

Abstract
Platelets are the sole source of EGF in circulation, yet how EGF is stored or released from stimulated cells is undefined. In fact, we found platelets did not store EGF, synthesized as a single 6-kDa domain in pro-EGF, but rather expressed intact pro-EGF precursor on granular and plasma membranes. Activated platelets released high-molecular-weight (HMW)-EGF, produced by a single cleavage between the EGF and the transmembrane domains of pro-EGF. We synthesized a fluorogenic peptide encompassing residues surrounding the putative sessile arginyl residue and found stimulated platelets released soluble activity that cleaved this pro-EGF1020–1027 peptide. High throughput screening identified chymostatins, bacterial peptides with a central cyclic arginyl structure, as inhibitors of this activity. In contrast, the matrix metalloproteinase/TACE (tumor necrosis factor-α-converting enzyme) inhibitor GM6001 was ineffective. Stimulated platelets released the soluble protease ADAMDEC1, recombinant ADAMDEC1 hydrolyzed pro-EGF1020–1027, and this activity was inhibited by chymostatin and not GM6001. Biotinylating platelet surface proteins showed ADAMDEC1 hydrolyzed surface pro-EGF to HMW-EGF that stimulated HeLa EGF receptor (EGFR) reporter cells and EGFR-dependent tumor cell migration. This proteolysis was inhibited by chymostatin and not GM6001. Metabolizing pro-EGF Arg1023 to citrulline with recombinant polypeptide arginine deiminase 4 (PAD4) abolished ADAMDEC1-catalyzed pro-EGF1020–1027 peptidolysis, while pretreating intact platelets with PAD4 suppressed ADAMDEC1-, thrombin-, or collagen-induced release of HMW-EGF. We conclude that activated platelets release ADAMDEC1, which hydrolyzes pro-EGF to soluble HMW-EGF, that HMW-EGF is active, that proteolytic cleavage of pro-EGF first occurs at the C-terminal arginyl residue of the EGF domain, and that proteolysis is the regulated and rate-limiting step in generating soluble EGF bioactivity from activated platelets.
Keywords: ADAM, epidermal growth factor (EGF), growth factor, metalloprotease, platelet
Introduction
EGF is the prototypical growth-promoting cytokine (1, 2) but also has well established roles in stimulating tumor-initiating stem cells and tumorigenesis (3). Platelets receive immunoreactive EGF from megakaryocytes during thrombopoiesis (4), with submaxillary glands and kidney the major sources of EGF message (5, 6). Activated platelets (7–9) are the sole source of EGF in the circulation (10–13). Cytokines, growth factors, and small molecules released from activated platelets promote growth and migration of vascular cells (1, 10), and EGF accounts for a portion of the salutary effects of this material on endothelial cell proliferation (14), macrophage activation in atherosclerotic plaques (15), and vessel remodeling (16). However, EGF release from platelets is significantly delayed relative to degranulation (7, 9, 17), leaving EGF storage and release from its only source in the circulation uncharacterized.
EGF is synthesized as a single 6-kDa domain within its ∼140-kDa glycosylated pro-EGF precursor (Fig. 1A), with the EGF domain separated from the single transmembrane domain by a short 10-residue spacer sequence (1, 2). Indeed, all EGF family members are synthesized as single domains of their proproteins, although there is no conserved sequence defining the amino or carboxyl proteolytic sites that flank the growth factor domains. In general, the P1 sessile residues (cleavage occurs between the P1 and P1′ residues) in these growth factor precursors tend to be small or hydrophobic (1, 2, 18). These types of residues are preferred by the matrix metalloproteinase family, and membrane-bound ADAM102 and ADAM17 (TACE) of the ADAM subfamily of matrix metalloproteinases proteolyze and solubilize most pro-EGF family members (19–21). However pro-EGF itself is a poor substrate for these ADAM proteases.
Figure 1.

Platelets express pro-EGF, not EGF, and release high-molecular-weight EGF after activation. A, schematic representation of the structural features of 1201-residue pro-EGF (UniProtKP-P01133), high-molecular-weight-EGF (HMW-EGF), and 6-kDa EGF. Also shown are sequences surrounding the sessile Arg972 and Arg1023 bonds whose protolysis releases 6-kDa EGF (2). The EGF sequence is separated by 10 residues from the single transmembrane domain. B, density gradient separation of platelet components. Quiescent platelets were homogenized and centrifuged to density equilibrium in a sucrose gradient, with fractions collected from low to high density. Protein in individual fractions was solubilized by SDS, resolved by gradient gel electrophoresis, transferred, and immunoblotted for total EGF, ADAMDEC1, or P-selectin as described under “Experimental Procedures.” n = 3. C, platelets display EGF immunoreactivity on their surface. Washed human platelets were fixed with formaldehyde and stained with anti-EGF or isotype-matched non-immune antibody, and fluorescence of Alexa Fluor 488-conjugated secondary antibody was assessed by flow cytometry (n = 3) in a platelet gate defined by forward and side scatter. D, stimulated platelets release soluble HMW-EGF over time. Washed platelets were treated with buffer, thrombin (0.2 units/ml), ADP (5 μm), or the PAR1 thrombin receptor agonist TRAP6 (30 μm) for the stated times before platelets were cleared by centrifugation. Supernatant protein was precipitated with cold acetone, and total EGF was resolved and detected by Western blotting. n = 3. E, stimulated platelets release only HMW-EGF. Washed platelets were treated with the stated agonists up to 3 h with buffer, collagen (2 μg/ml), ADP (5 μm), TRAP6 (30 μm), or 20 ng/ml EGF before immunoreactive EGF in the supernatant was detected by Western blotting EGF in a gradient gel as in the preceding panel. Fully processed EGF was present in the supernatants only after the addition of recombinant EGF as an agonist. n = 3. F, platelet-derived immunoreactive EGF is primarily HMW-EGF. Supernatants from thrombin-activated platelets (after FPR-CMK inhibition of thrombin activity) were divided by filtration through a 50-kDa cut-off filter, with the retained material diluted with Hanks' buffer to the original volume. HeLa cells, as EGFR reporters, were stimulated with identical volumes of these two fractions for the stated times before the HeLa cell proteins were solubilized and resolved by SDS-PAGE. EGFR phosphorylated on tyrosine 1068 was detected by Western blotting. n = 3. G, stimulated platelets release only a small portion of their total pro-EGF. Platelets were stimulated with the stated agonists for the stated times before soluble material was recovered by centrifugation, denatured in SDS, and resolved by electrophoresis in a 4–20% gradient gel before EGF immunoreactivity was detected by Western blotting. n = 3.
Incompletely understood events activate membrane-bound proteases that then become competent to solubilize membrane-bound pro-EGF family members by cleavage between the cytokine domain and a spacer sequence that separates the EGF family member from the transmembrane domain. There is a single soluble member of the ADAM family, ADAMDEC1 (a disintegrin and metalloproteinase domain-like protein decysin-1) (22, 23), that would overcome the stearic difficulty posed by the juxtamembrane cleavage of a membrane-bound growth factor precursor by a membrane-bound protease, but this enzyme has a mutated Zn+2-binding site (23) and displays only dampened proteolytic activity (24).
Pro-EGF differs from its family members in that the P1 residue for both the N-and C-terminal cleavage is the positively charged residue arginine (2). In fact, the first purification of EGF by Cohen and co-workers in the 1960s and 1970s found both low and high-molecular-weight forms of EGF (25) along with a co-purifying arginyl esterase that was “postulated to function in the enzymatic liberation of EGF from a precursor” (26). The identity of this protein remains obscure, but because platelets are the source of EGF in the circulation, then platelets or their megakaryocyte precursors should contain a protease to process pro-EGF to active growth factor whether or not it is the previously purified arginyl esterase. The reduced proteome of platelets contains just four ADAM protease family members including ADAM10 and ADAM17 (27, 28), suggesting that one of these four proteases hydrolyzes the sessile arginyl bonds of pro-EGF to form active cytokine.
We determined how platelets store and release EGF to find that platelets do not actually store fully processed and soluble EGF but, rather, expressed the pro-EGF precursor on their surface and in granules. Activated platelets released soluble ADAMDEC1 that proteolyzed surface pro-EGF at the appropriate sessile arginyl residue in the spacer sequence to generate soluble high-molecular-weight (HMW)-EGF. HMW-EGF was an effective ligand for its EGF receptor (ErbB1, Her1) and promoted migration and invasion of untransformed head-and-neck tumor cells.
Results
Platelets express membrane-bound pro-EGF and release HMW-EGF after activation
Platelets are the primary source of EGF in the circulation, but how stimulated platelets release this EGF to their environment is undefined. We used density gradient centrifugation of platelet lysates and Western blotting to determine which platelet compartments contain EGF and in what form. Immunoblotting showed that platelets contained full-length pro-EGF in plasma membrane and granule fractions identified by the marker P-selectin (CD62P) (Fig. 1B). Pro-EGF is a single pass transmembrane protein, and flow cytometry confirmed immunoreactive EGF was displayed on the surface of quiescent platelets (Fig. 1C).
We found unstimulated platelets released little immunoreactive EGF over time but that HMW-EGF appeared in the media after stimulation by either the weak agonist ADP, the strong agonist thrombin, or the PAR1 (the primary platelet thrombin receptor)-specific peptide agonist TRAP6 (Fig. 1D). Ultracentrifugation showed that ∼95% of the EGF released from activated platelets was soluble protein not associated with microparticles (not shown). Recombinant 6-kDa EGF itself induced release of HMW-EGF from platelets that was equivalent to that released by ADP or collagen (Fig. 1E). This experiment also shows, because 6-kDa EGF was only detected after the addition of recombinant EGF, that we could have captured and detected this small protein in platelet supernatants had it been present. Thus, stimulated platelets released only HMW-EGF, even hours after stimulation, and not fully processed EGF.
We confirmed the primary EGFR agonist released from activated platelets was a high-molecular-weight protein by size exclusion filtration. We divided the material released from activated platelets into material > or <50 kDa by filtration to find that the majority of the activity released from thrombin-activated platelets was high-molecular-weight material (Fig. 1F). We also determined what portion of platelet pro-EGF was released after activation to find that stimulated platelets release only a small portion of their total pro-EGF (Fig. 1G).
Stimulated platelets release pro-EGF proteolytic activity
Platelets express EGF receptor (EGFR, not shown), and we found that thrombin-stimulated platelets released agonists to their supernatants that subsequently induced EGFR autophosphorylation in naïve platelets (Fig. 2A). Release of EGFR agonistic activity was evident by 5 min of stimulation and remained elevated by 10 min relative to material recovered from unstimulated platelets. Solubilization of EGFR agonists from pro-EGF displayed on the platelet surface over time suggests extracellular proteolysis, so we determined whether platelets released a soluble protease using a TNFα sequence suitable for screening ADAM family members (29). We found activated platelets released hydrolytic activity(ies), which cut-off filters indicated had an apparent size between 35 and 100 kDa (not shown). We sought a way to implicate this activity in EGF solubilization by performing a high throughput screen of the Lopac library of biologically active compounds using this peptide substrate to find that chymostatin inhibited the platelet-derived peptidase (not shown).
Figure 2.
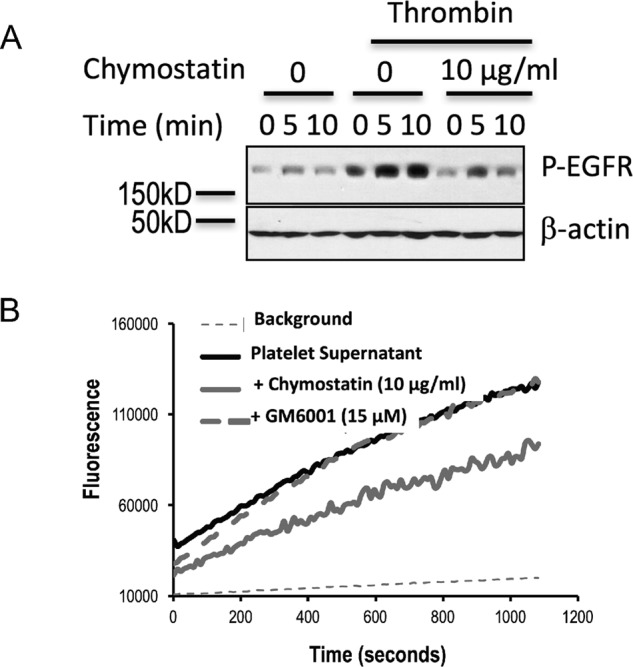
Thrombin-stimulated platelets release a chymostatin-inhibitable pro-EGF hydrolytic activity and EGF bioactivity. A, chymostatin suppresses release of active EGF from thrombin-stimulated platelets. Platelets were preincubated with or without chymostatin (10 μg/ml, 2 h) and simulated with thrombin (0.05 units) for 16 h before thrombin was irreversibly inhibited with FPR-CMK, and platelets were cleared by centrifugation. HeLa EGFR reporter cells were then incubated with the resulting cell-free media for the stated times before phospho-EGFR and total β-actin was assessed by Western blotting. n = 2. B, platelets release a pro-EGF endopeptidase. Hydrolysis of fluorogenic pro-EGF1020–1027 peptide carboxymethylfluorescein-Trp-Glu-Leu-Arg-His-Ala-Gly-His-Lys(DABCYL)-Gly over time by supernatants from thrombin-stimulated platelets in the presence of chymostatin or GM6001. n = 4.
The externally disposed EGF domain in pro-EGF is separated from the transmembrane domain by a 10-residue spacer sequence (Fig. 1A), limiting the region for proteolytic solubilization. We thus synthesized a fluorogenic pro-EGF peptide centered on the physiologic Arg1023 cleavage site in pro-EGF. This peptide included the final three carboxyl residues of the EGF domain, the sessile residue Arg1023, and then the three initial residues of the spacer sequence. We included a fluorescent amino carboxylmethylfluorescein group and a carboxyl DABCYL function to internally quench peptide fluorescence. We found supernatants of activated platelets released a peptidolytic activity that hydrolyzed this fluorogenic pro-EGF peptide (Fig. 2B). This activity, like TNFα peptide hydrolysis, was not inhibited by the matrix metalloproteinase/ADAM inhibitor GM6001 even at a concentration more than twice that necessary to fully block platelet ADAM17 (TACE) (30). We also found that chymostatin reduced release of biologically active EGF from naïve platelets that had been treated with supernatants of thrombin-activated platelets (after irreversible inactivation of thrombin) (Fig. 2A). This chymostatin inhibition of EGFR ligand release from activated platelets confirms that activation-dependent proteolysis occurs at the time of EGFR ligand release.
Activated platelets release the protease ADAMDEC1
Platelets express four ADAM protease family members including 53-kDa ADAMDEC1 (27, 28), but ADAMDEC1 is the only soluble (23) protease in this family. We found ADAMDEC1 floated in the granule and plasma membrane fractions during density gradient centrifugation (Fig. 1B), localizing with pro-EGF. Little ADAMDEC1 was released from quiescent platelets or those stimulated with the weak agonist ADP, but stimulation with thrombin led to the rapid and time-dependent release of ADAMDEC1 into the media (Fig. 3A). ADAMDEC1 was also released in response to the TRAP6 peptide, showing the PAR1 receptor alone, although less efficient than thrombin, was a sufficient stimulus to induce release of this protease.
Figure 3.
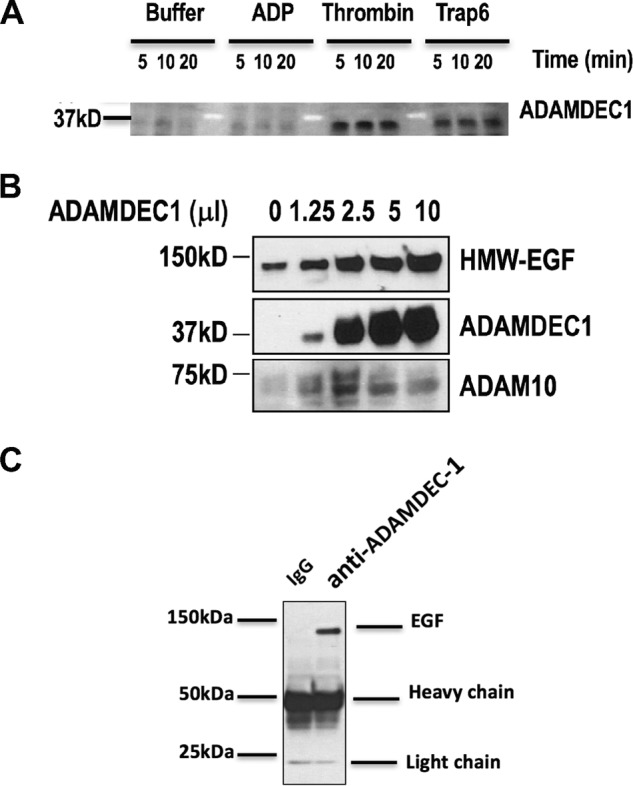
Platelets release ADAMDEC1 that associates with HMW-EGF. A, activated platelets release ADAMDEC1. Washed human platelets were stimulated or not with the stated agonists for the stated times, the cells were cleared by centrifugation, and the supernatants were immunoblotted for ADAMDEC1. n = 3. B, recombinant ADAMDEC1 hydrolyzes high-molecular-weight EGF from platelets. The stated volumes of media concentrated 10-fold from CHO cells transfected with ADAMDEC1-FLAG were incubated with freshly isolated quiescent platelets and incubated (20 min) before the cells were cleared by centrifugation, and soluble proteins acetone-precipitated, resolved by SDS-PAGE, and Western-blotted for EGF (top), ADAMDEC1 (middle), or ADAM10 (bottom). n = 3. C, ADAMDEC1 physically associates with HMW-EGF. Recombinant ADAMDEC1-FLAG was incubated (30 min) with quiescent platelets, the cells were removed by centrifugation, and nonspecific proteins were cleared with protein G beads. These supernatants were incubated with anti-ADAMDEC1 or non-immune antibody, the antibody was captured by protein G beads, and protein was then eluted by SDS and resolved by gradient SDS-PAGE before immunoblotting for EGF. n = 2.
We determined whether ADAMDEC1 was an EGF sheddase by treating quiescent platelets with increasing concentrations of recombinant ADAMDEC1 and recovering solubilized material after this exposure. We found (Fig. 3B) ADAMDEC1 induced release of HMW-EGF and that the amount of HMW-EGF in the media corresponded to the level of ADAMDEC1 in the incubation. Conversely, the abundance of the family member ADAM10, which is abundant in platelets (27, 28), did not correlate to EGF release.
The original purification of EGF shows it physically associates with an uncharacterized arginyl esterase (26) presumed to proteolyze pro-EGF at the N- and/or C-terminal arginyl residues of the EGF domain (31). This is an unusually strong interaction between a protease and its protein target, suggesting that other proteases hydrolyzing pro-EGF may also form a tight complex with this substrate. To test this we immunoprecipitated recombinant ADAMDEC1 after incubation with quiescent platelets. We found, by blotting the recovered proteins with anti-EGF antibody, that recombinant ADAMDEC1 co-immunoprecipitated with the HMW-EGF it solubilized from quiescent platelets (Fig. 3C).
ADAMDEC1 is a chymostatin-sensitive pro-EGF protease
We determined whether recombinant ADAMDEC1 proteolyzed the fluorogenic pro-EGF peptide, like the activity released from stimulated platelets, to find that it did so, and that this peptidolysis also was inhibited by increasing concentrations of chymostatin (Fig. 4A). Also like the activity(ies) released from activated platelets, this hydrolysis was not inhibited by GM6001. We next determined whether ADAMDEC1 was able to act on the pro-EGF displayed on the platelet surface to generate soluble EGFR agonistic activity. To do this, we treated quiescent platelets with increasing concentrations of recombinant ADAMDEC1, cleared the cells by centrifugation, concentrated the resulting supernatants over 10-kDa cutoff filters, and then diluted this material into media overlaying HeLa EGFR reporter cells. The reporter cells were lysed after increasing amounts of time before EGFR tyrosine autophosphorylation was determined by Western blotting. This experiment showed that ADAMDEC1 solubilized EGFR agonists from resting platelets in a concentration-dependent way and that the HMW-EGF solubilized by ADAMDEC1 was an active EGFR agonist (Fig. 4B). ADAMDEC1 solubilization of active EGFR ligand, like platelet-derived activity, was sensitive to chymostatin but not the potent ADAM inhibitor GM6001 (Fig. 4C).
Figure 4.

Recombinant ADAMDEC1 releases active HMW-EGF from quiescent platelets in a chymostatin-dependent way. A, ADAMDEC1 hydrolyzes a fluorogenic pro-EGF peptide and is inhibited by chymostatin. The fluorogenic pro-EGF1020–1027 peptide was incubated with recombinant ADAMDEC1 with increasing concentrations of chymostatin or 25 μm GM6001 during peptidolysis. n = 3. *, p < 0.05. B, recombinant ADAMDEC1 solubilized active EGF from platelets. Quiescent platelets were treated (30 min) with the stated amounts of chimeric ADAMDEC1-FLAG before platelets were cleared by centrifugation. The resulting supernatants were incubated with HeLa cells for the stated times before HeLa cell phospho-EGFR was assessed by Western blotting with a mix of five EGFR phosphotyrosine (top) or β-actin (bottom) antibodies. n = 2. ADAMDEC1 itself did not stimulate HeLa EGFR (not shown). C, chymostatin, but not GM6001, inhibits ADAMDEC1 solubilization of active EGF from platelets. Platelets were treated (30 min) with buffer or ADAMDEC1-FLAG in the presence of chymostatin (10 μg/ml) or the matrix metalloproteinase inhibitor GM6001 (25 μm), and the supernatants were cleared of platelets by centrifugation and then incubated with HeLa cells for the stated times. HeLa cell EGFR tyrosine phosphorylation or β-actin content was assessed as in the preceding panel. n = 3. D, recombinant ADAMDEC1 releases biotinylated EGF from the platelet surface. Platelet surface proteins were labeled with an impermeant biotin derivitization reagent, washed, and incubated (30 min) with the stated volume of media from CHO cells or CHO cells transfected with ADAMDEC1-FLAG in the absence or presence of 10 μg/ml chymostatin. Material in the platelet supernatant was captured with avidin beads, stripped with SDS before the recovered material was resolved by SDS-PAGE, and immunoblotted for EGF. n = 2.
Pro-EGF localized in at least two pools in platelets, so to determine whether exogenous ADAMDEC1 hydrolyzes plasma membrane-associated pro-EGF, we biotinylated the surface proteins of quiescent platelets. These cells were then treated with ADAMDEC1 in the presence or absence of chymostatin before recovering solubilized biotinylated proteins by streptavidin capture and then Western blotting these proteins for EGF. These data confirm that surface pro-EGF is accessible to ADAMDEC1 (Fig. 4D) and that the hydrolytic product of the externally facing pro-EGF precursor is HMW-EGF.
ADAMDEC1 proteolyzes pro-EGF at the proposed C-terminal sessile arginyl bond to form HMW-EGF
The polypeptide arginine deiminase (PAD) released by Porphyromonas gingivalis inactivates 6-kDa EGF by metabolizing the C-terminal Arg1023 of fully processed EGF to a citrulline residue (32). This Arg1023 residue is the proposed proteolytic site in pro-EGF that yields HMW-EGF (33) and so was the central residue in the fluorogenic pro-EGF1020–1027 peptide. We, therefore, determined whether recombinant neutrophil PAD4 would target this arginyl residue in the context of the pro-EGF sequence. We found pretreating the fluorogenic peptide with increasing amounts of PAD4 inhibited and ultimately abolished subsequent ADAMDEC1 hydrolysis of the peptide (Fig. 5A). ADAMDEC1 hydrolysis of the pro-EGF peptide thus required Arg1023 and so only cleaves pro-EGF at its physiologic hydrolytic site.
Figure 5.
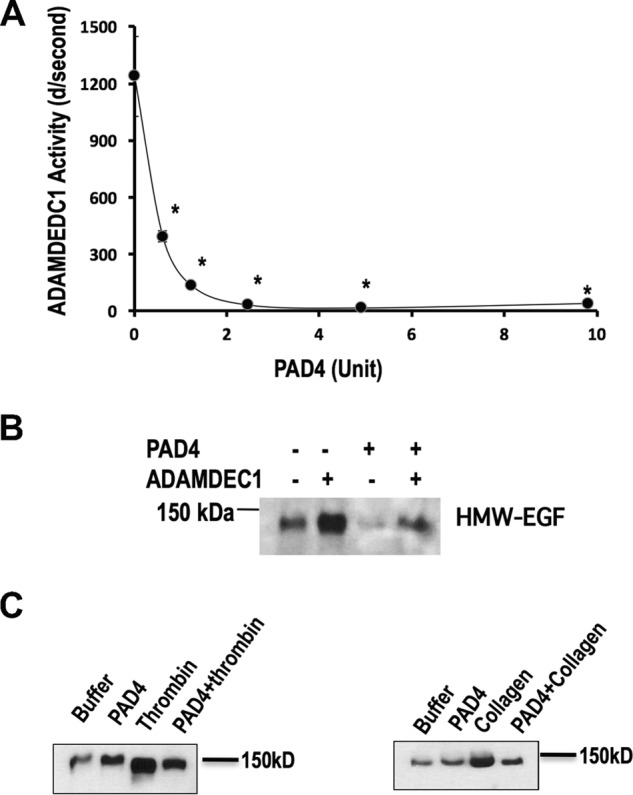
ADAMDEC1 required pro-EGF Arg1023 for peptidolysis and pro-EGF hydrolysis. A, ADAMDEC1 cleavage of pro-EGF1020–1027 peptide requires Arg1023. Fluorogenic pro-EGF1020–1027 peptide was preincubated (1 h) with the stated amount of recombinant PAD4 activity per 100-μl reaction before the addition of recombinant ADMADEC1 before the initial hydrolytic rates of the pro-EGF1020–1027 fluorogenic peptide were assessed by fluorimetry. n = 3. *, p < 0.05. B, PAD4 pretreatment suppressed ADAMDEC1 proteolysis of platelet pro-EGF. Washed platelets were treated with recombinant PAD4 (13 μg/ml, 45 min), recovered by centrifugation, and then treated with recombinant ADAMDEC1 (20 min) before cells were cleared by high speed centrifugation. Supernatant proteins were acetone-precipitated, resolved by gradient SDS-PAGE, and immunoblotted for EGF. C, PAD4 pretreatment suppressed HMW-EGF release from stimulated platelets. Washed platelets were treated with a recombinant PAD4 (50 μg/ml of a new lot, 45 min), recovered by centrifugation, and then stimulated with 0.05 units/ml thrombin or 2 μg/ml collagen for 5 min. The cells were then cleared by high speed centrifugation, and proteins in the remaining supernatant were acetone-precipitated, resolved by gradient SDS-PAGE, and immunoblotted for EGF as in the proceeding panel. n = 2.
We determined whether arginyl residues of pro-EGF were critical for ADAMDEC1-induced release of HMW-EGF from platelets. We pretreated unstimulated platelets with recombinant PAD4 before incubation with recombinant ADAMDEC1 to find this impaired release of HMW-EGF (Fig. 5B).
We then tested whether recognition of arginyl residues contributes to proteolysis of platelet pro-EGF by endogenous proteolytic activity. We pretreated quiescent platelets with recombinant PAD4 and then stimulated the cells, or not, with either a low concentration of thrombin or soluble collagen. We found PAD4 pretreatment reduced HMW-EGF release in response to each agonist (Fig. 5C), so arginyl recognition also aids stimulated pro-EGF proteolysis by platelet-derived activity(ies).
ADAMDEC1 proteolysis of platelet pro-EGF promotes tumor cell migration
Head and neck squamous cell carcinomas (HNSCC), like many tumors, require EGFR activation and signaling (34), with EGFR expression correlating with progression and survival (35). Accordingly, the humanized monoclonal anti-EGFR antibody Cetuximab® is the first therapy found to increase survival of these patients (36). The H1 subline of the poorly differentiated HNSCC SAS cell line is highly mobile (37), but we found was unable to penetrate through Matrigel® if this barrier was depleted of growth factors (Fig. 6A). Supernatants from thrombin-stimulated platelets (after FPR-CMK inhibition of residual thrombin activity), however, markedly enhanced penetration of these tumor cells through the Matrigel®/filter barrier. Invasion in response to these supernatants from activated platelets was abolished by inhibition of SAS-H1 EGFR tyrosine kinase by AG1478. The selective and irreversible EGFR inhibitor CL387785 (38), the clinically used anti-EGFR humanized monoclonal antibody Cetuximab, and sequestration with anti-EGF antibody also proved effective inhibitors of SAS-H1 tumor cell penetration into and through the Matrigel® barrier. As anticipated, these inhibitors also suppressed EGFR signaling induced by material released from thrombin-stimulated platelets as shown by their suppression of the autophosphorylation of EGFR in HeLa reporter cells (Fig. 6B). Thus, stimulated platelets release a functional EGF agonist(s) that alters HNSCC tumor cell migration through the EGFR to augment tumor cell invasion (39).
Figure 6.
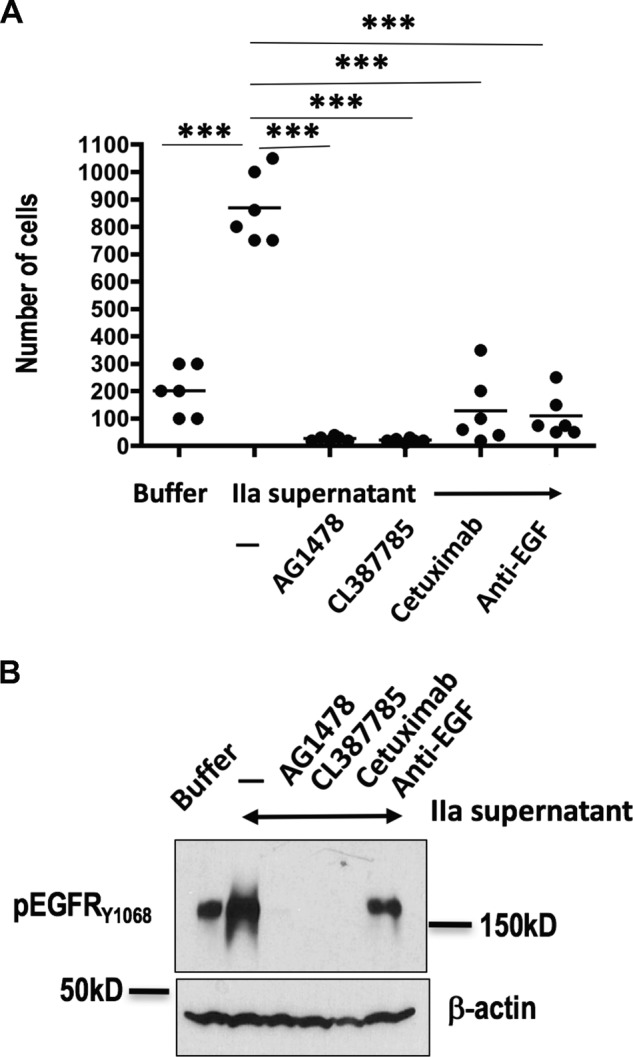
Platelet-derived HMW-EGF is functional. A, platelet-derived HMW-EGF promotes tumor cell invasion and migration. Washed platelets were incubated with thrombin (0.05 units/ml, 37°) overnight and cleared by centrifugation before supernatants were concentrated by filtration. Supernatants or buffer was added to SAS-H1 head-and-neck tumor cells in the upper well of protein-depleted Matrigel® Bio-Coat PET membrane chambers wells along with EGFR inhibitors (AG1478, 20 μm; CL387785, 20 μm; humanized anti-EGFR monoclonal antibody Cetuximab, 2 μg/ml), or anti-EGF antibody (2 μg/ml) as shown. Cells that had migrated through the barrier after 48 h were solubilized with trypsin and enumerated with a hemocytometer (n = 2 with triplicate wells with data combined for analysis by one-way non-parametric analysis of variance Kruskal-Wallis testing with post-hoc Dunn's testing). ***, p < 0.0001. B, EGFR ligand released from thrombin-activated platelets is EGF. HeLa cells were stimulated (10 min) with buffer or concentrated supernatants of thrombin-stimulated platelets after pretreatment (20 min) with EGFR inhibitors or anti-EGF at concentrations used in the preceding panel. Recovered HeLa cells were lysed and their proteins solubilized in SDS and then separated by electrophoresis before detecting anti-EGFR phosphorylated on tyrosine 1068 by Western blotting. n = 2.
Discussion
Platelets are the only cells in the circulation that contain EGF (10–12) and indeed are a primary source of this growth factor outside kidney (5, 6). How platelets store and release this EGF, however, remains opaque as its release is significantly delayed relative to the rapid and complete release of the proteins and small molecules stored in platelet alpha and dense granules (7, 9, 11, 17). We elucidate a basis for hysteresis in EGF release by finding that platelets do not store processed, soluble EGF but rather contain its membrane-bound precursor pro-EGF. Fractionating platelet lysates in density gradients showed a portion of pro-EGF did localize to granule fractions but also that pro-EGF was associated with light, cholesterol-containing plasma membrane fractions. Flow cytometry of intact cells showed the presence of an externally disposed pool of pro-EGF, while biotinylation of the platelet surface shows this pool contributes, although it may not be the sole source, to platelet-derived EGF.
Prior work (12, 40) establishes that serum primarily contains HMW-EGF and, because plasma lacks immunoreactive EGF, the serum HMW-EGF is platelet-derived. In accordance with this, we observed stimulated platelets released immunoreactive EGF and that this growth factor was exclusively HMW-EGF rather than fully processed 6 kDa EGF. Solubilization of membrane pro-EGF to HMW-EGF reflects proteolysis at a juxtamembrane site, and we found stimulated platelets released a protease to their environment with the capacity to hydrolyze exofacial pro-EGF to soluble HMW-EGF. Notably, the time-dependent release of HMW-EGF represented only a small portion of the total pro-EGF of platelets, both highlighting the difference in its release from material stored in granules and showing that platelets entrapped in thrombi can function as a protracted source of vascular EGF. We also found that this platelet-derived HMW-EGF is a functional EGFR ligand (41) and was competent to stimulating tumor cell migration and invasion through a Matrigel® barrier.
The seven functional members of the EGF family (1, 42) are expressed as membrane-bound pro-proteins that are proteolyzed to soluble, active factors (18), with the proteases ADAM10 and ADAM17 able to catalyze this event for six family members (21). In contrast, ADAM17 appears unable to solubilize pro-EGF (33), whereas ADAM10 did not contribute to stimulated release of a chimeric EGF-alkaline phosphatase reporter in an ex vivo model system (21). Notably, the N- and C-terminal sessile residues of the EGF domain in pro-EGF are arginyl residues, which is unique in the pro-EGF family (18), and neither ADAM10 nor ADAM17 preferentially cleave after arginyl residues (43). In accordance with this, we found the small molecule GM6001, which effectively inhibits platelet ADAM10 (44) and platelet ADAM17 (30) enzymatic activity, did not reduce ADAMDEC1-catalyzed pro-EGF peptidolytic activity or its pro-EGF proteolytic activity that released active EGFR ligand. Prior work does, however, suggest that some member of the zinc metalloproteinase family is responsible for stimulated shedding of the pro-EGF ectodomain (33).
ADAMDEC1 is a member of the ADAM metalloproteinase family but an aspartyl residue substitutes for an invariant histidyl residue of the consensus zinc-binding active site of matrix metalloproteinases (22, 23). This aspartyl substitution can yield a functional enzyme (23), but ultimately ADAMDEC1 is a poorly performing general protease that lacks a defined physiologic role (24, 45). A single nucleotide polymorphism in its gene does, however, correlate with a propensity for thrombosis (46), while its genetic ablation shows the enzyme aids intestinal immunity (47).
Platelets express a thousand copies of ADAMDEC1 per cell (27, 28), and because its gene lacks the transmembrane domain of the ADAM family, it is predicted to be (22, 23), and is (24, 45), soluble. Accordingly, Western blotting (above) and mass spectrometry (28) show ADAMDEC1 was released from platelets after stimulation. This ADAMDEC1 has access to platelet pro-EGF because it solubilized biotinylated surface pro-EGF and because recombinant enzyme co-immunoprecipitated with the HMW-EGF it solubilized from platelet membranes. This unusually robust protease-substrate interaction is consistent with the original purification of EGF by Cohen and coworkers (25) resulting in the 1986 Nobel prize for Physiology or Medicine that recovered low- and high-molecular-weight forms of EGF with the latter including a co-purifying binding protein (26). This 30-kDa binding protein displayed arginine esterase activity and was “postulated to function in the enzymatic liberation of active EGF from an inactive precursor” (26). Although this enzyme is unlikely to be ADAMDEC1, as submaxillary glands are not known to express this protease (48), it does suggest EGF can form a stable interaction with arginyl-directed esterases.
We found both platelet-derived material and recombinant ADAMDEC1 hydrolyzed active pro-EGF from platelet membranes, so ADAMDEC1 can function as an EGF sheddase. We then sought to determine whether ADAMDEC1 is a physiologically relevant EGF sheddase by screening the Lopac chemical library for inhibitors. This identified chymostatin, a mixture of related bacterial peptides featuring a central cyclic arginyl structure that inhibited the hydrolysis of a general ADAM family substrate (29) by activity released from activated platelets. The effectiveness of this competitive inhibitor will vary with substrate abundance, but chymostatin, with its highly restricted inhibitory profile (49), proved to effectively inhibit ADAMDEC1 hydrolysis of a pro-EGF peptide encompassing the sessile Arg1023 residue and suppress HMW-EGF release from platelets treated with recombinant ADAMDEC1. There should be, however, an additional element necessary for ADAMDEC1 proteolysis as members of the matrix metalloproteinase family are synthesized as inactive proproteins regulated by a cysteine switch but also a range of other molecules (50). We do not know whether, or how, platelet ADAMDEC1 becomes proteolytically competent after platelet activation. We also have not established whether ADAMDEC1 and pro-EGF physically interact in unactivated platelets, so we do not know why platelets do not hydrolyze their pro-EGF over time and then primarily store soluble EGF.
Arginine deimination can modify the Arg1023 residue in the context of EGF sequence because P. gingivalis PAD inactivates EGF by catabolizing this residue to a citrulline residue (32). Catabolism of this residue by recombinant neutrophil PAD4 showed this nascent C-terminal residue of the EGF domain was essential for ADAMDEC1 hydrolysis of the fluorogenic pro-EGF peptide and that in its absence ADAMDEC1 did not proteolyze other sites in the peptide. We then used recombinant PAD4 to pretreat intact platelets to determine whether recombinant ADAMDEC1 required the presence of the pro-EGF Arg1023 residue to solubilize pro-surface EGF. We observed that PAD4 pretreatment effectively suppressed subsequent ADAMDEC1-induced HMW-EGF release. Activated or dying neutrophils release PAD4 (51–54), suggesting the potential for a novel mode of neutrophil-platelet interaction in inflammation able to dampen vascular remodeling and tumor cell vasculogenesis.
Our data show the slow step in EGF release from activated platelets is proteolysis of pro-EGF by, at least in part, released ADAMDEC1 and not simply delayed release of stored, fully processed soluble growth factor. Platelet-derived HMW-EGF effectively stimulated HNSSC tumor cell penetration through a Matrigel® gel barrier and then their migration through a transwell filter, defining a way for activated platelets to contribute to tumorigenesis. This source of EGF will, as well, contribute to endothelial cell proliferation (14), macrophage migration (15), and vasculature maintenance (16). Platelet activation and initiation of thrombus formation are rapid events, with release of stored ADP and newly synthesize thromboxane A2 augmenting platelet recruitment into developing thrombi (55). In contrast, EGF expression is delayed relative to release by degranulation of materials stored in platelet granules. Activated platelets in thrombi do, however, communicate with vascular cells over prolonged times as shown by platelet IL-1β synthesis and release over hours in occluded carotid arteries (56). Similarly, HMW-EGF solubilization over prolonged times from activated platelets entrapped in thrombi is positioned to contribute to normal wound repair but also to tumorigenesis through EGFR stimulation of normal vascular and epithelial cells as well as tumor and tumor stem cells (57).
Experimental procedures
Chemicals and reagents
Endotoxin-free human serum albumin (25% solution) was from Baxter Healthcare. Anti-EGF, anti-pEGFR, and horseradish peroxidase-conjugated secondary antibody were from Cell Signaling Technology (Danvers MA). EGF was obtained either from R&D Systems (Minneapolis MN) or Cell Signaling Technology, ADAMDEC1 or ADAMDEC1-FLAG cDNA was from Origene (Rockville MD), and CL387785 was from Selleckchem (Houston, TX). PAD4 protein was from Cayman Chemical (Ann Arbor MI). Cetuximab (Bristol-Meyers Squibb) was excess material remaining from clinical procedures. Protease inhibitor mix was from Roche Diagnostics, with sulfo-NHS-biotin supplied by Thermo (Waltham, MA). Media and sterile filtered Hanks' balanced salt solution were prepared by the Cleveland Clinic LRI media preparation core. Size filters (50 kDa) were from EMD Millipore (Billerica, MA). The thrombin inhibitor Phe-Pro-Arg-chloromethyl ketone (FPR-CMK) was from Hemalogic Technologies (Essex Junction MA). The fluorogenic pro-EGF1020–1027 peptide carboxymethylfluorescein-Trp-Glu-Leu-Arg-His-Ala-Gly-His-Lys (DABCYL)-Gly was synthesized and analyzed by the Cleveland Clinic Molecular Technology core. The pro-TNFα peptide DABCYL-Leu-Ala-Gln-Ala-Homophe-Arg-Ser-Lys-(5-carboxyfluorescein) was from Enzo Life Science (Farmingdale NY). BioCoat growth factor-reduced Matrigel® invasion chambers were from Corning (Tewksbury MA). Thrombin, AG1478, PGE1, SDS, chymostatin (a mixture of A, B, and C peptides), GM6001, the PAR1 peptide TRAP6, and all other reagents were from Sigma.
Platelet preparation
Human blood was drawn into acid-citrate-dextrose and centrifuged (200 × g, 20 min) without braking to obtain platelet-rich plasma. This protocol was approved by the Cleveland Clinic Institutional Review Board. Purified platelets were prepared from this platelet-rich plasma as in the past (58), where platelet-rich plasma was filtered through 2 layers of 5-μm mesh (BioDesign) to remove nucleated cells and recentrifuged (520 × g, 30 min) in the presence of 100 nm PGE1. The pellet was resuspended in 50 ml PIPES/saline/glucose (5 mm PIPES, 145 mm NaCl, 4 mm KCl, 50 μm Na2HPO4, 1 mm MgCl2, and 5.5 mm glucose) containing 100 nm PGE1. These cells were centrifuged (520 × g, 30 min), and the recovered washed platelets were resuspended in 0.5% human serum albumin in Hanks' balanced salt solution.
Western blotting
Washed platelets (4 × 108/ml) were treated or not with the stated agonists, and the cells were lysed, solubilized in SDS, and resolved by SDS-PAGE gels for Western blotting. For some experiments, soluble proteins were acetone-precipitated before Western blotting. The proteins were resolved by a SDS-PAGE in gradient 4–20% cross-linked gels, and the proteins were transferred to PVDF and probed with the stated primary antibody overnight and then probed with immunoreactive horseradish peroxidase-conjugated secondary antibody. For some experiments, quiescent platelets were biotinylated with membrane-impermeant sulfo-NHS-biotin and incubated 30 h with concentrated control or ADAMDEC1-transfected CHO cell medium in the presence or absence of 10 μg/ml chymostatin. In this case solubilized material was recovered by avidin chromatography before Western blotting for EGF.
ADAMDEC1 peptidolytic activity
Recombinant ADAMDEC1 concentrated from stably transfected CHO cell supernatants was assayed using a carboxymethylfluorescein-pro-EGF1020–1027-DABCYL self-quenched fluorogenic peptide (CF-WELRHAGHKDABCYLG). Hydrolysis was assessed by fluorescence after excitation at 485 nm, and emission collected at 530 nm. For some experiments, the fluorogenic pro-EGF1020–1027 peptide was incubated (1 h) with recombinant PAD4 before the hydrolytic reaction.
EGFR activity assay
HeLa reporter cells, or naïve platelets, were treated with material released from stimulated platelets or platelets treated with recombinant ADAMDEC1, the media were removed, and the cells were lysed in radioimmune precipitation assay buffer before cellular proteins were resolved by SDS-PAGE and immunoblotted with a mix of five phosphotyrosine EGFR, anti-pEGFR Tyr1068, or other antibodies as stated. Material released from thrombin-stimulated platelets was treated with FPRCK (phenyl-prolyl-arginyl-chloromethyl ketone) to inactivate thrombin before the addition to naïve cells.
SAS-H1 cell invasion and migration
SAS-H1 cells were suspended (1 × 105 cells/ml) in DMEM-F-12 medium containing 0.1% BSA and any stated inhibitors before 0.5 ml of the suspension was added to the upper well of a rehydrated Matrigel®/membrane insert. The lower chamber contained 0.75 ml of either thrombin-activated platelet supernatant or control medium. Cells in the lower chamber were quantified using a hemocytometer after 48 h of incubation.
Expression of data and statistics
Experiments were performed at least three times with cells from different donors, and all assays were performed in triplicate unless otherwise stated. The standard errors of the mean from all experiments are presented as error bars. Figures and statistical analyses were generated with Prism4 (GraphPad Software). A value of p ≤ 0.05 was considered statistically significant.
Author contributions
R. C. conducted all the experiments, analyzed the results, and aided in manuscript writing. G. J. contributed reagents, conceived of experimental approaches, and participated in writing this manuscript. T. M. M. conceived the project, obtained project funding, designed experiments, and was the primary element in manuscript preparation.
Acknowledgments
We appreciate the help of Dr. E. Poptic and the Molecular Screening Core, and we greatly appreciate the help of Dr. S. Bandyopadhyay and W.-Z. Shen in the Molecular Biotechnology core for synthesis and purification of the fluorogenic pro-EGF peptide. We also thank our many blood donors. The Clinical and Translational Science Collaborative of Cleveland, 4UL1TR000439, supported the inhibitor screen under the CTSC Core Utilization Pilot Grant Program.
1 This work was supported in part by National Institutes of Health Grants R01 HL130090 (to T. M. M.) and R01 DE025284 (to G. J.). This work was also supported by a Cleveland Clinic Innovators Award and the Lerner Research Institute Center-of-Excellence for Cancer-associated Thrombosis. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ADAM
- a disintegrin and metalloproteinase
- ADAMDEC1
- a disintegrin and metalloproteinase domain-like protein decysin-1
- DABCYL
- 4-((4-(dimethylamino)phenyl)azo)benzoic acid
- EGFR
- EGF receptor
- HNSCC
- head and neck squamous cell carcinomas
- PAD4
- polypeptide arginine deiminase 4
- HMW
- high molecular weight
- FPR-CMK
- Phe-Pro-Arg-chloromethyl ketone
- TACE
- tumor necrosis factor-α-converting enzyme.
References
- 1. Dreux A. C., Lamb D. J., Modjtahedi H., and Ferns G. A. (2006) The epidermal growth factor receptors and their family of ligands: their putative role in atherogenesis. Atherosclerosis 186, 38–53 [DOI] [PubMed] [Google Scholar]
- 2. Harris R. C., Chung E., and Coffey R. J. (2003) EGF receptor ligands. Exp. Cell Res. 284, 2–13 [DOI] [PubMed] [Google Scholar]
- 3. Mazzoleni S., Politi L. S., Pala M., Cominelli M., Franzin A., Sergi Sergi L., Falini A., De Palma M., Bulfone A., Poliani P. L., and Galli R. (2010) Epidermal growth factor receptor expression identifies functionally and molecularly distinct tumor-initiating cells in human glioblastoma multiforme and is required for gliomagenesis. Cancer Res. 70, 7500–7513 [DOI] [PubMed] [Google Scholar]
- 4. Ben-Ezra J., Sheibani K., Hwang D. L., and Lev-Ran A. (1990) Megakaryocyte synthesis is the source of epidermal growth factor in human platelets. Am. J. Pathol. 137, 755–759 [PMC free article] [PubMed] [Google Scholar]
- 5. Rall L. B., Scott J., Bell G. I., Crawford R. J., Penschow J. D., Niall H. D., and Coghlan J. P. (1985) Mouse prepro-epidermal growth factor synthesis by the kidney and other tissues. Nature 313, 228–231 [DOI] [PubMed] [Google Scholar]
- 6. Bell G. I., Fong N. M., Stempien M. M., Wormsted M. A., Caput D., Ku L. L., Urdea M. S., Rall L. B., and Sanchez-Pescador R. (1986) Human epidermal growth factor precursor: cDNA sequence, expression in vitro and gene organization. Nucleic Acids Res. 14, 8427–8446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kiuru J., Viinikka L., Myllylä G., Pesonen K., and Perheentupa J. (1991) Cytoskeleton-dependent release of human platelet epidermal growth factor. Life Sci. 49, 1997–2003 [DOI] [PubMed] [Google Scholar]
- 8. Savage A. P., Chatterjee V. K., Gregory H., and Bloom S. R. (1986) Epidermal growth factor in blood. Regul. Pept. 16, 199–206 [DOI] [PubMed] [Google Scholar]
- 9. Durante C., Agostini F., Abbruzzese L., Toffola R. T., Zanolin S., Suine C., and Mazzucato M. (2013) Growth factor release from platelet concentrates: analytic quantification and characterization for clinical applications. Vox Sang. 105, 129–136 [DOI] [PubMed] [Google Scholar]
- 10. Oka Y., and Orth D. N. (1983) Human plasma epidermal growth factor/β-urogastrone is associated with blood platelets. J. Clin. Invest. 72, 249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pesonen K., Viinikka L., Myllylä G., Kiuru J., and Perheentupa J. (1989) Characterization of material with epidermal growth factor immunoreactivity in human serum and platelets. J. Clin. Endocrinol. Metab. 68, 486–491 [DOI] [PubMed] [Google Scholar]
- 12. Aybay C., Karakus R., and Yucel A. (2006) Characterization of human epidermal growth factor in human serum and urine under native conditions. Cytokine 35, 36–43 [DOI] [PubMed] [Google Scholar]
- 13. Joh T., Itoh M., Katsumi K., Yokoyama Y., Takeuchi T., Kato T., Wada Y., and Tanaka R. (1986) Physiological concentrations of human epidermal growth factor in biological fluids: use of a sensitive enzyme immunoassay. Clin. Chim. Acta 158, 81–90 [DOI] [PubMed] [Google Scholar]
- 14. Bertrand-Duchesne M. P., Grenier D., and Gagnon G. (2010) Epidermal growth factor released from platelet-rich plasma promotes endothelial cell proliferation in vitro. J. Periodontal Res. 45, 87–93 [DOI] [PubMed] [Google Scholar]
- 15. Lamb D. J., Modjtahedi H., Plant N. J., and Ferns G. A. (2004) EGF mediates monocyte chemotaxis and macrophage proliferation and EGF receptor is expressed in atherosclerotic plaques. Atherosclerosis 176, 21–26 [DOI] [PubMed] [Google Scholar]
- 16. Schreier B., Gekle M., and Grossmann C. (2014) Role of epidermal growth factor receptor in vascular structure and function. Curr. Opin. Nephrol. Hypertens. 23, 113–121 [DOI] [PubMed] [Google Scholar]
- 17. Su C. Y., Kuo Y. P., Nieh H. L., Tseng Y. H., and Burnouf T. (2008) Quantitative assessment of the kinetics of growth factors release from platelet gel. Transfusion 48, 2414–2420 [DOI] [PubMed] [Google Scholar]
- 18. Sanderson M. P., Dempsey P. J., and Dunbar A. J. (2006) Control of ErbB signaling through metalloprotease mediated ectodomain shedding of EGF-like factors. Growth Factors 24, 121–136 [DOI] [PubMed] [Google Scholar]
- 19. Sunnarborg S. W., Hinkle C. L., Stevenson M., Russell W. E., Raska C. S., Peschon J. J., Castner B. J., Gerhart M. J., Paxton R. J., Black R. A., and Lee D. C. (2002) Tumor necrosis factor-α-converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J. Biol. Chem. 277, 12838–12845 [DOI] [PubMed] [Google Scholar]
- 20. Lee D. C., Sunnarborg S. W., Hinkle C. L., Myers T. J., Stevenson M. Y., Russell W. E., Castner B. J., Gerhart M. J., Paxton R. J., Black R. A., Chang A., and Jackson L. F. (2003) TACE/ADAM17 processing of EGFR ligands indicates a role as a physiological convertase. Ann. N.Y. Acad. Sci. 995, 22–38 [DOI] [PubMed] [Google Scholar]
- 21. Sahin U., Weskamp G., Kelly K., Zhou H. M., Higashiyama S., Peschon J., Hartmann D., Saftig P., and Blobel C. P. (2004) Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 164, 769–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mueller C. G., Rissoan M. C., Salinas B., Ait-Yahia S., Ravel O., Bridon J. M., Briere F., Lebecque S., and Liu Y. J. (1997) Polymerase chain reaction selects a novel disintegrin proteinase from CD40-activated germinal center dendritic cells. J. Exp. Med. 186, 655–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bates E. E., Fridman W. H., and Mueller C. G. (2002) The ADAMDEC1 (decysin) gene structure: evolution by duplication in a metalloprotease gene cluster on chromosome 8p12. Immunogenetics 54, 96–105 [DOI] [PubMed] [Google Scholar]
- 24. Lund J., Olsen O. H., Sørensen E. S., Stennicke H. R., Petersen H. H., and Overgaard M. T. (2013) ADAMDEC1 Is a metzincin metalloprotease with dampened proteolytic activity. J. Biol. Chem. 288, 21367–21375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Taylor J. M., Cohen S., and Mitchell W. M. (1970) Epidermal growth factor: high and low molecular weight forms. Proc. Natl. Acad. Sci. U.S.A. 67, 164–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taylor J. M., Mitchell W. M., and Cohen S. (1974) Characterization of the binding protein for epidermal growth factor. J. Biol. Chem. 249, 2188–2194 [PubMed] [Google Scholar]
- 27. Burkhart J. M., Vaudel M., Gambaryan S., Radau S., Walter U., Martens L., Geiger J., Sickmann A., and Zahedi R. P. (2012) The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 120, e73–e82 [DOI] [PubMed] [Google Scholar]
- 28. Wijten P., van Holten T., Woo L. L., Bleijerveld O. B., Roest M., Heck A. J., and Scholten A. (2013) High precision platelet releasate definition by quantitative reversed protein profiling: brief report. Arterioscler. Thromb. Vasc. Biol. 33, 1635–1638 [DOI] [PubMed] [Google Scholar]
- 29. Moss M. L., and Rasmussen F. H. (2007) Fluorescent substrates for the proteinases ADAM17, ADAM10, ADAM8, and ADAM12 useful for high-throughput inhibitor screening. Anal. Biochem. 366, 144–148 [DOI] [PubMed] [Google Scholar]
- 30. Rabie T., Strehl A., Ludwig A., and Nieswandt B. (2005) Evidence for a role of ADAM17 (TACE) in the regulation of platelet glycoprotein V. J. Biol. Chem. 280, 14462–14468 [DOI] [PubMed] [Google Scholar]
- 31. Hirata Y., and Orth D. N. (1979) Conversion of high molecular weight human epidermal growth factor (hEGF)/urogastrone (UG) to small molecular weight hEGF/UG by mouse EGF-associated arginine esterase. J. Clin. Endocrinol. Metab. 49, 481–483 [DOI] [PubMed] [Google Scholar]
- 32. Pyrc K., Milewska A., Kantyka T., Sroka A., Maresz K., Kozieł J., Nguyen K. A., Enghild J. J., Knudsen A. D., and Potempa J. (2013) Inactivation of epidermal growth factor by Porphyromonas gingivalis as a potential mechanism for periodontal tissue damage. Infect. Immun. 81, 55–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Le Gall S. M., Auger R., Dreux C., and Mauduit P. (2003) Regulated cell surface pro-EGF ectodomain shedding is a zinc metalloprotease-dependent process. J. Biol. Chem. 278, 45255–45268 [DOI] [PubMed] [Google Scholar]
- 34. Roskoski R., Jr. (2014) The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 79, 34–74 [DOI] [PubMed] [Google Scholar]
- 35. Ang K. K., Andratschke N. H., and Milas L. (2004) Epidermal growth factor receptor and response of head-and-neck carcinoma to therapy. Int. J. Radiat. Oncol. Biol. Phys. 58, 959–965 [DOI] [PubMed] [Google Scholar]
- 36. Markovic A., and Chung C. H. (2012) Current role of EGF receptor monoclonal antibodies and tyrosine kinase inhibitors in the management of head and neck squamous cell carcinoma. Expert Rev. Anticancer Ther. 12, 1149–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Muramatsu H., Kogawa K., Tanaka M., Okumura K., Nishihori Y., Koike K., Kuga T., and Niitsu Y. (1995) Superoxide dismutase in SAS human tongue carcinoma cell line is a factor defining invasiveness and cell motility. Cancer Res. 55, 6210–6214 [PubMed] [Google Scholar]
- 38. Discafani C. M., Carroll M. L., Floyd M. B. Jr., Hollander I. J., Husain Z., Johnson B. D., Kitchen D., May M. K., Malo M. S., Minnick A. A. Jr, Nilakantan R., Shen R., Wang Y. F., Wissner A., and Greenberger L. M. (1999) Irreversible inhibition of epidermal growth factor receptor tyrosine kinase with in vivo activity by N-[4-[(3-bromophenyl)amino]-6-quinazolinyl]-2-butynamide (CL-387,785). Biochem. Pharmacol. 57, 917–925 [DOI] [PubMed] [Google Scholar]
- 39. Ohnishi Y., Lieger O., Attygalla M., Iizuka T., and Kakudo K. (2008) Effects of epidermal growth factor on the invasion activity of the oral cancer cell lines HSC3 and SAS. Oral Oncol. 44, 1155–1159 [DOI] [PubMed] [Google Scholar]
- 40. Hwang D. L., Lev-Ran A., Yen C. F., and Sniecinski I. (1992) Release of different fractions of epidermal growth factor from human platelets in vitro: preferential release of 140-kDa fraction. Regul. Pept. 37, 95–100 [DOI] [PubMed] [Google Scholar]
- 41. Mroczkowski B., Reich M., Chen K., Bell G. I., and Cohen S. (1989) Recombinant human epidermal growth factor precursor is a glycosylated membrane protein with biological activity. Mol. Cell. Biol. 9, 2771–2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bazley L. A., and Gullick W. J. (2005) The epidermal growth factor receptor family. Endocr. Relat. Cancer 12, S17–S27 [DOI] [PubMed] [Google Scholar]
- 43. Tucher J., Linke D., Koudelka T., Cassidy L., Tredup C., Wichert R., Pietrzik C., Becker-Pauly C., and Tholey A. (2014) LC-MS based cleavage site profiling of the proteases ADAM10 and ADAM17 using proteome-derived peptide libraries. J. Proteome Res 13, 2205–2214 [DOI] [PubMed] [Google Scholar]
- 44. Mezyk-Kopeć R., Bzowska M., Stalińska K., Chełmicki T., Podkalicki M., Jucha J., Kowalczyk K., Mak P., and Bereta J. (2009) Identification of ADAM10 as a major TNF sheddase in ADAM17-deficient fibroblasts. Cytokine 46, 309–315 [DOI] [PubMed] [Google Scholar]
- 45. Lund J., Troeberg L., Kjeldal H., Olsen O. H., Nagase H., Sørensen E. S., Stennicke H. R., Petersen H. H., and Overgaard M. T. (2015) Evidence for restricted reactivity of ADAMDEC1 with protein substrates and endogenous inhibitors. J. Biol. Chem. 290, 6620–6629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Berger M., Moscatelli H., Kulle B., Luxembourg B., Blouin K., Spannagl M., Lindhoff-Last E., and Schambeck C. M. (2008) Association of ADAMDEC1 haplotype with high factor VIII levels in venous thromboembolism. Thromb. Haemost. 99, 905–908 [DOI] [PubMed] [Google Scholar]
- 47. O'Shea N. R., Chew T. S., Dunne J., Marnane R., Nedjat-Shokouhi B., Smith P. J., Bloom S. L., Smith A. M., and Segal A. W. (2016) Critical role of the disintegrin metalloprotease ADAM-like Decysin-1 (ADAMDEC1) for intestinal immunity and inflammation. J. Crohns Colitis 10, 1417–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Uhlén M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C. A., et al. (2015) Proteomics: tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- 49. Umezawa H., Aoyagi T., Morishima H., Kunimoto S., and Matsuzaki M. (1970) Chymostatin, a new chymotrypsin inhibitor produced by actinomycetes. J. Antibiot. 23, 425–427 [DOI] [PubMed] [Google Scholar]
- 50. Yamamoto K., Murphy G., and Troeberg L. (2015) Extracellular regulation of metalloproteinases. Matrix Biol. 44, 255–263 [DOI] [PubMed] [Google Scholar]
- 51. Fadini G. P., Menegazzo L., Rigato M., Scattolini V., Poncina N., Bruttocao A., Ciciliot S., Mammano F., Ciubotaru C. D., Brocco E., Marescotti M. C., Cappellari R., Arrigoni G., Millioni R., Vigili de Kreutzenberg S., Albiero M., and Avogaro A. (2016) NETosis delays diabetic wound healing in mice and humans. Diabetes 65, 1061–1071 [DOI] [PubMed] [Google Scholar]
- 52. Wong S. L., Demers M., Martinod K., Gallant M., Wang Y., Goldfine A. B., Kahn C. R., and Wagner D. D. (2015) Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 21, 815–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Umeda N., Matsumoto I., Kawaguchi H., Kurashima Y., Kondo Y., Tsuboi H., Ogishima H., Suzuki T., Kagami Y., Sakyu T., Ishigami A., Maruyama N., and Sumida T. (2016) Prevalence of soluble peptidylarginine deiminase 4 (PAD4) and anti-PAD4 antibodies in autoimmune diseases. Clin. Rheumatol. 35, 1181–1188 [DOI] [PubMed] [Google Scholar]
- 54. Wang S., and Wang Y. (2013) Peptidylarginine deiminases in citrullination, gene regulation, health and pathogenesis. Biochim. Biophys. Acta 1829, 1126–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Offermanns S. (2006) Activation of platelet function through G protein-coupled receptors. Circ. Res. 99, 1293–1304 [DOI] [PubMed] [Google Scholar]
- 56. Brown G. T., Narayanan P., Li W., Silverstein R. L., and McIntyre T. M. (2013) Lipopolysaccharide stimulates platelets through an IL-1β autocrine loop. J. Immunol. 191, 5196–5203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bodnar R. J. (2013) Epidermal growth factor and epidermal growth factor receptor: the yin and yang in the treatment of cutaneous wounds and cancer. Adv. Wound Care (New Rochelle) 2, 24–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Brown G. T., and McIntyre T. M. (2011) Lipopolysaccharide signaling without a nucleus: kinase cascades stimulate platelet shedding of proinflammatory IL-1β-rich microparticles. J. Immunol. 186, 5489–5496 [DOI] [PMC free article] [PubMed] [Google Scholar]