

Abstract
The gp120 subunit of the HIV-1 envelope (Env) protein is heavily glycosylated at ∼25 glycosylation sites, of which ∼7–8 are located in the V1/V2 and V3 variable loops and the others in the remaining core gp120 region. Glycans partially shield Env from recognition by the host immune system and also are believed to be indispensable for proper folding of gp120 and for viral infectivity. Previous attempts to alter glycosylation sites in Env typically involved mutating the glycosylated asparagine residues to structurally similar glutamines or alanines. Here, we confirmed that such mutations at multiple glycosylation sites greatly diminish viral infectivity and result in significantly reduced binding to both neutralizing and non-neutralizing antibodies. Therefore, using an alternative approach, we combined evolutionary information with structure-guided design and yeast surface display to produce properly cleaved HIV-1 Env variants that lack all 15 core gp120 glycans, yet retain conformational integrity and multiple-cycle viral infectivity and bind to several broadly neutralizing antibodies (bNAbs), including trimer-specific antibodies and a germline-reverted version of the bNAb VRC01. Our observations demonstrate that core gp120 glycans are not essential for folding, and hence their likely primary role is enabling immune evasion. We also show that our glycan removal approach is not strain restricted. Glycan-deficient Env derivatives can be used as priming immunogens because they should engage and activate a more divergent set of germlines than fully glycosylated Env. In conclusion, these results clarify the role of core gp120 glycosylation and illustrate a general method for designing glycan-free folded protein derivatives.
Keywords: human immunodeficiency virus (HIV), N-linked glycosylation, protein design, protein folding, vaccine development, germline antibody binding, immune evasion, native trimer, viral infectivity
Introduction
Human immunodeficiency virus 1 (HIV-1) is the causative agent of acquired immunodeficiency syndrome (AIDS). The Env3 glycoprotein of HIV-1 is composed of two polypeptide chains, the receptor-binding soluble subunit gp120 and the transmembrane fusion-active subunit gp41, which is present on the viral surface as a trimer of heterodimers (Fig. 1) (1). The gp120 glycoprotein is the primary viral component that is exposed on the virion surface and is the main target for neutralizing antibodies (2–4). The structure of gp120 can be subdivided into three distinct parts, the inner domain, the outer domain (OD), and the bridging sheet (Fig. 1A) (5). The OD of the HIV-1 Env glycoprotein gp120 is an important target for vaccine design as it contains the binding site for primary cellular receptor CD4 (CD4bs) and a number of conserved epitopes for various bNAbs, such as VRC01, VRC-PG04, NIH45–46, 3BNC60, b12, PGT128, and 2G12 (6–12). However, the virus has evolved various mechanisms such as a high mutation rate, conformational flexibility (13), and extensive glycan coverage of the surface (14, 15) to evade a neutralizing antibody response (16). As a result, when recombinant gp120 is used as an immunogen, the antibodies generated are often directed to immunodominant epitopes, some of which are present in variable loops, and the resulting sera have limited breadth of neutralization (17–21).
Figure 1.
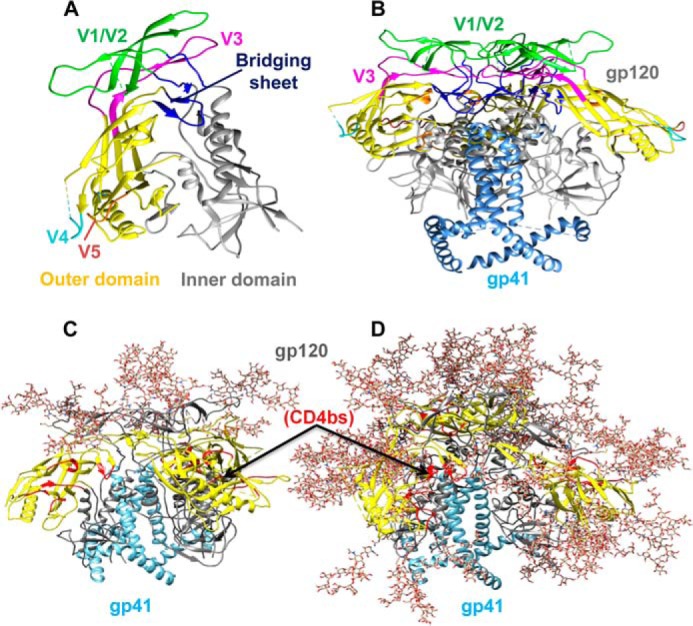
Schematic representations showing the structural organization of gp120 monomer and gp120-gp41 trimer. A, structurally, gp120 can be broadly subdivided into the inner domain, outer domain, and bridging sheet regions that are shown in gray, yellow, and dark blue, respectively. The V1/V2, V3, truncated V4, and truncated V5 variable loops are shown in green, magenta, cyan, and brown, respectively (PDB code 4NCO (142)). B, gp120-gp41 trimer is composed of surface-exposed gp120 (multicolored as in A) and membrane-anchored gp41 (light blue) (PDB code 4NCO (142)). The viral and cellular membranes are located at the bottom and top of the structure, respectively. C, gp120 subunit is shown in gray except the outer domain region (yellow) and the primary receptor, CD4-binding site (CD4bs) (red). gp120 is one of the most heavily glycosylated viral proteins known in nature. All potential N-linked glycosylation sites in the gp120 subunit are modeled with representative oligomannose glycans. gp41 subunit (blue) is involved in the viral fusion process. D, glycans are modeled only at the glycosylation sites present in the variable loops of gp120 and not at the glycosylation sites present in the core gp120 region. Trimeric envelope lacking core gp120 glycans should allow better exposure of conserved CD4bs (red) while retaining important glycan-dependent neutralization epitopes in the variable loop region. The gp120 glycan coverage in C and D is modeled on the gp120-gp41 trimeric structure (PDB code 3J5M (143)) using Glyprot software (http://www.glycosciences.de/glyprot/) (Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site) (144). gp41 glycans are not shown.
Protein N-linked glycosylation of Asn residues is an important modification found in all three domains of life (22). In eukaryotes, N-glycans mediate interactions of protein substrates with the protein quality control machinery in the endoplasmic reticulum. N-Glycans also directly affect protein folding, enhance stability, and prevent aggregation of proteins (23). Glycans constitute ∼50% of the molecular mass of gp120 (24). A large number of conserved epitopes on gp120 are often shielded as the OD is heavily glycosylated (Fig. 1C) (24). This masking of conserved epitopes is one of the several factors that has impeded the development of an Env-derived immunogen capable of eliciting bNAbs against HIV-1 (25). All the glycans on the viral surface are entirely derived from the host machinery. Analysis of HIV-1 gp120 shows that highly variable regions are often found adjacent to N-linked glycosylation sites. A potential role for N-linked glycans of HIV-1 Env is to facilitate the accumulation of silent mutations in the region of the gp120 surface that is occluded by glycans (26). Thus, a glycan array forms a non-immunogenic cloak protecting the underlying protein surface (27–42). On average, gp120 contains ∼25 putative N-linked glycosylation sites of which ∼4 are located in the inner domain, ∼7–8 in the V1/V2 and V3 variable loops, and the rest in the outer domain of gp120 (43–45). Apart from N-glycosylation, HIV-1 Env can also be modified with O-glycosylation, but this modification is less well characterized. A mass spectrometric study was able to identify only a single O-linked glycosylation at residue Thr-499 in the C5 region of recombinant gp120 (46). However, a recent study reported that O-linked glycosylation is absent even at residue Thr-499 in the context of HIV-1 virions (47). In this study, we therefore focus exclusively on N-linked glycosylation.
Glycan synthesis begins in the endoplasmic reticulum of host cells by transfer of N-linked oligomannose precursors to the amide side chain of asparagine residues in the sequon NX(T/S) (X ≠Proline) (48–50). It is widely believed that proper glycosylation is essential for correct folding of gp120 (34). However, no single glycosylation site is completely conserved in all HIV-1 isolates. Recently, all glycosylation sites in gp120 were individually mutated (44). Consistent with the lack of complete conservation, there was relatively little effect observed upon mutating individual sites both in the context of a monomer (44) and a trimer (51). However, effects of deletions of multiple glycans on Env folding have not been well studied. The ability to modulate glycan coverage is highly desirable, as it can be used to ensure selective exposure of conserved regions or unmasking of important neutralization epitopes (Fig. 1D).
Glycosylation is asserted to be essential for viral infectivity (45, 52–55). Most prior studies that examine the effect of glycan removal on viral infectivity were performed with single-site mutants (45, 54). Whenever multiple glycosylation sites were mutated in combination, it resulted in the loss of viral infectivity, which led to the hypothesis that a high level of glycan coverage is essential for maintaining infectivity of the virion (55). Apart from glycosylation, another factor affecting the generation of an effective immune response against HIV-1 is that the bNAbs against HIV-1 show a very high level of affinity maturation (57, 58). Germline (GL) sequence-reverted variants of these bNAbs fail to recognize mature Env (58). Thus, immunogens based on mature Env will likely not be able to activate the target germline B cells (59, 60). Env-based immunogens that bind well to precursors of bNAbs, as well as to the corresponding mature antibodies, may help to elicit such bNAbs upon vaccination (59–65).
We have previously described an Escherichia coli expressed outer domain fragment ODEC and showed that it can be refolded from inclusion bodies in vitro. This fragment consists of residues 255–474 of gp120. It lacks 67 V1/V2 and 32 V3 variable loop residues but contains the 12-residue V4 variable loop. Because of the absence of glycosylation machinery in E. coli, the ODEC fragment was glycan-free. It bound CD4 weakly, and in rabbits it elicited sera with measurable neutralization of some tier 1 viruses from both subtype B and C primary isolates (66). The successful refolding of ODEC in vitro suggested that glycosylation might not be essential for correct folding of OD in vivo. In this study, using mutations derived from a global sequence analysis of HIV-1 Env and subsequent optimization of electrostatic interactions, we show that glycosylation is not required for in vivo folding of an isolated OD fragment, or OD in the context of core gp120 that lacks V1/V2 and V3 variable loops, when these molecules are displayed on the surface of the yeast Saccharomyces cerevisiae. We further show here that virions with such mutations retain infectivity even in the absence of all glycans from core gp120, as tested in a single-round pseudoviral assay and in a multiple-cycle viral assay with infectious viruses derived from full-length HIV-1 molecular clones. In contrast, Env derivatives with the widely used and structurally similar Asn → Gln mutations at the same glycosylation sites fail to rescue viral infectivity. We also demonstrate that recognition of the germline-reverted version of bNAb VRC01 increases substantially with the progressive loss of glycans from JRFL pseudoviruses and that the glycan-free OD immunogens bind to mature as well as a germline-reverted VRC01 with nanomolar affinity. Such immunogens are important tools to test the usefulness of germline targeting in HIV-1 vaccine design.
Results
Design of gp120 OD fragments with mutations at multiple glycosylation sites
Our previous observation that a glycan-free OD could fold, bind bNAbs, and elicit immune response contradicted some earlier reports pointing to the importance of glycosylation in gp120 folding (34, 54, 66). Notably, the earlier studies on HIV-1 glycosylation involved mutating Asn to either Ala or to structurally similar Gln (45, 54, 55), without taking into account the effects that these mutations could have on the protein structure and stability. Substitutions with amino acids other than Gln and Ala have generally not been tested at these glycosylation sites. Therefore, a more rational approach to identify more suitable mutations at glycosylation sites seemed necessary. Methods for stabilizing mutations can be classified as rational structure-based methods, directed evolution-based methods, and semi-rational sequence frequency-based methods (67). Because it is often difficult to predict stabilizing mutations solely using rational structure-based methods and also because a huge amount of sequence information is available for HIV-1, we decided to make use of an amino acid frequency-based approach to identify potentially suitable mutations at glycosylation sites. The approach was then augmented by the use of structure-based and directed evolution methods. We hypothesized that by selecting naturally occurring substitutions at the glycosylation sites, it might be possible to retain significant Env stability and function in the absence of glycosylation. To examine the effect of glycan removal on the in vivo folding of OD, various ODEC (66) derivatives were designed (Table 1). The choice of mutations was based on a multiple sequence alignment of HIV-1 sequences from the HIV-1 Env Sequence Compendium 2010 (68). The methodology used for the design of glycan-free Env derivatives is described in Fig. 2. Briefly, amino acid propensities at all the potential N-linked glycosylation site(s) (PNGS) across all clades of HIV-1 were calculated. It was found that none of the glycosylation sites in the OD is 100% conserved. Based on the above calculations, Asn at each glycosylation site was mutated to the second most frequent amino acid in the multiple sequence alignment. Residues that were hydrogen-bonded to the Asn were identified using the program HBPLUS (69), and they were similarly mutated to the second most frequent amino acid to avoid unsatisfied H-bonding groups. Finally, models of the mutant constructs were built using the program MODELLER (70). Electrostatic calculations (71) on the wild-type and mutant OD constructs confirmed that the mutations had not introduced unfavorable electrostatic interactions. Next, a yeast codon-optimized (YCO) version of ODEC was synthesized (ODYCO), where all the 14 PNGS (NX(T/S)) were retained, whereas in the construct ΔG14-ODYCO, Asn residues at all 14 PNGS were mutated. It was likely that mutating all 14 glycosylation sites simultaneously could adversely affect the folding of the OD molecule. Therefore, in the construct ΔG4-ODYCO, only four Asn residues (276, 386, 392, and 463) proximal to the primary receptor-binding site (CD4bs) were mutated to allow increased exposure of this highly conserved region while causing minimal structural perturbation (Fig. 3). A ΔG14-ODYCO variant with the D368R mutation was also synthesized as a control to confirm that the binding of ΔG14-ODYCO to VRC01 bNAb is specific. The D368R mutation is known to reduce the binding of gp120 molecule with CD4 and CD4bs antibodies (72). Table 1 and the “Experimental procedures” provide a detailed description of all OD constructs. Mutations introduced at PNGS in the OD constructs are listed in supplemental Table S1.
Table 1.
Nomenclature and description of HIV-1 gp120 outer domain fragment constructs based on HXBc2 strain
| No. | Construct name | Description |
|---|---|---|
| 1 | ODEC | HIV-1 gp120 outer domain (OD) fragment from HXBc2 strain was codon-optimized for expression in E. coli with all 14 PNGS intact. This fragment consists of residues 255–474 of gp120 with 11 designed mutations to prevent aggregation. It lacks V1/V2 and V3 variable loops but retains V4 loop residues (66) |
| 2 | ΔG14-ODECa | An ODEC variant lacking all 14 PNGS |
| 3 | ODYCO | The same amino acid sequence as ODEC but codon-optimized for expression in yeast (S. cerevisiae). All 14 PNGS are intact. YCO stands for yeast codon-optimized |
| 4 | ΔG4-ODYCO | Partially glycan-free ODYCO lacking four PNGS (at residues 276, 386, 392, and 463) proximal to the CD4-binding region |
| 5 | ΔG14-ODYCO | Completely glycan-free derivative of ODYCO. Devoid of all 14 outer domain PNGS |
| 6 | ΔG14-ODYCO-D368R | ΔG14-ODYCO with a D368R mutation known to decrease the binding of gp120 with CD4-binding site antibodies |
a ΔG in a construct's name indicates that it lacks certain glycosylation sites, whereas the numeral followed by ΔG indicates the total number of glycosylation sites mutated. The background in which each construct is made is also indicated in the construct's name. This scheme of nomenclature is followed throughout the paper.
Figure 2.
Methodology to design glycan-free HIV-1 envelope derivatives.
Figure 3.

Schematic representations of the glycosylation site mutants of OD fragments. A, structure of WT core gp120 (PDB code 1G9M). The OD fragment from residues 255 to 474 is shown in yellow; the CD4-binding site (CD4bs) is shown in red; and the rest of the protein is shown in gray. OD constructs (B–E) represent models of ODEC (all 14 PNGS are intact but still glycan-free due to expression in E. coli) (66), ODYCO (all 14 PNGS are intact, same amino acid sequence as ODEC but codon-optimized for expression in yeast, glycosylated due to expression in S. cerevisiae), ΔG14-ODYCO (devoid of all 14 PNGS and therefore glycan-free despite expression in S. cerevisiae), and ΔG4-ODYCO (four PNGS close to CD4bs mutated in ODYCO background, partially glycosylated), respectively. Asn residues at PNGS in C and E are shown as green spheres. Asn residues at PNGS mutated to other amino acids are shown in blue in D. These constructs are further described in Table 1.
Yeast surface display and binding studies of OD-based glycosylation variants
All the above OD fragments were cloned in the yeast surface display vector pPNLS under a galactose-inducible promoter, and their expression on the yeast surface as well as binding to the bNAb VRC01 was examined using FACS. Yeast was used as an initial screening system to study the effects of glycosylation on the in vivo folding of OD and core gp120. Yeast surface display allows for simultaneous rapid screening of a large number of mutants, alleviating the need to express and purify the individual clones (73). The expression on the yeast surface has previously been shown to be directly correlated with the folding efficiency of the protein (74). All OD-based glycosylation variants showed comparable expression on the yeast surface, indicating that partial or even complete removal of glycans does not adversely affect the folding of the OD (supplemental Fig. S1A). To further confirm the conformational integrity of surface-displayed OD molecules, binding interactions with the bNAb VRC01 were monitored using FACS as described previously (73, 75). Fully glycosylated OD molecule (ODYCO) bound VRC01 with a KD greater than 300–400 nm. The ΔG4-ODYCO molecule lacking four glycans near the CD4bs showed ∼10-fold higher affinity for VRC01, whereas ΔG14-ODYCO lacking all 14 N-linked glycans bound VRC01 with ∼30-fold higher affinity (KD of ∼10 nm) than ODYCO (Table 2). This significant increase in affinity for VRC01 shows that removal of glycans did not impact the folding of OD but instead resulted in a better exposure of the VRC01 epitope. VRC01 affinity for these deglycosylated variants is comparable with that of the fully glycosylated core gp120 (Table 2). A ΔG14-ODYCO variant with the D368R mutation was used as a control to show that the binding to VRC01 is indeed specific. The D368R mutation is known to reduce the binding of gp120 to CD4 and CD4bs antibodies (72). As expected, the ΔG14-ODYCO-D368R mutant showed a greater than 70-fold decrease (KD >700 nm) in affinity for VRC01 relative to the base construct ΔG14-ODYCO, demonstrating the specificity of VRC01 binding (Table 2). Importantly, these constructs did not show measurable binding with the non-neutralizing monoclonal antibody (mAb) b6 as they lack certain regions of the b6 epitope (data not shown) (66). Representative FACS histograms for the binding of the VRC01 antibody with a yeast displayed OD glycan mutant are shown in supplemental Fig. S2. Titration curves for VRC01 binding with various OD constructs are shown in supplemental Fig. S3.
Table 2.
KD values for the binding of various OD fragments displayed on the yeast surface to VRC01 antibody, determined by FACS titrations
KD indicates equilibrium dissociation constant; S.D. is standard deviation for the data from two independent experiments.
| Construct name | KD ±S.D. |
|---|---|
| nm | |
| ODYCO (fully glycosylated) | >300–400 |
| ΔG4-ODYCO (partially glycan-free) | 27 ± 3 |
| ΔG14-ODYCO (completely glycan-free) | 10 ± 1 |
| ΔG14-ODYCO-D368Ra (completely glycan-free negative control) | >700 |
| WT core gp120 (positive control) | 24.3 ± 0.4 |
a D368R mutation is known to decrease the binding of gp120 with CD4-binding site antibodies such as VRC01.
These results show that the isolated gp120 outer domain can be expressed on the yeast surface in a well folded conformation, and glycosylation is not essential for the folding of the gp120 outer domain either in vitro or in a eukaryotic system. This is a significant result as it shows that it is possible to design and produce OD-based immunogens that are devoid of glycosylation. Such constructs should allow better exposure of epitopes normally shielded by glycans and thus might act as better immunogens for eliciting VRC01-like broadly neutralizing antibodies.
ΔG14-ODEC purification, biophysical characterization, and binding studies
ΔG14-ODYCO is completely devoid of glycosylation sites. Thus, it was possible to express and purify it from E. coli for further characterization. An E. coli codon-optimized version (ΔG14-ODEC) was cloned in pET-28a(+) vector with an N-terminal His tag. The protein was expressed in E. coli BL21(DE3) cells and purified on an nickel-nitrilotriacetic acid affinity matrix after resolubilization from inclusion bodies. The protein yield was about 1–2 mg/liter of culture. SDS-PAGE studies confirmed that the protein was at least 90% pure (supplemental Fig. S4). The CD spectrum of the protein (supplemental Fig. S5A) showed that it has significant secondary structure. The fluorescence spectrum (supplemental Fig. S5B) of the protein showed an expected red shift and a change in emission intensity upon denaturation, indicating that the protein is likely to be folded with a burial of some tryptophan residues in the native state. The observed mass of ΔG14-ODEC in ESI-MS analysis was found to be 22,886.34 Da indicating that all six cysteines in ΔG14-ODEC are oxidized (supplemental Fig. S6). Native ΔG14-ODEC eluted from a C5 analytical reverse-phase column largely as a single peak (supplemental Fig. S7A), thereby proving that it is largely a homogeneous species in solution and not a mixture of different disulfide-bonded isomers. The denatured, reduced protein eluted at a different acetonitrile concentration than the native protein, re-confirming that native ΔG14-ODEC is folded and oxidized. ΔG14-ODEC contains six cysteines that are expected to form three disulfide bonds resulting in a calculated mass of 22,886.25 Da. Using tandem mass spectrometric analysis, two disulfides (296–331 and 385–418) were assigned native connectivities (supplemental Table S2). As two disulfides were already assigned, the third disulfide (378–445) is assigned automatically. These data indicate that the outer-domain disulfides can have native connectivities in the absence of glycosylation.
Analytical gel filtration (supplemental Fig. S7B) showed that ΔG14-ODEC is primarily a monomer in solution and elutes at the same position as the base construct ODEC. The elution time is longer than expected for a monomer, suggestive of some interaction between the protein and the Superdex carbohydrate matrix (76). ESI-MS of the eluted peak confirmed that it had the expected mass of 22,886 Da (supplemental Fig. S6), ruling out degradation of the protein as the cause for the longer than expected elution time.
SPR binding studies
ΔG14-ODEC is completely devoid of glycosylation sites, the inner domain, a part of the bridging sheet, and V1/V2 and V3 loops and is ∼5 times smaller than the full-length glycosylated gp120 molecule. Nonetheless, it is capable of binding the soluble 4-domain CD4 (sCD4) and the bNAbs b12 and VRC01 with affinities comparable with that of full-length gp120, as demonstrated by SPR (Table 3 and supplemental Fig. S8). Binding of ΔG14-ODEC to a germline-reverted version of VRC01 antibody (GL-VRC01, encoded by IGHV1–2*02 VH gene with a mature CDRH3 region and IGKV3–20*01 VL gene) was also characterized. ΔG14-ODEC bound GL-VRC01 with a KD of ∼10 nm (Table 3 and supplemental Fig. S8D), and as expected, full-length WT gp120 did not show any binding to GL-VRC01 (Table 3 and supplemental Fig. S8F, curve 5). Competition binding experiments demonstrate that the binding of ΔG14-ODEC with GL-VRC01 antibody is specific as it decreases in the presence of mature VRC01 antibody in a concentration-dependent manner (supplemental Fig. S8E). Amino acid sequence comparison of mature VRC01 and GL-VRC01 is shown in supplemental Fig. S9. Both ODEC (all PNGS intact) and ΔG14-ODEC (all 14 PNGS mutated) are bacterially expressed and therefore lack glycans. Even then, mutations at the putative glycosylation sites in ODEC resulted in improved binding of ΔG14-ODEC to VRC01, b12, and sCD4 by ∼8-, 10-, and 15-fold, respectively, relative to ODEC (Table 3). A comparison of all kinetic parameters for binding is given in Table 3.
Table 3.
Kinetic parameters for binding of bNAb VRC01, bNAb b12, sCD4, and GL-VRC01 with full-length WT gp120, ODEC, and ΔG14-ODEC as determined by SPR studies
kon is association rate; koff is dissociation rate; KD is equilibrium dissociation constant determined using SPR; S.D. is standard deviation for the data from two independent experiments; bNAb is broadly neutralizing antibody; GL is germline. ODEC and ΔG14-ODEC were purified from E. coli.
| Ligand | Analyte | kon | koff | KD ±S.D. |
|---|---|---|---|---|
| m−1 s−1 | s−1 | nm | ||
| VRC01 | WT gp120 | 3.3 × 104 | 8.7 × 10−4 | 26 ± 5 |
| ODEC | 3.7 × 104 | 1.3 × 10−2 | 359 ± 55 | |
| ΔG14-ODEC | 5.6 × 104 | 3.83 × 10−3 | 47 ± 17 | |
| b12 | WT gp120 | 3.5 × 104 | 1.7 × 10−3 | 49 ± 5 |
| ODEC | 1.7 × 104 | 2.7 × 10−3 | 153 ± 17 | |
| ΔG14-ODEC | 1.1 × 105 | 1.8 × 10−3 | 16 ± 1 | |
| sCD4 | WT gp120 | 2.3 × 104 | 3.3 × 10−4 | 15 ± 4 |
| ODEC | 1.9 × 104 | 4.2 × 10−3 | 219 ± 11 | |
| ΔG14-ODEC | 1.1 × 105 | 1.5 × 10−3 | 15 ± 3 | |
| GL-VRC01 | WT gp120 | –a | –a | –a |
| ODEC | 3.3 × 105 | 1.3 × 10−3 | 4 ± 1 | |
| ΔG14-ODEC | 1.4 × 105 | 1.2 × 10−3 | 9 ± 2 |
a No detectable binding was noted.
Design, yeast cell-surface expression, and binding studies of core gp120 glycosylation site mutants from JRFL isolate
After demonstrating that glycosylation is not required for the folding of the isolated outer domain fragment in vivo and in vitro, we extended our inquiry into the context of a larger Env construct and examined the effects of glycan removal on the core gp120 molecule from JRFL strain. As described under “Experimental procedures,” core gp120 retains well structured core region of the gp120 glycoprotein but lacks highly flexible V1/V2 and V3 variable loops, making it a simpler system to manipulate and analyze as compared with full-length gp120. Core gp120 from JRFL strain contains 15 PNGS out of which 13 PNGS are present in the outer domain and two in the inner domain. Out of 13 OD PNGS, two are very close to the interface of inner domain and outer domain. WT core gp120 (in which none of the PNGS were mutated), ΔG15 core gp120 (all 15 PNGS were mutated), and ΔG4 core gp120 (four PNGS near CD4bs at residues 276, 386, 392, and 463 were mutated) (Table 4 and Fig. 4) were expressed on the yeast cell surface under a galactose-inducible promoter. Mutations in core gp120 derivatives were designed in a similar manner as for the OD-based constructs from the HXBc2 strain described above and in Fig. 2. Apart from the mutations at PNGS, three additional mutations (K336Q, H363Q, and K240T) were introduced in the gp120 constructs to avoid unfavorable electrostatic interactions (71). Mutations introduced at PNGS in the JRFL gp120 constructs are listed in supplemental Table S3. Expression of WT core gp120 and ΔG4 core gp120 was comparable on the yeast cell surface, indicating that removal of four glycans near the CD4bs did not adversely affect the folding of the protein (supplemental Fig. S1B). On the yeast surface, ΔG4 core gp120 binds to VRC01 with ∼10-fold, to b12 with ∼3-fold, and to b6 with ∼17-fold higher affinity as compared with WT core gp120. However, both molecules showed a similar affinity toward recombinant CD4D12 (KD ∼25 nm) (Table 5, CD4D12 and b12 binding affinities are in Table 5 footnote). It is possible that the smaller two-domain CD4 molecule CD4D12 (∼22 kDa) experiences less steric constraints from glycans near the CD4-binding site than larger IgG molecules (∼150 kDa). Next, we tested the effect of removal of these four glycans on full-length gp120. Upon re-introduction of V1/V2 and V3 loop regions in ΔG4 core gp120 (E168K ΔG4 full-length gp120, Table 4), the molecule bound to the cell-surface receptor CD4D12, bNAbs b12, VRC01, and the non-neutralizing antibody b6 with comparable affinity to WT core gp120 (Table 5, CD4D12 and b12 binding affinities in Table 5 footnote) demonstrating that the full-length gp120 lacking four glycans near the CD4bs molecule can maintain the conformational integrity of the CD4bs. This is in contrast to a previous report, which failed to detect any binding with anti-gp120 antibodies when loop regions are incorporated into the core molecule and displayed on the yeast surface (77). Expectedly, E168K ΔG4 full-length gp120 did not bind the bNAb PG9, which recognizes a glycan-dependent quaternary epitope in the V1/V2 loop (data not shown). The lack of PG9 binding is consistent with the fact that the yeast cell surface-expressed gp120 is not expected to form trimers due to the absence of gp41 subunit. The absence of PG9 binding could also be due to the differences in glycosylation between yeast and mammalian cells. In contrast to ΔG4 core gp120, ΔG15 core gp120 (which lacks all 15 glycans) was not expressed well on the yeast cell surface (supplemental Fig. S1B), did not bind b12, b6, and CD4 (data not shown), and bound VRC01 with a significantly reduced binding affinity (Table 5). Hence, it can be inferred that removal of all the glycans from core gp120 has an adverse effect on the folding of the molecule. To identify the glycosylation sites adversely affecting core gp120 folding and generate minimally glycosylated gp120 molecules, we gradually removed glycosylation sites from core gp120 as described below.
Table 4.
Nomenclature and description of HIV-1 gp120 constructs based on JRFL strain
| No. | Construct name | Description |
|---|---|---|
| 1 | WT core gp120 | Wild-type JRFL gp120 lacking V1/V2 and V3 loops. It contains residues 31–127-GAG-195–297-GAG-330–507 |
| 2 | ΔG15 core gp120 | Core gp120 devoid of all 15 PNGS |
| 3 | ΔG4 core gp120 | ΔG4 core gp120 contains all PNGS except for four sites (at residues 276, 386, 392 and 463) near the CD4-binding region |
| 4 | ΔG11 core gp120 | This construct contains only four PNGS: two in the inner domain of gp120 (at residues 88 and 241) and two at the interface of the inner and outer domains of gp120 (at residues 262 and 276) |
| 5 | ΔG12a core gp120 | An additional PNGS (N88Q) removed in ΔG11 core gp120 background, now contains three PNGS |
| 6 | ΔG12b core gp120 | An additional PNGS (N241K) removed in ΔG11 core gp120 background now contains three PNGS |
| 7 | ΔG12c core gp120 | An additional PNGS (N262D) removed in ΔG11 core gp120 background now contains three PNGS |
| 8 | ΔG12d core gp120 | An additional PNGS (N276D) removed in ΔG11 core gp120 background now contains three PNGS |
| 9 | ΔG13 core gp120 | Contains only two PNGS at inner domain residues 88 and 241 |
| 10 | ΔG14a core gp120 | Contains only one PNGS at residue inner domain residue 241. Mutation N88G at inner domain position 88 in this construct was selected using yeast library screening |
| 11 | ΔG14b core gp120 | Contains only one PNGS at inner domain residue 88. Mutation N241K at inner domain position 241 in this construct was selected using yeast library screening |
| 12 | ΔG15F core gp120 | Devoid of all 15 core gp120 PNGS. Mutations at inner domain positions 88 and 241 in this construct were selected using yeast library screening |
| 13 | E168K ΔG4 full-length gp120 | A ΔG4 core gp120 variant that contains the V1/V2 and V3 loop regions and the E168K mutation in the V1 loop in order to introduce PG9 antibody epitope |
Figure 4.

Illustration of the glycosylation site mutants of V1/V2 and V3 loop-deleted JRFL gp120 (core gp120). The CD4-binding site (CD4bs) epitope is shown in red, and Asn residues at PNGS are shown as green spheres. OD fragment from residues 255 to 474 is shown in yellow, and the rest of the protein is in gray. Mutated amino acids in each construct are shown in blue (PDB code 2B4C) (56). Individual mutants or mutant libraries at PNGS were screened using yeast surface display and FACS. Additional details of mutants are given in Table 4.
Table 5.
KD values for the binding of yeast cell surface displayed WT core gp120 and its PNGS mutants with various CD4-binding site antibodies as determined by FACS titrations
KD is equilibrium dissociation constant determined using FACS; S.D. is standard deviation for the data from two independent experiments; b12/VRC01 is broadly neutralizing antibodies against HIV-1; b6, non-neutralizing anti HIV-1 antibody.
| Construct | Mutations |
KD ± S.D. (nm)a |
|
|---|---|---|---|
| VRC01 | b6 | ||
| WT core gp120 | 24.3 ± 0.4 | 1.75 ± 0.2 | |
| ΔG15 core gp120 | all 15 PNGS mutated | >500 | –b |
| ΔG4 core gp120 | N276D + N386D + N392S + N463D | 2.33 ± 0.3 | 0.1 ± 0.006 |
| E168K ΔG4 full-length gp120c | E168K + N276D + N386D + N392S + N463D | 20.5 ± 5.3 | 2.8 ± 0.7 |
| ΔG11 core gp120 | 11 PNGS mutated (*). PNGS at positions 88, 241, 262, and 276 retained. | 17 ± 0.25 | 0.4 ± 0.03 |
| ΔG12a core gp120 | (*) + N88Q | 43.5 ± 5 | 1.87 ± 0.7 |
| ΔG12b core gp120 | (*) + N241K | 94.2 ± 19.2 | 1.82 ± 0.7 |
| ΔG12c core gp120 | (*) +N262D | 26 ± 5.3 | 0.3 ± 0.04 |
| ΔG12d core gp120 | (*) + N276D | 2.8 ± 0.9 | 0.3 ± 0.07 |
| ΔG13 core gp120 | (*) + N262D + N276D | 4.9 ± 0.7 | 0.5 ± 0.1 |
| ΔG14a core gp120 | (*) + N262D + N276D + N88G | 4.2 ± 0.25 | 0.35 ± 0.006 |
| ΔG14b core gp120 | (*) + N262D + N276D + N241H | 4.9 ± 1.8 | 0.45 ± 0.01 |
a KD values for the binding of WT core gp120, ΔG4 core gp120, E168K ΔG4 full-length gp120, and ΔG11 core gp120 with CD4D12 (two-domain CD4) were found to be 27 ± 1.9, 22 ± 0.4, 18.2 ± 2, and 32 ± 2.3 nm, respectively; whereas KD values for binding with bNAb b12 were found to be 1.2 ± 0.6, 0.4 ± 0.06, 11.2 ± 6.9, and 0.6 ± 0.3 nm, respectively, as determined by FACS.
b – indicates no detectable binding.
c This contains V1/V2 and V3 variable loops, all other gp120 constructs lack these.
Design, yeast cell-surface expression, and binding of ΔG11 core gp120 (core gp120 devoid of eleven outer domain glycans) and its single-site mutants
As described above, the studies with ΔG14-ODYCO showed that outer domain glycans are not necessary for the folding of the isolated OD fragment in vivo (Table 2 and supplemental Fig. S3). Therefore, we characterized the construct ΔG11 core gp120, wherein 11 glycosylation sites from the outer domain were mutated simultaneously (Fig. 4 and Table 4). This construct retained only four out of 15 glycosylation sites present in JRFL core gp120: two in the inner domain of gp120 (at residues 88 and 241) and two at the interface of the inner and outer domains of gp120 (at residues 262 and 276). These four PNGS were not mutated in the initial constructs as we expected one or more of these to be critical for core gp120 folding due to their structural positioning. ΔG11 core gp120 expression on the yeast cell surface was comparable with WT core gp120 (supplemental Fig. S1C). This construct bound CD4D12, b12, VRC01, and b6 with affinities comparable with WT core gp120 (Table 5, CD4D12 and b12 binding affinities in Table 5 footnote). Correct folding of ΔG11 core gp120 in the absence of 11 OD glycans combined with the lack of folding of core gp120 lacking all 15 glycans indicates that one or more of the four remaining glycans are important for gp120 folding. To determine which of the four remaining glycans are important for the proper folding and activity of the core gp120 molecule, these four glycosylation sites were individually mutated resulting in the following constructs: ΔG12a core gp120 (N88Q), ΔG12b core gp120 (N241K), ΔG12c core gp120 (N262D), and ΔG12d core gp120 (N276D) (Fig. 4 and Table 4 describe these constructs in greater detail). Cell-surface expression of all four constructs was found to be similar (supplemental Fig. S1D). However, the former two constructs bound b6 with ∼6-fold lower and VRC01 with much lower affinity than ΔG12c core gp120 and ΔG12d core gp120 (Table 5). Hence, another mutant ΔG13 core gp120 was made, which contains only two glycans at residues 88 and 241 in the inner domain (Fig. 4 and Table 4). ΔG13 core gp120 retained surface expression similar to ΔG11 core gp120 (supplemental Fig. S1E), ΔG13 core gp120 bound b6 with similar affinity, and VRC01 with ∼3-fold improved affinity as compared with ΔG11 core gp120 (Table 5) indicating that glycan removal from positions 88 and 241 is affecting proper folding of core gp120, resulting in reduced mAb binding.
Site-directed saturation mutagenesis and screening of mutants at Asn-88 and Asn-241 positions
To examine whether any other residues at positions 88 and 241 can compensate for the loss of glycans and improve antibody binding, both positions were individually randomized to all other residues except Asn. The Asn-88 position was randomized in ΔG12a core gp120 background, whereas Asn-241 position was randomized in ΔG12b core gp120 background (Table 4). Thus, two yeast-surface display libraries with 19 mutants in each were made and screened by FACS for binding to the conformation-specific mAb b6 to select properly folded core gp120 mutants (supplemental Fig. S10). We used b6 rather than VRC01 because b6 binds with much higher affinity to monomeric gp120 than VRC01 and thus allows screening of folded variants at low antibody concentrations (61, 78). Two mutants N88G and N241H were isolated after three rounds of sorting. These mutants show surface expression and binding with b12, b6, and VRC01 comparable with that of ΔG11 core gp120 (data not shown). These two mutations were incorporated individually and in combination in the ΔG13 core gp120 background. The resulting mutants, ΔG14a core gp120 and ΔG14b core gp120, contain only one glycan each at either residue 241 or 88, respectively (Table 4 and Fig. 4). ΔG15F core gp120 is devoid of all 15 core gp120 glycans and contains the N88G and N241H mutations selected through yeast library screening instead of N88Q and N241K mutations that are present in ΔG15 core gp120 (Table 4). VRC01 and b6 affinities with ΔG14a core gp120 and ΔG14b core gp120 are comparable with ΔG13 core gp120 (Table 5). However, ΔG15F core gp120 shows poor surface expression (supplemental Fig. S1E) and reduction in bNAb binding (data not shown). Representative FACS histograms and titration curves for the binding of mAbs with yeast surface-displayed ΔG14a core gp120 glycosylation site mutant are shown in supplemental Figs. S11 and S12, respectively. Both Asn-88 and Asn-241 belong to the inner domain of gp120, which interacts with gp41 (79–81). Because gp41 is absent in the present constructs, we hypothesized that removal of inner domain glycans may result in the exposure of hydrophobic surface and aggregation of core gp120. In summary, when displayed on the yeast surface, we found that core gp120 with just a single glycan either at residue number 88 or 241 still retained the ability to bind VRC01 and is likely to be properly folded.
Mammalian cell-surface display of WT gp160 and its glycosylation site mutants from JRFL isolate
Following yeast-surface display of glycosylation mutants in the context of monomeric core gp120 as described above, we examined the effect of glycan removal in the context of native trimeric Env on the mammalian cell surface. WT gp160, ΔG11 gp160, ΔG12 gp160, ΔG13 gp160, ΔG14 gp160, and ΔG15F gp160 were displayed on the mammalian cell surface (Fig. 5) (see Table 6 and under “Experimental procedures” for more details of gp160 constructs). All variants of ΔG11 gp160, including ΔG15F gp160, which lacks all 15 core gp120 glycans, bound conformation-specific CD4bs bNAbs b12 and VRC01 with apparent affinity similar to that of WT gp160, indicating that removal of all the glycans from core gp120 did not disrupt the native fold of oligomeric Env on the mammalian cell surface (Fig. 6). A native trimer-specific bNAb PG9 bound ΔG11 gp160 with an affinity similar to that of WT gp160 indicating that the native Env trimer can form in the absence of most outer domain glycans. Removal of additional glycans from ΔG11 gp160 resulted in an ∼2-fold decrease in PG9 interaction (Fig. 6). In contrast to monomeric gp120 variants displayed on the yeast surface, trimeric envelope molecules are not expected to show significant binding with the non-neutralizing antibody b6 if they are properly cleaved and well folded (82, 83). Similar to WT gp160, all the ΔG11 gp160 variants showed poor binding with the non-neutralizing antibody b6, indicating limited exposure of this non-neutralizing epitope in the context of glycan-deficient trimeric envelope molecules (Fig. 6). We have previously shown that a cleavage-defective Env (SEKS gp160) shows higher binding with anti-gp120 polyclonal sera as compared with WT gp160, indicating increased exposure of non-neutralizing epitopes in SEKS gp160 (82). To determine whether removal of core gp120 glycans also results in such increased exposure of non-neutralizing epitopes, we examined the binding of various glycan-deficient gp160 constructs with anti-gp120 polyclonal sera. Binding of anti-gp120 polyclonal sera with glycan-deficient Envs was found to be comparable with WT gp160 (supplemental Fig. S13). This is expected only if glycan removal does not cause a significant increase in the exposure of non-neutralizing epitopes because gp120 antisera consist primarily of non-neutralizing antibodies that do not bind well to properly folded Env trimers (83). As expected, unlike all the glycan-deficient gp160 derivatives described above, WT Env bound 2G12 and PGT128, whose epitopes are glycan-dependent (Fig. 6) (9, 11, 84). Reintroduction of the Asn-332 PNGS into the ΔG15F gp160 background (devoid of all 15 core gp120 glycans) resulted in the recovery of PGT128 binding (Fig. 6), illustrating the usefulness of these glycan-deficient envelopes in epitope mapping studies. We also monitored the binding of an anti-gp41 bNAb (2F5) with WT gp160 and its glycosylation site variants and found that 2F5 binding is largely retained in the absence of all core gp120 glycans (Fig. 6).
Figure 5.

Schematic representation of JRFL gp160 glycosylation site mutants. PNGS in variable loops (V1/V2 and V3), in the core gp120 region, and in the gp41 subunit are indicated by green, red, and blue asterisks, respectively. The gp120 sequence is from residue 31 to 511, and the cytoplasmic tail truncated gp41 is from residues 512 to 712. A, WT gp160 retains all 15 PNGS located in the core gp120 region. B, in ΔG11 gp160 construct, asparagines at most OD PNGS were rationally mutated. C–F, four remaining PNGS, two at the interface of the inner and outer domains of gp120 (at residues 262 and 276) and two in the inner domain of gp120 (at residues 88 and 241), were sequentially mutated in the ΔG11 gp160 background to finally generate a mutant lacking all 15 core gp120 glycans (ΔG15F gp160). G, in ΔGQ11 gp160 construct, asparagines at 11 outer domain glycosylation sites were mutated to glutamine. This ΔG11 gp160 variant was designed as a control to understand the importance of rationally designed mutations for glycan removal. H, gp120-gp41 cleavage site REKR was mutated to SEKS in WT gp160 background to generate a cleavage-defective gp160 variant (SEKS gp160). Mutated core gp120 glycosylation sites are indicated by replacement of red asterisks with single letter amino acid codes for the residues introduced in place of asparagines. E168K mutation was introduced in all JRFL gp160 constructs to confer binding of the trimer-specific, broadly neutralizing antibodies PG9 and PG16. Additional details of gp160 mutants are given in Table 6.
Table 6.
Nomenclature and description of HIV-1 gp160 constructs
All gp160 constructs were expressed in mammalian cells.
| No. | Construct name | Description |
|---|---|---|
| 1a | WT gp160 | Full-length WT gp160 from the JRFL strain with all PNGS intact. It contains 15 PNGS in the core gp120 region |
| 2 | ΔG11 gp160 | A WT gp160 variant lacking 11 OD PNGS. It contains only four core gp120 PNGS: two at the interface of the inner and outer domains of gp120 (at residues 262 and 276) and two in the inner domain of gp120 (at residues 88 and 241) |
| 3 | ΔG12 gp160 | Asn-276 glycan removed in ΔG11 gp160 background. |
| 4 | ΔG13 gp160 | Devoid of all OD PNGS including two (Asn-262 and Asn-276) present at the interface of inner and outer domains of gp120 |
| 5 | ΔG14 gp160 | Devoid of all OD PNGS + mutation at inner domain PNGS position 88 (N88G) |
| 6 | ΔG15F gp160 | Devoid of all 15 core gp120 glycans. Mutations at inner domain positions 88 (N88G) and 241 (N241H) in this construct were selected using yeast library screening of core gp120 |
| 7 | T332N ΔG15F gp160 | ΔG15F gp160 with T332N mutation to reintroduce PGT128 bNAb epitope |
| 8 | ΔGQ11 gp160 | A variant of ΔG11 gp160 wherein Asn residues at 11 OD PNGS were mutated to structurally similar Gln instead of rational mutations |
| 9 | SEKS gp160 | Cleavage defective JRFL gp160. Furin cleavage site REKR is mutated to SEKS in WT gp160 background |
| 10b | Q842 WT gp160 | WT gp160 from subtype A strain Q842env.d16 |
| 11 | Q842 ΔG13 gp160 | Gln-842 gp160 variant lacking 13 OD PNGS |
| 12 | QH343 WT gp160 | WT gp160 from subtype A strain QH343.21M.ENV.B5 |
| 13 | QH343 ΔG12 gp160 | QH343 gp160 variant lacking 12 OD PNGS |
| 14 | DU WT gp160 | WT gp160 from subtype A strain DU422.1 |
| 15 | DU ΔG10 gp160 | DU gp160 variant lacking 10 OD PNGS |
| 16 | CAP WT gp160 | WT gp160 from subtype A strain CAP45.2.00G3 |
| 17 | CAP ΔG12 gp160 | CAP gp160 variant lacking 12 OD PNGS |
a Constructs 1–9 are based on the JRFL isolate. All JRFL gp160 molecules have the E168K mutation. The E168K mutation confers PG9/PG16 (trimer-specific bNAbs) binding on JRFL Env. All JRFL gp160 constructs have a cytoplasmic tail deletion (ΔCT) that is known to improve expression on the mammalian surface. Mutations introduced in JRFL gp160 constructs were the same as in core gp120 constructs and are described in supplemental Table S3.
b Constructs 10–17 are based on different HIV-1 strains from clade A and clade C HIV-1 reference panel of Env clones. These construct names bear the initial of the strain from which they are derived. A variable number of OD glycosylation sites are present in different strains, and the nomenclature reflects the number of PNGS mutations in each construct. Mutations introduced in these gp160 variants are described in supplemental Table S4.
Figure 6.
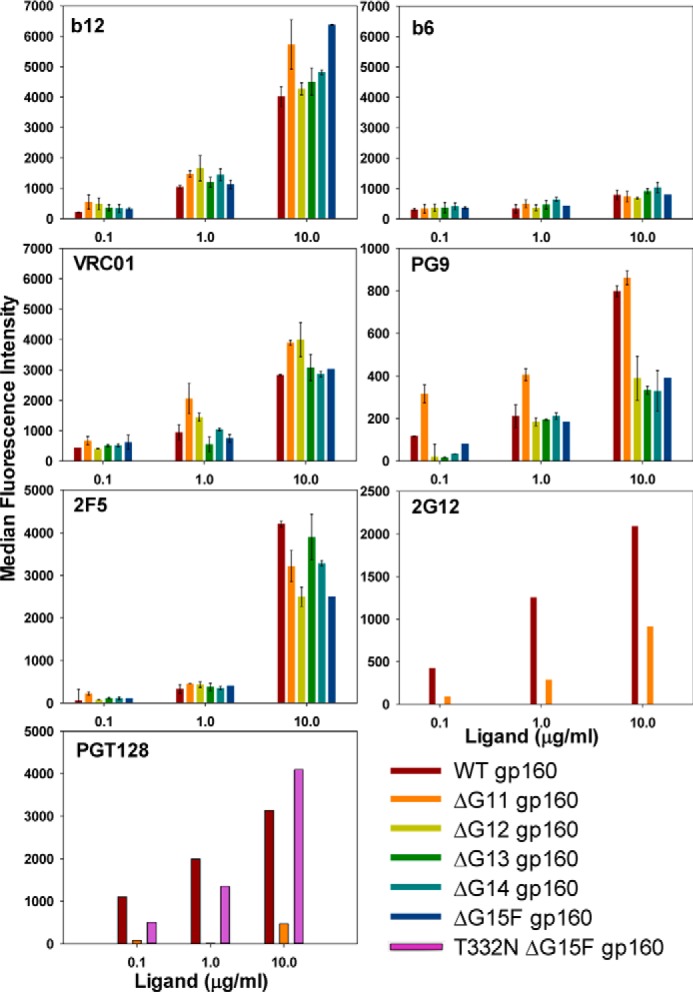
FACS-based mammalian cell-surface staining plots of JRFL cleavage-competent WT gp160 and its glycan derivatives for binding to various anti-HIV-1 antibodies. ΔG11 gp160 (devoid of 11 outer domain glycans) binds to the anti-gp120-neutralizing monoclonal antibodies b12, VRC01, PG9, and anti-gp41 mAb 2F5 similar to the WT gp160, and like the WT it binds poorly to the non-neutralizing mAb b6, confirming that it is properly folded. It does not bind to the neutralizing mAb PGT128 as it lacks the Asn-332 glycan. Other glycan-deficient derivatives of JRFL gp160 also show a binding pattern similar to ΔG11 gp160. Reintroduction of the Asn-332 glycan in ΔG15F gp160 (bottom panel) restores PGT128 binding. Error bars represent standard deviation for the data from two independent experiments.
Pseudoviral infectivity and neutralization assays
To assess the effect of glycan removal on HIV-1 viral infectivity, JRFL Env glycan mutants were incorporated in pseudoviruses. Pseudoviral concentrations were normalized after measuring p24 levels, and equal concentrations were used for infectivity assays. All these pseudotyped viruses having glycosylation-deficient Env showed significant infection in TZM-bl cells (Fig. 7). Pseudoviruses having ΔG13 gp160 Env with only two glycosylation sites in the core gp120 region showed infectivity levels similar to that of WT gp160 Env-containing pseudoviruses (Fig. 7). Further removal of the remaining two glycans from the inner domain of core gp120 resulted in a reduction in the infectivity. However, a significant amount of infectivity was retained even after the removal of all core gp120 glycans from Env (Fig. 7). These results demonstrate that all core gp120 glycans are dispensable for an infectious virion, in the context of a single-round pseudoviral infectivity assay. Neutralization sensitivity of JRFL pseudoviruses containing either WT or a glycan-deficient Env was measured against VRC01, GL-VRC01, PG9, and the non-neutralizing CD4bs antibody b6. Sequential removal of glycans increased neutralization sensitivity for both VRC01 (Fig. 8A) and GL-VRC01 (Fig. 8B and Table 7) and also resulted in detectable neutralization with b6 (Fig. 8D and Table 7), probably due to better exposure of the CD4bs. WT, ΔG13, and ΔG15F pseudoviruses show neutralization of ∼37, 50, and 80%, respectively, with b6 antibody at a concentration of 2 μg/ml. Although the GL-VRC01 curves showed clear pre-transition baselines and a distinct transition, the b6 data had a larger scatter and no clear baseline and thus could not be fit to obtain an IC50 estimate. A previous study suggests that mutating glycosylation sites at residue positions 295, 332, and 339 decreases the sensitivity to PG9 neutralization (85). In agreement with the previous study, PG9 neutralization was found to be sensitive to the loss of PNGS (85) with a 5–6-fold increase in IC50 value on the removal of 13 out of 15 PNGS (Fig. 8C and Table 7) in core gp120. Thus, the decreased neutralization sensitivity of glycan-deficient pseudoviruses to PG9 could be due to the loss of glycans at these positions rather than altered Env conformation.
Figure 7.
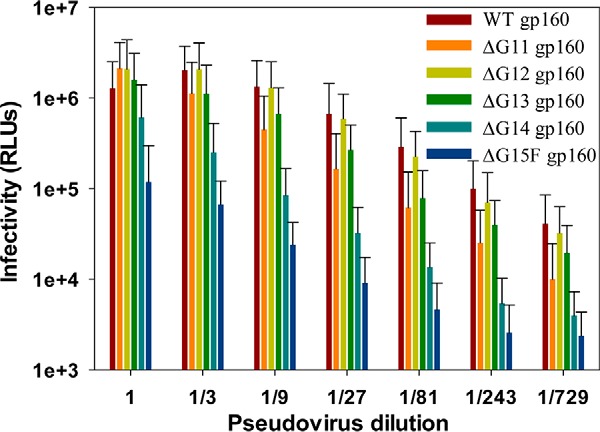
Pseudoviral infectivity assay. Comparison of infectivity for pseudoviruses containing WT or differentially glycosylated JRFL gp160 Env using the TZM-bl assay, which gives the readout of infectivity as relative luminescence units (RLUs). The relative amount of pseudotyped virus in cell supernatants was normalized following a p24 assay, and an equal amount of each pseudovirus was used for the infectivity assay. Mutant pseudoviruses lacking some or all core gp120 glycans retained significant infectivity. Error bars represent standard deviation for the data from two independent experiments.
Figure 8.

Pseudoviral neutralization assays. Neutralization activity of VRC01 (A), germline-reverted VRC01 (GL-VRC01) (B), PG9 (C), and b6 (D) antibodies against JRFL-E168K pseudoviruses with either WT envelope or PNGS mutant envelopes. Lines represent sigmoidal logistic, four parameters fit using Sigma PlotTM software. In the case of non-neutralizing antibody b6, the data could not be fit, so no lines are shown. Removal of glycans resulted in a significantly increased neutralization sensitivity for VRC01 (A) and GL-VRC01 (B) antibodies. Pseudoviruses containing WT gp160 failed to show neutralization with GL-VRC01, indicating that glycans restrict interaction with this germline antibody. Error bars represent standard deviation for the data from two independent experiments.
Table 7.
Comparison of neutralization activity of VRC01, GL-VRC01, and PG9 antibodies against JRFL E168K pseudoviruses with differentially glycosylated gp160 Env
IC50 indicates antibody concentration required to inhibit pseudoviral activity by 50%; S.D. is standard deviation for the data from two independent experiments.
| Pseudovirus Env | IC50 ±S.D. (μg/ml) |
||
|---|---|---|---|
| VRC01 | GL-VRC01 | PG9 | |
| WT gp160 | 0.0061 ± 0.001 | a | 0.004 ± 0.001 |
| ΔG11 gp160 | 0.00073 ± 0.001 | 2.95 ± 0.12 | 0.02 ± 0.003 |
| ΔG13 gp160 | 0.00074 ± 0.001 | 0.19 ± 0.05 | 0.02 ± 0.003 |
| ΔG15F gp160 | 0.0004 ± 0.001 | 0.64 ± 0.07 | 0.58 ± 0.5 |
a There was no detectable neutralization until 10 μg/ml.
Significance of rationally designed mutations at glycosylation sites in maintaining Env conformation
In most earlier studies that probed the contribution of glycosylation to HIV-1 Env function (45, 54, 55), Asn residues at PNGS were mutated to uncharged Gln/Ala residues. The rationale for selection of these substitutions is not clear. In contrast, in this study, we rationally mutated Asn residues at PNGS as described in Fig. 2 and could achieve JRFL gp160 folding even in the absence of all 15 core gp120 glycans (Figs. 6–8). To demonstrate that the mutations selected through our method are superior to the widely used Gln mutation, which is structurally similar to Asn, we designed a JRFL gp160 derivative where all Asn residues at the outer domain glycosylation sites, except 262 and 276 which interact with the inner domain, were mutated to Gln residues. This construct was named ΔGQ11 gp160 (see Fig. 5 and Table 6 for a complete description of gp160 constructs). ΔGQ11 gp160, rationally designed ΔG11 gp160, WT gp160, and cleavage-defective SEKS gp160 were displayed on the mammalian cell surface, and their binding with CD4bs targeting bNAb VRC01 was monitored using FACS (Fig. 9). SEKS gp160 was used as a negative control because it lacks significant binding with cleavage-dependent, trimer-specific, broadly neutralizing antibodies and is known to show substantial binding with non-neutralizing antibodies (82, 83, 86). WT gp160 and ΔG11 gp160 (rationally mutated) showed comparable binding to VRC01 (Fig. 9). In contrast, Asn → Gln mutations at 11 outer domain glycosylation sites (ΔGQ11 gp160) resulted in a significant reduction in the binding of bNAb VRC01 (Fig. 9). A properly cleaved native-like trimer binds CD4bs-directed neutralizing antibodies such as b12 considerably better than non-neutralizing ones such as b6 (82, 83). Thus a high ratio of b12:b6 binding indicates the presence of native-like trimers. Rationally mutated ΔG11 gp160 showed the best b12:b6 binding ratios among all the constructs tested (Fig. 9). The minimal binding observed with non-neutralizing antibodies targeting CD4i (17b), V3 (447–52D), C1/C5 (C11), and gp41 cluster I (Asp-49) epitopes further indicated that removal of OD glycans from gp160 does not expose non-neutralizing epitopes or significantly perturb the conformation of the native Env trimer (Fig. 9). A recently discovered bNAb (PGT151) targets the gp120-gp41 interface region and selectively binds to properly cleaved native trimers. Binding with PGT151 is considered as a good marker for the presence of the native HIV-1 Env trimer as it shows minimal binding with cleavage-defective Envs (86, 87). ΔG11 gp160 and WT gp160 showed similar binding to PGT151 bNAb, confirming that both constructs are properly cleaved and maintain a native-like trimeric structure (Fig. 9). In contrast, binding of ΔGQ11 gp160 with PGT151 bNAb was comparable with that of cleavage-defective SEKS gp160, which cannot form native functional trimers, indicating that mutations chosen by our method are superior to Asn → Glu substitutions in maintaining the native trimeric structure (Fig. 9). A representative FACS histogram overlay for the binding of PGT151 antibody is shown in supplemental Fig. S14. A similar binding pattern was observed with V1/V2 loop targeting trimer-specific antibodies PG16 (88) and PGT145 (Fig. 9) (11). A marginal decrease in the binding of PGT151 and PG9 with ΔG11 gp160 relative to WT gp160 could be attributed to the fact that binding of both these trimer-specific antibodies is dependent on the presence of certain core gp120 glycans (45, 80, 86, 90), some of which are absent in our designs. For example, a recent cryo-EM structure for the trimeric Env in complex with PGT151 antibody clearly demonstrates that Asn-241, Asn-262, Asn-276, and Asn-448 glycans from the core gp120 region significantly contribute to the PGT151 epitope (80).
Figure 9.
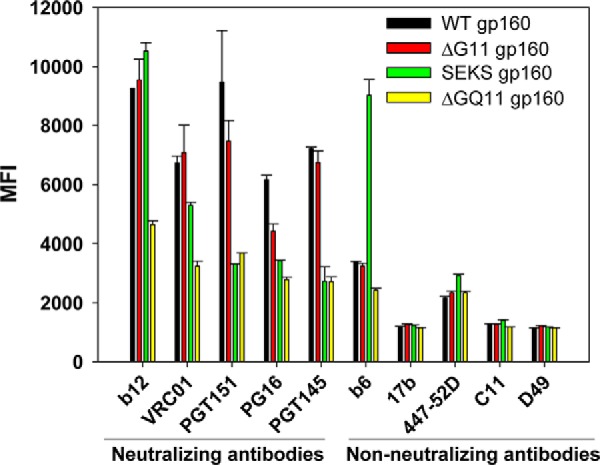
Effect of rationally designed versus Asn → Gln glycosylation site mutations on HIV-1 Env conformation probed by binding of anti-HIV-1 neutralizing and non-neutralizing mAbs to mammalian cell surface-expressed HIV-1 Env variants. WT gp160 and ΔG11 gp160 show similar binding profiles to CD4bs targeting neutralizing mAbs b12 and VRC01 as well as to the trimer-specific neutralizing antibodies PG16, PGT145, and PGT151. In contrast to the designed mutations, conventional Asn → Gln mutations at outer domain glycosylation sites (ΔGQ11 gp160) result in a significantly reduced binding of all neutralizing antibodies tested. Binding of ΔGQ11 gp160 with PGT145 and PGT151 is comparable with that of cleavage-defective SEKS gp160. A representative FACS histogram overlay for the binding of PGT151 antibody is shown in supplemental Fig. S14. A marginal decrease in the PGT151 and PG16 binding of rationally designed ΔG11 gp160 relative to WT gp160 could be attributed to the fact that binding of both these trimer-specific antibodies is dependent on the presence of certain core gp120 glycans (56, 86, 90). Both WT gp160 and ΔG11 gp160 show minimal binding to a number of different non-neutralizing mAbs with epitopes in diverse regions of the Env structure. In contrast, the cleavage-defective SEKS gp160 molecule shows high binding with the non-neutralizing mAb b6 but binds poorly to the trimer-specific neutralizing mAbs PGT145 and PGT151. All mAbs were used at a concentration of 1 μg/ml. Error bars represent standard deviation for the data from two independent experiments.
As described above, the ΔG11 gp160 and ΔG13 gp160 pseudoviruses lacking outer domain glycans retain WT-like infectivity. In contrast, consistent with mammalian cell-surface display data (Fig. 9), the ΔGQ11 gp160 pseudoviruses show a drastic reduction in infectivity, indicating that our designed mutations are superior to Asn → Gln mutations for maintaining pseudoviral infectivity in the absence of OD glycosylation (Fig. 10). As expected, pseudoviruses with cleavage-defective SEKS gp160 Env were not infective due to the lack of a properly cleaved, native-like trimeric envelope required for infection (Fig. 10). This negative control demonstrates the specificity of the binding and neutralization experiments.
Figure 10.
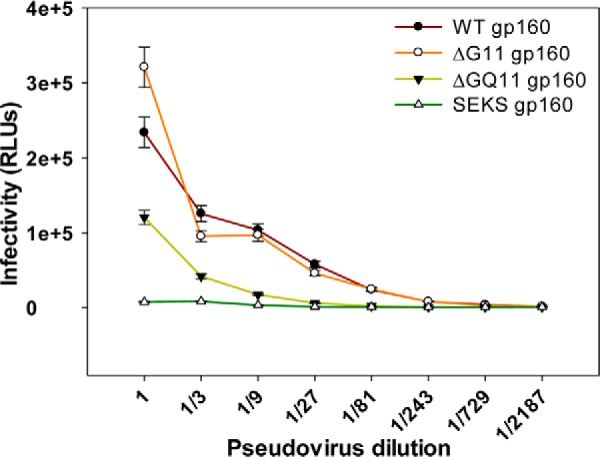
Effect of rationally designed versus conventional Asn → Gln glycosylation site mutations on pseudoviral infectivity. Infectivity of pseudoviruses containing WT or differentially glycosylated JRFL gp160 Env was measured using a TZM-bl assay. The relative amount of pseudotyped virus in cell supernatants was normalized following a p24 assay, and equal amounts of each pseudovirus were used for the infectivity assay. Pseudoviruses with ΔG11 gp160 Env retain WT-like infectivity, and pseudoviruses displaying ΔGQ11 gp160 show a drastic reduction in infectivity. As expected, pseudoviruses incorporating cleavage-defective SEKS gp160 Env show loss of infectivity because they lack a properly cleaved, native-like trimeric envelope. Error bars represent standard deviation for the data from two independent experiments. RLUs, relative luminescence units.
Replication fitness of glycan-deficient HIV-1 viruses in a multiple-cycle infectivity assay
Pseudoviral infectivity assays (Fig. 7) show that all core gp120 glycans are dispensable for producing an infectious virion. However, TZM-bl cell-based pseudoviral infectivity experiments are single-cycle infection assays. It is still possible that removal of core gp120 glycans might affect the replication fitness of infectious viruses derived from full-length HIV-1 molecular clones in a multiple-round infection study. To assess the effect of glycan removal on viral replication fitness, pLAI-JRFL (91, 92)-based full-length molecular clones (MCs) of Env glycan mutants were generated. Concentrations of infectious viral stocks were normalized by measuring p24 antigen levels. Equal viral amounts were first used in a single-cycle TZM-bl infectivity assay. In agreement with pseudoviral infectivity assays, infectivity of WT gp160, ΔG13 gp160, and ΔG15F gp160 Env-containing molecular clones in TZM-bl cells was found to be comparable (Fig. 11).
Figure 11.
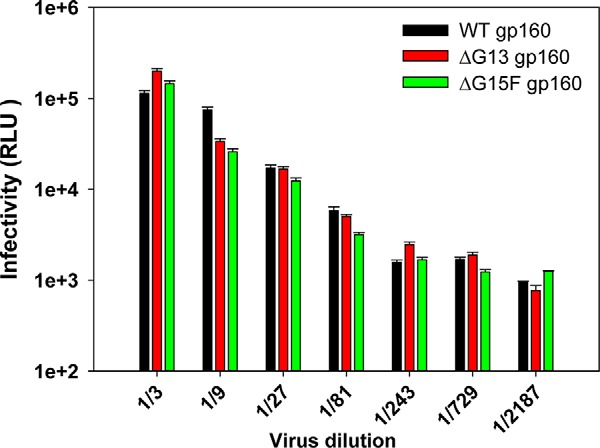
Single-round viral infectivity assay in TZM-bl cells. Comparison of infectivity for full-length viral clones containing WT or differentially glycosylated JRFL gp160 Env using the TZM-bl assay. This assay gives the readout of infectivity as relative luminescence units (RLUs). The relative amount of virus in cell supernatants was normalized following a p24 assay, and an equal amount of each virus was used for the infectivity assay. Mutant viruses lacking either all outer domain glycans (ΔG13 gp160) or all core gp120 glycans (ΔG13 gp160) retained infectivity comparable with that of viruses having WT gp160 Env. Error bars represent standard deviation for the data from two independent experiments.
Replication fitness of these infectious viruses was subsequently tested in a multiple-cycle infectivity assay in the HUT-R5 cell line (93–95). Infected cells were split every 3rd day, and virus replication was quantitated by measuring the HIV-1 p24 antigen released in culture supernatants at different time points, post-infection. In agreement with the single-round viral infection studies, glycan-deficient full-length viruses were infectious in multiple-cycle assays as well. All the full-length molecular clones of HIV-1 that were tested, viz. ΔG13 (devoid of all OD glycans), ΔG15F (lacks all core gp120 glycans) and WT exhibit high p24 concentrations even after 21 days post-infection, indicating productive virus replication and infection in this multicycle assay (Fig. 12). In fact, removal of core gp120 glycans was shown to enhance infectivity relative to WT (Fig. 12).
Figure 12.
Multicycle viral infectivity assay in HUT-R5 cells. HIV-1 viruses derived from full-length molecular clones incorporating WT (black), ΔG13 (red), or ΔG15F (green) gp160 were produced from 293T cells and used to infect HUT-R5 cells. Infected cells were split every 3rd day, and virus replication was quantitated by measuring the HIV-1 p24 antigen released in the culture supernatants at different time points post-infection. In agreement with the single-round pseudoviral infection studies, glycan-deficient full-length viruses were found to be infectious in the multiple-cycle assay as well. All the viruses are capable of productive replication. The glycan-deficient viruses exhibit higher p24 levels and faster replication kinetics than the WT, confirming that core gp120 glycans are not essential for HIV-1 viral infectivity.
Mammalian cell surface display of WT and corresponding ΔG11 gp160s from subtype A and subtype C HIV-1 strains
HIV-1 Env glycoprotein shows very high sequence diversity. HIV-1 subtype A is mainly found in Africa; subtype B is most prevalent in North America and Europe; and subtype C is commonly found in Africa and Asia (96). All the glycan-deficient OD fragment constructs described above were made from subtype B, HXBc2 strain, and all the gp120 and gp160 constructs described above were based on the subtype B, JRFL strain. To test whether our approach of glycan removal is generally applicable, we designed outer domain glycan-deficient Env derivatives for four additional HIV-1 strains, two from subtype A and two from subtype C (see Table 6 for a complete description of these constructs). These outer domain glycan-deficient Env derivatives were designed using the frequency-based approach as described above for the subtype B constructs (Fig. 2). The complete list of glycosylation site mutations introduced in subtype A and subtype C sequences is given in supplemental Table S4. The supplemental Table S4 suggests that a number of mutated glycosylation sites are unique to each strain. To examine the effect of glycan removal on the native Env structure, WT and glycan-deficient subtype A and subtype C constructs were displayed on the mammalian cell surface, and their binding with various bNAbs was monitored using FACS (supplemental Fig. S15). All these gp160 constructs could bind trimer and cleavage-specific bNAb PGT 151, similar to their corresponding WT gp160s (supplemental Fig. S15A). Binding with a trimer and V1/V2 loop-specific bNAb PG16 was also found to be similar for glycan-deficient and WT gp160 variants (supplemental Fig. S15B). We also monitored binding of these molecules with a CD4bs targeting bNAb VRC01 and found that the VRC01 epitope is maintained in the absence of most OD glycans for subtype A and C Envs also (supplemental Fig. S15C). Thus, our approach for glycan removal is not strain-restricted and in principle can be applied to other glycoproteins.
Discussion
Glycan-mediated surface coverage plays an important role in immune evasion by HIV-1 (37). Glycosylation is essential for HIV-1 infectivity (37, 45, 54) and is also widely believed to be essential for the folding of gp120. Early evidence for a glycan requirement for gp120 folding came from a study in which non-glycosylated forms of gp120 were generated either by deletion of the signal sequence of gp120 or by synthesis in the presence of tunicamycin. This non-glycosylated gp120 failed to bind CD4, whereas enzymatic removal of glycans from gp120 had no effect on CD4 binding. This led to the conclusion that N-linked glycosylation is essential for the proper folding of gp120 (34). It is not known how many of the ∼25 glycans present on the gp120 surface are essential for folding. This is an important question as these glycans mask various conserved epitopes on the viral surface, prevent recognition of germline-reverted bNAbs, and thus impede the development of an effective Env-based vaccine. In addition, several recently isolated bNAbs bind to specific glycans on gp120 (11, 86, 97–99). If the importance of multiple PNGS in maintaining the structural and functional integrity of gp120 could be determined, this information can be used to modulate coverage of Env by glycans.
The isolated OD fragment is an important target for vaccine design (25, 60, 61). We demonstrate that all 14 glycans are dispensable for folding in the context of the OD fragment. It is important to note that apart from complete lack of glycosylation, ΔG14-ODEC is also devoid of the inner domain, part of the bridging sheet, and V1/V2/V3 loops and has a 5-fold lower molecular weight than full-length gp120. However, it can still bind the b12, VRC01, and CD4 ligands with affinities comparable with that of full-length gp120. These results are encouraging as in a previous study the WT ODEC molecule, which binds CD4bs ligands weakly, was able to elicit sera in rabbits with measurable cross-clade neutralization of some easy to neutralize tier 1 viruses (66).
Broadly neutralizing antibodies against HIV-1 show a very high level of affinity maturation, and it has been proposed that targeting the germline precursors of these bNAbs is a pre-requisite for generating such bNAbs against HIV-1 (57, 61, 100–103). Germline-reverted bNAbs typically do not bind recombinant gp120s for most HIV-1 strains or neutralize the corresponding pseudoviruses (60, 62, 104, 105). Epitope design targeting a particular germline can potentially activate the corresponding B-cell precursors for affinity maturation (60). Recently, it was demonstrated that the VRC01-class precursor naive B cells can be isolated from HIV-1 uninfected donors using a germline-targeting OD-based immunogen, again indicating the possible usefulness of germline-targeting immunogens in eliciting VRC01-type antibodies (103). Various immunization strategies are currently being tested in transgenic mouse models to further understand the usefulness of germline-targeting immunogens in directing affinity maturation toward neutralizing antibody response (106–109). gp120 glycans have been shown to contribute to lack of binding to GL-VRC01 (62). As bacterially expressed ODEC and ΔG14-ODEC fragments are devoid of all glycans, binding of these to GL-VRC01 was monitored using SPR. Both these molecules bound GL-VRC01 with ∼5–10 nm KD, whereas glycosylated full-length WT gp120 failed to show any detectable binding. These molecules are therefore important tools to test the usefulness of germline-targeting in HIV-1 vaccine design.
In the context of core gp120 from the JRFL isolate, we found that 14 of the 15 glycosylation sites are dispensable for proper folding. The gp120 outer domain is the primary target for non-glycan-dependent anti-HIV-1 broadly neutralizing antibodies. However, non-outer domain glycosylation sites in core gp120 were also mutated in this study to identify dispensable inner domain glycans. This information could be useful in the future for the purification of Env-derived immunogens from low-cost expression systems where glycosylation is either absent or differs from mammalian systems. In the recent past, a number of new bNAbs targeting the gp120-gp41 interface have been discovered, for example PGT151 and 8ANC195 (80, 86, 87, 110). Such antibodies often interact with glycans from the inner domain (80); thus, removal of glycans from the inner domain while retaining proper conformation can help in mapping and targeting such epitopes. In this study, inner domain glycosylation sites Asn-88 and Asn-241 were found to be the least dispensable for proper folding of gp120. It is interesting to note that these two potential N-linked glycosylation sites are among the most conserved in core gp120 (44). Recent cryo-EM and crystal structures of glycosylated native HIV-1 Env trimers show that Asn-88 and Asn-241 glycans are in close proximity to the gp120-gp41 interface (80, 81). The Asn-88 glycan shields the conserved MPER region, whereas the Asn-241 glycan limits access to the PGT151 epitope (80).
A number of bNAbs target specific glycans on the V1/V2 and V3 loop regions. Among them, PG9 and PG16 are the founder members of this class of bNAbs (85, 98, 99, 105, 111). Hence, we used ΔG11 core gp120 as a starting point to re-introduce important neutralization epitopes present in the V1/V2 and V3 variable loops and expressed the ΔG11 gp160 molecule and its PNGS variants on the mammalian cell surface. Similar to native WT gp160, glycan-deficient gp160 constructs bound well with multiple bNAbs, including b12, VRC01, PG9, PG16, PGT145, PGT151, and 2F5, and poorly to non-neutralizing mAbs b6, 17b, 447–52D, C11, and D49 (Figs. 6 and 9), which have epitopes located in diverse regions of the Env structure (82). The binding of PGT151 and PGT145 bNAbs to glycan-deficient Envs is particularly interesting because PGT145 is considered to bind only native-like trimers (11), whereas PGT151 selectively binds to properly cleaved native trimers (86, 87). The low-affinity binding of glycan-deficient gp160 constructs with b6 shows that removal of all glycans from core gp120 does not expose non-neutralizing epitopes or perturb the conformation of the native Env trimer significantly (Fig. 6). However, increased neutralization with b6 was observed in a more sensitive pseudoviral neutralization assay, indicating that glycan removal at least partially relieves some steric constraints for the approach of various CD4bs antibodies. This can possibly distract the immune system from neutralizing epitopes because charged and bulky mutations introduced at PNGS can be more immunogenic (112). A number of gp160 mutants described in this study retain all glycans in the variable loops of gp120 but are glycan-deficient in the core gp120 region and therefore should allow better exposure of conserved CD4bs while retaining important glycan-dependent neutralization epitopes in the variable loop region (Fig. 1D).
Reintroduction of the Asn-332 glycosylation site into ΔG15F gp160 (lacking all 15 PNGS) led to a recovery of PGT128 binding (Fig. 6). It has been shown that viruses lacking the glycan at the Asn-332 position acquired this glycan to escape certain strain-specific antibodies and thus became sensitive to the bNAb PGT128 (97). Our result is a proof of principle that these deglycosylated molecules can be used to reintroduce glycans at desired positions to generate or map epitopes for glycan-dependent bNAbs. Such knock-in glycan mutants in a largely glycan-free background can be used as complementary tools to knock out mutants. They can be useful in determining the minimal glycosylation requirement for a particular antibody epitope. Furthermore, minimally glycosylated Env mutants are likely to be more homogeneous in their chemical composition and would be useful for mass spectrometry and cryo-EM-based approaches to probe Env structure, stability, and function. They can also be useful in determining the exact glycan chemistry required for interaction with a particular glycan-dependent antibody, as they will facilitate mass spectrometric glycan analysis due to a reduced background.
In the current designs, ∼70–90% of the Asn residues at core gp120 glycosylation sites for Envs from subtypes A, B, and C were rationally substituted with charged or polar amino acids, and the resulting molecules were well folded. In contrast, in most of the earlier studies (45, 54, 55) Asn residues at PNGS were mutated to uncharged Gln/Ala residues. It is likely that simultaneously mutating multiple glycosylation sites with non-optimal amino acids could have led to the loss of infectivity in the previous studies. Our results indeed suggest that Asn → Gln mutations at glycosylation sites are not optimal for proper cleavage, for maintaining a native trimeric structure, and for retaining infectivity. In contrast, mutations identified by us could rescue Env folding and viral infectivity in both single- and multiple-cycle assays in the absence of core gp120 glycans. However, the widely used Asn → Gln mutations do not support even single-cycle infection in TZM-bl cells, indicating the usefulness of the glycan removal method employed in this study. Indeed sequence analysis reveals that Asn → Gln substitutions are quite rare at HIV-1 Env PNGS in natural isolates and occur at ∼10-fold lower frequency than the substitutions (Ser, Thr, Asp, and Lys) selected in this study (http://www.hiv.lanl.gov). The present data conclusively demonstrate that glycans in core gp120 are not directly involved in maintaining the infectivity of the HIV-1 virion, and hence their primary role is likely to be in immune evasion.
Whereas VRC01 class antibodies arise from a restricted set of germline precursors, other CD4bs antibodies are less restricted (113, 114). Hence, it might be useful to have priming immunogens that activate a large number of diverse antibody precursors. The molecules described in this study retain native-like Env conformation despite extensive glycan removal (as far as five major epitopes targeted by bNAbs are concerned, see Fig. 13). Thus, these molecules can potentially be used in DNA immunizations as priming immunogens because they can likely bind to a large number of germline precursors, including those of bNAbs. This can be followed by boosting with glycosylated Env molecules to elicit cross-reactive antibodies. This line of approach is supported by recent molecular dynamics studies on crystal structures of fully glycosylated trimeric Env molecules (81). Whereas glycan removal may also result in exposure of non-neutralizing epitopes, the use of appropriately glycosylated or hyper-glycosylated boosting immunogens can be used to focus the immune response to desired neutralizing epitopes. In addition, combinations of the molecules reported in this study with fully glycosylated WT Env in various prime/boost regimes will be helpful in expanding our understanding regarding the role of glycosylation in modulating the gp120-directed immune response (115). It has previously been shown (116) that removal of glycans from the surface of influenza hemagglutinin results in an enhanced neutralization response, and in another study, we found that a bacterially expressed glycan-free fragment bound b12 and when combined with a gp120 boost induced a broad and potent neutralizing antibody response against a limited number of tier 2 as well as tier 3 HIV-1 viruses (117).
Figure 13.
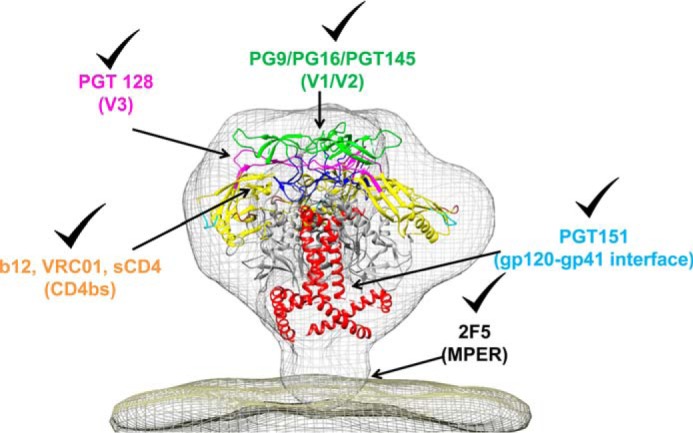
Probing the conformational integrity of mammalian cell surface displayed HIV-1 native trimer using antibodies targeting important neutralization epitopes. gp120-gp41 trimer (PDB code 4NC0 (142)) is fit into the model of the cryo-EM density of the HIV-1 spike (gray mesh) (89). gp120 is multicolored, and gp41 is shown in red. As shown by checkmarks, antibodies targeting five major conformational epitopes: CD4bs (b12 and VRC01), V1/V2 loop (PG9, PG16, and PGT145), V3 loops (PGT128), gp120-gp41 interface (PGT151), and MPER region (2F5) bound various glycan-deficient gp160s and WT gp160 with comparable affinities indicating that native-like trimeric structure can be maintained in the absence of core gp120 glycans.
A single virus known as the transmitted/founder (T/F) virus is generally responsible for the establishment of around 70–80% of heterosexually transmitted HIV-1 infections (118). T/F viruses differ substantially from chronic strains in having shorter variable loops and a lower level of glycosylation (119–121). Lower glycan coverage helps the T/F virus in the establishment of initial infection, possibly by increasing affinity for various mucosal receptors (122). Using mass spectrometry, it was shown that more than 50% potential N-linked glycosylation sites in T/F viruses are either variably glycosylated or completely unutilized (123). Recently, it has been shown that elicitation of sera with broad neutralization is dependent on early Env characteristics such as a shorter V1 domain and decreased glycosylation (124). Thus, it has been proposed that vaccine design should focus on generating immunogens with T/F virus-like properties to expose conserved epitopes in order to elicit broad neutralization (124–127). However, this approach is largely unexplored, in part due to the difficulty in generating glycan-deficient envelopes. A number of immunogens described here lack V1/V2 and V3 loops and/or are glycan-deficient and show high affinity for CD4bs ligands. As a result, they are close mimics of the T/F virus envelope. Pseudoviruses generated in this study show greatly increased neutralization sensitivity for germline VRC01 with progressive loss of glycans. Our results, coupled with the studies listed above, support the idea that limited glycosylation might play an important role in HIV-1 infection establishment as well as in the evolution of the immune response against the virus.
In summary, using a large number of OD fragments, core gp120, full-length gp120, and gp160 Env mutants in a variety of expression systems such as E. coli, yeast, and mammalian cells, we were able to show that gp120 folding can be largely made independent of glycosylation and such glycan-deficient virions retain infectivity and replication fitness. We employed a combination of frequency-based rational and random mutagenesis methods to achieve this goal. N-Linked glycosylation has been shown to accelerate protein folding and improve stability by native state stabilization and decreasing the activation barrier for folding (104, 128). Glycosylation is also known to prevent protein aggregation by reducing the interaction between surface-exposed hydrophobic patches (129, 130). A large number of mutations identified by us were charged mutations, possibly preventing aggregation in a manner similar to that of glycosylation. The ability to engineer glycan-free protein derivatives allows for protein expression in inexpensive, prokaryotic, or yeast systems where glycosylation is absent or different from mammalian cells, but it has been difficult to achieve previously (131). The methodology outlined in this work can in principle be used to probe the role of glycans in the stability and folding of any glycosylated protein and to construct glycan-free protein variants.
Experimental procedures
Construct descriptions
OD-based constructs from HXBc2 strain
The OD sequences used in this study were derived from the previously described ODEC molecule (66), which was based on the subtype B CXCR4-tropic HXBc2 strain. Various glycosylation site mutants of ODEC were designed (Table 1). Descriptions of these OD constructs are provided in Fig. 3 and Table 1.
Core gp120-based constructs from JRFL isolate
The gp120 sequences used in this work were all derived from the subtype B, CCR5-tropic JRFL isolate. Gene sequences were codon-optimized for expression in S. cerevisiae. All gp120 glycosylation site mutants were made in the core gp120 background except one (Table 4). Core gp120 lacks V1/V2 (residues 128–194) and V3 (residues 298–329) loops, which are replaced by three residue amino acid (GAG) linkers. The core gp120 sequence contains the following residues 31–127-GAG-195–297-GAG-330–507, unless stated otherwise. The WT core gp120 construct retains all 15 PNGS of the core region of the JRFL isolate. A description of the various core gp120-based mutants is provided in Table 4.
gp160-based trimeric Env constructs from JRFL isolate
Construct WT gp160 consists of the gp160 nucleotide sequence from the JRFL isolate, harboring the E168K mutation. Descriptions of the various gp160 mutant constructs are provided in Fig. 5 and Table 6. The glycosylation mutations introduced in the JRFL gp160 variants were identical to the mutations introduced in the core gp120 glycosylation site mutants described above and are listed in supplemental Table S3.
To understand the importance of rational mutations at glycosylation sites, we designed a JRFL gp160 derivative, wherein 11 Asn residues in the outer domain glycosylation sites were mutated to structurally similar Gln residues. This construct was named ΔGQ11 gp160 (Table 6, no. 8). A cleavage-defective JRFL gp160 construct (SEKS gp160) was also made, where the gp120-gp41 cleavage site region REKR was mutated to SEKS using site-directed mutagenesis (Table 6, no. 9, and Fig. 5) (83). All JRFL gp160 constructs have a cytoplasmic tail deletion (ΔCT), which is known to improve expression on the mammalian surface (132–134). JRFL gp160 constructs were cloned into the pSVIII vector (83) for mammalian cell-surface expression studies.
Cloning of OD and gp120 glycosylation site mutants into yeast surface display vector
Yeast codon-optimized genes for ODYCO, ΔG14-ODYCO, ΔG4-ODYCO, ΔG14-ODYCO-D368R, WT core gp120, ΔG15 core gp120, ΔG4 core gp120, E168K ΔG4 full-length gp120, and ΔG11 core gp120 having ∼50-bp homology at each end with SfiI-digested pPNLS vector were synthesized by GenScript (135). These gene products were PCR-amplified with pPNLS-specific primers and individually recombined into SfiI-digested linear pPNLS vector by yeast homologous recombination in the EBY100 strain of S. cerevisiae (73, 75, 135). All other gp120 mutants described in Table 4 were also cloned in a similar manner.
Yeast-surface display of glycosylation variants
pPNLS vector contains an AGA2p fusion at the N terminus of the surface-displayed protein along with two epitope tags, HA (YPYDVPDYA) and c-Myc (EQKLISEEDL) for detection (135). All constructs were displayed on the cell surface of S. cerevisiae strain EBY100 using a standard protocol (75). Briefly, EBY100 cells were transformed with plasmids, and colonies were grown in glucose-containing liquid SDCAA media (pH 4.0) until mid-log phase at 30 °C, followed by induction in galactose-containing SGCAA (pH 4.0) media at 20 °C for 16–36 h.
Strains, antibodies, and reagents
Monoclonal antibodies IgG-b12, IgG-b6, IgG-C11, IgG-D49, IgG-17b, IgG-PGT151, IgG-PGT145, IgG-PGT128, gp120, and sCD4 were obtained from the Neutralizing Antibody Consortium of the International AIDS Vaccine Initiative (IAVI), New York. IgG-447-52D was obtained from AIDS Reagent Program, National Institutes of Health. VRC01 and germline VRC01 antibody plasmids were transfected into HEK-293T mammalian cells, and antibodies were purified using protein G affinity chromatography. Antibodies PG9 and PG16 were kindly provided by Dr. S. Phogat. The HUT-R5 T-cell line was a kind gift from Dr. Ritu Gaur (South Asian University, New Delhi, India). Two-domain CD4 protein with a C-terminal FLAG tag for detection (CD4D12-FLAG) was purified from E. coli as described previously (136).
Flow cytometry analysis for proteins displayed on yeast surface
Surface expression of yeast-displayed proteins was monitored using chicken anti-c-Myc antibody (1:300 dilution) (Life Technologies, Inc.) as the primary antibody and anti-chicken Alexa Fluor 488 (1:300 dilution) as the secondary antibody. Binding to various ligands was determined using an anti-gp120 antibody (b12/b6/VRC01/PG9) as a primary antibody and goat anti-human PE (1:100 dilution) (Sigma) as the secondary antibody. All labeling was done for 30 min at 4 °C. After each step, cells were washed with labeling buffer (0.5% BSA in PBS (pH 7.4)). Cells were analyzed using Accuri C6 cytometer (BD Accuri Cytometers, Ann Arbor, MI), Canto II cytometer (BD Biosciences), or an ARIA III instrument (BD Biosciences). Sorting was performed on an ARIA III instrument (BD Biosciences). For monitoring the interaction with CD4D12-FLAG, yeast cells were first labeled with CD4D12-FLAG protein followed by incubation with mouse anti-FLAG antibody (1:300 dilution) (Sigma). Secondary labeling was performed with rabbit anti-mouse Alexa Fluor 633 (Life Technologies, Inc.) (1:1200 dilution). Binding affinities of yeast surface-displayed proteins with various anti-HIV-1 antibodies were determined as described previously (75).
Protein purification
An E. coli, codon-optimized version of the ΔG14-ODEC gene was synthesized and cloned into the pET28a(+) vector (Novagen) between the NdeI and BamHI sites and contained an N-terminal His tag. E. coli BL21(DE3) cells transformed with the plasmid were grown in 1 liter of Luria-Broth (LB) at 37 °C until an A600 of 0.6. Cells were then induced with 1 mm isopropyl β-d-1-thiogalactopyranoside and grown for another 6–8 h at 37 °C. Cells were harvested at 3500 × g and resuspended in 35 ml of PBS (pH 7.4) containing 100 μm PMSF and 0.2% Triton X-100. The cell suspension was lysed by sonication on ice and centrifuged at 15,000 × g. The supernatant was discarded, and the pellet was washed with 35 ml of 0.2% Triton X-100, PBS (pH 7.4), and subjected to centrifugation at 15,000 × g. The pellet was solubilized in 40 ml of 8 m guanidine hydrochloride (GdnHCl) in PBS (pH 7.4) overnight at room temperature. The solution was centrifuged at 15,000 × g for 30 min. The supernatant was bound to 3 ml of Ni-NTA beads (GE Healthcare) and washed twice with 25 ml of 50 mm imidazole containing 8 m GdnHCl in PBS (pH 7.4), and finally, the denatured protein was eluted with 8 m GdnHCl in PBS (pH 7.4) containing 500 mm imidazole at room temperature.
The first two elution fractions (each 5 ml) were pooled together and then rapidly diluted 10-fold with 0.5 m arginine hydrochloride in PBS containing 1 mm EDTA at 4 °C to reduce the denaturant concentration from 8 to 0.8 m. The resulting solution was again concentrated back to the original volume using an Amicon concentration cell. The concentrated solution from the first two elution fractions was combined (total 10 ml) and dialyzed once against 1 liter of 0.5 m arginine hydrochloride in PBS containing 1 mm EDTA at 4 °C to remove the denaturant (GdnHCl) and then dialyzed extensively (three times) against PBS, 1 mm EDTA (using 2 liters of buffer each time) at 4 °C to remove arginine hydrochloride. The dialyzed protein was concentrated to a final concentration of 0.2 mg/ml, flash-frozen in liquid nitrogen, and stored in aliquots at −80 °C. The yield was determined by densitometry analysis from SDS-PAGE using standard proteins of known concentrations.
Far-UV CD and fluorescence spectroscopy
Circular dichroism (CD) spectra were recorded on a Jasco J-715C spectropolarimeter flushed with nitrogen gas. The concentration of protein sample was 5 μm, and the buffer used was PBS (pH 7.4). Measurements were recorded in a 1-mm path length quartz cuvette with a scan rate of 50 nm/min, a response time of 4 s, and a bandwidth of 2 nm. Each spectrum was an average of three scans. Mean residue ellipticities were calculated as described previously (137). Buffer spectra were also acquired under similar conditions and subtracted from protein spectra before analysis. All fluorescence spectra were recorded at 25 °C on a Jasco FP-6300 spectrofluorimeter. The concentration of protein used was 1 μm either in PBS (pH 7.4) or in the presence of 4 m GdnHCl in PBS (pH 7.4). The excitation was at 280 nm, and emission was recorded from 300 to 400 nm. The excitation and emission slit widths were 3 and 5 nm, respectively. Each spectrum was an average of three consecutive scans. Buffer spectra were also acquired under similar conditions and subtracted.
Gel filtration analysis
Approximately 50 μg of protein was analyzed under non-denaturing conditions by gel filtration chromatography in PBS (pH 7.4) at room temperature on a Superdex-75 analytical gel filtration column. As a control, ODEC having almost the same mass as ΔG14-ODEC (∼23 kDa) (66) was used to determine the expected position of the monomeric peak.
HPLC
20 μg of native protein in PBS (pH 7.4) was injected into a Discovery C5 analytical column (150 × 4.6 mm, 5-μm particle size) (Supelco), and for reduced samples, 20 μg of protein was incubated with 4 m GdnHCl in PBS (pH 7.4) and 5 mm DTT at 37 °C, prior to injection. Proteins were eluted with a gradient of 5–95% acetonitrile containing 0.1% formic acid at a flow rate of 2%/min.
Mass spectrometric analysis to determine disulfide connectivity
In solution, digestion of ΔG14-ODEC was performed under non-reducing conditions using proteomics grade trypsin (Sigma) as per the manufacturer's protocol. The peptides were separated on an Agilent Zorbax C18 RP-HPLC column and fragmented by collision-induced dissociation on Bruker maXis ImpactTM mass spectrometer. Ion masses were analyzed both manually and by using the MS2DB+ software (138) to determine the disulfide connectivities. The molecular mass of the uncleaved protein was also determined to ensure that all cysteines were oxidized.
SPR experiments
All SPR experiments were performed with a Biacore 2000 or Biacore 3000 (Biacore, Uppsala, Sweden) optical biosensor at 25 °C. 700–1300 resonance units of either 4-domain soluble CD4 (sCD4), mAb b12, VRC01, or GL-VRC01 were attached by standard amine coupling to the surface of a research-grade CM5 chip. A sensor surface (without CD4 or any antibody) that had been activated and deactivated served as a negative control for each binding interaction. Different concentrations of full-length WT gp120 from the YU2 strain of HIV-1, ΔG14-ODEC, or ODEC were run across each sensor surface in a running buffer of PBS (pH 7.4) containing 0.005% P20 surfactant. Protein concentrations ranged from 1 μm to 25 nm. For competition experiments, 250 nm ΔG14-ODEC was pre-incubated with 125 or 500 nm VRC01 before passing over the chip surface. Both association and dissociation were measured at a flow rate of 30 μl/min. In all cases, the sensor surface was regenerated between binding reactions by one to two washes with 4 m MgCl2 for 10–30 s at 30 μl/min. Each binding curve was corrected for nonspecific binding by subtraction of the signal obtained from the negative control flow cell. The kinetic parameters were obtained by fitting the data to a simple 1:1 Langmuir interaction model by using BIA EVALUATION 3.1 software as described previously (117).
Generation of saturation mutagenesis library at positions Asn-88 and Asn-241
The glycosylation sites at amino acid positions 88 and 241 in core gp120 sequence were individually randomized using a combination of random primers containing NNR and BNT codons to avoid asparagine at these positions (N = A/T/G/C, R = A/G, and B = C/G/T). The Asn-88 position was randomized in ΔG12a core gp120 background, and the Asn-241 position was randomized in ΔG12b core gp120 background (Table 4). The mutated gp120 genes for both the libraries were generated using overlap PCR. These mutant genes were then introduced into the yeast display vector pPNLS using homologous recombination in the EBY100 strain of S. cerevisiae to generate mutant libraries for positions 88 and 241. Both these libraries were screened and sorted to isolate core gp120 mutants with improved surface expression and b6 mAb binding. First, second, and third rounds of sorting were done with 100 and 10 nm and 500 pm b6, respectively. Those clones that showed improved binding with 500 pm b6 as compared with the corresponding N88Q or N241K starting mutants were sequenced to identify more suitable substitutions at Asn-88 and Asn-241 positions.
Mammalian cell surface display of WT and variants of ΔG11 gp160 from the JRFL isolate
One day prior to transfection, 2.5 × 106 HEK-293T cells were seeded in a T75 culture flask. On the following day, cells were transfected with pSVIII expression plasmids encoding wild-type and mutant Envs and pSVIIItat using either PolyFect (Qiagen) or PEI (Sigma) according to the manufacturer's instructions. For mammalian cell display analysis of JRFL strain-based constructs, the cytoplasmic tail of gp160 was deleted. This has been shown previously to enhance cell-surface expression of Env oligomers (132–134). FACS staining was performed as described previously (32). 48 h post-transfection, the cells were harvested with PBS containing 5 mm EDTA and washed with FACS buffer (PBS, 5% FBS and 0.02% sodium azide). The harvested cells (4 × 105) were stained with the desired antibody for 1 h at 4 °C. The antibody/cell mixture was washed with FACS buffer followed by incubation with anti-human PE (Sigma) (1:100 dilution) for 1 h at 4 °C. The cells were again washed with the FACS buffer and analyzed on a FACS analyzer (BD Accuri).
Pseudovirus generation and single-round infectivity assay
HIV-1 Env pseudoviruses were generated as described previously (139, 140). Briefly, HEK-293T cells were co-transfected with a plasmid containing the virus backbone (pSG3Δenv) and a plasmid (pSVIII) for different envelope derivatives in 1:3 molar ratio using PolyFect transfection reagent (Qiagen) or PEI reagent (Sigma). The relative amounts of p24 protein in pseudoviral stocks were estimated using HIV-1 p24 fourth generation Microlisa kit (J. Mitra & Co. Pvt. Ltd.) according to the manufacturer's protocol, and this was used to normalize pseudoviral stock concentrations used for the infectivity assay. Supernatants containing pseudoviruses were harvested 48 h post-transfection and tested for infectivity, using a luciferase-based assay in TZM-bl cells as described previously (139, 140). Briefly, dilutions of pseudoviruses were mixed with TZM-bl cells, plated in 96-well flat bottom plates, and incubated at 37 °C in a CO2 incubator. 48 h post-transfection, 100 μl of media was removed from each well, and 80 μl of Britelite plus reagent (PerkinElmer Life Sciences) was added. Plates were left at room temperature for 2 min for complete lysis of cells. Luminescence was measured in a Victor X2 luminometer (PerkinElmer Life Sciences). The infectivity was reported on the basis of relative luminescence units (RLUs) at various dilutions of pseudoviruses.
Neutralization assay
Pseudoviruses having glycan-deficient Env were tested for neutralization with mature bNAb VRC01, germline VRC01 (GL-VRC01), mature bNAb PG9, and the non-neutralizing CD4bs mAb b6 as described previously (139, 140). Pseudoviruses were incubated with various dilutions of antibody at 37 °C for 1 h. After incubation, TZM-bl cells were added to pseudoviruses in 96-well plates and further incubated for 48 h at 37 °C in a CO2 incubator. The reduction in infectivity in the presence and absence of an antibody was quantitated by the luciferase assay using Britelite plus reagent as mentioned and described previously (139, 140).
Generation of full-length HIV-1 molecular clones and multiple-round infectivity assay
MCs pLAI-ΔG13 gp160 and pLAI-ΔG15F gp160 were generated by replacement of the Env region of the pLAI-JRFL plasmids (91, 92) with the corresponding glycan-deficient Env (see Table 6 for ΔG13 and ΔG15F gp160 description). MCs were produced in HEK-293T cells as described above for pseudoviral production, by transfection of pLAI-JRFL, pLAI-ΔG13 gp160, or pLAI-ΔG15F gp160 plasmid using PEI. HUT-R5 cells were infected with these MCs to analyze spreading multiple-cycle HIV-1 infection as described previously (93–95). HUT-R5 cells refer to the human T-cell line HUT-78, which is stably transduced with CCR5 co-receptor. These cells were propagated in RPMI 1640 medium supplemented with 10% FBS. For multiple-cycle viral replication assays, 5 × 106 HUT-R5 cells were infected with 5 ng of HIV-1 p24 equivalent stocks of MCs using DEAE-dextran. Following infection, cells were split in a 1:2 ratio every 3rd day, and virus replication was quantitated by measuring the HIV-1 p24 antigen released in the culture supernatants at different time points post-infection. The relative amounts of p24 protein were estimated using HIV-1 p24 fourth generation Microlisa kit (J. Mitra & Co. Pvt. Ltd.) using the manufacturer's protocol in combination with absolute viral copy number estimation by RT-PCR. Virus-containing supernatants were also tested for infectivity, using a luciferase-based assay in TZM-bl cells as described previously (139, 140).
Outer domain glycan-deficient gp160 constructs from HIV-1 subtypes A and C
To understand the effect of glycan removal on non-subtype B HIV-1 Env, two sequences each from subtype A (Q842env.d16 and QH343.21M.ENV.B5) and subtype C (DU422.1 and CAP45.2) virus reference panels of Env molecular clones were selected (Table 6, nos. 10–17). WT subtype A genes were available in the pCI-neo vector cloned between MluI and NotI sites, and WT subtype C genes were available in the pcDNA3.1D/V5-His TOPO® vector (AIDS Reagent Program, National Institutes of Health). Glycosylation sites in these strains were identified, and Asn residues at all outer domain glycosylation sites, except 262 and 276, which interact with the inner domain, were mutated to the second most frequent amino acid at that site in the HIV-1 multiple sequence alignment using the approach described above for the subtype B-based constructs (Fig. 2). These constructs are described in Table 6, and a list of glycosylation site mutations for each strain is given in supplemental Table S4. Genes for the glycan-deficient outer domain for each strain having a 33-bp homology at each end with the adjoining gp160 sequence were synthesized by GenScript and supplied in pUC vectors. Glycan-deficient OD regions were PCR-amplified from the corresponding pUC vectors. Vectors containing gp160 sequences were amplified using inverse PCR. Primers were designed to amplify the gp160 region, excluding the OD region along with the vector backbone. This generated a gapped vector containing a gp160 sequence except the OD region. In vitro Gibson assembly reaction was performed to recombine the gapped vectors and corresponding glycan-deficient ODs (141). Recombined products were transformed into E. coli and plasmids were purified. All glycan-deficient gp160 constructs were further confirmed by sequencing.
Mammalian cell surface display of WT and corresponding glycan-deficient gp160 mutants from subtype A and subtype C strains
WT gp160s and OD glycan-deficient gp160s from Gln-842, QH343, DU422, and CAP45.2 strains were displayed on the surface of HEK-293T cells, and their binding with various bNAbs targeting trimeric and monomeric epitopes was monitored using FACS as described above for the JRFL-based gp160 derivatives. Unlike JRFL-based constructs, these subtype A and C gp160s do not have the cytoplasmic tail deletion.
Author contributions
U. R. and P. S. designed the immunogens, carried out yeast surface display, mammalian surface display, and FACS experiments. U. R. and S. K. did pseudoviral experiments. U. R. and R. Datta did multiple-cycle viral assays. U. R. performed purification, biophysical characterization, and SPR binding studies for proteins purified from E. coli. A. A. K., S. D., and R. Das contributed to yeast surface display, cloning, and protein purification studies, respectively. J. R. M. provided resources. U. R., P. S., and R. V. wrote the original draft; U. R. and R. V. edited the draft. R. V. provided overall supervision.
Supplementary Material
Acknowledgments
Monoclonal antibodies b12, b6, PGT128, PGT151, PGT145, 2G12, and 2F5 were obtained from the Neutralizing Antibody Consortium of IAVI, 447-52D from AIDS Reagent Program of the National Institutes of Health, and PG9 and PG16 from Dr. S. Phogat. The yeast display vector pPNLS was kindly provided by Dr. Dennis Burton. Dr. Udaykumar Ranga, Dr. Ritu Gaur, Uddhav Timilsina, and Disha Bhange are acknowledged for help with multicycle infectivity assays. We are thankful to the proteomics facility at Molecular Biophysics Unit, Indian Institute of Science, especially Sunita Prakash and Raghu Tadala for assistance with mass spectrometric data generation. The assistance of Nonavinakere Seetharam Srilatha is duly acknowledged for the SPR experiments. We thank Dr. Chetana Baliga and Dr. Shweta Karambelkar for their valuable suggestions.
This work was supported by National Institutes of Health Grant R01A1118366-01 from NIAID and Department of Biotechnology and Department of Science and Technology, Government of India, Grant BT/PR4791/MED/29/465/2012. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S15, Tables S1–S4, and References 1–4.
- Env
- envelope
- bNAb
- broadly neutralizing antibody
- OD
- outer domain
- CD4bs
- CD4-binding site
- GL
- germline
- PNGS
- potential N-linked glycosylation site
- YCO
- yeast codon-optimized
- T/F
- Transmitted/Founder
- GdnHCl
- guanidine hydrochloride
- RLU
- relative luminescence unit
- PDB
- Protein Data Bank
- PE
- phycoerythrin
- SPR
- surface plasmon resonance
- MC
- molecular clone.
References
- 1. Wyatt R., and Sodroski J. (1998) The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 280, 1884–1888 [DOI] [PubMed] [Google Scholar]
- 2. Overbaugh J., and Morris L. (2012) The antibody response against HIV-1. Cold Spring Harb. Perspect. Med. 2, a007039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Parren P. W., Gauduin M. C., Koup R. A., Poignard P., Fisicaro P., Burton D. R., and Sattentau Q. J. (1997) Relevance of the antibody response against human immunodeficiency virus type 1 envelope to vaccine design. Immunol. Lett. 57, 105–112 [DOI] [PubMed] [Google Scholar]
- 4. Shcherbakov D. N., Bakulina A. Y., Karpenko L. I., and Ilyichev A. A. (2015) Broadly neutralizing antibodies against HIV-1 as a novel aspect of the immune response. Acta Naturae 7, 11–21 [PMC free article] [PubMed] [Google Scholar]
- 5. Kwong P. D., Wyatt R., Robinson J., Sweet R. W., Sodroski J., and Hendrickson W. A. (1998) Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393, 648–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burton D. R., Pyati J., Koduri R., Sharp S. J., Thornton G. B., Parren P. W., Sawyer L. S., Hendry R. M., Dunlop N., and Nara P. L. (1994) Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science 266, 1024–1027 [DOI] [PubMed] [Google Scholar]
- 7. Corti D., Langedijk J. P., Hinz A., Seaman M. S., Vanzetta F., Fernandez-Rodriguez B. M., Silacci C., Pinna D., Jarrossay D., Balla-Jhagjhoorsingh S., Willems B., Zekveld M. J., Dreja H., O'Sullivan E., Pade C., et al. (2010) Analysis of memory B cell responses and isolation of novel monoclonal antibodies with neutralizing breadth from HIV-1-infected individuals. PLoS ONE 5, e8805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou T., Georgiev I., Wu X., Yang Z. Y., Dai K., Finzi A., Kwon Y. D., Scheid J. F., Shi W., Xu L., Yang Y., Zhu J., Nussenzweig M. C., Sodroski J., Shapiro L., et al. (2010) Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science 329, 811–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Trkola A., Purtscher M., Muster T., Ballaun C., Buchacher A., Sullivan N., Srinivasan K., Sodroski J., Moore J. P., and Katinger H. (1996) Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J. Virol. 70, 1100–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Scheid J. F., Mouquet H., Ueberheide B., Diskin R., Klein F., Oliveira T. Y., Pietzsch J., Fenyo D., Abadir A., Velinzon K., Hurley A., Myung S., Boulad F., Poignard P., Burton D. R., et al. (2011) Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science 333, 1633–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Walker L. M., Huber M., Doores K. J., Falkowska E., Pejchal R., Julien J. P., Wang S. K., Ramos A., Chan-Hui P. Y., Moyle M., Mitcham J. L., Hammond P. W., Olsen O. A., Phung P., Fling S., et al. (2011) Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477, 466–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu X., Zhou T., Zhu J., Zhang B., Georgiev I., Wang C., Chen X., Longo N. S., Louder M., McKee K., O'Dell S., Perfetto S., Schmidt S. D., Shi W., Wu L., et al. (2011) Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science 333, 1593–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kwong P. D., Doyle M. L., Casper D. J., Cicala C., Leavitt S. A., Majeed S., Steenbeke T. D., Venturi M., Chaiken I., Fung M., Katinger H., Parren P. W., Robinson J., Van Ryk D., Wang L., et al. (2002) HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 420, 678–682 [DOI] [PubMed] [Google Scholar]
- 14. Dacheux L., Moreau A., Ataman-Onal Y., Biron F., Verrier B., and Barin F. (2004) Evolutionary dynamics of the glycan shield of the human immunodeficiency virus envelope during natural infection and implications for exposure of the 2G12 epitope. J. Virol. 78, 12625–12637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fenouillet E., Gluckman J. C., and Jones I. M. (1994) Functions of HIV envelope glycans. Trends Biochem. Sci. 19, 65–70 [DOI] [PubMed] [Google Scholar]
- 16. Grundner C., Pancera M., Kang J. M., Koch M., Sodroski J., and Wyatt R. (2004) Factors limiting the immunogenicity of HIV-1 gp120 envelope glycoproteins. Virology 330, 233–248 [DOI] [PubMed] [Google Scholar]
- 17. Connor R. I., Korber B. T., Graham B. S., Hahn B. H., Ho D. D., Walker B. D., Neumann A. U., Vermund S. H., Mestecky J., Jackson S., Fenamore E., Cao Y., Gao F., Kalams S., Kunstman K. J., et al. (1998) Immunological and virological analyses of persons infected by human immunodeficiency virus type 1 while participating in trials of recombinant gp120 subunit vaccines. J. Virol. 72, 1552–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Flynn N. M., Forthal D. N., Harro C. D., Judson F. N., Mayer K. H., Para M. F., and rgp120 HIV Vaccine Study Group (2005) Placebo-controlled phase 3 trial of a recombinant glycoprotein 120 vaccine to prevent HIV-1 infection. J. Infect. Dis. 191, 654–665 [DOI] [PubMed] [Google Scholar]
- 19. Gilbert P. B., Peterson M. L., Follmann D., Hudgens M. G., Francis D. P., Gurwith M., Heyward W. L., Jobes D. V., Popovic V., Self S. G., Sinangil F., Burke D., and Berman P. W. (2005) Correlation between immunologic responses to a recombinant glycoprotein 120 vaccine and incidence of HIV-1 infection in a phase 3 HIV-1 preventive vaccine trial. J. Infect. Dis. 191, 666–677 [DOI] [PubMed] [Google Scholar]
- 20. Grundner C., Li Y., Louder M., Mascola J., Yang X., Sodroski J., and Wyatt R. (2005) Analysis of the neutralizing antibody response elicited in rabbits by repeated inoculation with trimeric HIV-1 envelope glycoproteins. Virology 331, 33–46 [DOI] [PubMed] [Google Scholar]
- 21. Graham B. S., McElrath M. J., Connor R. I., Schwartz D. H., Gorse G. J., Keefer M. C., Mulligan M. J., Matthews T. J., Wolinsky S. M., Montefiori D. C., Vermund S. H., Lambert J. S., Corey L., Belshe R. B., Dolin R., et al. (1998) Analysis of intercurrent human immunodeficiency virus type 1 infections in phase I and II trials of candidate AIDS vaccines. AIDS Vaccine Evaluation Group, and the Correlates of HIV Immune Protection Group. J. Infect. Dis. 177, 310–319 [DOI] [PubMed] [Google Scholar]
- 22. Schwarz F., and Aebi M. (2011) Mechanisms and principles of N-linked protein glycosylation. Curr. Opin. Struct. Biol. 21, 576–582 [DOI] [PubMed] [Google Scholar]
- 23. Helenius A., and Aebi M. (2001) Intracellular functions of N-linked glycans. Science 291, 2364–2369 [DOI] [PubMed] [Google Scholar]
- 24. Leonard C. K., Spellman M. W., Riddle L., Harris R. J., Thomas J. N., and Gregory T. J. (1990) Assignment of intrachain disulfide bonds and characterization of potential glycosylation sites of the type 1 recombinant human immunodeficiency virus envelope glycoprotein (gp120) expressed in Chinese hamster ovary cells. J. Biol. Chem. 265, 10373–10382 [PubMed] [Google Scholar]
- 25. Rathore U., Kesavardhana S., Mallajosyula V. V., and Varadarajan R. (2014) Immunogen design for HIV-1 and influenza. Biochim. Biophys. Acta 1844, 1891–1906 [DOI] [PubMed] [Google Scholar]
- 26. Scanlan C. N., Offer J., Zitzmann N., and Dwek R. A. (2007) Exploiting the defensive sugars of HIV-1 for drug and vaccine design. Nature 446, 1038–1045 [DOI] [PubMed] [Google Scholar]
- 27. Back N. K., Smit L., De Jong J. J., Keulen W., Schutten M., Goudsmit J., and Tersmette M. (1994) An N-glycan within the human immunodeficiency virus type 1 gp120 V3 loop affects virus neutralization. Virology 199, 431–438 [DOI] [PubMed] [Google Scholar]
- 28. Bolmstedt A., Sjölander S., Hansen J. E., Akerblom L., Hemming A., Hu S. L., Morein B., and Olofsson S. (1996) Influence of N-linked glycans in V4-V5 region of human immunodeficiency virus type 1 glycoprotein gp160 on induction of a virus-neutralizing humoral response. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 12, 213–220 [DOI] [PubMed] [Google Scholar]
- 29. Cole K. S., Steckbeck J. D., Rowles J. L., Desrosiers R. C., and Montelaro R. C. (2004) Removal of N-linked glycosylation sites in the V1 region of simian immunodeficiency virus gp120 results in redirection of B-cell responses to V3. J. Virol. 78, 1525–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Derdeyn C. A., and Hunter E. (2008) Viral characteristics of transmitted HIV. Curr. Opin. HIV AIDS 3, 16–21 [DOI] [PubMed] [Google Scholar]
- 31. Frost S. D., Wrin T., Smith D. M., Kosakovsky Pond S. L., Liu Y., Paxinos E., Chappey C., Galovich J., Beauchaine J., Petropoulos C. J., Little S. J., and Richman D. D. (2005) Neutralizing antibody responses drive the evolution of human immunodeficiency virus type 1 envelope during recent HIV infection. Proc. Natl. Acad. Sci. U.S.A. 102, 18514–18519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koch M., Pancera M., Kwong P. D., Kolchinsky P., Grundner C., Wang L., Hendrickson W. A., Sodroski J., and Wyatt R. (2003) Structure-based, targeted deglycosylation of HIV-1 gp120 and effects on neutralization sensitivity and antibody recognition. Virology 313, 387–400 [DOI] [PubMed] [Google Scholar]
- 33. Li M., Salazar-Gonzalez J. F., Derdeyn C. A., Morris L., Williamson C., Robinson J. E., Decker J. M., Li Y., Salazar M. G., Polonis V. R., Mlisana K., Karim S. A., Hong K., Greene K. M., Bilska M., et al. (2006) Genetic and neutralization properties of subtype C human immunodeficiency virus type 1 molecular env clones from acute and early heterosexually acquired infections in Southern Africa. J. Virol. 80, 11776–11790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li Y., Luo L., Rasool N., and Kang C. Y. (1993) Glycosylation is necessary for the correct folding of human immunodeficiency virus gp120 in CD4 binding. J. Virol. 67, 584–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McCaffrey R. A., Saunders C., Hensel M., and Stamatatos L. (2004) N-Linked glycosylation of the V3 loop and the immunologically silent face of gp120 protects human immunodeficiency virus type 1 SF162 from neutralization by anti-gp120 and anti-gp41 antibodies. J. Virol. 78, 3279–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Olofsson S., and Hansen J. E. (1998) Host cell glycosylation of viral glycoproteins–a battlefield for host defence and viral resistance. Scand. J. Infect. Dis. 30, 435–440 [DOI] [PubMed] [Google Scholar]
- 37. Reitter J. N., Means R. E., and Desrosiers R. C. (1998) A role for carbohydrates in immune evasion in AIDS. Nat. Med. 4, 679–684 [DOI] [PubMed] [Google Scholar]
- 38. Richman D. D., Wrin T., Little S. J., and Petropoulos C. J. (2003) Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc. Natl. Acad. Sci. U.S.A. 100, 4144–4149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wei X., Decker J. M., Wang S., Hui H., Kappes J. C., Wu X., Salazar-Gonzalez J. F., Salazar M. G., Kilby J. M., Saag M. S., Komarova N. L., Nowak M. A., Hahn B. H., Kwong P. D., and Shaw G. M. (2003) Antibody neutralization and escape by HIV-1. Nature 422, 307–312 [DOI] [PubMed] [Google Scholar]
- 40. Wu Z., Kayman S. C., Honnen W., Revesz K., Chen H., Vijh-Warrier S., Tilley S. A., McKeating J., Shotton C., and Pinter A. (1995) Characterization of neutralization epitopes in the V2 region of human immunodeficiency virus type 1 gp120: role of glycosylation in the correct folding of the V1/V2 domain. J. Virol. 69, 2271–2278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wyatt R., Kwong P. D., Desjardins E., Sweet R. W., Robinson J., Hendrickson W. A., and Sodroski J. G. (1998) The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 393, 705–711 [DOI] [PubMed] [Google Scholar]
- 42. Stansell E., and Desrosiers R. C. (2010) Functional contributions of carbohydrate on AIDS virus glycoprotein. Yale J. Biol. Med. 83, 201–208 [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang M., Gaschen B., Blay W., Foley B., Haigwood N., Kuiken C., and Korber B. (2004) Tracking global patterns of N-linked glycosylation site variation in highly variable viral glycoproteins: HIV, SIV, and HCV envelopes and influenza hemagglutinin. Glycobiology 14, 1229–1246 [DOI] [PubMed] [Google Scholar]
- 44. Pritchard L. K., Spencer D. I., Royle L., Bonomelli C., Seabright G. E., Behrens A. J., Kulp D. W., Menis S., Krumm S. A., Dunlop D. C., Crispin D. J., Bowden T. A., Scanlan C. N., Ward A. B., Schief W. R., et al. (2015) Glycan clustering stabilizes the mannose patch of HIV-1 and preserves vulnerability to broadly neutralizing antibodies. Nat. Commun. 6, 7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang W., Nie J., Prochnow C., Truong C., Jia Z., Wang S., Chen X. S., and Wang Y. (2013) A systematic study of the N-glycosylation sites of HIV-1 envelope protein on infectivity and antibody-mediated neutralization. Retrovirology 10, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Go E. P., Liao H. X., Alam S. M., Hua D., Haynes B. F., and Desaire H. (2013) Characterization of host-cell line specific glycosylation profiles of early transmitted/founder HIV-1 gp120 envelope proteins. J. Proteome Res. 12, 1223–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stansell E., Panico M., Canis K., Pang P. C., Bouché L., Binet D., O'Connor M. J., Chertova E., Bess J., Lifson J. D., Haslam S. M., Morris H. R., Desrosiers R. C., and Dell A. (2015) Gp120 on HIV-1 virions lacks O-linked carbohydrate. PLoS ONE 10, e0124784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Choi B. K., Bobrowicz P., Davidson R. C., Hamilton S. R., Kung D. H., Li H., Miele R. G., Nett J. H., Wildt S., and Gerngross T. U. (2003) Use of combinatorial genetic libraries to humanize N-linked glycosylation in the yeast Pichia pastoris. Proc. Natl. Acad. Sci. U.S.A. 100, 5022–5027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gagneux P., and Varki A. (1999) Evolutionary considerations in relating oligosaccharide diversity to biological function. Glycobiology 9, 747–755 [DOI] [PubMed] [Google Scholar]
- 50. Kornfeld R., and Kornfeld S. (1985) Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 54, 631–664 [DOI] [PubMed] [Google Scholar]
- 51. Behrens A. J., Vasiljevic S., Pritchard L. K., Harvey D. J., Andev R. S., Krumm S. A., Struwe W. B., Cupo A., Kumar A., Zitzmann N., Seabright G. E., Kramer H. B., Spencer D. I., Royle L., Lee J. H., et al. (2016) Composition and antigenic effects of individual glycan sites of a trimeric HIV-1 envelope glycoprotein. Cell Rep. 14, 2695–2706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Binley J. M., Ban Y. E., Crooks E. T., Eggink D., Osawa K., Schief W. R., and Sanders R. W. (2010) Role of complex carbohydrates in human immunodeficiency virus type 1 infection and resistance to antibody neutralization. J. Virol. 84, 5637–5655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Huang X., Jin W., Hu K., Luo S., Du T., Griffin G. E., Shattock R. J., and Hu Q. (2012) Highly conserved HIV-1 gp120 glycans proximal to CD4-binding region affect viral infectivity and neutralizing antibody induction. Virology 423, 97–106 [DOI] [PubMed] [Google Scholar]
- 54. Lee W. R., Syu W. J., Du B., Matsuda M., Tan S., Wolf A., Essex M., and Lee T. H. (1992) Nonrandom distribution of gp120 N-linked glycosylation sites important for infectivity of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. U.S.A. 89, 2213–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ohgimoto S., Shioda T., Mori K., Nakayama E. E., Hu H., and Nagai Y. (1998) Location-specific, unequal contribution of the N-glycans in simian immunodeficiency virus gp120 to viral infectivity and removal of multiple glycans without disturbing infectivity. J. Virol. 72, 8365–8370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Huang C. C., Tang M., Zhang M. Y., Majeed S., Montabana E., Stanfield R. L., Dimitrov D. S., Korber B., Sodroski J., Wilson I. A., Wyatt R., and Kwong P. D. (2005) Structure of a V3-containing HIV-1 gp120 core. Science 310, 1025–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Klein F., Diskin R., Scheid J. F., Gaebler C., Mouquet H., Georgiev I. S., Pancera M., Zhou T., Incesu R. B., Fu B. Z., Gnanapragasam P. N., Oliveira T. Y., Seaman M. S., Kwong P. D., Bjorkman P. J., and Nussenzweig M. C. (2013) Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell 153, 126–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hoot S., McGuire A. T., Cohen K. W., Strong R. K., Hangartner L., Klein F., Diskin R., Scheid J. F., Sather D. N., Burton D. R., and Stamatatos L. (2013) Recombinant HIV envelope proteins fail to engage germline versions of anti-CD4bs bNAbs. PLoS Pathog. 9, e1003106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dosenovic P., von Boehmer L., Escolano A., Jardine J., Freund N. T., Gitlin A. D., McGuire A. T., Kulp D. W., Oliveira T., Scharf L., Pietzsch J., Gray M. D., Cupo A., van Gils M. J., Yao K. H., et al. (2015) Immunization for HIV-1 broadly neutralizing antibodies in human Ig knockin mice. Cell 161, 1505–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jardine J. G., Ota T., Sok D., Pauthner M., Kulp D. W., Kalyuzhniy O., Skog P. D., Thinnes T. C., Bhullar D., Briney B., Menis S., Jones M., Kubitz M., Spencer S., Adachi Y., et al. (2015) HIV-1 vaccines. Priming a broadly neutralizing antibody response to HIV-1 using a germline-targeting immunogen. Science 349, 156–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jardine J., Julien J. P., Menis S., Ota T., Kalyuzhniy O., McGuire A., Sok D., Huang P. S., MacPherson S., Jones M., Nieusma T., Mathison J., Baker D., Ward A. B., Burton D. R., et al. (2013) Rational HIV immunogen design to target specific germline B cell receptors. Science 340, 711–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. McGuire A. T., Hoot S., Dreyer A. M., Lippy A., Stuart A., Cohen K. W., Jardine J., Menis S., Scheid J. F., West A. P., Schief W. R., and Stamatatos L. (2013) Engineering HIV envelope protein to activate germline B cell receptors of broadly neutralizing anti-CD4-binding site antibodies. J. Exp. Med. 210, 655–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kwong P. D., Mascola J. R., and Nabel G. J. (2013) Broadly neutralizing antibodies and the search for an HIV-1 vaccine: the end of the beginning. Nat. Rev. Immunol. 13, 693–701 [DOI] [PubMed] [Google Scholar]
- 64. Kulp D. W., and Schief W. R. (2013) Advances in structure-based vaccine design. Curr. Opin. Virol. 3, 322–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mouquet H., and Nussenzweig M. C. (2013) HIV: Roadmaps to a vaccine. Nature 496, 441–442 [DOI] [PubMed] [Google Scholar]
- 66. Bhattacharyya S., Rajan R. E., Swarupa Y., Rathore U., Verma A., Udaykumar R., and Varadarajan R. (2010) Design of a non-glycosylated outer domain-derived HIV-1 gp120 immunogen that binds to CD4 and induces neutralizing antibodies. J. Biol. Chem. 285, 27100–27110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lehmann M., and Wyss M. (2001) Engineering proteins for thermostability: the use of sequence alignments versus rational design and directed evolution. Curr. Opin. Biotechnol. 12, 371–375 [DOI] [PubMed] [Google Scholar]
- 68. Kuiken C. F. B., Leitner T., Apetrei C., Hahn B., Mizrachi I., Mullins J., Rambaut A., Wolinsky S., and Korber B. (eds) (2010) in HIV Sequence Compendium, 2010, Los Alamos National Laboratory, Los Alamos, NM, Theoretical Biology and Biophysics Group, Los Alamos, NM [Google Scholar]
- 69. McDonald I. K., and Thornton J. M. (1994) Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238, 777–793 [DOI] [PubMed] [Google Scholar]
- 70. Sali A., and Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 71. Ibarra-Molero B., Loladze V. V., Makhatadze G. I., and Sanchez-Ruiz J. M. (1999) Thermal versus guanidine-induced unfolding of ubiquitin. An analysis in terms of the contributions from charge-charge interactions to protein stability. Biochemistry 38, 8138–8149 [DOI] [PubMed] [Google Scholar]
- 72. Thali M., Olshevsky U., Furman C., Gabuzda D., Li J., and Sodroski J. (1991) Effects of changes in gp120-CD4 binding affinity on human immunodeficiency virus type 1 envelope glycoprotein function and soluble CD4 sensitivity. J. Virol. 65, 5007–5012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gai S. A., and Wittrup K. D. (2007) Yeast surface display for protein engineering and characterization. Curr. Opin. Struct. Biol. 17, 467–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shusta E. V., Kieke M. C., Parke E., Kranz D. M., and Wittrup K. D. (1999) Yeast polypeptide fusion surface display levels predict thermal stability and soluble secretion efficiency. J. Mol. Biol. 292, 949–956 [DOI] [PubMed] [Google Scholar]
- 75. Chao G., Lau W. L., Hackel B. J., Sazinsky S. L., Lippow S. M., and Wittrup K. D. (2006) Isolating and engineering human antibodies using yeast surface display. Nat. Protoc. 1, 755–768 [DOI] [PubMed] [Google Scholar]
- 76. Arakawa T., Ejima D., Li T., and Philo J. S. (2010) The critical role of mobile phase composition in size exclusion chromatography of protein pharmaceuticals. J. Pharm. Sci. 99, 1674–1692 [DOI] [PubMed] [Google Scholar]
- 77. Mata-Fink J., Kriegsman B., Yu H. X., Zhu H., Hanson M. C., Irvine D. J., and Wittrup K. D. (2013) Rapid conformational epitope mapping of anti-gp120 antibodies with a designed mutant panel displayed on yeast. J. Mol. Biol. 425, 444–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chen L., Kwon Y. D., Zhou T., Wu X., O'Dell S., Cavacini L., Hessell A. J., Pancera M., Tang M., Xu L., Yang Z. Y., Zhang M. Y., Arthos J., Burton D. R., Dimitrov D. S., et al. (2009) Structural basis of immune evasion at the site of CD4 attachment on HIV-1 gp120. Science 326, 1123–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pancera M., Zhou T., Druz A., Georgiev I. S., Soto C., Gorman J., Huang J., Acharya P., Chuang G. Y., Ofek G., Stewart-Jones G. B., Stuckey J., Bailer R. T., Joyce M. G., Louder M. K., et al. (2014) Structure and immune recognition of trimeric pre-fusion HIV-1 Env. Nature 514, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lee J. H., Ozorowski G., and Ward A. B. (2016) Cryo-EM structure of a native, fully glycosylated, cleaved HIV-1 envelope trimer. Science 351, 1043–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Stewart-Jones G. B., Soto C., Lemmin T., Chuang G. Y., Druz A., Kong R., Thomas P. V., Wagh K., Zhou T., Behrens A. J., Bylund T., Choi C. W., Davison J. R., Georgiev I. S., Joyce M. G., et al. (2016) Trimeric HIV-1-Env structures define glycan shields from clades A, B, and G. Cell 165, 813–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kesavardhana S., and Varadarajan R. (2014) Stabilizing the native trimer of HIV-1 Env by destabilizing the heterodimeric interface of the gp41 postfusion six-helix bundle. J. Virol. 88, 9590–9604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Pancera M., and Wyatt R. (2005) Selective recognition of oligomeric HIV-1 primary isolate envelope glycoproteins by potently neutralizing ligands requires efficient precursor cleavage. Virology 332, 145–156 [DOI] [PubMed] [Google Scholar]
- 84. Pejchal R., Doores K. J., Walker L. M., Khayat R., Huang P. S., Wang S. K., Stanfield R. L., Julien J. P., Ramos A., Crispin M., Depetris R., Katpally U., Marozsan A., Cupo A., Maloveste S., et al. (2011) A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science 334, 1097–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Doores K. J., and Burton D. R. (2010) Variable loop glycan dependency of the broad and potent HIV-1-neutralizing antibodies PG9 and PG16. J. Virol. 84, 10510–10521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Falkowska E., Le K. M., Ramos A., Doores K. J., Lee J. H., Blattner C., Ramirez A., Derking R., van Gils M. J., Liang C. H., Mcbride R., von Bredow B., Shivatare S. S., Wu C. Y., Chan-Hui P. Y., et al. (2014) Broadly neutralizing HIV antibodies define a glycan-dependent epitope on the prefusion conformation of gp41 on cleaved envelope trimers. Immunity 40, 657–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Blattner C., Lee J. H., Sliepen K., Derking R., Falkowska E., de la Peña A. T., Cupo A., Julien J. P., van Gils M., Lee P. S., Peng W., Paulson J. C., Poignard P., Burton D. R., Moore J. P., et al. (2014) Structural delineation of a quaternary, cleavage-dependent epitope at the gp41-gp120 interface on intact HIV-1 Env trimers. Immunity 40, 669–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Pejchal R., Walker L. M., Stanfield R. L., Phogat S. K., Koff W. C., Poignard P., Burton D. R., and Wilson I. A. (2010) Structure and function of broadly reactive antibody PG16 reveal an H3 subdomain that mediates potent neutralization of HIV-1. Proc. Natl. Acad. Sci. U.S.A. 107, 11483–11488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Liu J., Bartesaghi A., Borgnia M. J., Sapiro G., and Subramaniam S. (2008) Molecular architecture of native HIV-1 gp120 trimers. Nature 455, 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wibmer C. K., Moore P. L., and Morris L. (2015) HIV broadly neutralizing antibody targets. Curr. Opin. HIV AIDS 10, 135–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Leaman D. P., Kinkead H., and Zwick M. B. (2010) In-solution virus capture assay helps deconstruct heterogeneous antibody recognition of human immunodeficiency virus type 1. J. Virol. 84, 3382–3395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Peden K., Emerman M., and Montagnier L. (1991) Changes in growth properties on passage in tissue culture of viruses derived from infectious molecular clones of HIV-1LAI, HIV-1MAL, and HIV-1ELI. Virology 185, 661–672 [DOI] [PubMed] [Google Scholar]
- 93. Moore M. D., Chin M. P., and Hu W. S. (2009) HIV-1 recombination: an experimental assay and a phylogenetic approach. Methods Mol. Biol. 485, 87–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Timilsina U., Ghimire D., Timalsina B., Nitz T. J., Wild C. T., Freed E. O., and Gaur R. (2016) Identification of potent maturation inhibitors against HIV-1 clade C. Sci. Rep. 6, 27403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Ghimire D., Timilsina U., Srivastava T. P., and Gaur R. (2017) Insights into the activity of maturation inhibitor PF-46396 on HIV-1 clade C. Sci. Rep. 7, 43711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Buonaguro L., Tornesello M. L., and Buonaguro F. M. (2007) Human immunodeficiency virus type 1 subtype distribution in the worldwide epidemic: pathogenetic and therapeutic implications. J. Virol. 81, 10209–10219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Moore P. L., Gray E. S., Wibmer C. K., Bhiman J. N., Nonyane M., Sheward D. J., Hermanus T., Bajimaya S., Tumba N. L., Abrahams M. R., Lambson B. E., Ranchobe N., Ping L., Ngandu N., Abdool Karim Q., et al. (2012) Evolution of an HIV glycan-dependent broadly neutralizing antibody epitope through immune escape. Nat. Med. 18, 1688–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. McLellan J. S., Pancera M., Carrico C., Gorman J., Julien J. P., Khayat R., Louder R., Pejchal R., Sastry M., Dai K., O'Dell S., Patel N., Shahzad-ul-Hussan S., Yang Y., Zhang B., et al. (2011) Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature 480, 336–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Pancera M., Shahzad-Ul-Hussan S., Doria-Rose N. A., McLellan J. S., Bailer R. T., Dai K., Loesgen S., Louder M. K., Staupe R. P., Yang Y., Zhang B., Parks R., Eudailey J., Lloyd K. E., Blinn J., et al. (2013) Structural basis for diverse N-glycan recognition by HIV-1-neutralizing V1-V2-directed antibody PG16. Nat. Struct. Mol. Biol. 20, 804–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ma B. J., Alam S. M., Go E. P., Lu X., Desaire H., Tomaras G. D., Bowman C., Sutherland L. L., Scearce R. M., Santra S., Letvin N. L., Kepler T. B., Liao H. X., and Haynes B. F. (2011) Envelope deglycosylation enhances antigenicity of HIV-1 gp41 epitopes for both broad neutralizing antibodies and their unmutated ancestor antibodies. PLoS Pathog. 7, e1002200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Scharf L., West A. P. Jr, Gao H., Lee T., Scheid J. F., Nussenzweig M. C., Bjorkman P. J., and Diskin R. (2013) Structural basis for HIV-1 gp120 recognition by a germ-line version of a broadly neutralizing antibody. Proc. Natl. Acad. Sci. U.S.A. 110, 6049–6054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. West A. P. Jr., Diskin R., Nussenzweig M. C., and Bjorkman P. J. (2012) Structural basis for germ-line gene usage of a potent class of antibodies targeting the CD4-binding site of HIV-1 gp120. Proc. Natl. Acad. Sci. U.S.A. 109, E2083–E2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Jardine J. G., Kulp D. W., Havenar-Daughton C., Sarkar A., Briney B., Sok D., Sesterhenn F., Ereño-Orbea J., Kalyuzhniy O., Deresa I., Hu X., Spencer S., Jones M., Georgeson E., Adachi Y., et al. (2016) HIV-1 broadly neutralizing antibody precursor B cells revealed by germline-targeting immunogen. Science 351, 1458–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Liao H. X., Lynch R., Zhou T., Gao F., Alam S. M., Boyd S. D., Fire A. Z., Roskin K. M., Schramm C. A., Zhang Z., Zhu J., Shapiro L., NISC Comparative Sequencing Program, Mullikin J. C., Gnanakaran S., et al. (2013) Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature 496, 469–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Pancera M., McLellan J. S., Wu X., Zhu J., Changela A., Schmidt S. D., Yang Y., Zhou T., Phogat S., Mascola J. R., and Kwong P. D. (2010) Crystal structure of PG16 and chimeric dissection with somatically related PG9: structure-function analysis of two quaternary-specific antibodies that effectively neutralize HIV-1. J. Virol. 84, 8098–8110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Briney B., Sok D., Jardine J. G., Kulp D. W., Skog P., Menis S., Jacak R., Kalyuzhniy O., de Val N., Sesterhenn F., Le K. M., Ramos A., Jones M., Saye-Francisco K. L., Blane T. R., et al. (2016) Tailored immunogens direct affinity maturation toward HIV neutralizing antibodies. Cell 166, 1459–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Sok D., Briney B., Jardine J. G., Kulp D. W., Menis S., Pauthner M., Wood A., Lee E. C., Le K. M., Jones M., Ramos A., Kalyuzhniy O., Adachi Y., Kubitz M., MacPherson S., et al. (2016) Priming HIV-1 broadly neutralizing antibody precursors in human Ig loci transgenic mice. Science 353, 1557–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Steichen J. M., Kulp D. W., Tokatlian T., Escolano A., Dosenovic P., Stanfield R. L., McCoy L. E., Ozorowski G., Hu X., Kalyuzhniy O., Briney B., Schiffner T., Garces F., Freund N. T., Gitlin A. D., et al. (2016) HIV vaccine design to target germline precursors of glycan-dependent broadly neutralizing antibodies. Immunity 45, 483–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Tian M., Cheng C., Chen X., Duan H., Cheng H. L., Dao M., Sheng Z., Kimble M., Wang L., Lin S., Schmidt S. D., Du Z., Joyce M. G., Chen Y., DeKosky B. J., et al. (2016) Induction of HIV neutralizing antibody lineages in mice with diverse precursor repertoires. Cell 166, 1471–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Scharf L., Scheid J. F., Lee J. H., West A. P. Jr, Chen C., Gao H., Gnanapragasam P. N., Mares R., Seaman M. S., Ward A. B., Nussenzweig M. C., and Bjorkman P. J. (2014) Antibody 8ANC195 reveals a site of broad vulnerability on the HIV-1 envelope spike. Cell Rep. 7, 785–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Walker L. M., Phogat S. K., Chan-Hui P. Y., Wagner D., Phung P., Goss J. L., Wrin T., Simek M. D., Fling S., Mitcham J. L., Lehrman J. K., Priddy F. H., Olsen O. A., Frey S. M., Hammond P. W., et al. (2009) Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326, 285–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Onda M., Beers R., Xiang L., Nagata S., Wang Q. C., and Pastan I. (2008) An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc. Natl. Acad. Sci. U.S.A. 105, 11311–11316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Freund N. T., Horwitz J. A., Nogueira L., Sievers S. A., Scharf L., Scheid J. F., Gazumyan A., Liu C., Velinzon K., Goldenthal A., Sanders R. W., Moore J. P., Bjorkman P. J., Seaman M. S., Walker B. D., et al. (2015) A new glycan-dependent CD4-binding site neutralizing antibody exerts pressure on HIV-1 in vivo. PLoS Pathog. 11, e1005238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wibmer C. K., Gorman J., Anthony C. S., Mkhize N. N., Druz A., York T., Schmidt S. D., Labuschagne P., Louder M. K., Bailer R. T., Abdool Karim S. S., Mascola J. R., Williamson C., Moore P. L., Kwong P. D., and Morris L. (2016) Structure of an N276-dependent HIV-1 neutralizing antibody targeting a rare V5 glycan hole adjacent to the CD4-binding site. J. Virol. 90, 10220–10235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Pantophlet R., Wilson I. A., and Burton D. R. (2004) Improved design of an antigen with enhanced specificity for the broadly HIV-neutralizing antibody b12. Protein Eng. Des. Sel. 17, 749–758 [DOI] [PubMed] [Google Scholar]
- 116. Wang C. C., Chen J. R., Tseng Y. C., Hsu C. H., Hung Y. F., Chen S. W., Chen C. M., Khoo K. H., Cheng T. J., Cheng Y. S., Jan J. T., Wu C. Y., Ma C., and Wong C. H. (2009) Glycans on influenza hemagglutinin affect receptor binding and immune response. Proc. Natl. Acad. Sci. U.S.A. 106, 18137–18142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Bhattacharyya S., Singh P., Rathore U., Purwar M., Wagner D., Arendt H., DeStefano J., LaBranche C. C., Montefiori D. C., Phogat S., and Varadarajan R. (2013) Design of an Escherichia coli expressed HIV-1 gp120 fragment immunogen that binds to b12 and induces broad and potent neutralizing antibodies. J. Biol. Chem. 288, 9815–9825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Keele B. F., Giorgi E. E., Salazar-Gonzalez J. F., Decker J. M., Pham K. T., Salazar M. G., Sun C., Grayson T., Wang S., Li H., Wei X., Jiang C., Kirchherr J. L., Gao F., Anderson J. A., et al. (2008) Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U.S.A. 105, 7552–7557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Bunnik E. M., Euler Z., Welkers M. R., Boeser-Nunnink B. D., Grijsen M. L., Prins J. M., and Schuitemaker H. (2010) Adaptation of HIV-1 envelope gp120 to humoral immunity at a population level. Nat. Med. 16, 995–997 [DOI] [PubMed] [Google Scholar]
- 120. Derdeyn C. A., Decker J. M., Bibollet-Ruche F., Mokili J. L., Muldoon M., Denham S. A., Heil M. L., Kasolo F., Musonda R., Hahn B. H., Shaw G. M., Korber B. T., Allen S., and Hunter E. (2004) Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science 303, 2019–2022 [DOI] [PubMed] [Google Scholar]
- 121. Joseph S. B., Swanstrom R., Kashuba A. D., and Cohen M. S. (2015) Bottlenecks in HIV-1 transmission: insights from the study of founder viruses. Nat. Rev. Microbiol. 13, 414–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Nawaz F., Cicala C., Van Ryk D., Block K. E., Jelicic K., McNally J. P., Ogundare O., Pascuccio M., Patel N., Wei D., Fauci A. S., and Arthos J. (2011) The genotype of early-transmitting HIV gp120s promotes α(4)β(7)-reactivity, revealing α(4)β(7)+/CD4+ T cells as key targets in mucosal transmission. PLoS Pathog. 7, e1001301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Go E. P., Hewawasam G., Liao H. X., Chen H., Ping L. H., Anderson J. A., Hua D. C., Haynes B. F., and Desaire H. (2011) Characterization of glycosylation profiles of HIV-1 transmitted/founder envelopes by mass spectrometry. J. Virol. 85, 8270–8284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. van den Kerkhof T. L., Feenstra K. A., Euler Z., van Gils M. J., Rijsdijk L. W., Boeser-Nunnink B. D., Heringa J., Schuitemaker H., and Sanders R. W. (2013) HIV-1 envelope glycoprotein signatures that correlate with the development of cross-reactive neutralizing activity. Retrovirology 10, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. van Gils M. J., and Sanders R. W. (2013) Broadly neutralizing antibodies against HIV-1: templates for a vaccine. Virology 435, 46–56 [DOI] [PubMed] [Google Scholar]
- 126. Schiffner T., Sattentau Q. J., and Dorrell L. (2013) Development of prophylactic vaccines against HIV-1. Retrovirology 10, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Ping L. H., Joseph S. B., Anderson J. A., Abrahams M. R., Salazar-Gonzalez J. F., Kincer L. P., Treurnicht F. K., Arney L., Ojeda S., Zhang M., Keys J., Potter E. L., Chu H., Moore P., Salazar M. G., et al. (2013) Comparison of viral Env proteins from acute and chronic infections with subtype C human immunodeficiency virus type 1 identifies differences in glycosylation and CCR5 utilization and suggests a new strategy for immunogen design. J. Virol. 87, 7218–7233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Hanson S. R., Culyba E. K., Hsu T. L., Wong C. H., Kelly J. W., and Powers E. T. (2009) The core trisaccharide of an N-linked glycoprotein intrinsically accelerates folding and enhances stability. Proc. Natl. Acad. Sci. U.S.A. 106, 3131–3136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Sagt C. M., Kleizen B., Verwaal R., de Jong M. D., Müller W. H., Smits A., Visser C., Boonstra J., Verkleij A. J., and Verrips C. T. (2000) Introduction of an N-glycosylation site increases secretion of heterologous proteins in yeasts. Appl. Environ. Microbiol. 66, 4940–4944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Wu S. J., Luo J., O'Neil K. T., Kang J., Lacy E. R., Canziani G., Baker A., Huang M., Tang Q. M., Raju T. S., Jacobs S. A., Teplyakov A., Gilliland G. L., and Feng Y. (2010) Structure-based engineering of a monoclonal antibody for improved solubility. Protein Eng. Des. Sel. 23, 643–651 [DOI] [PubMed] [Google Scholar]
- 131. Kim Y. S., Bhandari R., Cochran J. R., Kuriyan J., and Wittrup K. D. (2006) Directed evolution of the epidermal growth factor receptor extracellular domain for expression in yeast. Proteins 62, 1026–1035 [DOI] [PubMed] [Google Scholar]
- 132. Berlioz-Torrent C., Shacklett B. L., Erdtmann L., Delamarre L., Bouchaert I., Sonigo P., Dokhelar M. C., and Benarous R. (1999) Interactions of the cytoplasmic domains of human and simian retroviral transmembrane proteins with components of the clathrin adaptor complexes modulate intracellular and cell-surface expression of envelope glycoproteins. J. Virol. 73, 1350–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. LaBranche C. C., Sauter M. M., Haggarty B. S., Vance P. J., Romano J., Hart T. K., Bugelski P. J., Marsh M., and Hoxie J. A. (1995) A single amino acid change in the cytoplasmic domain of the simian immunodeficiency virus transmembrane molecule increases envelope glycoprotein expression on infected cells. J. Virol. 69, 5217–5227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Si Z., Cayabyab M., and Sodroski J. (2001) Envelope glycoprotein determinants of neutralization resistance in a simian-human immunodeficiency virus (SHIV-HXBc2P 3.2) derived by passage in monkeys. J. Virol. 75, 4208–4218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Bowley D. R., Labrijn A. F., Zwick M. B., and Burton D. R. (2007) Antigen selection from an HIV-1 immune antibody library displayed on yeast yields many novel antibodies compared to selection from the same library displayed on phage. Protein Eng. Des. Sel. 20, 81–90 [DOI] [PubMed] [Google Scholar]
- 136. Saha P., Barua B., Bhattacharyya S., Balamurali M. M., Schief W. R., Baker D., and Varadarajan R. (2011) Design and characterization of stabilized derivatives of human CD4D12 and CD4D1. Biochemistry 50, 7891–7900 [DOI] [PubMed] [Google Scholar]
- 137. Varadarajan R., Sharma D., Chakraborty K., Patel M., Citron M., Sinha P., Yadav R., Rashid U., Kennedy S., Eckert D., Geleziunas R., Bramhill D., Schleif W., Liang X., and Shiver J. (2005) Characterization of gp120 and its single-chain derivatives, gp120-CD4D12 and gp120-M9: implications for targeting the CD4i epitope in human immunodeficiency virus vaccine design. J. Virol. 79, 1713–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Murad W., Singh R., and Yen T. Y. (2011) An efficient algorithmic approach for mass spectrometry-based disulfide connectivity determination using multi-ion analysis. BMC Bioinformatics 12, Suppl 1, S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Li M., Gao F., Mascola J. R., Stamatatos L., Polonis V. R., Koutsoukos M., Voss G., Goepfert P., Gilbert P., Greene K. M., Bilska M., Kothe D. L., Salazar-Gonzalez J. F., Wei X., Decker J. M., et al. (2005) Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J. Virol. 79, 10108–10125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Montefiori D. C. (2005) Evaluating neutralizing antibodies against HIV, SIV, and SHIV in luciferase reporter gene assays. Curr. Protoc. Immunol. Chapter 12, Unit 12.11 [DOI] [PubMed] [Google Scholar]
- 141. Gibson D. G., Young L., Chuang R. Y., Venter J. C., Hutchison C. A. 3rd., and Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 [DOI] [PubMed] [Google Scholar]
- 142. Julien J. P., Cupo A., Sok D., Stanfield R. L., Lyumkis D., Deller M. C., Klasse P. J., Burton D. R., Sanders R. W., Moore J. P., Ward A. B., and Wilson I. A. (2013) Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science 342, 1477–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Lyumkis D., Julien J. P., de Val N., Cupo A., Potter C. S., Klasse P. J., Burton D. R., Sanders R. W., Moore J. P., Carragher B., Wilson I. A., and Ward A. B. (2013) Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science 342, 1484–1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Bohne-Lang A., and von der Lieth C. W. (2005) GlyProt: in silico glycosylation of proteins. Nucleic Acids Res. 33, W214–W219 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.