

Abstract
CDK4 regulates G1/S phase transition in the mammalian cell cycle by phosphorylating retinoblastoma family proteins. However, the mechanism underlying the regulation of CDK4 activity is not fully understood. Here, we show that CDK4 protein is degraded by anaphase-promoting complex/cyclosome (APC/C) during metaphase-anaphase transition in HeLa cells, whereas its main regulator, cyclin D1, remains intact but is sequestered in cytoplasm. CDK4 protein reaccumulates in the following G1 phase and shuttles between the nucleus and the cytoplasm to facilitate the nuclear import of cyclin D1. Without CDK4, cyclin D1 cannot enter the nucleus. Point mutations that disrupt CDK4 and cyclin D1 interaction impair the nuclear import of cyclin D1 and the activity of CDK4. RNAi knockdown of CDK4 also induces cytoplasmic retention of cyclin D1 and G0/G1 phase arrest of the cells. Collectively, our data demonstrate that CDK4 protein is degraded in late mitosis and reaccumulates in the following G1 phase to facilitate the nuclear import of cyclin D1 for activation of CKD4 to initiate a new cell cycle in HeLa cells.
Keywords: cell cycle; cyclin D1; cyclin-dependent kinase (CDK); protein degradation; retinoblastoma protein (pRb, RB)
Introduction
CDK4 regulates G1/S phase transition in mammalian cells mainly by phosphorylating retinoblastoma family proteins (RBs)4 to release RB-bound E2F family transcription factors (1–3). D-type cyclins, including D1, D2, and D3, of which cyclin D1 has been extensively studied, are essential for the activation of CDK4 (1, 4). Dysfunction of CDK4 causes cells to be arrested at the G1 phase in ovarian cancer cells, whereas elevation of CDK4 activity results in overcoming of the G1 phase arrest and promotes proliferation of the cells (5–7). CDK4 protein is nuclear, whereas cyclin D1 may be cytoplasmic and relocated to the nucleus for CDK4 kinase activation (8–13). In G0 phase cells arrested by serum starvation, the protein level of cyclin D1 is low but elevates in response to growth factor stimulation (14). Degradation or cytoplasmic retardation of cyclin D1 reduces CDK4 activity (11, 14–16). Nuclear accumulation of cyclin D1 also regulates cell growth, migration, differentiation, and development in addition to cell cycle control (17–19). Overexpression of cyclin D1 is also linked with tumorigenesis (12, 20–22).
Although tremendous progress has been made in investigating the function of CDK4 during the G1/S phase transition, many of those results were obtained from studies based on G0 cells. In contrast, less attention has been given to the regulatory mechanisms controlling the fluctuation of CDK4 and cyclin D1 proteins and the activation of CDK4 in cycling cells (23), although deciphering these mechanisms is fundamentally important. In this work, we studied the mechanism regulating CDK4 activation in HeLa cells.
Results
CDK4 protein is degraded by APC/C during metaphase-anaphase (M-A) transition and reaccumulates in subsequent G1 phase
To investigate the protein levels of CDK4 and cyclin D1 during the M-G1 phase transition in cycling cells, we first performed immunofluorescence microscopy (IFM) in HeLa cells using specific antibodies. Surprisingly, we found that the protein level of CDK4 was decreased during the M-A transition, whereas that of cyclin D1 was unchanged (Fig. 1A). This result was confirmed in WI-38 and 3T3 cells (supplemental Fig. S1A). Then we reseeded mitotic HeLa cells collected by shake-off (Fig. 1B) and analyzed the protein levels of both cyclin D1 and CDK4 by Western blotting. We also observed that cyclin D1 protein level was stable during the G2/M/G1 phases (Fig. 1C), as reported previously (24). In contrast, CDK4 protein level was decreased, and the kinase activity of CDK4 was also impaired, as indicated by unphosphorylation of RB Ser-780 (pS780-RB), a preferential phosphorylation site for CDK4 (Fig. 1C) (25). We further analyzed synchronized HeLa cells at the G2/M border and the indicated time points after G2/M release. Strikingly, we found that CDK4 protein was degraded during the M-A transition, and the phosphorylation of RB Ser-780 was decreased accordingly (Fig. 1D and supplemental Fig. S1B). Interestingly, from middle to late G1 phase, the protein level of CDK4 was recovered gradually, and the phosphorylation of RB Ser-780 was increased (Fig. 1D and supplemental Fig. S1B). These results were also confirmed in 3T3 cells (supplemental Fig. S1C). When the mitotic cells were treated with proteasome inhibitor MG132, CDK4 protein was kept at a steady high level (supplemental Fig. S1B), suggesting that CDK4 protein was degraded in mitosis via a ubiquitin-proteasomal pathway. To define the timing of ubiquitination and degradation of CDK4 proteins, we transfected HeLa cells with non-degradable GFP-cyclin B1Δ90 (27, 28), which sustains CDK1 activity, and arrested cells at the anaphase-telophase transition (29), treated the cells with nocodazole, and then released them into fresh medium. We observed that the protein levels of endogenous CDK4 and cyclin B1 were high in prometaphase and low in anaphase-telophase, whereas exogenous GFP-cyclin B1Δ90 was kept stable (Fig. 1E). As analyzed by microscopy, we also found that CDK4 protein was decreased during the M-A transition, recovered, and was rapidly imported into the nucleus during the G1 phase, whereas cyclin D1 was stable in the same period in all of the filmed cells (Fig. 1F and supplemental Movies 1 and 2). In contrast, the protein levels of CDK4 and cyclin D1 and the kinase activity of CDK4 in quiescent-like cells were low but quickly increased upon serum stimulation (Fig. 1 (G and H) and supplemental Fig. S1D). Moreover, in an in vivo ubiquitination assay, by co-expressing His-ubiquitin and GFP-CDK4 in HeLa cells followed by synchronization of the cells at the M-A transition, we also detected multiple ladder-like bands above the main band of GFP-CDK4 (Fig. 1I), indicating that CDK4 was ubiquitinated during the M-A transition. To investigate which E3 ubiquitin ligase was responsible for the ubiquitination of CDK4, we found that CDK4 interacted with Cdc20, one of the APC/C activators (Fig. 1J). By knocking down Cdc20 and Cdh1 separately, two activators for APC/C activity (26), we found that Cdc20-activated APC/C was responsible for the ubiquitination of CDK4 (Fig. 1K and supplemental Fig. S1E). To confirm this result, by treating Cdc20-knockdown cells with cycloheximide (CHX), we measured CDK4 degradation kinetics. The results showed that, whereas the protein level of CDK4 was decreased and eventually disappeared from 4 to 8 h with the treatment of CHX in control cells, the protein level of CDK4 in Cdc20-knockdown cells treated with CHX was stable (Fig. 1L), indicating that Cdc20-activated APC/C did mediate the ubiquitination and degradation of CDK4. Taken together, these data demonstrate that CDK4 protein is degraded by APC/C during the M-A transition and reaccumulates in the following G1 phase, whereas cyclin D1 protein is stable during the M-G1 transition in HeLa cells.
Figure 1.

CDK4 is degraded by APC/C-Cdc20 during M-A transition and reaccumulates from early to mid-G1 phase. A, HeLa cells were immunostained with anti CDK4 and cyclin D1 antibodies. The early prometaphase, late prometaphase, metaphase, and anaphase/telophase cells are labeled 1, 2, 3, and 4, respectively, in a, whereas the G2, metaphase, anaphase, and telophase cells are labeled with 1′, 2′, 3′, and 4′ in b. B, HeLa cells were synchronized at the G1/S transition and were then released into fresh culture medium for 10 h. Mitotic cells that were in prometaphase were shaken off, reseeded, and stained with DAPI. C, the cell lysate of the reseeded mitotic and released cells was subjected to Western blotting. D, HeLa cells were arrested at the G2/M border using RO3306 (9 μm) and released. The lysates from these cells were analyzed by Western blotting. E, HeLa cells transfected with GFP-cyclin B1Δ90 were arrested at prometaphase and anaphase/telophase and analyzed by Western blotting. F, HeLa cells expressing GFP-CDK4 and GFP-cyclin D1 were processed for time-lapse microscopy. More than 20 cells were filmed in each of three repeated experiments, and representative movie images are shown. G, HeLa cells arrested at the quiescent-like phase were released into G1 phase by serum at the indicated time points. The cell lysates were analyzed by Western blotting. H, quiescent-like phase to G1 phase cells were fixed and immunostained. DNA was stained by DAPI. 200 cells were randomly counted at each time point of the three repeated experiments. Representative images are shown. I, HeLa cells were co-transfected with His-ubiquitin and GFP or GFP-CDK4 or were transfected with GFP-CDK4 alone. Transfected cells were synchronized at the G2/M border using RO3306 and then released to M-A transition. Lysate of cells was subjected to a pull-down assay using nickel-Sepharose beads. The isolated proteins were analyzed by Western blotting. J, HeLa cells were transfected with GFP-CDK4, and the total cell extract was used for immunoprecipitation with a GFP antibody and probed with Cdc20 and GFP antibodies. K, HeLa cells were separately transfected with negative control (NC), Cdc20, or Cdh1 RNAi double-stranded RNA followed by co-transfection with His-ubiquitin and GFP-CDK4. As negative controls, HeLa cells transfected with NC RNAi double-stranded RNA were transfected with GFP-CDK4 or co-transfected with His-ubiquitin and GFP. Transfected cells were synchronized at the G2/M border using RO3306 and then released to M-A transition. Lysates of cells were subjected to a pull-down assay using nickel-Sepharose beads followed by Western blotting using anti-His and -GFP antibodies. L, HeLa cells were separately transfected with negative control or Cdc20 RNAi double-stranded RNA for 72 h. The cells were then treated with or without 30 μg/ml CHX for the indicated number of hours, lysed, and probed with CDK4 and α-tubulin antibodies. Scale bars, 10 μm.
Slow nuclear import and fast export retain cyclin D1 in the cytoplasm in the absence of CDK4
Next, we investigated the subcellular localizations of CDK4 and cyclin D1 in HeLa cells. Through IFM using specific antibodies, we observed that cyclin D1 was extensively localized in the cytoplasm of the paired early G1 cells, whereas CDK4, although at a very low level, was located in the nucleus (Fig. 2A). Along with the progress of the cell cycle, CDK4 protein was accumulated gradually and imported into the nucleus. Coincidently, cyclin D1 protein was imported into the nucleus and co-localized with CDK4 (Fig. 2A). Through transient expression of GFP-cyclin D1 and treatment of leptomycin B (LMB), an effective inhibitor for nuclear exportin CRM1 (30), we also found that GFP-cyclin D1 was mainly localized in the cytoplasm of the paired G1 phase cells, even in the presence of LMB (Fig. 2B). Because it was reported that mutation at Thr-286 prevents cyclin D1 from binding to CRM1 and displays low nuclear export efficiency (11, 31), we generated GFP-cyclin D1T286A mutant and expressed it in HeLa cells. Interestingly, we found that this mutant also became localized in the cytoplasm within 24 h, although eventually it was imported to the nucleus in the presence of LMB by 72 h (Fig. 2B), suggesting that nuclear import of this mutant was also insufficient in early G1 phase. By linking a nuclear localization signal (NLS) to cyclin D1 (NLS-D1-GFP) (32), we also observed that only a fraction of these fusion proteins were in the nucleus and that, when treated with LMB, all of the NLS-D1-GFPs were localized to the nucleus (Fig. 2C). Furthermore, when linking an NLS to cyclin D1T286A-GFP (NLS-D1T286A-GFP), we found that all of the NLS-D1T286A-GFPs were localized in the nucleus despite the LMB treatment (Fig. 2C). Taken together, these results indicate that slow nuclear import and fast nuclear export retain cyclin D1 in the cytoplasm of the early G1 phase cells in the absence of CDK4.
Figure 2.

Slow nuclear import and fast export retain cyclin D1 in the cytoplasm in the absence of CDK4. A, mitosis (M)-arrested HeLa cells were released to interphase (I) at the indicated time points. Cyclin D1 and CDK4 were immunostained. The DNA was stained with DAPI. 200 pairs of the cells were randomly counted at each time point of three repeated experiments. Representative images are shown. B, HeLa cells were transfected with the wild-type GFP-cyclin D1 (GFP-D1) or mutant GFP-cyclin D1T286A (GFP-D1T286A) for 24 or 72 h. The cells were then treated with or without 10 ng/ml LMB for 4 h. C, HeLa cells transfected with the wild-type NLS-cyclin D1-GFP (NLS-D1-GFP) or mutant (NLS-D1T286A-GFP) were treated with or without 10 ng/ml LMB for 4 h. More than 200 cells each of three experiments were randomly counted. Representative images are shown. Scale bars, 10 μm.
CDK4 shuttles between the nucleus and cytoplasm and promotes the nuclear import of cyclin D1
Through separate expression or co-expression of CDK4 and cyclin D1 in HeLa cells, we investigated how these proteins are transported to the nucleus. We observed that, when expressed separately, CDK4 entered the nucleus efficiently, whereas cyclin D1 was retained in the cytoplasm. When co-expressed, both CDK4 and cyclin D1 were imported and co-localized in the nucleus (Fig. 3A). In comparison, when cyclin D1 was co-expressed with CDK1 or CDK2, cyclin D1 was retained in the cytoplasm, although both CDK1 and CDK2 entered the nucleus efficiently (Fig. 3A). These results indicate that CDK4 promotes the nuclear import of cyclin D1. Because CDK6 also binds to cyclin D1 directly, we co-expressed CDK6 with cyclin D1 and found that both cyclin D1 and CDK6 were imported into the nucleus (Fig. 3A and supplemental Fig. S2 (A and B)). These results were also confirmed by experiments done in HEK 293T and MCF-7 cells (supplemental Fig. S2C). When CDK4 was knocked down by RNA interference (RNAi), the nuclear localization of cyclin D1 was significantly decreased (Fig. 3, B–D). To understand whether the nuclear accumulation of cyclin D1 was due to CDK4-promoted nuclear import or CDK4-mediated nuclear retention, we linked an NLS to CDK4-Myc (NLS-CDK4-Myc) and expressed this fusion protein in HeLa cells. The results showed that the forced nuclear import of NLS-CDK4-Myc, which does not weaken the binding of CDK4 with cyclin D1, impaired the nuclear localization of cyclin D1 (Fig. 3, A, E, and F), indicating that disrupting nucleocytoplasmic shuttling of CDK4 reduced the nuclear import of cyclin D1. Taken together, these data demonstrate that CDK4 shuttles between nucleus and cytoplasm, and this shuttling facilitates the nuclear import of cyclin D1.
Figure 3.

CDK4 shuttles between the nucleus and the cytoplasm and promotes the nuclear accumulation of cyclin D1. A, HeLa cells were co-transfected with GFP- or Myc-tagged cyclin D1 and CDK4 for 24 h. Myc-tagged protein was immunostained. 200 cells were randomly counted in each of three repeated experiments. Representative images are shown. B, HeLa cells were transfected with CDK4 siRNA for 72 h and fractionated into nuclear and cytoplasmic fractions, followed by Western blotting. C and D, asynchronous HeLa cells co-transfected with control, CDK4 or CDK6 siRNA, and RFP-H2B for 72 h were stained using an anti-cyclin D1 antibody and DAPI (C), and the cells with cyclin D1 in the nucleus were counted (D). *, p < 0.05; **, p < 0.01 (Student's t test). E and F, HeLa cells co-transfected with GFP-cyclin D1 (GFP-D1) and NLS-CDK4-Myc for 24 h were visualized by IFM (E; scale bars, 10 μm) or a co-immunoprecipitation assay (F).
Direct binding of CDK4 with cyclin D1 is required for CDK4-mediated nuclear import of cyclin D1
Next, we tested how CDK4 mediates the nuclear translocation of cyclin D1. First, by co-expressing a CDK4 kinase-dead mutant CDK4D158N with cyclin D1 in HeLa cells, we observed that both CDK4D158N and cyclin D1 were imported into the nucleus (Fig. 4A), indicating that the kinase activity of CDK4 was not required for their nuclear import. In contrast, by co-expressing GFP-cyclin D1 with CDK4 fused with an NES sequence of PKI-α and a Myc tag (CDK4-NES-Myc), we observed that GFP-cyclin D1 was co-localized with this NES-containing CDK4 in cytoplasm. Interestingly, when the cells were treated with LMB, both GFP-cyclin D1 and CDK4-NES-Myc were co-localized in the nucleus (Fig. 4B). We also constructed another two mutants, CDK4K22A and CDK43A (K22A/R24A/D25A), that bind to cyclin D1 weakly (Fig. 4C) (33) and co-expressed each of them with cyclin D1. We observed that CDK4K22A, but not CDK43A, was able to partially target cyclin D1 to the nucleus (Fig. 4D).
Figure 4.

Direct binding is required for CDK4-mediated nuclear import of cyclin D1. A, HeLa cells co-transfected with GFP-cyclin D1 (GFP-D1) and CDK4K158N-Myc plasmids for 24 h were visualized by IFM. B, asynchronous HeLa cells co-transfected with wild-type GFP-cyclin D1 (GFP-D1) and CDK4-NES-Myc plasmids for 24 h and treated without or with 10 ng/ml LMB for 4 h were visualized by IFM. C, HeLa cells were transfected with GFP, wild-type GFP-CDK4, and mutant plasmids for 22 h. The cell lysates were immunoprecipitated with a GFP antibody, and the co-immunoprecipitated cyclin D1 was detected. D, asynchronous HeLa cells co-transfected with GFP-cyclin D1 and CDK4-Myc mutant for 24 h were visualized by IFM. E, HeLa cells transfected with the wild-type GFP-cyclin D1 or mutant plasmid for 24 h followed by a co-immunoprecipitation assay using an anti-GFP antibody. The co-immunoprecipitated CDK4 with cyclin D1 or mutants was detected. F, HeLa cells co-transfected with CDK4-Myc and wild-type GFP-cyclin D1 or mutant cyclin D1 for 24 h were visualized by IFM. G, HeLa cells transfected with NLS-NoLS-CDK4-Myc and GFP-cyclin D1 mutant for 24 h were visualized by IFM. The DNA in all microscopy images was stained with DAPI. More than 200 cells in each of three repeated experiments were randomly counted. Representative images are shown. Scale bars, 10 μm.
It has been reported that the residues Lys-112 and Lys-114 within the cyclin box of cyclin D1 are important for its interaction with CDK4, although it is ambiguous which one is pivotal (34, 35). In this work, we generated the mutants cyclin D1K112E, cyclin D1K114E, and D1K112E/K114E and co-expressed each of them with CDK4 in HeLa cells. Through co-immunoprecipitation assays, we found that cyclin D1K112E and cyclin D1K112E/K114E did not bind to CDK4, whereas cyclin D1K114E retained about 30% of binding capability with CDK4 compared with the wild type (Fig. 4E). Through IFM, we observed that only GFP-cyclin D1K114E was co-localized with CDK4 in the nucleus (Fig. 4F). We also generated a CDK4 construct containing an NLS and a nucleolus localization signal (NoLS) (NLS-NoLS-CDK4-Myc) and co-expressed it with each of the cyclin D1 mutants. We observed that only cyclin D1K114E was co-localized with NLS-NoLS-CDK4-Myc in the nucleus and the nucleoli, whereas most of cyclin D1K112E and cyclin D1K112E/K114E were retained in cytoplasm without co-localization with NLS-NoLS-CDK4-Myc (Fig. 4G). By establishing a tet-on system (36) to induce the expression of GFP-cyclin D1K112E/K114E by tetracycline and time-lapse microscopy, we also observed that GFP-cyclin D1K112E/K114E was persistently cytoplasmic throughout the cell cycle (supplemental Movie 3). Collectively, these results demonstrate that only Lys-112 on cyclin D1, but not Lys-114, is essential for direct binding with CDK4 and also that this direct binding is required for the nuclear import of cyclin D1. We further tested whether another part of the cyclin D1 molecule is involved in its nuclear import. By co-expressing the GFP- or Myc-tagged cyclin box, C and N terminus deletion mutants of cyclin D1 with CDK4, we found that, no matter whether the mutant without cyclin box was expressed alone or co-expressed with CDK4, this mutant remained in the cytoplasm (supplemental Fig. S2, D and E). In contrast, when the mutants with either C or N terminus deletion were expressed alone, they were retained in cytoplasm, but, once co-expressed with CDK4, they entered the nucleus (supplemental Fig. S2F), indicating that neither the N nor C terminus of cyclin D1 contributes to its nuclear import. Taken together, we conclude that the Lys-112 residue of cyclin D1 is required for the interaction of cyclin D1 with CDK4 and the nuclear import of cyclin D1 in HeLa cells.
Stable binding of D-type cyclins with CDK4 enhances their nuclear localization
Because physical interaction between CDK4 and cyclin D1 promotes the nuclear import of cyclin D1, we decided to test whether the stability of CDK4-cyclin D1 complex influences the subcellular localization of cyclin D1 in HeLa cells. Because P16INK4 disrupts the interaction by competing with cyclin D1 for binding with CDK4 and p21CIP1/p27KIP1 directly binds and stabilizes cyclin D1-CDK4 complex (37, 38), we investigated the influence of P16INK4 and p21CIP1/p27KIP1 on the complex stability of CDK4-cyclin D1 and the subcellular localization of cyclin D1. Through a co-immunoprecipitation assay, we found that the stability of cyclin D1-CDK4 complex was compromised in the cells expressing GFP-p16INK4. In contrast, the stability of the cyclin D1-CDK4 complex was strengthened in the cells expressing GFP-p21CIP1 or -p27KIP1 (Fig. 5A). IFM showed that cyclin D1 was mostly localized in the cytoplasm in GFP-p16INK4-expressing cells and in the nucleus without GFP-p16INK4 expression (Fig. 5B). On the contrary, cyclin D1 was localized exclusively in the nucleus in GFP-p21CIP1- or GFP-p27KIP1-expressing cells (Fig. 5B). Western blot analysis also confirmed that cyclin D1 was mainly cytoplasmic in p16INK4-expressing cells (Fig. 5C). Because the other two D-type cyclins, D2 and D3, can also bind to CDK4 directly, by co-expressing these proteins in HeLa cells, we tested whether D2 and D3 cyclins are also imported into the nucleus along with CDK4. We found that, as a control, cyclin E was persistently in the nucleus, whereas cyclin D2 and D3 were cytoplasmic when expressed alone and entered the nucleus when co-expressed with CDK4 (supplemental Fig. S3A). These results indicate that cyclins D1, D2, and D3 share a very similar mechanism for their nuclear import and retention. Taken together, we conclude that a stable binding between D-type cyclins and CDK4 enhances their nuclear localization in HeLa cells.
Figure 5.
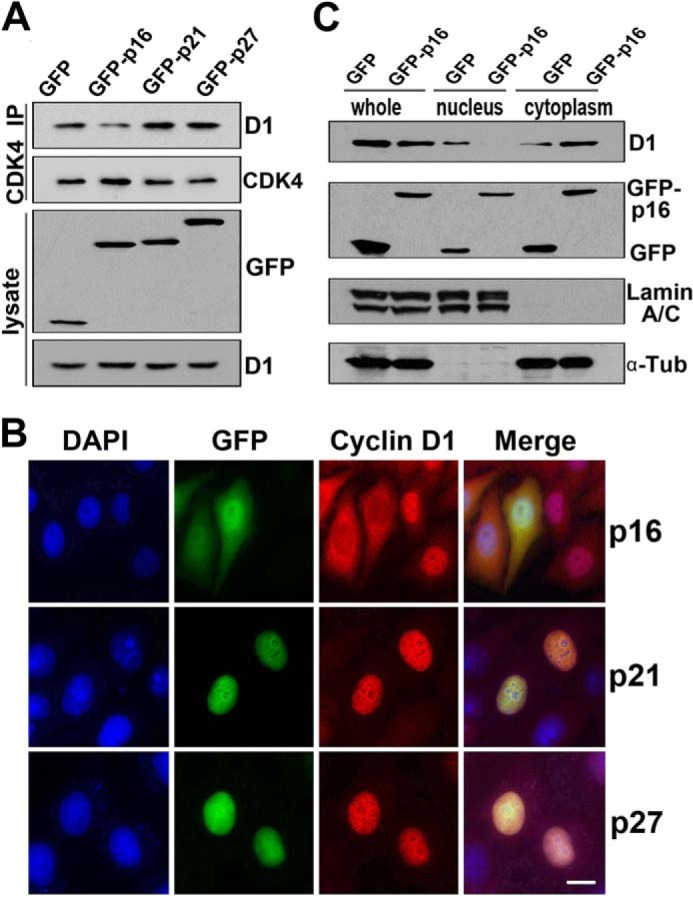
Stable binding of D-type cyclins with CDK4 enhances its nuclear localization, and CDK4/CDK6 knockdown results in cell-cycle arrest at the G1 phase. A, HeLa cells were transfected with GFP, GFP-p16, GFP-p21, or GFP-p27 for 24 h. The cell lysates were immunoprecipitated with anti-CDK4 antibody. The co-immunoprecipitated cyclin D1 was detected by Western blotting. B, HeLa cells transfected with GFP-tagged p16, p21, or p27 for 24 h were visualized by IFM. The DNA was stained with DAPI. More than 200 cells in each of three repeated experiments were randomly counted. Representative images are shown. C, HeLa cells were transfected with GFP or GFP-tagged p16 for 24 h. The nuclear and cytoplasmic extracts were analyzed by Western blotting. Scale bars, 10 μm.
CDK4/CDK6 knockdown results in cell-cycle arrest at the G1 phase
Knowing that accumulation of CDK4 in cells facilitates nuclear import of cyclin D1 and activation of CDK4 kinase in the nucleus to promote the M/G1/S transition, we wanted to verify whether knocking down CDK4 would arrest post-mitotic cells in G1 phase due to cytoplasmic retention of cyclin D1 and low CDK4 kinase activity. To do this, we knocked down CDK4 with siRNA and simultaneously expressed GFP-cyclin D1 in HeLa cells. The results showed that cyclin D1 could not be imported to the nucleus in CDK4-knockdown cells (Fig. 6 (A and B) and supplemental Movies 4 and 5). We also combined CDK4/6 knockdown with tet-on expression of wild-type GFP-cyclin D1 or mutant GFP-cyclin D1K112E/K114E. Through time-lapse microscopy, we confirmed that only a small portion of wild-type cyclin D1 was able to enter the nucleus, whereas the total proteins of mutant cyclin D1K112E/K114E were restrained in the cytoplasm in CDK4/CDK6-knockdown cells (Fig. 6C and supplemental Movies 6 and 7). We also examined the phosphorylation status of RB Ser-780 and found that CDK4 knockdown resulted in the accumulation of unphosphorylated RB at Ser-780 (supplemental Fig. S3B), indicating that the cells were arrested in the G1 phase (39). Taken together, these results demonstrate that, to initiate a new cell cycle, the post-mitotic HeLa cells need to accumulate CDK4 to gain the CDK4 kinase activity.
Figure 6.

CDK4/CDK6 knockdown results in cell-cycle arrest at G1 phase. A, HeLa cells were transfected with specific siRNA for 72 h to knock down CDK4 and CDK6 individually or simultaneously. Cell lysates were analyzed by Western blotting. B, GFP-cyclin D1 tet-on cells were transfected with specific double-stranded CDK4-knockdown siRNA and RFP-H2B for 64 h, treated with tetracycline for 8 h to induce GFP-cyclin D1 expression, and viewed by time-lapse microscopy. C, GFP-cyclin D1 and GFP-cyclin D1K112E/K114E tet-on cell lines were transfected with specific CDK4/CDK6 knockdown siRNA for 64 h, treated with tetracycline for 8 h to induce GFP-cyclin D1 (a) and GFP-cyclin D1K112E/K114E (b) expression, and viewed by time-lapse microscopy. More than 20 cells were filmed in each of three repeated experiments (a and b). Representative movie images are shown. Scale bars, 10 μm.
Discussion
Collectively, in this work, we show that periodic degradation of CDK4 is directly related to the activation of CDK4 in HeLa cells. The CDK4 protein level declines dramatically during metaphase-anaphase transition, and this decline results in a dramatic loss of CDK4 kinase activity in late M and early G1 phases. Importantly, the protein level of CDK4 increases during the following early and middle G1 phase, and accordingly, the kinase activity of CDK4 is recovered to enable the phosphorylation of RB again from middle to late G1 phase. Although we currently do not know why CDK4 is degraded in late mitosis, we suspect that this degradation may be crucial for a cell to decide to either continue cycling or arrest at G0 phase.
Cyclin D1 is regarded as a nuclear protein (40), although it is localized in the cytoplasm at certain stages of the cell cycle. Because cyclin D1 does not possess a recognized NLS, it may be co-imported into the nucleus with other factors. In this work, we have identified that CDK4 protein shuttles between the nucleus and cytoplasm and promotes the nuclear import of cyclin D1. CDK4 may enter the nucleus without cyclin D1, but cyclin D1 alone cannot enter the nucleus. Due to the degradation of CDK4 in mitosis, cyclin D1 will be retained in the cytoplasm in early G1 phase, although its protein level remains unchanged. Therefore, CDK4 reaccumulation after mitosis is also required for nuclear translocation of cyclin D1.
Based on our results and previously reported data, we propose a working model for the role of CDK4 in initiating a new cell cycle in HeLa cells (Fig. 7). In this model, CDK4 protein is degraded during the metaphase-anaphase transition in a proteasome-dependent manner, whereas cyclin D1 protein remains stable and is retained in the cytoplasm. CDK4 protein accumulates from the next early to mid-G1 phase and shuttles between the nucleus and the cytoplasm to facilitate the nuclear import of cyclin D1 and the M/G1/S transition (Fig. 7). When the divided cells lack nutrients, such as during serum starvation, they do not synthesize CDK4 and remain in the G0 phase. Accordingly, cyclin D1 is degraded to a basal level in these G0 cells. Once supplied with nutrients, these G0 cells begin to synthesize CDK4 and cyclin D1, as well as other essential factors, and initiate a new cell cycle starting from the G0 phase (G0/G1/S transition) (Fig. 7). In conclusion, this work reveals a CDK4 degradation/reaccumulation-dependent mechanism for initiating a new cell cycle in HeLa cells. Our findings may have important implications, potentially helping us to understand cell cycle control and tumorigenesis.
Figure 7.

A CDK4 reaccumulation-dependent model elucidating the regulation of a new cell-cycle initiation in HeLa cells. In HeLa cells, CDK4 is degraded during the M-A transition and reaccumulates in the following early G1 phase. The newly synthesized CDK4 shuttles between the nucleus and the cytoplasm and facilitates the nuclear transport of cyclin D1 to activate the kinase activity of CDK4. The activated nuclear CDK4 kinase then phosphorylates RB to release RB-bound E2F family transcription factors for the new cell-cycle initiation.
Materials and methods
Cell culture and transfection
HeLa, HEK 293T, MCF-7, WI-38, or NIH3T3 cell lines were maintained at 37 °C in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/ml streptomycin in the presence of 5% CO2. WI38 cells were cultured in Eagle's minimum essential medium with Earle's balanced salts containing 10% FBS and 1% non-essential amino acids in the presence of 5% CO2. To express exogenous proteins, the cells were transiently transfected via the calcium phosphate or liposome techniques according to the manufacturer's instructions.
Plasmids and antibodies
Cyclin D1 cDNA was cloned from MCF-7 cells by PCR with relevant oligonucleotides. Upstream sense primer contains a BglII site and the cyclin D1 initiation codon, whereas the downstream antisense prime is complementary to the 3′-end of the cyclin D1 sequence containing a SalI site but having a deleted cyclin D1 termination codon. The PCR product was gel-purified and digested with BglII and SalI restriction endonucleases before ligation to pcDNA3.1myc vector that had been digested with BamHI and XhoI restriction endonucleases and pEGFP-C1 vector that had been digested with BglII and SalI restriction endonucleases. Other genes were cloned by the same method with corresponding oligonucleotides. A point mutation was constructed by a PCR-based technique with relevant oligonucleotides. All constructs were verified by DNA sequence analysis (supplemental Table 1).
Anti-GFP antibody used for immunoprecipitation (4 μg/sample) was generated by immunizing rabbits with bacterially expressed recombinant GFP tagged with His. For immunofluorescence, mouse anti-cyclin D1 (sc-20044, Santa Cruz Biotechnology; 1:50) and anti-c-Myc (M4439, Sigma; 1:200) and rabbit anti-CDK4 (sc-260, Santa Cruz Biotechnology; 1:50) antibodies were used. Anti-lamin A/C antibody used for Western blotting (1:1,000) was generated by immunizing a rabbit with bacterially expressed recombinant lamin A/C. For Western blotting, mouse anti-GFP (M048-3, MBL; 1:3,000), anti-cyclin D1 (sc-20044, Santa Cruz Biotechnology; 1:500), anti-CDK4 (sc-23896, Santa Cruz Biotechnology; 1:500), anti-CDK6 (sc-7961, Santa Cruz Biotechnology; 1:500), anti-α-tubulin (T6199, Sigma; 1:2,000), anti-Cdh1 (sc-56312; Santa Cruz Biotechnology, 1:500), and anti-GAPDH (60004-1-Ig, Proteintech; 1:3,000) and rabbit anti-p286-cyclin D1 (ab62151, Abcam; 1:1,000), anti-Rb (sc-50, Santa Cruz Biotechnology; 1:500), anti-p780-Rb (555S, MBL; 1:1,000), anti-cyclin B1 (sc-752, Santa Cruz Biotechnology; 1:1,000), and anti-Cdc20 (A1231, ABclonal; 1:1,000) antibodies were used. All animal experiments were performed according to approved guidelines.
Western blotting and immunoprecipitation assay
HeLa or 3T3 cells were washed twice with calcium/magnesium-free PBS and lysed in lysis buffer (0.5% Nonidet P-40, 50 mm HEPES, 150 mm NaCl, 1 mm EDTA, 1 mm DTT, 1 mm NaF, 0.1 mm NaVO4, 0.2 mm PMSF, and 10 μg/ml protein inhibitors). GFP-tagged proteins were immunoprecipitated with an anti-GFP antibody. The protein samples were denatured and separated by 10% SDS-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. The membranes were probed with primary antibodies for 2 h at 37 °C, and the positive bands were detected by using the specific secondary antibody conjugated with horseradish peroxidase followed by a chemiluminescence detection system.
His6 tag pull-down assays
HeLa cells were seeded on 10-cm tissue culture plates and transfected with relevant siRNA and vectors. 40 h post-transfection, cells were treated with 10 μm MG132 for an additional 8 h. Treated cells were washed by PBS and lysed with 6 ml of lysis buffer (6 m guanidinium-HCl, 0.1 m Na2HPO4/NaH2PO4, 0.01 m Tris/HCl, pH 8.0, 5 mm imidazole, and 10 mm β-mercaptoethanol), rotating 30 min at room temperature. 75 μl of Ni2+-nitrilotriacetic acid-agarose beads (Qiagen) were then added, and lysates were rotated at room temperature for 4–5 h. Lysates were placed on the rack, kept standing for 5 min, and centrifuged at 2,000 rpm for 5 min at room temperature. The beads were roughly washed and rotated for 5 min at room temperature with 750 μl of wash buffer 1 (6 m guanidinium-HCl, 0.1 m Na2HPO4/NaH2PO4, 0.01 m Tris/HCl, pH 8.0, plus 10 mm β-mercaptoethanol); then the samples were kept standing for 5 min and centrifuged at 2,000 rpm for 5 min at room temperature. The pelleted beads were sequentially washed with wash buffer 2 (8 m urea, 0.1 m Na2HPO4/NaH2PO4, 0.01 m Tris/HCl, pH 8.0, 10 mm β-mercaptoethanol), wash buffer 3 (8 m urea, 0.1 m Na2HPO4/NaH2PO4, 0.01 m Tris/HCl, pH 6.3, 10 mm β-mercaptoethanol plus 0.2% Triton X-100), and wash buffer 4 (8 m urea, 0.1 m Na2HPO4/NaH2PO4, 0.01 m Tris/HCl, pH 6.3, 10 mm β-mercaptoethanol plus 0.1% Triton X-100). Finally, proteins were eluted by incubating the beads in 40 μl of elution buffer (500 mm imidazole, 0.15 m Tris/HCl, pH 6.7, 30% glycerol, 0.72 m β-mercaptoethanol, 5% SDS) for 20 min at room temperature. 35 μl of eluates were mixed with 2× loading buffer in a 1:1 ratio and analyzed by Western blot analysis.
RNAi
HeLa cells were transfected with siRNA especially designed to silence the CDK4 or CDK6 gene. Two oligomers of CDK4-specific siRNA were synthesized with the following sequences: 5′-GCAGCACUCUUAUCUACAU-3′ and 5′-GCAGAGAUGUUUCGUCGAA-3′. Two oligomers of CDK6-specific siRNA were synthesized with the following sequences: 5′-GCAGAAAUGUUUCGUAGAA-3′ and 5′-GCAAAGACCUACUUCUGAA-3′. 72 h after transfection, the cells were subjected to immunofluorescence or nuclear and cytoplasmic fractionation.
Cell-cycle synchronization
HeLa cells were synchronized at the G1/S phase transition by double thymidine blocks. Briefly, cells were treated with 2.5 mm thymidine for 16 h, released for 10 h, and incubated again for an additional 14 h in thymidine-containing medium. Cells blocked in the G1/S boundary were released for 8 h in complete medium and subsequently treated with 0.25 μg/ml nocodazole for an additional 6 h to obtain prometaphase cells. Prometaphase cells were then released and treated with 5 mm MG132 for 2 h to obtain metaphase cells. Quiescent G0 to G1 3T3 cells were achieved by serum starvation for 72 h and release as described previously (41).
IFM
HeLa, HEK 293T, MCF-7, or WI-38 cells grown on coverslips in Petri dishes were fixed with 4% paraformaldehyde for 15 min at room temperature and permeabilized with 0.2% Triton X-100 in PBS, pH 7.4, on ice for 2 min. The cells were sealed with Mowiol containing 1 μg/ml DAPI. For indirect immunofluorescence, cells were incubated with the respective primary antibodies for 1 h at 37 °C. After washing, the cells were successively incubated with the secondary antibodies conjugated with TRITC or FITC for 1 h at room temperature, and the cells were sealed with Mowiol containing 1 μg/ml DAPI.
Nuclear and cytoplasmic cell fractionation
HeLa cells were washed twice with cold low-penetration buffer (20 mm HEPES (pH 7.8), 5 mm potassium acetate, 0.5 mm MgCl2, 0.5 mm DTT, 1 mm PMSF, 20 units/ml aprotinin, 5 mg/ml leupeptin, and 0.4 mm NaF). After incubation on ice for 15 min, cells were collected with a scraper. 50 μl of cell suspension mix was incubated with 50 μl of 2× loading buffer (12.5 mm Tris, pH 6.8, 2% SDS, 2 mm DTT, 20% glycerine, 5% β-mercaptoethanol, and 0.2% bromphenol blue) to obtain whole-cell sample. The rest of the cell suspension was added to a Dounce homogenizer. Nuclei were pelleted at 8,000 rpm for 5 min, and the supernatant (cytoplasmic extracts) was centrifuged at 13,200 rpm for 5 min. Nuclei were washed three times with 1 ml of PBS and suspended in 50 μl of sample buffer to obtain nuclear sample.
Cell line generation and cell culture
Tet-on cell lines were generated according to the supplier's instructions (plasmids and drugs from Invitrogen) as we described previously (36). HeLa cells were transfected with pcDNA6/TR, and cultured in the presence of 10 μg/ml blasticidin for 2 weeks. Positive clones were selected for further transfection of pcDNA4/TO-GFP-tagged wild-type cyclin D1 and mutant cyclin D1K112E/K114E and cultured in the presence of 200 μg/ml zeocin for an additional 2 weeks. Again, positive clones were selected and cultured for use. GFP-tagged wild-type cyclin D1 and mutant cyclin D1K112E/K114E were expressed when 1 μg/ml tetracycline was added.
Living cell imaging and microscopy
HeLa cells were cultured on dishes with glass bottoms. The dynamics of the targeting proteins during the cell cycle were recorded by spinning disc microscopy with a heated culture chamber (37 °C, 5% CO2) and a Nikon ×60 (numeric aperture 1.43) Plan-apo oil objective.
Statistical analyses
HeLa cells transfected with double-stranded siRNA and RFP-H2B for 72 h were immunostained with antibody against cyclin D1. The cells with obviously less cyclin D1 in the nucleus were counted. Each experiment was repeated three times, and error bars in Fig. 3 represent S.D.
Author contributions
C. Z., Q. J., H. C., and X. X. conceptualized the study; H. C., X. X., G. W., B. Y., G. W., and G. X. performed the experiments; J. J. L., Q. J., and H. Y. Z. discussed the project and analyzed the data. C. Z., Q. J., H. C., and X. X. wrote the manuscript; all authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank the other members of the Zhang laboratory for helpful comments and critical discussion on this work.
This work was supported by National Natural Science Foundation of China (NSFC) Grants 31371365, 31520103906, and 31430051 and Ministry of Science and Technology of China Grants 2016YFA0100501 and 2016YFA0500201. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Table 1, Figs. S1–S3, and Movies 1–7.
- RB
- retinoblastoma protein
- APC/C
- anaphase-promoting complex/cyclosome
- M-A
- metaphase-anaphase
- IFM
- immunofluorescence microscopy
- CHX
- cycloheximide
- LMB
- leptomycin B
- NLS
- nuclear localization sequence
- NoLS
- nucleolus localization signal
- NES
- nuclear export sequence
- TRITC
- tetramethylrhodamine isothiocyanate.
References
- 1. Murray A. W. (2004) Recycling the cell cycle: cyclins revisited. Cell 116, 221–234 [DOI] [PubMed] [Google Scholar]
- 2. Nevins J. R. (2001) The Rb/E2F pathway and cancer. Hum. Mol. Genet. 10, 699–703 [DOI] [PubMed] [Google Scholar]
- 3. Dyson N. (1998) The regulation of E2F by pRB-family proteins. Genes Dev. 12, 2245–2262 [DOI] [PubMed] [Google Scholar]
- 4. Massagué J. (2004) G1 cell-cycle control and cancer. Nature 432, 298–306 [DOI] [PubMed] [Google Scholar]
- 5. Masamha C. P., and Benbrook D. M. (2009) Cyclin D1 degradation is sufficient to induce G1 cell cycle arrest despite constitutive expression of cyclin E2 in ovarian cancer cells. Cancer Res. 69, 6565–6572 [DOI] [PubMed] [Google Scholar]
- 6. Weng L. P., Brown J. L., and Eng C. (2001) PTEN coordinates G1 arrest by down-regulating cyclin D1 via its protein phosphatase activity and up-regulating p27 via its lipid phosphatase activity in a breast cancer model. Hum. Mol. Genet. 10, 599–604 [DOI] [PubMed] [Google Scholar]
- 7. Retzer-Lidl M., Schmid R. M., and Schneider G. (2007) Inhibition of CDK4 impairs proliferation of pancreatic cancer cells and sensitizes towards TRAIL-induced apoptosis via downregulation of survivin. Int. J. Cancer 121, 66–75 [DOI] [PubMed] [Google Scholar]
- 8. Alt J. R., Gladden A. B., and Diehl J. A. (2002) p21(Cip1) Promotes cyclin D1 nuclear accumulation via direct inhibition of nuclear export. J. Biol. Chem. 277, 8517–8523 [DOI] [PubMed] [Google Scholar]
- 9. Musgrove E. A., Caldon C. E., Barraclough J., Stone A., and Sutherland R. L. (2011) Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 11, 558–572 [DOI] [PubMed] [Google Scholar]
- 10. Diehl J. A., and Sherr C. J. (1997) A dominant-negative cyclin D1 mutant prevents nuclear import of cyclin-dependent kinase 4 (CDK4) and its phosphorylation by CDK-activating kinase. Mol. Cell. Biol. 17, 7362–7374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alt J. R., Cleveland J. L., Hannink M., and Diehl J. A. (2000) Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev. 14, 3102–3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dhar K. K., Branigan K., Parkes J., Howells R. E., Hand P., Musgrove C., Strange R. C., Fryer A. A., Redman C. W., and Hoban P. R. (1999) Expression and subcellular localization of cyclin D1 protein in epithelial ovarian tumour cells. Br. J. Cancer 81, 1174–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Holland T. A., Elder J., McCloud J. M., Hall C., Deakin M., Fryer A. A., Elder J. B., and Hoban P. R. (2001) Subcellular localisation of cyclin D1 protein in colorectal tumours is associated with p21(WAF1/CIP1) expression and correlates with patient survival. Int. J. Cancer 95, 302–306 [DOI] [PubMed] [Google Scholar]
- 14. Aggarwal P., Vaites L. P., Kim J. K., Mellert H., Gurung B., Nakagawa H., Herlyn M., Hua X., Rustgi A. K., McMahon S. B., and Diehl J. A. (2010) Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell 18, 329–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aggarwal P., Lessie M. D., Lin D. I., Pontano L., Gladden A. B., Nuskey B., Goradia A., Wasik M. A., Klein-Szanto A. J., Rustgi A. K., Bassing C. H., and Diehl J. A. (2007) Nuclear accumulation of cyclin D1 during S phase inhibits Cul4-dependent Cdt1 proteolysis and triggers p53-dependent DNA rereplication. Genes Dev. 21, 2908–2922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barbash O., Egan E., Pontano L. L., Kosak J., and Diehl J. A. (2009) Lysine 269 is essential for cyclin D1 ubiquitylation by the SCF(Fbx4/αB-crystallin) ligase and subsequent proteasome-dependent degradation. Oncogene 28, 4317–4325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pestell R. G., Albanese C., Reutens A. T., Segall J. E., Lee R. J., and Arnold A. (1999) The cyclins and cyclin-dependent kinase inhibitors in hormonal regulation of proliferation and differentiation. Endocr. Rev. 20, 501–534 [DOI] [PubMed] [Google Scholar]
- 18. Fantl V., Stamp G., Andrews A., Rosewell I., and Dickson C. (1995) Mice lacking cyclin D1 are small and show defects in eye and mammary gland development. Genes Dev. 9, 2364–2372 [DOI] [PubMed] [Google Scholar]
- 19. Sicinski P., Donaher J. L., Parker S. B., Li T., Fazeli A., Gardner H., Haslam S. Z., Bronson R. T., Elledge S. J., and Weinberg R. A. (1995) Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell 82, 621–630 [DOI] [PubMed] [Google Scholar]
- 20. Alao J. P. (2007) The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol. Cancer 6, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yu Q., Geng Y., and Sicinski P. (2001) Specific protection against breast cancers by cyclin D1 ablation. Nature 411, 1017–1021 [DOI] [PubMed] [Google Scholar]
- 22. Lu F., Gladden A. B., and Diehl J. A. (2003) An alternatively spliced cyclin D1 isoform, cyclin D1b, is a nuclear oncogene. Cancer Res. 63, 7056–7061 [PubMed] [Google Scholar]
- 23. Stacey D. W. (2010) Three observations that have changed our understanding of cyclin D1 and p27Kip1 in cell cycle control. Genes Cancer 1, 1189–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hitomi M., and Stacey D. (1999) Cyclin D1 production in cycling cells depends on ras in a cell-cycle-specific manner. Curr. Biol. 9, 1075–1084 [DOI] [PubMed] [Google Scholar]
- 25. Day P. J., Cleasby A., Tickle I. J., O'Reilly M., Coyle J. E., Holding F. P., McMenamin R. L., Yon J., Chopra R., Lengauer C., and Jhoti H. (2009) Crystal structure of human CDK4 in complex with a D-type cyclin. Proc. Natl. Acad. Sci. U.S.A. 106, 4166–4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Teixeira L. K., and Reed S. I. (2013) Ubiquitin ligases and cell cycle control. Annu. Rev. Biochem. 82, 387–414 [DOI] [PubMed] [Google Scholar]
- 27. Glotzer M., Murray A. W., and Kirschner M. W. (1991) Cyclin is degraded by the ubiquitin pathway. Nature 349, 132–138 [DOI] [PubMed] [Google Scholar]
- 28. Murray A. W., and Kirschner M. W. (1989) Cyclin synthesis drives the early embryonic cell cycle. Nature 339, 275–280 [DOI] [PubMed] [Google Scholar]
- 29. Zur A., and Brandeis M. (2001) Securin degradation is mediated by fzy and fzr, and is required for complete chromatid separation but not for cytokinesis. EMBO J. 20, 792–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nishi K., Yoshida M., Fujiwara D., Nishikawa M., Horinouchi S., and Beppu T. (1994) Leptomycin B targets a regulatory cascade of crm1, a fission yeast nuclear protein, involved in control of higher order chromosome structure and gene expression. J. Biol. Chem. 269, 6320–6324 [PubMed] [Google Scholar]
- 31. Diehl J. A., Zindy F., and Sherr C. J. (1997) Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 11, 957–972 [DOI] [PubMed] [Google Scholar]
- 32. Liu Q., Yu J., Zhuo X., Jiang Q., and Zhang C. (2010) Pericentrin contains five NESs and an NLS essential for its nucleocytoplasmic trafficking during the cell cycle. Cell Res. 20, 948–962 [DOI] [PubMed] [Google Scholar]
- 33. Coleman K. G., Wautlet B. S., Morrissey D., Mulheron J., Sedman S. A., Brinkley P., Price S., and Webster K. R. (1997) Identification of CDK4 sequences involved in cyclin D1 and p16 binding. J. Biol. Chem. 272, 18869–18874 [DOI] [PubMed] [Google Scholar]
- 34. Baker G. L., Landis M. W., and Hinds P. W. (2005) Multiple functions of D-type cyclins can antagonize pRb-mediated suppression of proliferation. Cell Cycle 4, 330–338 [PubMed] [Google Scholar]
- 35. Li Z., Jiao X., Wang C., Ju X., Lu Y., Yuan L., Lisanti M. P., Katiyar S., and Pestell R. G. (2006) Cyclin D1 induction of cellular migration requires p27(KIP1). Cancer Res. 66, 9986–9994 [DOI] [PubMed] [Google Scholar]
- 36. Fu J., Bian M., Liu J., Jiang Q., and Zhang C. (2009) A single amino acid change converts Aurora-A into Aurora-B-like kinase in terms of partner specificity and cellular function. Proc. Natl. Acad. Sci. U.S.A. 106, 6939–6944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. LaBaer J., Garrett M. D., Stevenson L. F., Slingerland J. M., Sandhu C., Chou H. S., Fattaey A., and Harlow E. (1997) New functional activities for the p21 family of CDK inhibitors. Genes Dev. 11, 847–862 [DOI] [PubMed] [Google Scholar]
- 38. Bagui T. K., Mohapatra S., Haura E., and Pledger W. J. (2003) P27Kip1 and p21Cip1 are not required for the formation of active D cyclin-cdk4 complexes. Mol. Cell. Biol. 23, 7285–7290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fry D. W., Bedford D. C., Harvey P. H., Fritsch A., Keller P. R., Wu Z., Dobrusin E., Leopold W. R., Fattaey A., and Garrett M. D. (2001) Cell cycle and biochemical effects of PD 0183812. A potent inhibitor of the cyclin D-dependent kinases CDK4 and CDK6. J. Biol. Chem. 276, 16617–16623 [DOI] [PubMed] [Google Scholar]
- 40. Baldin V., Lukas J., Marcote M. J., Pagano M., and Draetta G. (1993) Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 7, 812–821 [DOI] [PubMed] [Google Scholar]
- 41. Matsushime H., Quelle D. E., Shurtleff S. A., Shibuya M., Sherr C. J., and Kato J. Y. (1994) D-type cyclin-dependent kinase activity in mammalian cells. Mol. Cell. Biol. 14, 2066–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.