recapitulate the trends observed in the FA experiments (Fig. 3 (D–F) and Table 1). YbtE binds weakly to apo-ArCP (11.6 ± 1.9 μm) and interacts with holo- and SalNH-ArCP with similar affinities (2.7 ± 0.4 and 1.9 ± 0.2 μm, respectively). However, the thermodynamics of binding differ significantly (Table 1). The interaction between apo-ArCP and YbtE is endothermic, with an apparent value, ΔHapp = +0.62 kcal/mol. Thus, fewer favorable atomic interactions are present in the complex than in the isolated proteins, and their association is driven by a positive change in entropy of 24.6 cal/mol/K. In contrast, binding of YbtE to holo-ArCP is exothermic (−3.50 kcal/mol) with a positive change in entropy also contributing to this interaction, although to a lesser extent than with apo-ArCP (13.8 cal/mol/K). Finally, a lower change in enthalpy (−1.42 kcal/mol) and stronger positive change in entropy (21.5 cal/mol/K) contribute to the interaction between SalNH-ArCP and YbtE. Thus, although holo-ArCP and SalNH-ArCP have similar affinities for YbtE, substrate loading alters enthalpic and entropic contributions.
Table 1.
ITC-derived thermodynamic parameters of YbtE binding to all forms of ArCP
Binding constants (KD), enthalpy (ΔH), entropy (ΔS), and stoichiometry (n) were determined by fitting experimental data to a one-site binding model.
| KD | ΔH | ΔS | n | |
|---|---|---|---|---|
| μm | kcal/mol | cal/mol/K | ||
| Apo-ArCP | 11.6 ± 1.9 | 0.62 ± 0.04 | 24.6 ± 3.7 | 1.32 ± 0.05 |
| Holo-ArCP | 2.7 ± 0.4 | −3.50 ± 0.08 | 13.8 ± 3.5 | 1.09 ± 0.02 |
| SalNH-ArCP | 1.9 ± 0.2 | −1.42 ± 0.02 | 21.5 ± 2.5 | 0.96 ± 0.01 |
NMR titrations identify residues involved in binding to YbtE
To identify residues affected by the interaction with YbtE, we performed NMR titrations with apo-, holo-, and SalNH-ArCP. In these experiments, 15N2H-labeled ArCP was held at constant concentration, 2H-YbtE was titrated, and HN-HSQCs (41) were collected to obtain residue-specific information. The phosphopantetheinyl amide groups are detected for holo-ArCP but not for SalNH-ArCP, because the synthesized SalNH-pant is not 15N-labeled (see “Synthesis of SalNH-pant” in the supplemental materials).
Fig. 3 (G–I) illustrates the spectroscopic signatures of complex formation for all forms of ArCP. Comparison of apo-, holo-, and SalNH-ArCP titrations further demonstrates that YbtE preferentially binds to holo-ArCP and SalNH-ArCP over apo-ArCP. The signals in the apo-ArCP titration show large changes in chemical shift but small changes in intensity, indicative of fast-intermediate exchange on the NMR time scale. In contrast, signals of holo- and SalNH-ArCP broaden and disappear with minor shifts, characteristic of slow-intermediate exchange and a tighter interaction than that with apo-ArCP.
Joint analysis of signal intensities and positions was also performed to identify residues affected by binding. We calculated both the chemical shift perturbation (CSP = √((Δδ1H)2 + 0.2(Δδ15N)2), where Δδ denotes the change in chemical shift for the nucleus (42); Fig. 4, D–F) and the relative change in intensity at the first point in the titration (Rint = |(I1 − I0)/(C1 − C0)|, with C1 the concentration of YbtE at the first point, I1 the corresponding intensity, C0 = 0, and I0 the intensity in the absence of YbtE; Fig. 4, G–I). When displayed on structures, CSPs are represented by a color gradient, and Rint values are depicted by the thickness of a sausage, with a narrow line indicating no changes and the thickest line denoting the maximal change (Fig. 4, A–C).
Figure 4.

Spectroscopic perturbations induced by YbtE binding to apo-ArCP (A, D, G, and J), holo-ArCP (B, E, H, and K), and SalNH-ArCP (C, F, I, and L). A–C, CSPs and Rint plotted on the structures of ArCP. Global maxima (accounting for all forms) are used. CSPs are represented by a color gradient from blue (no CSP) to red (global maximum CSP in D–F), and Rint is represented by the thickness of the sausage, with the thickest line representing the maximum value in G–I. *, position of Ser52. D–F, CSPs representing shifts in HN-HSQCs of 1:0.125 ArCP/YbtE. For all forms, perturbations occur on α2 and α3, but they localize on the surface for apo-ArCP. G–I, relative change in intensity upon the addition of 0.125 eq of YbtE, Rint. Only specific residues are affected in apo-ArCP, but most residues in α2, α3, and loop 2 show dramatic changes in intensity for holo-ArCP and loaded ArCP. J–L, comparison between spectroscopic perturbations due to YbtE binding and a complex trapped in crystallographic studies for apo-SalNH (J), holo-SalNH (K), and SalNH-ArCP (L). Each form of ArCP (Protein Data Bank entries 5TTB (apo; this work), 2N6Y (holo), and 2N6Z (loaded)) was aligned onto EntB-ArCP in the EntE-EntB complex, Protein Data Bank entry 3RG2. For holo-ArCP (K), the phosphopantetheine group is shown in green when docked with ArCP and in black when extended between the N-terminal A(N) and C-terminal A(C) subdomains of EntE. The adenylate mimic used in the crystallographic study is shown in red spheres.
Spectroscopic perturbations affect α2 and α3 for all forms. However, whereas they localize on select surface residues in apo-ArCP, they extend to most residues of α2 and α3 and encompass loop 2 in holo-ArCP and loaded ArCP. Thus, although the same region of ArCP is involved, additional molecular events are at play in holo-ArCP and loaded ArCP.
Discussion
Little is known about whether and how NRPS catalytic domains discriminate between the various forms in which CPs exist. Over time, crystallography (8, 9, 13), NMR (12), and negative stain EM (8) have shown that NRPSs adopt a series of transient, well-defined domain/domain conformations during synthesis. However, it is still unclear whether and how NRPSs stabilize the relevant interaction in each catalytic step. Our results indicate that weak interactions between carrier proteins and their partner adenylation domains are altered by post-translational modifications. At the molecular level, these modifications lead to allosteric effects in the CP core, which are shown to be mediated by remodeling of a network of dynamic residues.
Binding studies of apo-ArCP provide a reference for protein recognition and subsequent analysis of the impacts of prosthetic groups. The residues involved in the interaction with YbtE localize on α2 and α3 (Fig. 4, A, D, and G). Further, ITC reveals that the endothermic interaction between apo-ArCP and YbtE is driven by entropy. Together with the observation that all residues are surface-exposed, we conclude that we are probing a weak but direct interaction between apo-ArCP and YbtE that is driven by desolvation.
The interaction between YbtE and holo-ArCP probably modulates transient interactions between PP and the ArCP protein core. Our previous studies (17) revealed that holo-ArCP exists in equilibrium between an undocked form, where PP is disordered, and a docked form, in which PP docks between α2 and α3 (green in Fig. 4K). ITC indicates that phosphopantetheinylation leads to an exothermic interaction with a large increase in affinity. Thus, more favorable intermolecular bonds are obtained following post-translational modification. Here, the PP arm may contribute by directly interacting with YbtE, by optimizing interactions between the protein cores, or both. Spectroscopic perturbations of the PP amide moieties of holo-ArCP reveal that PP communicates with YbtE through multiple interactions (Fig. 5). Indeed, for each PP amide moiety, a second signal becomes resolved during the titration, which may belong to a previously reported, yet uncharacterized, second conformation of (transiently) docked PP (17). Partial overlap with nearby core ArCP signals hinders interpretation of distal signals. However, the distinct behaviors of the two proximal signals during titration are indicative of two exchange regimes: (i) a fast exchange reflecting a weak interaction or a small change of chemical environment and (ii) a slow exchange indicating tighter interaction or a larger change in environment. In addition, other partially overlapped signals appear and disappear during the titration. Further, in contrast to apo-ArCP, spectroscopic perturbations are observed in most residues of α2 and α3 (Fig. 4, B, E, and H), alluding to a modulation of the interaction between ArCP and PP, possibly with fluctuations in the relative orientation of α2 and α3. These observations suggest that YbtE interacts with PP in at least two different manners and affects transient docking of PP with ArCP.
Figure 5.

Phosphopantetheinyl amide NMR signals from holo-ArCP show a complex response to binding by YbtE. A–E, proximal amide group (p in Fig. 1). A, two signals can be observed in free holo-ArCP (100 μm). As the concentration of YbtE is increased to 12.5 μm (B), 25 μm (C) 37.5 μm (D), and 50 μm (E), the signal marked with a blue plus shifts significantly and decreases in intensity, whereas the minor signal, marked with a black cross, shows little change in chemical shift. New signals also begin to appear, although it is not possible to assign them to either the major or minor PP signal. F–J, the distal group (d in Fig. 1) is also subject to an unusual perturbation, although spectral crowding hampers the interpretation. The question marks emphasize perturbations of ambiguous origin. The cross and plus symbols are not meant to be related with those used in A–E.
Our results serve as a point of comparison with crystal structures of CPs in complex with A domains, all of which show an A domain in the thioester conformation (Fig. 4, J–L) (8, 9, 34, 36, 37) with a phosphopantetheinyl arm extending between the N- and C-terminal subdomains (Fig. 4K, black PP). Here, A(N) interacts with α2 of the CP, and A(C) interacts with the N-terminal end of loop 1 (the region in yellow in Fig. 2, A and B). Whereas our NMR titrations show a strong response along α2, we saw little or no response for loop 1 in all forms. Thus, under our conditions, the thioester conformation of YbtE appears not to be selected upon complex formation with ArCP, regardless of its form. YbtE may be in an open or adenylation conformation (Fig. 1, D and E), or the orientation of A(C) may fluctuate. X-ray crystallography (5, 8, 9) and solution studies (6, 7) indicate that chemical substrates and adenylates alter the conformation of A(N) and A(C). Without them, ArCP/YbtE may transiently adopt the conformation in Fig. 4 with a defined A(N)/ArCP interaction but fleeting contacts between loop 1 and a dynamic A(C). Such a scenario may explain the behavior of holo-ArCP PP signals during titrations, because the prosthetic group may experience several A(C) interactions when extended between A(N) and A(C). Alternatively, for holo- (and loaded) ArCP, our results may reflect an encounter complex with YbtE, as seen in studies of related fatty acid synthase carrier proteins (43). Here, YbtE would bind to ArCP with a docked prosthetic group, as observed in the solution structure of free holo-ArCP (17), wherein PP would be sandwiched between ArCP and YbtE in the complex (Fig. 4K, green PP). Additional rearrangements needed to reach the catalytically active conformation would lead to the perturbations observed for PP and the ArCP core. Notably, whereas α3 is poorly positioned for interaction in the available structure, it displays a number of spectroscopic perturbations, even in apo-ArCP, which is free of prosthetic group dynamics. These perturbations may signal (transient) rearrangements in A(N) or ArCP or a subtle reorientation of ArCP when compared with Fig. 4K. Each binding model and associated variations in YbtE and prosthetic group dynamics can account for the observed differences in enthalpy-entropy balance between holo-forms and loaded forms, impeding further interpretation. NMR studies focusing on YbtE paired with experiments complementary to those employed here (e.g. cross-saturation transfer) will be needed to delineate between these models.
Having detected an influence of the ArCP prosthetic groups on its affinity toward YbtE, we studied the influence of these groups on the structure of the protein core itself. Comparison of the apo-structure with that of holo-ArCP and loaded ArCP (17) shows that post-translational modifications change the apparent conformation of loop 1 at locations remote from the modified serine (up to 20 Å away) and cause subtle reorientations of α3 with respect to α2 (Fig. 6G). Regions of loop 1, α2, and α3 are all involved in communicating with various catalytic partners, and structural changes or fluctuations thereof may impact domain affinities, as observed above. Given the dynamics of loop 1 (Fig. 2F), we rationalized that these apparent allosteric effects could be interpreted as modulations in protein dynamics.
Figure 6.

Modulation of protein dynamics imparted by covalent modifications of ArCP. A–C, 3D representation of significant variations resulting from phosphopantetheinylation (A), salicylate (Sal) attachment (B), and combined effect of S-salicylyl-phosphopantetheine (C). The thickness of the sausage reflects the magnitude of differences in order parameters (H–J, corresponding to A–C, respectively), with a thicker line emphasizing residues varying by more than one S.D. value from the mean, accounting for errors (shown with black dots in H–J). The color, from pale to dark, reports on changes in Rex. D–F, 3D representations of the dynamics of apo-ArCP (D), holo-ArCP (E), and loaded ArCP (F). G, apparent allosteric effect resulting from the modulation of dynamics shown in A–C. H–J, differences in order parameters, S2, used to generate the 3D representations in A–C. Dashed horizontal lines, limits for one S.D., used to emphasize residues with significant changes.
Fig. 6 (A–C) depicts the variations in ArCP dynamics imposed by chemical modifications of carrier proteins. The thickness of the sausage denotes changes in S2 (Fig. 6, H–J), and coloring reflects substantial changes in conformational fluctuations (supplemental Fig. S4). The structures and dynamics of each form of ArCP are shown in Fig. 6 (D–F), with apparent allosteric effects highlighted in Fig. 6G. Fig. 6 (A and H) highlights the impact of phosphopantetheinylation. Attachment of PP on Ser52 alters the picosecond-to-nanosecond dynamics of the preceding residues, affecting the structural fluctuations of αI and propagating to the center of loop 1, thereby affecting the swiveling of the N-terminal end of loop 1 (Fig. 2F) as well as the dynamics in vicinal loop 3 and α3. Interestingly, the heteronuclear NOEs of holo-DltC and a mutant of its apo-form also display differences in the region preceding the phosphopantetheinylation (and mutation) site and in loop 3, suggesting that the picosecond-to-nanosecond dynamics of this related system are altered by phosphopantetheinylation in a manner similar to that described for ArCP (44). αI is found to interact both with adenylation domains and PPTases (28, 34), and the N-terminal end of loop 1 can interact with A(C) subdomains. Thus, phosphopantetheinylation of a single residue in ArCP remodels a substantial fraction of the core protein dynamics, and its impact propagates to remote regions involved in domain communication.
Similarly, a network of dynamic residues helps to propagate the impact of thioester bond formation to the protein core (Fig. 6, B and I). Here, salicylate alters picosecond-to-nanosecond dynamics of its docking site, near αI, and in loop 2, which gains degrees of freedom in the new conformation adopted by PP in loaded ArCP. Like PP attachment, salicylate loading affects slow dynamics of the center of loop 1. Remarkably, the dynamics of the N-terminal end of loop 1 in holo-ArCP are unaltered upon substrate loading. In contrast, comparison of apo-ArCP with either holo-ArCP or loaded ArCP (Fig. 6, panels A and H and panels C and J, respectively) indicates that the dynamics of the N-terminal end of loop 1 are affected by attachment of the PP arm, regardless of its conformation in docked form or whether it harbors a substrate. This would point at a direct molecular communication between the first and last residues of loop 1. The presence of (small) changes in exchange rates observed for these residues (Fig. 6, A and C) supports such a scenario. The region defined by αI and the phosphopantetheinylation site stands out as a major sensor for post-translational modifications. This region encompasses the very motif used to identify CPs (45–48), with variants differentiated notably by the amino acid at position i − 1 from the PP site. ArCP and all acyl carrier proteins discussed belong to a family with aspartates at this position. Further studies with CPs harboring alternative motifs (e.g. with histidine or asparagine at i − 1) will highlight conserved and unique features in their dynamic profiles.
The results presented here, along with previous biochemical studies, paint a picture wherein covalent modifications to CPs and substrate binding by A domains work in concert to promote directionality to the set of protein/protein interactions necessary for priming and elongation. Previous ITC studies showed that the PPTase Sfp binds to apo-CPs with submicromolar affinity (28). PPTases would thus be able to outcompete A domains for binding to CPs (here, KD above tens of μm). Next, phosphopantetheinylation substantially increases the affinity of A domains for CPs, reflecting the role of holo-CPs as substrates for the loading reaction, although the thioester conformation of A domains is apparently not selected by holo-CPs alone. Instead, adenylates may be needed to reach this conformation and tighten domain association. Finally, substrate loading alters the nature of the interaction between CPs and A domains in a manner that may promote dissociation from the active site and shuttling to downstream catalytic domains.
Conclusion
We have presented the first quantitative investigation into the association between a carrier protein in all of its possible forms and a partner catalytic domain. We found that chemical modifications to CPs can modulate the affinity and thermodynamics of the interactions. Further, NMR titrations revealed that, in the absence of additional substrates, ArCP does not bind with YbtE with concomitant selection of the catalytically active thioester conformation, regardless of its form. Likewise, YbtE does not simply select for an extended conformation of the phosphopantetheinyl arm in holo-ArCP. Instead, PP is subject to multiple molecular events during binding, and its interaction with the ArCP protein core is modulated.
We identified allosteric effects imparted by post-translational modifications and discovered a network of dynamic residues responsive to these modifications. Because the network includes a conserved motif and binding sites and because similar dynamic profiles are seen in related carrier proteins, its preservation or alteration probably affects carrier protein functionality, prompting further studies. Our studies suggest that modulations of dynamics in the carrier protein fold may be sufficient to propagate the molecular impact of CP modifications and hence alter affinities toward partner domains, without resorting to dramatically different minor conformers that had been suggested (11) and later disputed (28, 44, 49).
Biological processes often rely on post-translational modifications to control protein interactions and function. Our results show that NRPSs utilize the chemical modifications inherent to NRPS synthesis to modulate the nature and strength of domain interactions in a manner that promotes efficient synthesis. Further, the work presented here lays the groundwork for understanding the role that additional substrates of adenylation domains, such as substrate, ATP, and substrate-adenylate, or catalytic domains, such as condensation or cyclization domains, play in modulating the complex set of interactions in which carrier proteins are involved and will inform future efforts to reengineer NRPS systems.
Experimental procedures
Expression and purification of ArCP
15N,13C-Labeled apo-ArCP and unlabeled and 15N-labeled apo- and holo-ArCP were expressed and purified as described previously (17, 50). For expression of 15N,2H-labeled apo- and holo-ArCP, recycled, distilled D2O was used in growth media, but protonated glucose was employed, leading to fractional deuteration. pD of media was adjusted to 6.8 prior to use.
NMR acquisitions
All NMR experiments were conducted on a 600-MHz Bruker AVANCEIII spectrometer equipped with a QCI cryoprobe at a temperature of 25 °C and in NMR buffer (50 mm N-(2-acetamido)-2-aminoethanesulfonic acid (ACES), pH 6.80 at 22 °C, 150 mm NaCl, 500 μm tris(2-carboxyethyl)phosphine (TCEP), 1 mm MgCl2).
Resonance assignments
15N,13C-ArCP backbone resonances were assigned as described previously with a 300 μm sample (50). Aliphatic resonances were assigned as described for holo-ArCP and loaded ArCP (17). Aromatic resonances were assigned with a 300 μm sample in D2O and hbCBcgcdHD (288 scans, 2048 (1H, 16.019 ppm at 4.696 ppm) × 26 (13C, 22 ppm at 30 ppm) complex points, 4 h 55 min), hbCBcgcdceHE (672 scans, 2048 (1H, 16.019 ppm at 4.696 ppm) × 26 (13C, 22 ppm at 30 ppm) complex points, 11 h 33 min), and HC-HSQC-NOESY (16 scans, 2048 (1H, 16.019 ppm at 4.696 ppm) × 100 (1H, 13 ppm at 4.696 ppm) × 50 (13C, 50.0003 ppm at 69.5 ppm) complex points, mixing time of 90 ms, 4 days 15 h 54 min). Distance constraints were obtained with the HC-HSQC-NOESY mentioned above and with a NOESY-HN-HSQCs (16 scans, 2048 (1H, 16.019 ppm at 4.696 ppm) × 40 (15N, 26 ppm at 117 ppm) × 120 (1H, 11 ppm at 4.696 ppm) complex points, mixing time of 90 ms, 4 days, 12 h) of 300 μm apo-ArCP in NMR buffer.
Structure calculation
NOESY cross-peaks were assigned with CARA (computer-aided resonance assignment) (51). 1128 unambiguous distance restraints and 141 angle constraints (from TALOS-N (torsion angle likelihood obtained from shift and sequence similarity)) (52, 53) were supplied for structure calculations using CYANA (54, 55) version 2.1. 100 structures were calculated using 50,000 steps. The CYANA target function was 1.98. There were no distance violations > 0.5 Å and no angle violations > 5° in the NMR ensemble. The average root mean square S.D. values were 0.38 Å (backbone atoms) and 0.98 Å (heavy atoms). The 20 structures with the lowest target function were chosen for water refinement in explicit solvent using CNS (56) and RECOORD (Recalculated Coordinate Database) (57) scripts. Structures were analyzed with PyMOL (58) or MOLMOL (59). Structure validation was performed using PSVS (protein structure validation software) (60).
15N relaxation
T1, T2, and {HN}-heteronuclear NOESY experiments were run on 250 μm samples with the following parameters: T1: 24 scans, 2048 (1H, 16.1092 ppm at 4.697 ppm) × 128 (15N, 26 ppm at 117 ppm) complex points, 3-s recycling delay, and relaxation delays of 0, 1.53, 0.510, and 1.02 s, collected in that order; T2: 16 scans, 2048 (1H 16.0192 ppm at 4.697 ppm) × 128 (15N, 26 ppm at 117 ppm) complex points, 4-s recycling delay, νCPMG = 999 Hz, and relaxation delays of 0, 0.140, 0.350, 0.210, 0.070, 0.280, and 0 (duplicate to verify the absence of aggregation) s collected in that order; {HN}-heteronuclear NOESY: 100 scans, 2048 (1H, 16.0192 ppm at 4.697 ppm) × 95 (15N, 26 ppm at 117 ppm) complex points, 5-s recycling delay for reference experiment and 2-s recycling delay followed by 3-s saturation by 120° 1H pulses every 5 ms for saturation experiment. Analysis was performed as described (17).
Expression and purification of YbtE
Unlabeled YbtE was expressed and purified as described previously (50). 2H-YbtE was expressed in M9 minimal medium (50) made with recycled, distilled D2O and protonated glucose. pD of medium was adjusted to 6.8 prior to use, and expression and purification were otherwise identical to unlabeled YbtE.
Expression and purification of enzymes from the CoA biosynthetic pathway: PanK, PPAT, and DPCK
Unless otherwise noted, the pH listed for each buffer is the pH at 4 °C. A plasmid encoding for PanK (courtesy of Dr. Craig Townsend) was transformed into Escherichia coli BL21 (DE3) cells and plated on Luria broth (LB) agar with kanamycin. A single colony was selected and added to 15 ml of LB with kanamycin and grown at 37 °C with shaking at 250 rpm overnight. The following day, 10 ml of the overnight culture was added to 1 liter of LB with kanamycin, and growth continued at 37 °C with shaking. At A600 = 0.6, cultures were placed in an ice bath and allowed to cool to 15 °C. Isopropyl 1-thio-β-d-galactopyranoside was then added to a final concentration of 1 mm, and growth continued at 16 °C with shaking for 16 h. Cells were harvested by centrifugation, and pellets were flash-frozen in liquid nitrogen. Pellets were stored at −80 °C until needed.
To begin purification, cell pellets were resuspended in 50 ml of lysis buffer (50 mm tris(hydroxymethyl)aminomethane (Tris), pH 8.0, 0.5 m NaCl, 30 mm imidazole, 200 μg/ml lysozyme, 2 μg/ml DNase I) and lysed using either a microfluidizer or French pressure cell. The lysate was clarified by centrifugation at 27,000 × g for 30 min at 4 °C. The supernatant was filtered and loaded onto a 5-ml HisTrap HP (GE Healthcare) column. The column was washed with 16 column volumes of His Buffer A (50 mm Tris, pH 8.0, 0.5 m NaCl, 30 mm imidazole) on an Aktapurifier (GE Healthcare) while collecting fractions. The column was eluted with a linear gradient from 0 to 100% His Buffer B (50 mm Tris, pH 8.0, 0.5 m NaCl, 0.5 m imidazole) over 25 column volumes while collecting fractions. Fractions were analyzed by SDS-PAGE, and fractions containing the desired protein were pooled and dialyzed against 2 liters of 20 mm Tris, pH 7.5, 0.1 m NaCl at 4 °C overnight.
The following day, the samples were concentrated to 2 ml or less and loaded onto a Superdex 200 16/60 pg size exclusion column that had been equilibrated with 20 mm Tris, pH 7.5, 0.1 m NaCl and run at maximum flow rate (1.2 ml/min) while collecting fractions. Fractions were analyzed by SDS-PAGE, and fractions containing pure target protein were pooled. Purification of PPAT and DPCK was performed in the same manner.
Removal of contaminating coenzyme A from PPAT and PanK
PPAT and PanK co-purify with coenzyme A, which produces contaminant holo-ArCP in samples of SalNH-ArCP, as evidenced by MALDI mass spectrometry. To remove coenzyme A, PPAT and PanK were buffer-exchanged into 20 mm Tris, pH 7.5, 0.1 m NaCl, 20 mm ATP by repeated concentration and dilution in this buffer until a 1000-fold exchange had been achieved. Under these conditions, ATP outcompetes CoA for binding with PPAT and PanK. Samples were then exchanged into 20 mm Tris, pH 7.5, 0.1 m NaCl, 10% glycerol (v/v) by repeated concentration and dilution until a 1,000-fold exchange had been achieved. DPCK was exchanged into the same buffer. Samples were then concentrated, aliquoted, flash-frozen in liquid nitrogen, and stored at −80 °C until needed. Concentrations were determined by UV-visible absorbance at 280 nm. Extinction coefficients used were 45,380 m−1 cm−1 (PanK), 8480 m−1 cm−1 (PPAT), and 16,960 m−1 cm−1 (DPCK). MALDI spectra confirmed that this procedure successfully prevents contamination with holo-ArCP.
Modification of apo-ArCP with nonhydrolyzable amide mimic
A 10-ml reaction in 100 mm Tris, pH 7.5 at 22 °C, 10 mm MgCl2, and 100 mm NaCl with 50 μm apo-ArCP, 150 μm SalNH-pant, 5 mm ATP, 500 nm Sfp, 500 nm PanK, 500 nm PPAT, and 500 nm DPCK was prepared and incubated at room temperature for 4 h. The sample was then concentrated and run on a Superdex 75 16/60 pg (GE Healthcare) size-exclusion column that had been equilibrated with ITC buffer (50 mm ACES, pH 6.80 at 22 °C, 150 mm NaCl, 1 mm MgCl2, 2 mm TCEP), and peak fractions were collected. Fractions were analyzed by SDS-PAGE, and completion of the reaction was confirmed using MALDI and HN-HSQC. HN-HSQCs of 15N samples were necessary to quantify traces of apo-ArCP, which are detected by MALDI. With the protocol presented above, no apo-ArCP is detected by NMR, and traces of apo-ArCP are estimated to be less than 1% (see also supplemental data).
ITC experiments
ITC buffer was used for all ITC experiments. To prepare YbtE for titrations, samples were exchanged >100-fold into freshly prepared ITC buffer by repeated concentration and dilution in a 30,000 molecular weight cut-off centrifugal filter (Millipore). Samples were then filtered through a 0.45-μm filter to remove any precipitate that had formed during concentration. A 5-μl sample was taken and diluted 5-fold into ITC buffer. 5 μl of this was then diluted 20-fold into 6.3 m guanidinium-HCl. 5 μl of ITC buffer was also diluted 20-fold into 6.3 m guanidinium-HCl to use as a blank. A280 was measured, and the concentration was calculated using an extinction coefficient of 52,370 m−1 cm−1. Samples were then diluted to the final working concentration and stored on ice until needed.
Samples of ArCP were exchanged > 100-fold into freshly prepared ITC buffer by repeated concentration and dilution in a 3000 molecular weight cut-off centrifugal filter. Samples were filtered through a 0.45-μm filter to remove any precipitate that had formed during concentration. A 5-μl sample was taken and diluted 20-fold into 6.3 m guanidinium-HCl. A blank was prepared as described above. A280 was measured, and the concentration was calculated using an extinction coefficient of 20,970 m−1 cm−1. Samples were diluted to the final working concentration and stored on ice until needed.
All samples were degassed prior to use. ITC experiments were performed using a VP-ITC Microcalorimeter. ArCP (40 μm) was in the cell, and YbtE (720 μm) was in the syringe, and the following settings were used: cell temperature, 27 °C; Reference power, 20 μcal/s; initial delay, 600 s; stirring speed, 300 rpm; feedback mode, High; equilibration options, Fast equil./Auto. The injection schedule was as follows: one 2-μl injection with 150-s delay followed by 24 12-μl injections with 300-s delays. Data were analyzed using Origin. Prior to fitting, the integrated heat from the final four points of each titration was averaged and subtracted from all points to account for the heat of dilution.
Cloning of ArCP_R16C
A cysteine was introduced into GB1-ArCP-14-93 using primers ArCP_14-93_R16C_For (5′-CACGAAAATCTTTATTTTCAAGGTACCGACAACTGCCACGCGGCTG-3′) and ArCP_14-93_R16C_Rev (5′-CAGCCGCGTGGCAGTTGTCGGTACCTTGAAAATAAAGATTTTCGTG-3′) using the Phusion High-Fidelity Master Mix with High Fidelity Buffer according to the protocol recommended by the manufacturer (New England Biolabs). The reaction contained 5% DMSO and used the following PCR protocol: one round of 30 s at 98 °C; 30 rounds of 10-s melting at 98 °C, 30-s annealing at 72 °C, 5-min extension at 72 °C; one round of 10-min extension at 72 °C. Colonies containing the proper mutation were identified by sequencing.
Expression and purification of ArCP_R16C was the same as for apo-ArCP-14-93 except that dialysis buffers contained 2 mm DTT, and the final size exclusion buffer was FA Labeling Buffer (10 mm HEPES, pH 7.5, 150 mm NaCl, 500 μm TCEP).
Fluorescent labeling of apo-ArCP_R16C
Purified apo-ArCP_R16C (5 mg) in FA Labeling Buffer was concentrated to a volume of 3 ml, and Triton X-100 was added to a final concentration of 1% (w/v). 1 mg of fluorescein-5-maleimide (Vector Laboratories) dissolved in 30 μl of DMSO was then added to apo-ArCP_R16C. Tubes were wrapped in parafilm and aluminum foil and rocked at 4 °C overnight.
The following day, β-mercaptoethanol was added to a final concentration of 10 mm, and rocking continued at 4 °C for 1 h. Excess dye was removed by repeated concentration and dilution in FA Labeling Buffer, and the sample was concentrated to 2 ml. The sample was then run on a Superdex 75 16/60 pg that had been equilibrated with ITC buffer. Peak fractions were analyzed by SDS-PAGE, and fractions containing ArCP were pooled. This pool was divided into three aliquots: one remained apo, one was phosphopantetheinylated as described (50), and the other was modified with a nonhydrolyzable amide mimic as described above.
Protein concentration and degree of labeling were calculated according to the manufacturer's protocols. Degree of labeling was found to be 0.282 (apo-ArCP), 0.352 (holo-ArCP), and 0.344 (SalNH-ArCP). Glycerol was added to 10% (w/v), and samples were flash-frozen in liquid nitrogen and stored at −80 °C until needed. After thawing and before use in titrations, samples were dialyzed against freshly prepared ITC buffer such that a >10,000-fold dilution of glycerol was achieved.
Fluorescence anisotropy
To prepare samples for fluorescence anisotropy titrations, YbtE was buffer-exchanged > 100-fold into freshly prepared ITC buffer by repeated concentration and dilution. The sample was filtered, and concentration was determined by diluting 5 μl 20-fold in 6.3 m guanidinium-HCl and measuring the A280. A series of 2× YbtE samples was then created from this stock. A 2× stock of fluorescently labeled ArCP was made by diluting concentrated samples to 200 nm in ITC buffer. Final samples were made by mixing 30 μl of 2× YbtE with 30 μl of 2× ArCP in 1.5-ml microcentrifuge tubes. Samples were briefly spun in a table top centrifuge and incubated in the dark for 20 min. Samples were then further mixed by pipetting, and 40 μl was added to a 384-well plate (Corning catalog no. 3575) and incubated in the dark for an additional 20 min before reading.
Measurements were performed on a Tecan M1000 Infinite PRO plate reader. The G-factor was determined using 1 nm fluorescein in 0.01 m NaOH, and the anisotropy readings were averaged across five wells. The excitation wavelength was 470 nm with 5-nm bandwidth. The emission wavelength was 518 nm with 8-nm bandwidth. 200 flashes were used for each point. Background fluorescence was averaged across 5 wells containing only ITC buffer. Data were analyzed using Prism.
NMR titrations of ArCP with YbtE
To prepare for titrations, ArCP and YbtE were both buffer-exchanged > 200-fold into NMR buffer. Samples were then concentrated and filtered. Concentration of YbtE and ArCP was determined as described for ITC experiments. For all experiments, ArCP was held at fixed concentration of 100 μm, and the concentration of YbtE was varied. All samples contained 10% D2O and DSS for referencing.
For apo-ArCP, spectra were collected in the following order described by the ratio of ArCP/YbtE: 1:0, 1:0.25, 1:0.375, 1:0.125. For holo-ArCP and SalNH-ArCP, spectra were collected in the following order described by the ratio of ArCP/YbtE: 1:0, 1:0.5, 1:0.25, 1:0.375, 1:0.125. Each HN-HSQC was collected with 32 scans and 1440 (1H, 16.11 ppm at 4.699 ppm) × 64 (15N, 26 ppm at 117 ppm) complex points for a total acquisition time of 1 h 20 min. All spectra were apodized and zero-filled to the nearest power of 2 in the 1H dimension and to 1024 points in the 15N dimension. Spectra were analyzed using SPARKY (61).
Author contributions
A. C. G. performed the experiments. A. C. G. and D. P. F. designed experiments, analyzed the results, and wrote the manuscript. D. J. M. synthesized SalNH-pant.
Supplementary Material
Acknowledgments
We thank Dr. Craig Townsend and Darcie Long for plasmids encoding PanK, PPAT, and DPCK and purification protocols; Dr. Scott Nichols and Emily Grasso for assistance with modifying apo-ArCP with SalNH-pant; Dr. L. Mario Amzel for helpful discussions about ITC results; Dr. Lou Charkoudian for helpful discussions; Bradley Harden for help with analyzing relaxation data; and Drs. Jungsan Sohn and Seamus Morrone for technical assistance and helpful discussions about fluorescence anisotropy experiments.
This work was supported in whole or in part by National Institutes of Health Grants R01 GM 104257 and S10 RR029191. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Tables S1–S3 and Figs. S1–S12.
The atomic coordinates and structure factors (code 5TTB) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- NRPS
- nonribosomal peptide synthetase
- PP
- phosphopantetheine
- HSQC
- heteronuclear single-quantum coherence
- ITC
- isothermal titration calorimetry
- FA
- fluorescence anisotropy
- CP
- carrier protein
- ArCP
- aryl carrier protein
- ACES
- N-(2-acetamido)-2-aminoethanesulfonic acid
- TCEP
- tris(2-carboxyethyl)phosphine
- PPTase
- phosphopantetheinyl transferase
- DPCK
- dephospho-coenzyme A kinase
- PanK
- pantothenate kinase
- PPAT
- phosphopantetheine adenylyltransferase
- SalNH-pant
- salicylate-loaded pantetheine
- SalNH-ArCP
- salicylate-loaded ArCP
- PltL
- pyrrole-loaded CP
- CSP
- chemical shift perturbation.
References
- 1. Walsh C. T. (2003) Antibiotics: Actions, Origins, Resistance, 1st Ed., ASM Press, Washington, D. C. [Google Scholar]
- 2. Crosa J. H., and Walsh C. T. (2002) Genetics and assembly line enzymology of siderophore biosynthesis in bacteria. Microbiol. Mol. Biol. Rev. 66, 223–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fischbach M. A., and Walsh C. T. (2006) Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem. Rev. 106, 3468–3496 [DOI] [PubMed] [Google Scholar]
- 4. Marahiel M. A., Stachelhaus T., and Mootz H. D. (1997) Modular peptide synthetases involved in nonribosomal peptide synthesis. Chem. Rev. 97, 2651–2674 [DOI] [PubMed] [Google Scholar]
- 5. Gulick A. M. (2009) Conformational dynamics in the acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem. Biol. 4, 811–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Khalil S., and Pawelek P. D. (2009) Ligand-induced conformational rearrangements promote interaction between the Escherichia coli enterobactin biosynthetic proteins EntE and EntB. J. Mol. Biol. 393, 658–671 [DOI] [PubMed] [Google Scholar]
- 7. Zettler J., and Mootz H. D. (2010) Biochemical evidence for conformational changes in the cross-talk between adenylation and peptidyl-carrier protein domains of nonribosomal peptide synthetases. FEBS J. 277, 1159–1171 [DOI] [PubMed] [Google Scholar]
- 8. Drake E. J., Miller B. R., Shi C., Tarrasch J. T., Sundlov J. A., Allen C. L., Skiniotis G., Aldrich C. C., and Gulick A. M. (2016) Structures of two distinct conformations of holo-non-ribosomal peptide synthetases. Nature 529, 235–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reimer J. M., Aloise M. N., Harrison P. M., and Schmeing T. M. (2016) Synthetic cycle of the initiation module of a formylating nonribosomal peptide synthetase. Nature 529, 239–242 [DOI] [PubMed] [Google Scholar]
- 10. Bruner S. D., Weber T., Kohli R. M., Schwarzer D., Marahiel M. A., Walsh C. T., and Stubbs M. T. (2002) Structural basis for the cyclization of the lipopeptide antibiotic surfactin by the thioesterase domain SrfTE. Structure 10, 301–310 [DOI] [PubMed] [Google Scholar]
- 11. Koglin A., Mofid M. R., Löhr F., Schäfer B., Rogov V. V., Blum M.-M., Mittag T., Marahiel M. A., Bernhard F., and Dötsch V. (2006) Conformational switches modulate protein interactions in peptide antibiotic synthetases. Science 312, 273–276 [DOI] [PubMed] [Google Scholar]
- 12. Frueh D. P., Arthanari H., Koglin A., Vosburg D. A., Bennett A. E., Walsh C. T., and Wagner G. (2008) Dynamic thiolation–thioesterase structure of a non-ribosomal peptide synthetase. Nature 454, 903–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tanovic A., Samel S. A., Essen L.-O., and Marahiel M. A. (2008) Crystal structure of the termination module of a nonribosomal peptide synthetase. Science 321, 659–663 [DOI] [PubMed] [Google Scholar]
- 14. Yonus H., Neumann P., Zimmermann S., May J. J., Marahiel M. A., and Stubbs M. T. (2008) Crystal structure of DltA: implications for the reaction mechanism of non-ribosomal peptide synthetase adenylation domains. J. Biol. Chem. 283, 32484–32491 [DOI] [PubMed] [Google Scholar]
- 15. Strieker M., Tanović A., and Marahiel M. A. (2010) Nonribosomal peptide synthetases: structures and dynamics. Curr. Opin. Struct. Biol. 20, 234–240 [DOI] [PubMed] [Google Scholar]
- 16. Bloudoff K., Rodionov D., and Schmeing T. M. (2013) Crystal structures of the first condensation domain of CDA synthetase suggest conformational changes during the synthetic cycle of nonribosomal peptide synthetases. J. Mol. Biol. 425, 3137–3150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goodrich A. C., Harden B. J., and Frueh D. P. (2015) Solution structure of a nonribosomal peptide synthetase carrier protein loaded with its substrate reveals transient, well-defined contacts. J. Am. Chem. Soc. 137, 12100–12109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Findlow S. C., Winsor C., Simpson T. J., Crosby J., and Crump M. P. (2003) Solution structure and dynamics of oxytetracycline polyketide synthase acyl carrier protein from Streptomyces rimosus. Biochemistry 42, 8423–8433 [DOI] [PubMed] [Google Scholar]
- 19. Weber T., Baumgartner R., Renner C., Marahiel M. A., and Holak T. A. (2000) Solution structure of PCP, a prototype for the peptidyl carrier domains of modular peptide synthetases. Structure 8, 407–418 [DOI] [PubMed] [Google Scholar]
- 20. Kim Y., and Prestegard J. H. (1990) Refinement of the NMR structures for acyl carrier protein with scalar coupling data. Proteins Struct. Funct. Genet. 8, 377–385 [DOI] [PubMed] [Google Scholar]
- 21. Holak T. A., Kearsley S. K., Kim Y., and Prestegard J. H. (1988) Three-dimensional structure of acyl carrier protein determined by NMR pseudoenergy and distance geometry calculations. Biochemistry 27, 6135–6142 [DOI] [PubMed] [Google Scholar]
- 22. Johnson M. A., Peti W., Herrmann T., Wilson I. A., and Wüthrich K. (2006) Solution structure of As11650, an acyl carrier protein from Anabaena sp. PCC 7120 with a variant phosphopantetheinylation-site sequence. Protein Sci. 15, 1030–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Arthur C. J., Williams C., Pottage K., Płoskoń E., Findlow S. C., Burston S. G., Simpson T. J., Crump M. P., and Crosby J. (2009) Structure and malonyl CoA-ACP transacylase binding of Streptomyces coelicolor fatty acid synthase acyl carrier protein. ACS Chem. Biol. 4, 625–636 [DOI] [PubMed] [Google Scholar]
- 24. Parris K. D., Lin L., Tam A., Mathew R., Hixon J., Stahl M., Fritz C. C., Seehra J., and Somers W. S. (2000) Crystal structures of substrate binding to Bacillus subtilis holo-(acyl carrier protein) synthase reveal a novel trimeric arrangement of molecules resulting in three active sites. Structure 8, 883–895 [DOI] [PubMed] [Google Scholar]
- 25. Sharma A. K., Sharma S. K., Surolia A., Surolia N., and Sarma S. P. (2006) Solution structures of conformationally equilibrium forms of holo-acyl carrier protein (PfACP) from Plasmodium falciparum provides insight into the mechanism of activation of ACPs. Biochemistry 45, 6904–6916 [DOI] [PubMed] [Google Scholar]
- 26. Drake E. J., Nicolai D. A., and Gulick A. M. (2006) Structure of the EntB multidomain nonribosomal peptide synthetase and functional analysis of its interaction with the EntE adenylation domain. Chem. Biol. 13, 409–419 [DOI] [PubMed] [Google Scholar]
- 27. Haslinger K., Redfield C., and Cryle M. J. (2015) Structure of the terminal PCP domain of the non-ribosomal peptide synthetase in teicoplanin biosynthesis. Proteins 83, 711–721 [DOI] [PubMed] [Google Scholar]
- 28. Tufar P., Rahighi S., Kraas F. I., Kirchner D. K., Löhr F., Henrich E., Köpke J., Dikic I., Güntert P., Marahiel M. A., and Dötsch V. (2014) Crystal structure of a PCP/Sfp complex reveals the structural basis for carrier protein posttranslational modification. Chem. Biol. 21, 552–562 [DOI] [PubMed] [Google Scholar]
- 29. Jaremko M. J., Lee D. J., Opella S. J., and Burkart M. D. (2015) Structure and substrate sequestration in the pyoluteorin type II peptidyl carrier protein PltL. J. Am. Chem. Soc. 137, 11546–11549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lipari G., and Szabo A. (1982) Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 104, 4546–4559 [Google Scholar]
- 31. Mandel A. M., Akke M., and Palmer A. G. 3rd (1995) Backbone dynamics of Escherichia coli ribonuclease HI: correlations with structure and function in an active enzyme. J. Mol. Biol. 246, 144–163 [DOI] [PubMed] [Google Scholar]
- 32. Walker O., Varadan R., and Fushman D. (2004) Efficient and accurate determination of the overall rotational diffusion tensor of a molecule from 15N relaxation data using computer program ROTDIF. J. Magn. Reson. 168, 336–345 [DOI] [PubMed] [Google Scholar]
- 33. Berlin K., Longhini A., Dayie T. K., and Fushman D. (2013) Deriving quantitative dynamics information for proteins and RNAs using ROTDIF with a graphical user interface. J. Biomol. NMR 57, 333–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sundlov J. A., Shi C., Wilson D. J., Aldrich C. C., and Gulick A. M. (2012) Structural and functional investigation of the intermolecular interaction between NRPS adenylation and carrier protein domains. Chem. Biol. 19, 188–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Haslinger K., Brieke C., Uhlmann S., Sieverling L., Süssmuth R. D., and Cryle M. J. (2014) The structure of a transient complex of a nonribosomal peptide synthetase and a cytochrome P450 monooxygenase. Angew. Chem. Int. Ed. Engl. 53, 8518–8522 [DOI] [PubMed] [Google Scholar]
- 36. Mitchell C. A., Shi C., Aldrich C. C., and Gulick A. M. (2012) Structure of PA1221, a nonribosomal peptide synthetase containing adenylation and peptidyl carrier protein domains. Biochemistry 51, 3252–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sundlov J. A., and Gulick A. M. (2013) Structure determination of the functional domain interaction of a chimeric nonribosomal peptide synthetase from a challenging crystal with noncrystallographic translational symmetry. Acta Crystallogr. D Biol. Crystallogr. 69, 1482–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Samel S. A., Schoenafinger G., Knappe T. A., Marahiel M. A., and Essen L. O. (2007) Structural and functional insights into a peptide bond-forming bidomain from a nonribosomal peptide synthetase. Structure 15, 781–792 [DOI] [PubMed] [Google Scholar]
- 39. Lee W., and Krishna N. R. (1992) Influence of conformational exchange on the 2D NOESY spectra of biomolecules existing in multiple conformations. J. Magn. Reson. 98, 36–48 [Google Scholar]
- 40. Worthington A. S., and Burkart M. D. (2006) One-pot chemo-enzymatic synthesis of reporter-modified proteins. Org. Biomol. Chem. 4, 44–46 [DOI] [PubMed] [Google Scholar]
- 41. Bodenhausen G., and Ruben D. J. (1980) Natural abundance nitrogen-15 NMR by enhanced heteronuclear spectroscopy. Chem. Phys. Lett. 69, 185–189 [Google Scholar]
- 42. Williamson M. P. (2013) Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 73, 1–16 [DOI] [PubMed] [Google Scholar]
- 43. Nguyen C., Haushalter R. W., Lee D. J., Markwick P. R. L., Bruegger J., Caldara-Festin G., Finzel K., Jackson D. R., Ishikawa F., O'Dowd B., McCammon J. A., Opella S. J., Tsai S.-C., and Burkart M. D. (2014) Trapping the dynamic acyl carrier protein in fatty acid biosynthesis. Nature 505, 427–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zimmermann S., Pfennig S., Neumann P., Yonus H., Weininger U., Kovermann M., Balbach J., and Stubbs M. T. (2015) High-resolution structures of the d-alanyl carrier protein (Dcp) DltC from Bacillus subtilis reveal equivalent conformations of apo- and holo-forms. FEBS Lett. 589, 2283–2289 [DOI] [PubMed] [Google Scholar]
- 45. De Crécy-Lagard V., Marlière P., and Saurin W. (1995) Multienzymatic non ribosomal peptide biosynthesis: identification of the functional domains catalysing peptide elongation and epimerisation. C. R. Acad. Sci. III 318, 927–936 [PubMed] [Google Scholar]
- 46. Weber T., and Marahiel M. A. (2001) Exploring the domain structure of modular nonribosomal peptide synthetases. Structure 9, R3–R9 [DOI] [PubMed] [Google Scholar]
- 47. Stachelhaus T., Hüser A., Marahiel M. A. (1996) Biochemical characterization of peptidyl carrier protein (PCP), the thiolation domain of multifunctional peptide synthetases. Chem. Biol. 3, 913–921 [DOI] [PubMed] [Google Scholar]
- 48. Mofid M. R., Finking R., and Marahiel M. A. (2002) Recognition of hybrid peptidyl carrier proteins/acyl carrier proteins in nonribosomal peptide synthetase modules by the 4′-phosphopantetheinyl transferases AcpS and Sfp. J. Biol. Chem. 277, 17023–17031 [DOI] [PubMed] [Google Scholar]
- 49. Kittilä T., Mollo A., Charkoudian L. K., and Cryle M. J. (2016) New structural data reveal the motion of carrier proteins in nonribosomal peptide synthesis. Angew. Chem. Int. Ed. Engl. 55, 9834–9840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Goodrich A. C., and Frueh D. P. (2015) A nuclear magnetic resonance method for probing molecular influences of substrate loading in nonribosomal peptide synthetase carrier proteins. Biochemistry 54, 1154–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Keller R. (2004) The Computer Aided Resonance Assignment Tutorial, Cantina Verlag, Goldau, Switzerland [Google Scholar]
- 52. Shen Y., Delaglio F., Cornilescu G., and Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shen Y., and Bax A. (2013) Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 56, 227–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Güntert P., Mumenthaler C., and Wüthrich K. (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273, 283–298 [DOI] [PubMed] [Google Scholar]
- 55. Güntert P. (2004) Automated NMR structure calculation with CYANA. Methods Mol. Biol. 278, 353–378 [DOI] [PubMed] [Google Scholar]
- 56. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., and Warren G. L. (1998) Crystallography & NMR System: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 57. Nederveen A. J., Doreleijers J. F., Vranken W., Miller Z., Spronk C. A. E. M., Nabuurs S. B., Güntert P., Livny M., Markley J. L., Nilges M., Ulrich E. L., Kaptein R., Bonvin A. M. J. J. (2005) RECOORD: a recalculated coordinate database of 500+ proteins from the PDB using restraints from the BioMagResBank. Proteins Struct. Funct. Genet. 59, 662–672 [DOI] [PubMed] [Google Scholar]
- 58. DeLano W. L. (2002) The PyMOL Molecular Graphics System, version 1.7.2, Schrödinger, LLC, New York [Google Scholar]
- 59. Koradi R., Billeter M., and Wüthrich K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 51–55 [DOI] [PubMed] [Google Scholar]
- 60. Bhattacharya A., Tejero R., and Montelione G. T. (2007) Evaluating protein structures determined by structural genomics consortia. Proteins 66, 778–795 [DOI] [PubMed] [Google Scholar]
- 61. Goddard T. D., and Kneller D. G.. SPARKY 3, University of California, San Francisco [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.