

Abstract
Cytokines, including receptor activator of nuclear factor κB ligand (RANKL) and TNF, induce increased osteoclast (OC) formation and bone loss in postmenopausal osteoporosis and inflammatory arthritides. RANKL and TNF can independently induce OC formation in vitro from WT OC precursors via TNF receptor-associated factor (TRAF) adaptor proteins, which bind to their receptors. Of these, only TRAF6 is required for RANKL-induced osteoclastogenesis in vitro. However, the molecular mechanisms involved remain incompletely understood. Here we report that RANKL induced the formation of bone-resorbing OCs from TRAF6−/− OC precursors when cultured on bone slices but not on plastic. The mechanisms involved increased TNF production by TRAF6−/− OC precursors resulting from their interaction with bone matrix and release of active TGFβ from the resorbed bone, coupled with RANKL-induced autophagolysosomal degradation of TRAF3, a known inhibitor of OC formation. Consistent with these findings, RANKL enhanced TNF-induced OC formation from TRAF6−/− OC precursors. Moreover, TNF induced significantly more OCs from mice with TRAF3 conditionally deleted in myeloid lineage cells, and it did not inhibit RANKL-induced OC formation from these cells. TRAF6−/− OC precursors that overexpressed TRAF3 or were treated with the autophagolysosome inhibitor chloroquine formed significantly fewer OCs in response to TNF alone or in combination with RANKL. We conclude that RANKL can enhance TNF-induced OC formation independently of TRAF6 by degrading TRAF3. These findings suggest that preventing TRAF3 degradation with drugs like chloroquine could reduce excessive OC formation in diseases in which bone resorption is increased in response to elevated production of these cytokines.
Keywords: bone, osteoclast, TNF receptor-associated factor (TRAF), TGF-β, TNF, RANKL
Introduction
Osteoclasts (OCs)3 are multinucleated cells that are responsible for generalized and localized bone loss in common conditions such as postmenopausal osteoporosis, periodontitis, and rheumatoid arthritis (1). RANKL is a member of the TNF superfamily that mediates terminal OC differentiation in cooperation with M-CSF (1). RANKL induces OC formation through its receptor, RANK, which transduces signals by recruiting adaptor molecules, such as the TNF receptor-associated factor (TRAF) family of proteins (2). TRAFs 1, 2, 3, 5, and 6 are recruited to the cytoplasmic tail of RANK (3–5), but of these, only TRAF6 is required for RANKL-induced OC formation based on the observation that mice generated to be deficient in TRAF6 by three independent groups have marked osteopetrosis (6–8). Interestingly, OC numbers are normal in the bones of one line of these TRAF6−/− mice (6), and no or few OCs are present in the bones of the other two lines (7, 8); however, OC precursors (OCPs) from all three lines of mice fail to form OCs in response to RANKL when they are cultured on plastic in vitro (6–8). However, some studies have reported OC formation occurring independently of RANKL-RANK-TRAF6 signaling in response to treatment of RANKL−/−, RANK−/−, or TRAF6−/− OCPs with TNF administered in combination with TGFβ1 in vitro (9) but not with either of these cytokines on their own (9, 10), although others have reported that TGFβ1 (11) and TNF (12–15) can induce OC formation on their own in the absence of RANKL or RANK.
TRAF6 recruits TGFβ-activated kinase 1 (TAK1), a member of the MAPK kinase family (16), to the cytoplasmic tail of RANK via TAK1-associated binding protein 2 (TAB2), which acts as a bridge between TRAF6 and TAK1 (17). Within this complex, TRAF6 acts as an E3 ubiquitin ligase, and its ubiquitination of TAK1 leads to TAK1-mediated phosphorylation of MAP kinases and inhibitory κB kinase (18), which are essential initial steps for OC formation (19, 20). TRAF6 and TAK1 are also recruited to the TGFβ receptor to mediate non-canonical TGFβ signaling in a variety of cell types (21, 22).
TRAF3 is recruited to the RANKL and TNF receptors, but unlike TRAF6, it inhibits OC formation in unstimulated OCPs by constitutively ubiquitinating NF-κB-inducing kinase (NIK) and inducing proteasomal degradation of NIK (15, 23), whereas overexpression of TRAF3 in OCPs inhibits RANKL-induced OC formation (23). TNF increases the levels of TRAF3 in OCPs as well as those of the downstream NF-κB non-canonical signaling inhibitor p100 as an important mechanism to limit inflammation-mediated osteoclastogenesis (15). TNF can also limit RANKL-induced OC formation in vitro by increasing TRAF3 levels in OCPs (15). However, RANKL reduces TRAF3 levels in OCPs by inducing its autophagolysosomal degradation, thus allowing NIK to mediate osteoclastogenesis. By this mechanism, RANKL-induced degradation of TRAF3 enhances TNF-induced osteoclastogenesis (15) because TNF alone has a rather limited ability to induce OC formation (15, 24). Thus, there are complex interactions between RANKL and TNF signaling that can affect TRAF levels in OCPs and, thus, OC formation.
Here we further investigated the role of TRAF6 and TRAF3 in TNF- and RANKL-induced OC formation and activation and examined whether TRAF6 is involved in the degradation of TRAF3. We found that TNF can induce OC formation from TRAF6−/− OCPs on plastic and bone and that RANKL or TGFβ enhances TNF-induced bone resorption by these cells. In addition, when TRAF6−/− OCPs are cultured on bone slices, they secrete TNF, resulting in increased TRAF3 expression. Treatment of TRAF6−/− OCPs on bone slices with RANKL causes TRAF3 degradation, resulting in OC formation and bone resorption. RANKL-induced OC formation from TRAF6−/−OCPs is prevented by overexpression of TRAF3 or treatment of the cells with chloroquine, an autophagolysosome inhibitor, which prevents TRAF3 degradation.
Results
RANKL enhances OC formation and resorption induced by TNF from TRAF6−/− OCPs
The line of TRAF6−/− mice we used was reported to have a few TRAP+ monocytes but no multinucleated OCs in their osteopetrotic bones and died within 2 weeks after birth (7). However, we observed TRAP+ multinucleated OCs on trabecular surfaces of this line of TRAF6−/− mice, although the numbers were ∼4-fold lower than in WT mice (Fig. 1A). We have reported that TNF alone, like RANKL, can induce OC formation from WT OCPs in vitro in the presence of M-CSF (15). To determine whether either of these cytokines is responsible for the formation of the small numbers of OCs we observed in the TRAF6−/− mice in vivo, we cultured OCPs from TRAF6−/− and WT littermate mouse spleens with either RANKL or TNF. Similar to previous reports (6, 7, 9), RANKL failed to induce any OCs from the TRAF6−/− cells on plastic plates (Fig. 1B). However, we found that TNF induced OC formation from these TRAF6−/− spleen cells without the addition of TGFβ1, and the number was similar to that derived from WT littermate cells (Fig. 1B). In addition, although RANKL failed to induce osteoclastogenesis from these TRAF6−/− OCPs, surprisingly, it enhanced TNF-induced OC formation (Fig. 1B). Because mice of this line of TRAF6−/− die before they are 2 weeks old, the spleen cells we used for OC culture could have undergone some pathological changes by the time the mice were 10–12 days old and thus affect their OC differentiation potential. To address this possibility, we treated WT spleen cells with M-CSF and siRNA to silence TRAF6 mRNA in OCPs and found that the TRAF6 siRNA reduced RANKL- but not TNF-induced OC formation compared with scrambled siRNA (supplemental Fig. 1), similar to our findings with TRAF6−/− OCPs. Importantly, OCs induced by TNF alone from the TRAF6−/− OCPs formed bone resorption pits in vitro, and the pit area was similar to that formed by WT cells (Fig. 1C). OCPs from either WT or TRAF6−/− mice cultured with M-CSF alone did not form resorption pits on bone slices (supplemental Fig. 2A), consistent with the fact that M-CSF alone is unable to complete OC differentiation, although it is required along with RANKL for OCP differentiation into OCs (25).
Figure 1.

TNF and TNF+RANKL induce OC formation from TRAF6−/− OCPs cultured on plastic. A, representative sections of tibiae from 12-day-old WT (+/+) and TRAF6 KO (−/−) mice stained for TRAP activity (red staining). OC numbers (Oc.N) in the tibial secondary ossification centers and metaphyses were counted. **, p < 0.01 for −/− mice (n = 7) versus +/+ mice (n = 5). B, 2 × 105 spleen cells from 12-day-old +/+ and −/− mice were treated with M-CSF for 2 days to recruit OCPs, followed by treatment with TNF (T, 20 ng/ml) or RANKL (R, 10 ng/ml) for 3 days in 96-well plates. TRAP+ OC numbers and areas (Oc.Ar) were counted. **, p < 0.01 versus the respective RANKL-treated cells; ##, p < 0.01 versus the respective TNF-treated cells; n = 4/group. C, OCPs were cultured with TNF and M-CSF for 10 days on bone slices in 96-well plates. The bone slices were fixed with 10% formalin, and the cells on them were brushed off followed by 0.1% toluidine blue staining. Resorption pit areas were counted. All in vitro experiments were repeated at least twice with similar results.
TRAF6−/− OCPs form osteoclasts and bone resorption pits in response to RANKL through the effects of TNF and TGFβ
We reported previously that OCPs produce IL-1β as a result of their interaction with bone matrix (26). We cultured TRAF6−/− splenocytes on bone slices to determine whether RANKL induces OC formation through a similar cytokine-mediated mechanism. Unexpectedly, we found that RANKL induced OC formation from TRAF6−/− cells cultured on bone slices (Fig. 2A), although the numbers of OCs were 3- to 4-fold lower than those from WT OCPs, similar to the reduced numbers in bone sections in vivo. Interestingly, OCs were also present on the plastic around the slices (Fig. 2A), and although the area available for them on which to form was less than the area of the slices, they occupied a larger area overall because they were much larger than the OCs that formed on the slices, which is typical for these culture conditions. Importantly, OCs induced by RANKL from TRAF6−/− spleen cells were able to resorb bone, although the mean resorption pit area was much less than that from WT cells (Fig. 2B). OCPs from either WT or TRAF6−/− mice cultured with M-CSF alone on bone slices did not form OCs on bone slices or on the plastic around the bone slices (supplemental Fig. 2B), excluding the possibility that TRAF6−/− OCPs could differentiate into OCs through an autocrine mechanism.
Figure 2.

RANKL induces OC formation from TRAF6−/− OCPs cultured on bone slices. 2 × 105 spleen cells from 7- to 11-day-old WT (+/+) and TRAF6−/− mice were seeded on bone slices in 96-well plates and cultured with RANKL+M-CSF for 9 days. A, TRAP+ OCs are shown on bone slices (left panel) and on the plastic around the bone slices (right panel) in the same well. TRAP-positive osteoclast numbers (Oc.N) and areas (Oc.Ar) on the bone slices and on the plastic around the slices were counted and expressed per slice or per well after removal of the slices. B, the bone slices were brushed to remove cells and stained with 0.1% toluidine blue to highlight resorption pits (left panel). Pit areas were counted and normalized to slice area. **, p < 0.01 versus values for WT cells. All experiments were repeated at least twice with similar results.
TNF, IL-1β, and TGFβ are candidate cytokines that could be produced by TRAF6−/− OCPs or released from bone matrix to induce TRAF6−/− OCP differentiation into mature OCs in cooperation with RANKL because OCPs are known to produce TNF and IL-1β (26, 27), and TGF-β is released from bone matrix and activated during bone resorption (28). However, neither IL-1β (10 ng/ml, a dose that effectively mediated OC formation from c-Fos-expressing OCPs (26)) nor TGF-β1 (2 ng/ml, a dose that mediated OC formation together with TNF, Fig. 4C) induced OC formation from TRAF6−/− OCPs when they were added along with RANKL on plastic culture plates (Fig. 3A). We next added inhibitors of TNF or IL-1β or a TGF-β antibody to TRAF6−/− OCPs cultured on bone slices and found that the TNF inhibitor and the TGFβ Ab, but not the IL-1 receptor antagonist (0.3 μg/ml, a dose that effectively inhibited OC formation from c-Fos-expressing OCPs on bone slices (26)), significantly reduced RANKL-induced OC formation from TRAF6−/− OCPs on bone slices (Fig. 3B).
Figure 4.

TRAF3 induced by TNF in OCPs is degraded by RANKL to enhance OC differentiation independently of TRAF6. A, spleen cells from 11-day-old WT and TRAF6−/− mice were cultured with M-CSF in 60-mm dishes for 3 days to recruit OCPs, which were then treated with the indicated cytokines for 8 h. Cell lysates were subjected to Western blot analysis of TRAF3, TRAF6, and β-actin. B, the autophagolysosomal inhibitor CQ or PBS was added to cultures similar to those in A along with cytokines for 8 h. Protein levels of TRAF3 and β-actin in the cell lysates were analyzed by Western blotting. C, 2 × 105 WT or TRAF6−/− spleen cells were cultured with M-CSF for 2 days in 96-well plates to recruit OCPs, followed by treatment with the indicated cytokines and CQ or PBS for an additional 3 days to generate OCs. OC numbers (Oc.N) were counted after TRAP staining (bottom panel). **, p < 0.01 between vehicle- and CQ-treated cells from WT and TRAF6−/− mice. All experiments were repeated at least twice with similar results. T, TNF, 20 ng/ml; R, RANKL, 10 ng/ml; Tβ1, TGFβ1, 1 ng/ml.
Figure 3.
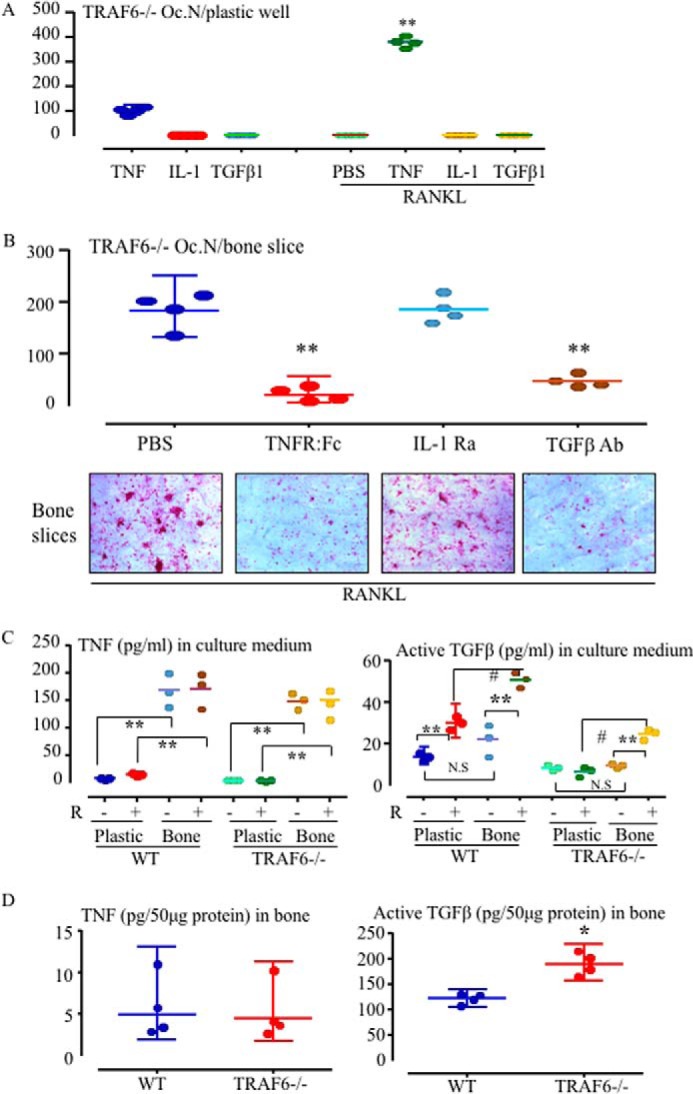
Either TNF or TGFβ1 mediates RANKL-induced OC formation from TRAF6−/− OCPs on bone. A, TRAF6−/− spleen cells were cultured with TNF, IL-1 (10 ng/ml), or TGFβ1 (2 ng/ml) or with each of them in combination with RANKL plus M-CSF in 96-well plastic plates for 5 days to generate OCs. TRAP+ OCs were counted after TRAP staining. **, p < 0.01 versus cells treated with TNF alone. Oc.N, OC number. B, spleen cells from 11-day-old WT and TRAF6−/− mice were seeded on bone slices in 96-well plates and cultured with RANKL and M-CSF plus the TNF inhibitor TNFR:Fc, an IL-I receptor antagonist (IL-1Ra), or a TGFβ-neutralizing Ab for 5 days. TRAP+ OCs were counted on bone slices after TRAP staining. **, p < 0.01 versus PBS-treated culture. C, 2 × 105 spleen cells from 11-day-old WT and TRAF6−/− mice were seeded in 96-well plates on plastic or on bone slices and treated with PBS (P) or RANKL (R) plus M-CSF for 5 days. Levels of TNF (left panel) and the active form of TGFβ (right panel) in the culture media were tested by ELISA. **, p < 0.01 versus the respective level in medium from cells cultured on plastic (n = 3/group); #, p < 0.05; N.S, no significant difference between the two groups. D, the lower extremities of 10-day-old TRAF6−/− and WT mice were homogenized in 1 ml of tissue protein extraction reagent containing a proteasome inhibitor mixture. 50 μg of tissue lysate protein from each sample was used to test the levels of TNF (left panel) and the active form of TGFβ1 (right panel) by ELISA (n = 4). All experiments were repeated at least twice with similar results.
Monocytes and macrophages are major producers of TNF (29, 30), and they also express TGFβ (31, 32). Thus, we investigated whether TRAF6−/− OCPs produce more TNF and/or TGFβ when they are cultured on bone slices or whether RANKL promotes the production of or release of TGFβ from bone matrix to induce OCP differentiation. We treated the cells with PBS or RANKL for 5 days, by which time mature OCs had formed. TNF levels in the media from both knockout and WT OCPs cultured on bone slices with PBS were significantly higher than those from cells cultured on plastic, but the levels were not affected significantly by the addition of RANKL (Fig. 3C, left panel). TNF protein levels in the lysates of bone tissue from 10-day-old TRAF6−/− and WT mice were similar and were around the lower limit of detection (Fig. 3D, left panel). We also measured levels of the active form of TGFβ in the conditioned medium in these experiments and found that they were higher in RANKL-treated WT cells cultured on plastic than in PBS controls (Fig. 3C, right panel), presumably because the OCs activated latent TGFβ in the culture medium. The levels of active TGFβ in media from cultures of WT OCPs treated with RANKL on bone slices for 5 days were significantly higher than those of OCPs cultured for 5 days with PBS on plastic or on bone slices (Fig. 3C, right panel). Similarly, the levels of active TGFβ in media from TRAF6−/− OCPs treated with RANKL on bone slices were higher than those from TRAF6−/− OCPs cultured on bone slices without RANKL or treated with RANKL on plastic. Interestingly, the levels of active TGFβ in bone samples from TRAF6−/− mice were significantly higher than those in bone from WT mice (Fig. 3D, right panel).
TRAF3 limits OC formation induced by TNF from TRAF6−/− OCPs
NF-κB plays a central role in OC differentiation by sequentially activating c-Fos followed by NFATc1 (24). TRAF6 is essential for activation of IL-1-induced canonical NF-κB signaling (33), but it negatively regulates TNF-induced canonical NF-κB signaling (34). Consistent with this, we found that neither IL-1β nor TGFβ1 induced IκBα phosphorylation, whereas TNF-induced IκBα phosphorylation was enhanced in TRAF6−/− cells compared with the change in WT cells (supplemental Fig. 4). TRAF3 associates with NIK to prevent NIK from inducing the processing of p100 to p52, a key event in non-canonical NF-κB activation (35). TRAF6 functions as an E3 ligase that regulates Akt ubiquitination and activation (36), and thus it could ubiquitinate TRAF3 and affect its levels in OCs and OCPs. However, we found that TRAF3 protein levels in the bones of 10-day-old TRAF6−/− mice were similar to those of WT mice, as assessed by Western blotting using the bone lysates shown in Fig. 3D (data not shown). We reported previously that TNF increases TRAF3 protein levels in WT OCPs and that TRAF3 overexpression limits TNF- and RANKL-induced OC formation (15, 23). In addition, RANKL induces TRAF3 autophagolysosomal degradation to promote OC formation (23). To investigate whether the TNF-induced increase in TRAF3 levels is dependent on TRAF6, we cultured spleen cells from TRAF6−/− and WT littermate mice with M-CSF to recruit OCPs, followed by treatment with TNF, RANKL, or TGFβ1 or combinations of them. The basal levels of TRAF3 were similar in WT and TRAF6−/− OCPs. As expected, RANKL reduced, whereas TNF markedly increased, TRAF3 protein levels, and addition of RANKL reduced TNF-induced TRAF3 levels in WT OCPs (Fig. 4A and supplemental Fig. 3A, p < 0.01). Similarly, RANKL also reduced, whereas TNF increased, TRAF3 protein levels in TRAF6−/− OCPs (Fig. 4A and supplemental Fig. 3A, p < 0.01) to an extent similar to that seen in WT cells. In contrast, TGFβ1 did not significantly affect basal or TNF-induced increased TRAF3 protein levels in either WT or TRAF6−/− cells (Fig. 4A and supplemental Fig. 3A). The absence of TRAF6 protein in TRAF6−/− cells confirmed the genotype of the knock-out mice (Fig. 4, A and B).
We reported previously that chloroquine (CQ), an autophagolysosome inhibitor, limits RANKL-induced OC formation (23). Interestingly, CQ elevated basal TRAF3 levels and prevented the reduction in TRAF3 levels in response to RANKL to a similar extent in WT and TRAF6−/− OCPs pretreated with TNF (Fig. 4B). Consistent with these findings, CQ completely blocked OC formation from TRAF6−/− OCPs induced by TNF alone or in combination with either RANKL or TGFβ1 (Fig. 4C). TNF+TGFβ1 induced similar numbers of OCs as TNF+RANKL from TRAF6−/− OCPs (298 ± 25 versus 274 ± 46/well, p > 0.05) (Fig. 4C). Furthermore, TRAF6−/− OCPs formed OCs earlier in response to TNF+TGFβ1 (3 days) compared with other treatments (4 days, data not shown). In addition, most of the TRAF6−/− OCs induced by TNF plus either RANKL or TGFβ1 formed actin rings, similar to WT OCs induced by RANKL (Fig. 5).
Figure 5.

TRAF6−/− OCs forms actin ring similar to WT OCs. 7-day-old WT and TRAF6−/− spleen cells were cultured with M-CSF plus the indicated cytokines for 5 days when mature OCs were observed under inverted microscopy. The cells were fixed with 10% neutral formalin for 20 min and stained with Alexa Fluor 488 phalloidin for 30 min with DAPI counterstaining. Representative images of phalloidin- and DAPI-stained and merged images are shown. Arrows indicate actin rings in the cell membrane of OCs.
To investigate whether TRAF3 is a key inhibitor of TNF-induced OC formation in TRAF6−/− mice, we first used OCPs from LyMcre-TRAF3f/f mice with deletion of TRAF3 specifically in myeloid cells (called TRAF3 cKO) (23). RANKL induced more OCs from TRAF3 cKO OCPs than from WT OCPs (Fig. 6A), as we reported previously (23). Importantly, TNF alone induced more OCs with a significantly higher area from TRAF3 cKO OCPs than from WT littermate cells, and TNF did not inhibit RANKL-induced osteoclastogenesis from TRAF3 cKO OCPs (Fig. 6A). Finally, we infected TRAF6−/− and WT OCPs with either GFP or TRAF3 retroviral constructs and found that overexpression of TRAF3 significantly reduced OC formation induced by RANKL, TNF, and RANKL+TNF from WT OCPs (Fig. 6B). Importantly, it blocked osteoclastogenesis induced by TNF or RANKL+TNF from TRAF6−/− OCPs (Fig. 6B).
Figure 6.

TRAF3 limits OC differentiation in the absence of TRAF6. A, BM cells from 2-month-old TRAF3f/f-LysMcre (TRAF3 conditional knock out in myeloid cells, −/−) mice and their WT (+/+) littermates were cultured with RANKL (R), TNF (T), TGFβ1 (Tβ1), or combinations of them in the presence of M-CSF for 5 days. TRAP staining was performed to evaluate OC numbers (Oc.N) and area (Oc.Ar). *, p < 0.05; **, p < 0.01 between cKO and WT cells. Western blotting was performed on cell lysates from PBS- and TNF-treated WT and TRAF3f/f-LysMcre OCPs and shows the absence of TRAF3 in the cells from TRAF3f/f-LysMcre (bottom right panel). B, 2 × 105 WT or TRAF6−/− spleen cells were cultured in 96-well plates with M-CSF for 2 days and then infected with GFP or TRAF3 retroviral supernatant. 1 day after infection, cytokines were added to the cultures for 3 days to generate OCs. OC numbers were counted after TRAP staining (top right panel). *, p < 0.05 versus its respective level in GFP-infected cells. TRAF3 protein levels were assessed in OCPs infected with GFP or TRAF3 retroviruses by Western blotting (bottom right panel) to confirm overexpression of TRAF3. All experiments were repeated at least twice with similar results. T, TNF, 20 ng/ml; R, RANKL, 10 ng/ml; Tβ1, TGFβ1, 1 ng/ml.
Discussion
Here we provide the first report that RANKL or TNF alone, without pretreatment with cytokines other than M-CSF, can induce the formation of bone-resorbing OCs in vitro independently of TRAF6 expression when TRAF6−/− OCPs are cultured on bone slices. These findings complement our observation of multinucleated osteoclasts in bone sections from the line of mice we studied, which had ∼4-fold fewer OCs than WT mice and whose OCPs formed 3- to 4-fold fewer OCs in response to TNF in vitro. Our detection of multinucleated OCs in bone sections from these mice differs from the findings of Naito et al. (7), who generated the mice and observed only a few weakly positive TRAP+ mononuclear cells in bone sections. This discrepancy could reflect differences in tissue processing and TRAP staining procedures. Naito et al. (7) did not observe the formation of OCs or resorption pits from TRAF6−/− OCPs cultured with WT calvarial cells and 1,25(OH)2 vitamin D3 or with M-CSF and RANKL, which we observed in our experiments. This could also reflect differences in culture conditions. For example, we used slices of bovine cortical bone, whereas Naito et al. (7) used dentine slices. Although these tissues have many similarities, they are laid down by different cell types and have differences in their proteomic makeup (37).
Kim et al. (9) studied the OC forming potential of TRAF6−/− OCPs using a different line of mice they had generated. OCPs from those knock-out mice, like the OCPS we studied, formed a few OCs in vitro in response to co-stimulation with TGFβ and TNF in the presence of M-CSF. However, M-CSF and RANKL did not induce any OCs from spleen cells from those TRAF6−/− mice unless they were pretreated with M-CSF and TGFβ for 3 days. However, those OCs did not form resorption pits on dentine slices. The authors did not explore the molecular mechanisms whereby pretreatment of TRAF6−/− spleen cells with M-CSF and TGFβ facilitated RANKL induction of osteoclastogenesis in their studies.
Our mechanistic studies have revealed that TRAF6−/− OCPs, like WT OCPs, increase their expression of TNF when they are cultured with M-CSF on bone slices, similar to the increased expression of IL-1 that we reported previously by WT OCPs as a consequence of their interaction with bone matrix proteins, including osteopontin, dentin sialoprotein, and bone sialoprotein (26). RANKL enhanced TNF-induced OC formation from TRAF6−/− OCPs mainly through degradation of TRAF3, which limits TNF-induced OC formation (15). Although TNF increased TRAF3 levels, it induced similar numbers of OCs from WT and TRAF6−/− OCPs, and addition of RANKL to these cultures increased the numbers of OCs further, consistent with RANKL degrading TRAF3. Indeed, when we added RANKL to TRAF6−/− or WT OCPs on bone slices, OCs formed resorption pits without addition of TGFβ. In addition, this RANKL-induced resorption increased the release of active TGFβ from the bone matrix into the culture medium from both TRAF6−/− and WT OCPs without affecting TNF levels in the culture medium.
TGFβ is produced by many cell types, including platelets (38), macrophages (32), and T cells (39), and is secreted along with its binding protein as a latent complex (40). It is thus maintained in an inactive state in the bloodstream (and in serum added to culture medium) and is stored in the extracellular bone matrix (41). Transient acidification can activate TGFβ by cleaving the latency-associated peptide and latent TGFβ-binding protein (42). Thus, TGFβ synthesized by osteoblasts (43) and released from bone slices during resorption can be activated in the acidic environment under the OC ruffled border membrane (44) and, thereafter, can exert its biological functions. A TGFβ antibody reduced RANKL-induced OC formation from TRAF6−/− OCPs cultured on bone slices, and TGFβ increased TNF-induced OC formation from WT and TRAF6−/−OCPs (Fig. 4C). However, different from reports by others that TGFβ can induce OC formation from human peripheral blood mononuclear cells and RAW cells without addition of RANKL, TNF, or IL-1 (11), we found that TGFβ did not induce any OC formation on its own from WT or TRAF6−/− OCPs (data not shown) and did not affect RANKL-induced OC formation from WT OCPs (Figs. 4C and 6A), suggesting that it enhances OC formation through a mechanism that is activated by TNF but not RANKL.
TGFβ receptors recruit TRAF6 and TAK1 to mediate downstream non-canonical signaling in which TRAF6 acts as an ubiquitin ligase that regulates Akt ubiquitination and activation in 293T cells (36) and thus could degrade TRAF3. However, TRAF3 levels were similar in WT and TRAF6−/− OCPs treated with TGFβ plus TNF or RANKL, indicating that TRAF6 is not activated by TGFβ or RANKL to degrade TRAF3. Several reports indicate that TRAF3 is degraded through the ubiquitin-proteasome pathway (45–47) by E3 ubiquitin ligases, including Peli1 (45) and Triad3A (46). In addition, cIAP1/2, as E3 ligases (47), cooperate with TRAF2 and TRAF3 to inhibit non-canonical NF-κB activation by degrading NF-κB-inducing kinase (48–51). Upon CD40L stimulation, cIAP1/2 in combination with TRAF2 induces the Lys-48-linked polyubiquitination and degradation of TRAF3 in B cells and, by this mechanism, activates NF-κB and MAPKs (52). Transgenic mice expressing a mutated form of NIK that lacks the TRAF3 binding domain and cannot be degraded are osteoporotic because of increased non-canonical NF-κB signaling-mediated osteoclastic resorption (53), highlighting the negative regulatory role of TRAF3 on osteoclastogenesis. TRAF6 is unlikely to affect the activity of the E3 ubiquitin ligases described above, and we did not examine their expression levels because the basal and stimulated TRAF3 protein levels in TRAF6−/− OCPs were very similar to those in WT OCPs.
We and others have reported that TNF can induce OC differentiation independently of RANKL signaling (15, 54, 55), but this is limited because it also increases the levels of inhibitory proteins, including p100 (15), TRAF3 (15), IRF-8 (55), and RBP-jκ (54). We recently reported that TNF inhibits RANKL-induced OC formation from M-CSF-induced M2-like macrophages but not from macrophages previously polarized to M1 cells by TNF (56). TRAF3 limits TNF-induced osteoclastogenesis because it prevents the processing of non-canonical NF-κB p100 to p52 (15). Our report that TRAF3 deletion specifically in myeloid progenitors resulted in early-onset osteoporosis associated with increased OC formation in mice indicates the important role of TRAF3 in maintaining bone mass by inhibiting bone resorption (23). In this report, we provide additional evidence to confirm that TRAF3 directly inhibits TNF-induced OC formation, an event that is independent of TRAF6. The number and area of OCs induced by TNF alone from OCPs from TRAF3 cKO mice were significantly higher than those from WT littermate cells, and TNF did not inhibit RANKL-induced osteoclastogenesis from OCPs from the TRAF3 cKO mice (Fig. 6A). Importantly, overexpression of TRAF3 significantly reduced OC formation induced by TNF or RANKL+ TNF from WT and TRAF6−/− OCPs (Fig. 6B), and prevention of TRAF3 degradation by chloroquine inhibited OC formation from these cells. However, TGFβ did not reduce TNF-induced TRAF3 protein levels (Fig. 4A), suggesting that TGFβ enhancement of TNF-induced osteoclastogenesis is not through degradation of TRAF3. Further studies will be required to determine whether TGFβ signaling enhances TNF-induced OC formation by affecting the levels of other inhibitory proteins, such as p100, IRF-8, or RBP-jκ.
The TRAF6−/− mice we studied had marked osteopetrosis despite the presence of OCs, normal levels of TNF in their bones, and increased levels of active TGFβ, which can induce their OCPs to form OCs in vitro. This may reflect the limited ability of TNF to induce OC formation under physiological conditions because of its parallel induction of strong inhibitors (15, 54, 55). However, TNF plays a major role mediating inflammation and joint destruction in rheumatoid and psoriatic arthritis (57–59) because it induces OC formation directly (15) and indirectly through stimulation of RANKL expression by accessory cells, including osteoblastic, synovial, and immune cells (60–64). Overall, our findings provide additional evidence that TRAF3 is an important negative regulator of OC formation and suggest that therapeutic approaches to prevent its degradation with drugs, such as chloroquine, should inhibit OC formation and bone loss in a broad array of common osteolytic bone diseases.
Experimental procedures
Reagents
Recombinant murine M-CSF, RANKL, TNF, TGFβ1 (which was treated with acid to activate it before use, following the instructions of the manufacturer), IL-1, IL-1Ra, and a TGFβ pan-specific Ab were purchased from R&D Systems (Minneapolis, MN). The TNF receptor fusion protein Etanercept was from Amgen (Thousand Oaks, CA) (26). TRAF3 and TRAF6 Abs for Western blotting were purchased from Santa Cruz Biotechnology, and anti-actin Ab was from Sigma. Ammonium chloride (NH4Cl) solution was purchased from STEMCELL Technologies. Chloroquine was purchased from Sigma. Alexa Fluor-488 phalloidin was purchased from Thermo Fisher Scientific.
Animals
The TRAF6−/− mice were generated originally on a C57Bl6 background by Naito et al. (7), and they die within a few days after birth. When TRAF6+/− mice were crossed to a 129 background, the −/− mice survived for ∼12–14 days (65, 66). We used these mice in our studies because this allowed us to harvest cells from these slightly older animals more effectively than from the original mice. Spleen cells from 7- to 12-day-old TRAF6−/− and WT littermates were used for in vitro OC cultures and related experiments. Mice with TRAF3 conditionally knocked out (TRAF3 cKO) in OC lineage cells were generated by crossing TRAF3f/f with LysMcre mice, as we reported previously (23). All animal procedures were conducted using procedures approved by the University of Rochester Committee for Animal Resources.
Osteoclastogenesis
The culture procedure was modified from our previous reports (15, 24, 26). Briefly, spleens were meshed in a 40-μm nylon cell strainer (BD Falcon) using 5 ml of minimum essential medium-α containing 5% FBS. The cells were incubated in NH4Cl solution for 15 min at room temperature to lyse red blood cells. 2 × 105 cells were seeded in each well in 96-well plates with 5 ng/ml M-CSF for 2 days, and then RANKL (10 ng/ml) or TNF (20 ng/ml) or both with or without different inhibitors was added. The cells were cultured for 2–4 days, when mature OCs typically are observed under inverted microscopy. The cells were then fixed with 10% neutral phosphate-buffered formalin for 10 min and stained for TRAP activity. TRAP+ cells with three or more nuclei were considered to be mature OCs. Bone resorption was assessed by culturing OCPs on bovine cortical bone slices in 96-well plates for 7–9 days, as we described previously (15, 26). The slices were fixed with 10% neutral formalin for 10 min, followed by TRAP staining to evaluate OC formation. OCs were then removed from the bone slices by brushing, and the slices were stained with 0.1% toluidine blue to visualize resorption pits. The mean pit area (square millimeters per slice) was measured as described previously (15, 26).
Assessment of actin ring formation
Cultured mature OCs were fixed with 10% neutral formalin for 10 min and then incubated with Alexa Fluor 488 phalloidin for 30 min, followed by DAPI counterstaining. The actin rings were observed under fluorescent microscopy at ×10 magnification.
Overexpression of TRAF3
Spleen cells, prepared as above, were seeded in 96-well plates to evaluate OC formation, or 1.2 × 106 cells were cultured in 60-mm dishes for 3 days for Western blotting. Cells were cultured with M-CSF for 2 days to recruit OCPs, and then fresh culture medium containing M-CSF, 2 μg/ml Polybrene, and ¼ volume of GFP or TRAF3 retroviral supernatant prepared from Plat-E packaging cells (15, 23, 24, 26) was added to infect the cells with these retroviruses. After 24 h, RANKL and/or TNF was added for 48–60 h to generate OCs or to prepare as cell lysate for Western blotting.
Western blot analysis
Cultured cells were lysed with M-PER mammalian protein extraction reagent (Thermo Scientific) containing a protease inhibitor mixture (Sigma). Lysates (10–20 μg) were loaded in 10% SDS-PAGE gels and transferred onto polyvinylidene difluoride membranes. Following blocking in 5% milk, membranes were incubated overnight at 4 °C with mouse TRAF3, TRAF6, or β-actin Ab. After washing, the membranes were incubated with horseradish peroxidase-linked secondary Ab (Bio-Rad). The membranes were exposed to ECL substrate, and signals were analyzed using a Bio-Rad imaging system.
ELISA
Culture media from WT or TRAF6−/− cells grown on plastic or bone slices in 96-well plates were collected when OCs could be seen on the culture wells or on the plastic around bone slices using an inverted microscope. The lower extremities of 10-day-old TRAF6−/− and WT mice were homogenized in 1 ml tissue protein extraction reagent (Thermo Fisher) containing a proteasome inhibitor mixture (Sigma-Aldrich). The concentrations of TNF in the culture media and bone lysates containing 50 μg of protein were measured using a mouse ELISA Ready-SET-Go!® (eBioscience, San Diego, CA) kit, according to the instructions of the manufacturer. Similarly, TGFβ concentrations in the media and bone tissue lysates were measured using ELISA Ready-SET-Go!®. To test the baseline level of the active form of TGFβ, 100 μl of culture medium or bone tissue lysate containing 50 μg of protein were directly loaded into a well of the plate. To test the total level (including the latent form) of TGFβ, 50 μl of culture medium or bone tissue lysate containing 50 μg of protein was first treated with 10 μl of 1 n HCl for 10 min to process the latent to the active form of TGFβ, followed by neutralization with 10 μl of 1 n NaOH.
Statistics
All results are given as the mean ± S.D. Comparisons between two groups were analyzed using Student's two-tailed unpaired t test, and those among three or more groups using one-way analysis of variance and Dunnett's post hoc multiple comparisons. p < 0.05 was considered statistically significant. Each experiment was repeated at least twice with similar results.
Author contributions
Z. Y. conceived and coordinated the study, performed and analyzed the experiments shown in Figs. 2, 3, and 6B, and wrote the paper. W. L. performed and analyzed the experiments shown in Figs. 4A, 5, and 6A. R. D. performed and analyzed the experiments shown in Fig. 4, B and C. Y. L. provided technical assistance and contributed to Fig. 1. L. L. performed and analyzed the experiments shown in supplemental Fig. 4 and the Fig. 6A Western blots. B. F. B. supervised the study and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Bryant G. Darnay (University of Texas M. D. Anderson Cancer Center, Houston, TX 77030) for providing the TRAF6−/− mice.
This work was supported by NIAMS, National Institutes of Health Grant R01AR43510 (to B. F. B.) and NIA, National Institutes of Health Grant R01AG049994 (to B. F. B. and Z. Y.). This work was also supported by NIAMS Grants P30 AR069655 and 1S10RR027340 (to. B. F. B) from the National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. 1–5.
- OC
- osteoclast
- RANKL
- receptor activator of nuclear factor κB ligand
- TRAF
- TNF receptor-associated factor
- OCP
- osteoclast precursor
- NIK
- NF-κB-inducing kinase
- Ab
- antibody/antibodies
- CQ
- chloroquine
- cKO
- conditional knockout
- TRAP
- tartrate-resistant acid phosphatase.
References
- 1. Boyce B. F. (2013) Advances in osteoclast biology reveal potential new drug targets and new roles for osteoclasts. J. Bone Miner. Res. 28, 711–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gravallese E. M., Galson D. L., Goldring S. R., and Auron P. E. (2001) The role of TNF-receptor family members and other TRAF-dependent receptors in bone resorption. Arthritis Res. 3, 6–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim H. H., Lee D. E., Shin J. N., Lee Y. S., Jeon Y. M., Chung C. H., Ni J., Kwon B. S., and Lee Z. H. (1999) Receptor activator of NF-κB recruits multiple TRAF family adaptors and activates c-Jun N-terminal kinase. FEBS Lett. 443, 297–302 [DOI] [PubMed] [Google Scholar]
- 4. Wong B. R., Josien R., Lee S. Y., Vologodskaia M., Steinman R. M., and Choi Y. (1998) The TRAF family of signal transducers mediates NF-κB activation by the TRANCE receptor. J. Biol. Chem. 273, 28355–28359 [DOI] [PubMed] [Google Scholar]
- 5. Galibert L., Tometsko M. E., Anderson D. M., Cosman D., and Dougall W. C. (1998) The involvement of multiple tumor necrosis factor receptor (TNFR)-associated factors in the signaling mechanisms of receptor activator of NF-κB, a member of the TNFR superfamily. J. Biol. Chem. 273, 34120–34127 [DOI] [PubMed] [Google Scholar]
- 6. Lomaga M. A., Yeh W. C., Sarosi I., Duncan G. S., Furlonger C., Ho A., Morony S., Capparelli C., Van G., Kaufman S., van der Heiden A., Itie A., Wakeham A., Khoo W., Sasaki T., et al. (1999) TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 13, 1015–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Naito A., Azuma S., Tanaka S., Miyazaki T., Takaki S., Takatsu K., Nakao K., Nakamura K., Katsuki M., Yamamoto T., and Inoue J. (1999) Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells 4, 353–362 [DOI] [PubMed] [Google Scholar]
- 8. Kobayashi T., Walsh P. T., Walsh M. C., Speirs K. M., Chiffoleau E., King C. G., Hancock W. W., Caamano J. H., Hunter C. A., Scott P., Turka L. A., and Choi Y. (2003) TRAF6 is a critical factor for dendritic cell maturation and development. Immunity 19, 353–363 [DOI] [PubMed] [Google Scholar]
- 9. Kim N., Kadono Y., Takami M., Lee J., Lee S. H., Okada F., Kim J. H., Kobayashi T., Odgren P. R., Nakano H., Yeh W. C., Lee S. K., Lorenzo J. A., and Choi Y. (2005) Osteoclast differentiation independent of the TRANCE-RANK-TRAF6 axis. J. Exp. Med. 202, 589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Novack D. V., Yin L., Hagen-Stapleton A., Schreiber R. D., Goeddel D. V., Ross F. P., and Teitelbaum S. L. (2003) The IκB function of NF-κB2 p100 controls stimulated osteoclastogenesis. J. Exp. Med. 198, 771–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Itonaga I., Sabokbar A., Sun S. G., Kudo O., Danks L., Ferguson D., Fujikawa Y., and Athanasou N. A. (2004) Transforming growth factor-β induces osteoclast formation in the absence of RANKL. Bone 34, 57–64 [DOI] [PubMed] [Google Scholar]
- 12. Kobayashi K., Takahashi N., Jimi E., Udagawa N., Takami M., Kotake S., Nakagawa N., Kinosaki M., Yamaguchi K., Shima N., Yasuda H., Morinaga T., Higashio K., Martin T. J., and Suda T. (2000) Tumor necrosis factor α stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J. Exp. Med. 191, 275–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Azuma Y., Kaji K., Katogi R., Takeshita S., and Kudo A. (2000) Tumor necrosis factor-α induces differentiation of and bone resorption by osteoclasts. J. Biol. Chem. 275, 4858–4864 [DOI] [PubMed] [Google Scholar]
- 14. Fuller K., Murphy C., Kirstein B., Fox S. W., and Chambers T. J. (2002) TNFα potently activates osteoclasts, through a direct action independent of and strongly synergistic with RANKL. Endocrinology 143, 1108–1118 [DOI] [PubMed] [Google Scholar]
- 15. Yao Z., Xing L., and Boyce B. F. (2009) NF-κB p100 limits TNF-induced bone resorption in mice by a TRAF3-dependent mechanism. J. Clin. Invest. 119, 3024–3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ninomiya-Tsuji J., Kishimoto K., Hiyama A., Inoue J., Cao Z., and Matsumoto K. (1999) The kinase TAK1 can activate the NIK-I κB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 398, 252–256 [DOI] [PubMed] [Google Scholar]
- 17. Besse A., Lamothe B., Campos A. D., Webster W. K., Maddineni U., Lin S. C., Wu H., and Darnay B. G. (2007) TAK1-dependent signaling requires functional interaction with TAB2/TAB3. J. Biol. Chem. 282, 3918–3928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adhikari A., Xu M., and Chen Z. J. (2007) Ubiquitin-mediated activation of TAK1 and IKK. Oncogene 26, 3214–3226 [DOI] [PubMed] [Google Scholar]
- 19. Ruocco M. G., Maeda S., Park J. M., Lawrence T., Hsu L. C., Cao Y., Schett G., Wagner E. F., and Karin M. (2005) IκB kinase (IKK)β, but not IKKα, is a critical mediator of osteoclast survival and is required for inflammation-induced bone loss. J. Exp. Med. 201, 1677–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ruocco M. G., and Karin M. (2007) Control of osteoclast activity and bone loss by IKK subunits: new targets for therapy. Adv. Exp. Med. Biol. 602, 125–134 [DOI] [PubMed] [Google Scholar]
- 21. Sorrentino A., Thakur N., Grimsby S., Marcusson A., von Bulow V., Schuster N., Zhang S., Heldin C. H., and Landström M. (2008) The type I TGF-β receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 10, 1199–1207 [DOI] [PubMed] [Google Scholar]
- 22. Yamashita M., Fatyol K., Jin C., Wang X., Liu Z., and Zhang Y. E. (2008) TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-β. Mol. Cell 31, 918–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xiu Y., Xu H., Zhao C., Li J., Morita Y., Yao Z., Xing L., and Boyce B. F. (2014) Chloroquine reduces osteoclastogenesis in murine osteoporosis by preventing TRAF3 degradation. J. Clin. Invest. 124, 297–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamashita T., Yao Z., Li F., Zhang Q., Badell I. R., Schwarz E. M., Takeshita S., Wagner E. F., Noda M., Matsuo K., Xing L., and Boyce B. F. (2007) NF-κB p50 and p52 regulate receptor activator of NF-κB ligand (RANKL) and tumor necrosis factor-induced osteoclast precursor differentiation by activating c-Fos and NFATc1. J. Biol. Chem. 282, 18245–18253 [DOI] [PubMed] [Google Scholar]
- 25. Boyce B. F., and Xing L. (2007) Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res. Ther. 9, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yao Z., Xing L., Qin C., Schwarz E. M., and Boyce B. F. (2008) Osteoclast precursor interaction with bone matrix induces osteoclast formation directly by an interleukin-1-mediated autocrine mechanism. J. Biol. Chem. 283, 9917–9924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wei S., Kitaura H., Zhou P., Ross F. P., and Teitelbaum S. L. (2005) IL-1 mediates TNF-induced osteoclastogenesis. J. Clin. Invest. 115, 282–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tang Y., Wu X., Lei W., Pang L., Wan C., Shi Z., Zhao L., Nagy T. R., Peng X., Hu J., Feng X., Van Hul W., Wan M., and Cao X. (2009) TGF-β1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 15, 757–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baer M., Dillner A., Schwartz R. C., Sedon C., Nedospasov S., and Johnson P. F. (1998) Tumor necrosis factor α transcription in macrophages is attenuated by an autocrine factor that preferentially induces NF-κB p50. Mol. Cell. Biol. 18, 5678–5689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Agbanoma G., Li C., Ennis D., Palfreeman A. C., Williams L. M., and Brennan F. M. (2012) Production of TNF-α in macrophages activated by T cells, compared with lipopolysaccharide, uses distinct IL-10-dependent regulatory mechanism. J. Immunol. 188, 1307–1317 [DOI] [PubMed] [Google Scholar]
- 31. Khalil N., Bereznay O., Sporn M., and Greenberg A. H. (1989) Macrophage production of transforming growth factor β and fibroblast collagen synthesis in chronic pulmonary inflammation. J. Exp. Med. 170, 727–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Toossi Z., Hirsch C. S., Hamilton B. D., Knuth C. K., Friedlander M. A., and Rich E. A. (1996) Decreased production of TGF-β 1 by human alveolar macrophages compared with blood monocytes. J. Immunol. 156, 3461–3468 [PubMed] [Google Scholar]
- 33. Cao Z., Xiong J., Takeuchi M., Kurama T., and Goeddel D. V. (1996) TRAF6 is a signal transducer for interleukin-1. Nature 383, 443–446 [DOI] [PubMed] [Google Scholar]
- 34. Funakoshi-Tago M., Kamada N., Shimizu T., Hashiguchi Y., Tago K., Sonoda Y., and Kasahara T. (2009) TRAF6 negatively regulates TNFα-induced NF-κB activation. Cytokine 45, 72–79 [DOI] [PubMed] [Google Scholar]
- 35. Boyce B. F., Xiu Y., Li J., Xing L., and Yao Z. (2015) NF-κB-mediated regulation of osteoclastogenesis. Endocrinology Metabolism 30, 35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang W. L., Wang J., Chan C. H., Lee S. W., Campos A. D., Lamothe B., Hur L., Grabiner B. C., Lin X., Darnay B. G., and Lin H. K. (2009) The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 325, 1134–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Salmon C. R., Tomazela D. M., Ruiz K. G., Foster B. L., Paes Leme A. F., Sallum E. A., Somerman M. J., and Nociti F. H. Jr. (2013) Proteomic analysis of human dental cementum and alveolar bone. J. Proteomics 91, 544–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pihusch V., Pihusch M., Penovici M., Kolb H. J., Hiller E., and Pihusch R. (2005) Transforming growth factor β-1 released from platelets contributes to hypercoagulability in veno-occlusive disease following hematopoietic stem cell transplantation. Thromb. Res. 116, 233–240 [DOI] [PubMed] [Google Scholar]
- 39. Kullberg M. C., Hay V., Cheever A. W., Mamura M., Sher A., Letterio J. J., Shevach E. M., and Piccirillo C. A. (2005) TGF-β1 production by CD4+ CD25+ regulatory T cells is not essential for suppression of intestinal inflammation. Eur. J. Immunol. 35, 2886–2895 [DOI] [PubMed] [Google Scholar]
- 40. Miyazono K., Olofsson A., Colosetti P., and Heldin C. H. (1991) A role of the latent TGF-β 1-binding protein in the assembly and secretion of TGF-β 1. EMBO J. 10, 1091–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shi M., Zhu J., Wang R., Chen X., Mi L., Walz T., and Springer T. A. (2011) Latent TGF-β structure and activation. Nature 474, 343–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Taylor A. W. (2009) Review of the activation of TGF-β in immunity. J. Leukocyte Biol. 85, 29–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oursler M. J. (1994) Osteoclast synthesis and secretion and activation of latent transforming growth factor β. J. Bone Miner. Res. 9, 443–452 [DOI] [PubMed] [Google Scholar]
- 44. Oreffo R. O., Mundy G. R., Seyedin S. M., and Bonewald L. F. (1989) Activation of the bone-derived latent TGF β complex by isolated osteoclasts. Biochem. Biophys. Res. Commun. 158, 817–823 [DOI] [PubMed] [Google Scholar]
- 45. Xiao Y., Jin J., Chang M., Chang J. H., Hu H., Zhou X., Brittain G. C., Stansberg C., Torkildsen Ø., Wang X., Brink R., Cheng X., and Sun S. C. (2013) Peli1 promotes microglia-mediated CNS inflammation by regulating Traf3 degradation. Nat. Med. 19, 595–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nakhaei P., Mesplede T., Solis M., Sun Q., Zhao T., Yang L., Chuang T. H., Ware C. F., Lin R., and Hiscott J. (2009) The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS Pathog. 5, e1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bertrand M. J., Lippens S., Staes A., Gilbert B., Roelandt R., De Medts J., Gevaert K., Declercq W., and Vandenabeele P. (2011) cIAP1/2 are direct E3 ligases conjugating diverse types of ubiquitin chains to receptor interacting proteins kinases 1 to 4 (RIP1–4). PLoS ONE 6, e22356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Abbas S., Zhang Y. H., Clohisy J. C., and Abu-Amer Y. (2003) Tumor necrosis factor-α inhibits pre-osteoblast differentiation through its type-1 receptor. Cytokine 22, 33–41 [DOI] [PubMed] [Google Scholar]
- 49. Liao G., Zhang M., Harhaj E. W., and Sun S. C. (2004) Regulation of the NF-κB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J. Biol. Chem. 279, 26243–26250 [DOI] [PubMed] [Google Scholar]
- 50. Vallabhapurapu S., Matsuzawa A., Zhang W., Tseng P. H., Keats J. J., Wang H., Vignali D. A., Bergsagel P. L., and Karin M. (2008) Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nat. Immunol. 9, 1364–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zarnegar B. J., Wang Y., Mahoney D. J., Dempsey P. W., Cheung H. H., He J., Shiba T., Yang X., Yeh W. C., Mak T. W., Korneluk R. G., and Cheng G. (2008) Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 9, 1371–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Matsuzawa A., Tseng P. H., Vallabhapurapu S., Luo J. L., Zhang W., Wang H., Vignali D. A., Gallagher E., and Karin M. (2008) Essential cytoplasmic translocation of a cytokine receptor-assembled signaling complex. Science 321, 663–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yang C., McCoy K., Davis J. L., Schmidt-Supprian M., Sasaki Y., Faccio R., and Novack D. V. (2010) NIK stabilization in osteoclasts results in osteoporosis and enhanced inflammatory osteolysis. PLoS ONE 5, e15383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhao B., Grimes S. N., Li S., Hu X., and Ivashkiv L. B. (2012) TNF-induced osteoclastogenesis and inflammatory bone resorption are inhibited by transcription factor RBP-J. J. Exp. Med. 209, 319–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhao B., Takami M., Yamada A., Wang X., Koga T., Hu X., Tamura T., Ozato K., Choi Y., Ivashkiv L. B., Takayanagi H., and Kamijo R. (2009) Interferon regulatory factor-8 regulates bone metabolism by suppressing osteoclastogenesis. Nat. Med. 15, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhao Z., Hou X., Yin X., Li Y., Duan R., Boyce B. F., and Yao Z. (2015) TNF Induction of NF-κB RelB enhances RANKL-induced osteoclastogenesis by promoting inflammatory macrophage differentiation but also limits it through suppression of NFATc1 expression. PLoS ONE 10, e0135728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Keffer J., Probert L., Cazlaris H., Georgopoulos S., Kaslaris E., Kioussis D., and Kollias G. (1991) Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 10, 4025–4031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Feldmann M. (2002) Development of anti-TNF therapy for rheumatoid arthritis. Nat. Rev. Immunol. 2, 364–371 [DOI] [PubMed] [Google Scholar]
- 59. Mease P. J. (2002) Tumour necrosis factor (TNF) in psoriatic arthritis: pathophysiology and treatment with TNF inhibitors. Ann. Rheum. Dis. 61, 298–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Atkins G. J., Kostakis P., Pan B., Farrugia A., Gronthos S., Evdokiou A., Harrison K., Findlay D. M., and Zannettino A. C. (2003) RANKL expression is related to the differentiation state of human osteoblasts. J. Bone Miner. Res. 18, 1088–1098 [DOI] [PubMed] [Google Scholar]
- 61. Danks L., Komatsu N., Guerrini M. M., Sawa S., Armaka M., Kollias G., Nakashima T., and Takayanagi H. (2016) RANKL expressed on synovial fibroblasts is primarily responsible for bone erosions during joint inflammation. Ann. Rheum. Dis. 75, 1187–1195 [DOI] [PubMed] [Google Scholar]
- 62. Takayanagi H., Kim S., Matsuo K., Suzuki H., Suzuki T., Sato K., Yokochi T., Oda H., Nakamura K., Ida N., Wagner E. F., and Taniguchi T. (2002) RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-β. Nature 416, 744–749 [DOI] [PubMed] [Google Scholar]
- 63. Kawai T., Matsuyama T., Hosokawa Y., Makihira S., Seki M., Karimbux N. Y., Goncalves R. B., Valverde P., Dibart S., Li Y. P., Miranda L. A., Ernst C. W., Izumi Y., and Taubman M. A. (2006) B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. Am. J. Pathol. 169, 987–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Meednu N., Zhang H., Owen T., Sun W., Wang V., Cistrone C., Rangel-Moreno J., Xing L., and Anolik J. H. (2016) Production of RANKL by memory B cells: a link between B cells and bone erosion in rheumatoid arthritis. Arthritis Rheumatology 68, 805–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lamothe B., Besse A., Campos A. D., Webster W. K., Wu H., and Darnay B. G. (2007) Site-specific Lys-63-linked tumor necrosis factor receptor-associated factor 6 auto-ubiquitination is a critical determinant of I κ B kinase activation. J. Biol. Chem. 282, 4102–4112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lamothe B., Webster W. K., Gopinathan A., Besse A., Campos A. D., and Darnay B. G. (2007) TRAF6 ubiquitin ligase is essential for RANKL signaling and osteoclast differentiation. Biochem. Biophys. Res. Commun. 359, 1044–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.