

Abstract
Whereas genomes can be rapidly sequenced, the functions of many genes are incompletely or erroneously annotated because of a lack of experimental evidence or prior functional knowledge in sequence databases. To address this weakness, we describe here a model-enabled gene search (MEGS) approach that (i) identifies metabolic functions either missing from an organism's genome annotation or incorrectly assigned to an ORF by using discrepancies between metabolic model predictions and experimental culturing data; (ii) designs functional selection experiments for these specific metabolic functions; and (iii) selects a candidate gene(s) responsible for these functions from a genomic library and directly interrogates this gene's function experimentally. To discover gene functions, MEGS uses genomic functional selections instead of relying on correlations across large experimental datasets or sequence similarity as do other approaches. When applied to the bioluminescent marine bacterium Vibrio fischeri, MEGS successfully identified five genes that are responsible for four metabolic and transport reactions whose absence from a draft metabolic model of V. fischeri caused inaccurate modeling of high-throughput experimental data. This work demonstrates that MEGS provides a rapid and efficient integrated computational and experimental approach for annotating metabolic genes, including those that have previously been uncharacterized or misannotated.
Keywords: bacterial metabolism, computational biology, functional genomics, metabolism, systems biology, constraint-based model, enzyme selection, genome annotation, genome-scale modeling
Introduction
The development of next-generation sequencing technologies has generated thousands of genome sequences. These are primarily annotated by a combination of bioinformatics methods that are both fast and can be applied genome-wide. Homology-based bioinformatics methods (e.g. BLAST) assume similar sequences share similar functions. Structure-based methods (1, 2) and genomic context-based methods (e.g. conserved operon, gene fusions, and gene co-occurrence across genomes (3–5)) can be utilized to infer functions that are difficult to annotate using BLAST alone. These bioinformatics methods are often used in conjunction with high-throughput experimental data (including gene expression, protein-protein interactions, mass spectrometry, RNAi, and mutant fitness) to suggest gene functions based on connections between genes with known and unknown functions. For example, recent studies have used correlations in transposon mutant-fitness scores across multiple experimental conditions to improve genome annotations (6). Despite the power of these bioinformatics methods, and the increasing availability of high-throughput data, 40–60% of newly sequenced genes still lack assigned functions (7–9). In addition, although bioinformatics methods can quickly predict specific gene functions, biochemical characterization must be still performed separately to validate those predictions. In fact, a majority of the gene functions assigned have no experimental evidence. For example, as of August 2016, only ∼27% of the entries in the UniProtKB Swiss-Prot knowledgebase contain experimental evidence at the protein or transcript level (10). Direct experimental testing of gene functions that are proposed bioinformatically or based on high-throughput experiments is needed to reduce the high rate of incomplete and incorrect annotations (8, 11, 12). Such assessment is important because functions incorrectly assigned to gene sequences enter databases that are subsequently used to assign functions to new sequences. As a result, errors that are hard both to identify and to correct will propagate.
Consequently, it is crucial to develop approaches that quickly identify missing and/or erroneously assigned gene functions and to provide fast and direct experimental validation for the correct function. Such goals can be achieved by combining genome-scale metabolic modeling with experimental techniques. Genome-scale metabolic models are developed primarily based on genome annotations obtained from bioinformatics tools. Current model-based algorithms, including GapFill, SMILEY, and GrowMatch, can use cell culture data to pinpoint knowledge gaps caused by missing or incorrectly called gene functions; however, these algorithms cannot identify candidate genes for these functions (13–15). More recent algorithms, including PHiller-GC, Model SEED, ADOMETA, and MIRAGE, identify missing metabolic reactions and candidate genes that might be responsible for catalyzing them (16–19), but these algorithms require additional data such as annotated sequences from other organisms and/or expensive gene-expression datasets that might not be available. Importantly, all of these current model-based approaches still do not provide direct experimental validation of the function of the candidate gene. Here, we propose a high-throughput model-enabled gene search (MEGS)2 method that rapidly identifies functions for unannotated or misannotated genes. The metabolic modeling procedures in MEGS quickly generate a list of missing or erroneous functions in genome annotations derived from bioinformatics tools and design functional selection experiments (experiments where only strains that gain an essential function from a genomic library are able to grow) to select for genes with these functions. Subsequent functional selection experiments identify the responsible gene(s) from a genomic library and provide fast and direct experimental evidence for the gene's function. In contrast to metagenomic functional selections, which have been used to identify ribulose-bisphosphate carboxylase/oxygenase, DNA polymerase, and antibiotic-resistance genes (20–22), MEGS' functional selections are based on knowledge gaps identified by metabolic modeling. By using genomic functional selections, MEGS does not rely on sequence similarity or genomic context to find gene functions and, as such, can be used to discover functions for previously uncharacterized groups of genes. As such, MEGS complements existing bioinformatics tools to improve genome annotations. MEGS was successfully used to identify the enzymatic and transport functions of five genes in Vibrio fischeri, which were subsequently confirmed by a combination of experiments involving complementation, mutant growth phenotyping, qPCR analysis, and enzyme assays.
Results
Overview of MEGS
MEGS involves three steps that combine metabolic modeling and experimentation (Fig. 1). First, a genome-scale metabolic model of an organism of interest is developed, and physiological experiments are performed to validate the model. Computational tools (14, 15) are then used to pinpoint metabolic function(s) that are missing from the model but are needed to resolve model data discrepancies. The physiological experiments suggest these missing functions occur, but the discrepancies between model predictions and experiments indicate that the functions are absent from current genome annotations. For example, the V. fischeri model originally lacked genes involved in catabolism of both d-xylose and mannitol; however, only genes involved in mannitol catabolism were identified by the model as missing because V. fischeri grew on mannitol (but not d-xylose) as a sole carbon source. Second, a recipient strain (derived from a well-characterized organism, e.g. Escherichia coli) and selective medium (e.g. a minimal medium supplemented with a single carbon source) are designed such that the recipient strain can only grow in the selective medium if the gene(s) presumptively encoding the missing metabolic function (from the organism of interest) is transferred to the recipient strain. Such pairs of recipient strains and selection media can be computationally designed for a reaction of interest using a forced coupling algorithm (23). Third, a genomic functional selection experiment is performed to locate the gene(s) in the genome that are responsible for the missing metabolic function and provide direct evidence for the gene's function. For this last step, a genomic library of the organism of interest is created by inserting random genomic DNA fragments into plasmids. This plasmid library is then transformed into the recipient strain. Gene(s) responsible for the missing metabolic function can then be identified by sequencing plasmids that complement growth of recipient strains in the selective medium. The discovered genes can then be further characterized and added to the model to improve predictions. As more experimental data are generated, any new model data discrepancies that arise can be used to drive additional MEGS cycles.
Figure 1.

Overview of MEGS. In the first step, a metabolic model for an organism of interest is constructed. Target reactions, which are either missing from the model or assigned to the wrong genes, are inferred from discrepancies between model predictions and experimental measurements. A recipient strain (derived from a well-characterized organism, e.g. E. coli) and selective medium are then designed for each target reaction. Such recipient strains can only grow in the selective medium if they acquire heterologous enzymes that catalyze the target reaction. Finally, a genomic library is created, and the recipient strain is used to select for genes capable of catalyzing the target reactions, because such genes will enable growth of the recipient strain in the selective medium. The discovered genes can then be further characterized and added to the model to improve predictions.
MEGS applied to discover V. fischeri gene functions
In this work, we applied MEGS to discover and characterize several enzyme- and transporter-encoding genes of V. fischeri ES114, which is a bioluminescent marine bacterium that forms a symbiotic relationship in the light-emitting organ of the Hawaiian bobtail squid, Euprymna scolopes (24). Its metabolic capabilities are representative of many other marine bacteria, including both beneficial and pathogenic members of the Vibrio genus, and are thus of particular interest (25). From the KEGG (Kyoto Encyclopedia of Genes and Genomes)-annotated genome, we reconstructed a genome-scale metabolic model for V. fischeri ES114, named iVF846 (supplemental Excel file S1). Reactions and metabolites from an E. coli model, iJO1366 (26), were transferred into the draft model of iVF846 when orthologs to E. coli metabolic genes were found in V. fischeri. The draft iVF846 model was then curated based on the following: (i) data and information reported in the literature, and (ii) new growth-phenotyping experiments using Biolog plates, a method for individually testing the ability to metabolize 96 different carbon sources using a microtiter dish format. To facilitate model curation, a modified version of the SMILEY (14) algorithm (see under “Experimental procedures”) was used to identify missing enzymatic or transport reactions that, if added to the model, would resolve discrepancies between model predictions and experimental growth phenotypes of V. fischeri ES114 wild type and mutants. This analysis identified that V. fischeri was missing an annotated aspartate 1-decarboxylase (encoded by panD in E. coli), which caused the draft model to predict no growth either in LBS or in a V. fischeri defined minimal medium (DMM) (Fig. 2a). The growth-phenotyping experiments were performed to identify sole carbon sources that support growth of V. fischeri (see under “Experimental procedures”). These results (supplemental Excel file S2) were compared with model-predicted sole carbon sources using flux-balance analysis (FBA) (27), and discrepancies were found for mannitol and N-acetylneuraminate. The modified SMILEY algorithm predicted that mannitol 1-phosphate 5-dehydrogenase (encoded by mtlD in E. coli) and an N-acetylneuraminate transporter (encoded by nanT in E. coli) were missing from the draft model (Fig. 2, b and c). Finally, FBA was used to predict essential V. fischeri genes in LBS medium, and gene essentiality predictions were compared with a recent transposon insertion study (supplemental Table S1) (28). One false-positive prediction was for glutamine synthase (VF_0098), where the model predicted the gene was essential but experimentally it was found to be non-essential. Based on this discrepancy, the modified SMILEY algorithm predicted the draft model was missing glutamine transporter(s) (Fig. 2d).
Figure 2.

Pathways missing reactions and genes in V. fischeri. a, aspartate 1-decarboxylase (panD in E. coli), involved in pantothenate and coenzyme A biosynthesis, is missing in V. fischeri. b, mannitol 1-phosphate 5-dehydrogenase (mtlD in E. coli), involved in mannitol catabolism, is missing in V. fischeri. c, transporters for N-acetylneuraminate (nanT in E. coli) (c) and glutamine (glnHPQ in E. coli) (d) are missing in V. fischeri.
To experimentally identify the V. fischeri genes encoding the missing aspartate 1-decarboxylase, and mannitol 1-phosphate 5-dehydrogenase, as well as transporters for N-acetylneuraminate and glutamine, a specific E. coli recipient strain and selective medium were designed for each missing metabolic function (supplemental Table S2). The iJO1366 metabolic model was used to demonstrate in silico that the growth of each E. coli recipient strain requires the specified missing metabolic function in selective medium (Fig. 3, a–d). Here, recipient E. coli strains were used because their metabolism is well characterized, and knock-out mutant collections are available (29, 30). For clarity, we will refer to the well-characterized E. coli genes using their gene symbols and V. fischeri genes using their locus tags. The locus tags of the E. coli genes are listed in supplemental Table S2. A 53-gigabase pair V. fischeri genomic library with ∼12,000-fold genome coverage was created and transformed into the recipient strains: ΔpanD, ΔmtlD, ΔnanT, and ΔglnP. After growth selection in each recipient strain's selection media, we found that plasmids expressing VF_0892, VF_A0062, and VF_0668 rescued ΔpanD, ΔmtlD, and ΔglnP, respectively, and plasmids independently expressing either VF_0924 or VF_1172 rescued ΔglnP.
Figure 3.

Growth coupling of a recipient strain to a missing metabolic function in selective medium. Growth dependence for each recipient strain in selective medium was calculated using iJO1366 (26). Feasible combinations of growth rate and a missing metabolic enzyme are shown in blue. In all cases, cell growth is not zero only when there is flux through the reaction on the y axis (because no non-trivial solutions exist on the x axis). The flux limits of oxygen and carbon uptake rates were set at 10 mmol/g dry weight (gDW) per h. a, aspartate 1-decarboxylase activity (associated with panD in iJO1366) is coupled to growth of a ΔpanD mutant in glucose minimal medium. b, mannitol 1-phosphate 5-dehydrogenase activity (associated with mtlD in iJO1366) is coupled to growth of a ΔmtlD mutant in mannitol minimal medium. c, N-acetylneuraminate transport (ACNAMt2pp associated with nanT in iJO1366) is coupled to growth of a ΔnanT mutant in N-acetylneuraminate minimal medium. d, glutamine transport (GLNabcpp associated with glnHPQ in iJO1366) is coupled to growth in a double knock-out ΔglnPΔansB in glutamine minimal medium. The ansB was not deleted experimentally because it has a low activity with glutamine (66), and ΔglnP mutants (67, 68) were previously shown to be unable to grow on glutamine.
Functional complementation in recipient strains
We individually cloned VF_0892, VF_A0062, VF_0668, VF_0924, and VF_1172 into an empty vector because plasmids in the genomic library can contain fragments that encode more than one gene. All transformed single-gene plasmids enabled growth of recipient strains in the corresponding selective medium (Fig. 4, a–d). This result demonstrated that these genes complemented recipient-strain growth and thus are functionally equivalent to the analogous E. coli genes. In the experiments with the glutamine transporter, glutamine was supplied as the sole carbon source. Wild-type E. coli grows poorly on glutamine as a sole carbon source due to low uptake rates (31). Overexpression of either VF_0924 or VF_1172 in the E. coli ΔglnP resulted in a faster growth rate compared with the wild-type E. coli strain expressing an empty-vector control (Fig. 4d).
Figure 4.
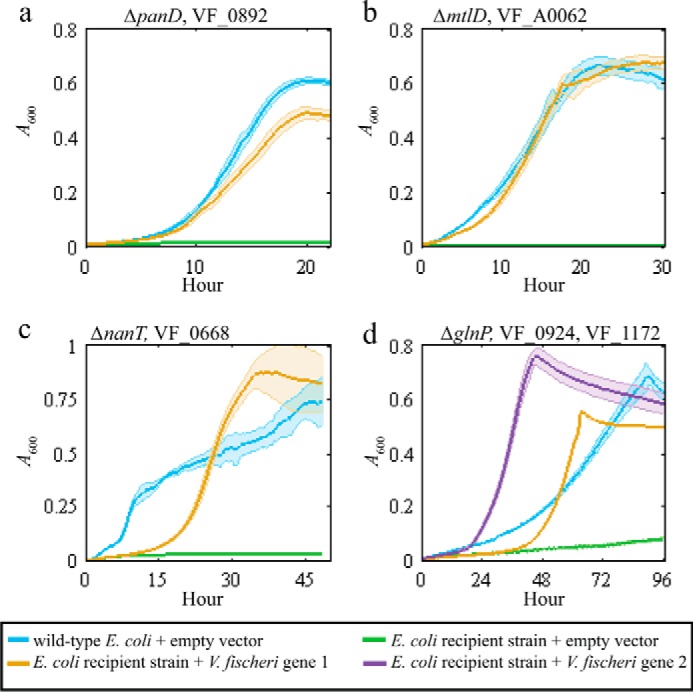
Plasmids expressing the MEGS-discovered V. fischeri genes enabled growth of recipient strains. Each panel shows the growth curves for the recipient strain listed at the top of each panel. Each solid line represents A600 over time, and the associated colored transparent shade indicates the range of standard deviations across biological replicates (n = 3). The selective medium used for each panel was a MOPS minimal medium supplemented with 20 mm glucose (a), 20 mm mannitol (b), 20 mm N-acetylneuraminate (c), and 20 mm glutamine and vitamin supplements (0.05 mm thiamine, 0.05 mm niacinamide, and 20 nm biotin) (d). Blue lines represent growth of the parent BW25113 with an empty pZE21MCS vector. Green lines represent growth of the listed recipient strains (ΔpanD, ΔmtlD, ΔnanT, or ΔglnP) with an empty pZE21MCS vector. Yellow and purple lines represent growth of recipient strains that contain pZE21MCS plasmids expressing the listed V. fischeri gene. d, yellow line represents ΔglnP + pZEVF0924 strain, and the purple line represents ΔglnP + pZEVF1172.
Functional complementation in the organism of interest, V. fischeri
The ΔVF_0892, ΔVF_A0062, or ΔVF_0668 V. fischeri knock-out mutants did not grow in minimal medium (similarly to the selective medium, where DMM instead of MOPS minimal medium was used); however, the V. fischeri knock-out mutants were complemented by plasmids expressing wild-type copies of VF_0892, VF_A0062, or VF_0668, respectively (Fig. 5, a–c). In addition, the ΔVF_0892 mutant could grow in glucose minimal media if supplemented with pantothenate and/or β-alanine (supplemental Fig. S1). Better growth of this mutant was observed with addition of 10 mm pantothenate compared with 10 mm β-alanine, possibly due to a slower β-alanine uptake. However, the transporter(s) in V. fischeri for β-alanine and pantothenate are, as yet, unknown. The ΔVF_0924ΔVF_1172 double mutant still grew in DMM supplemented with glutamine as the sole carbon source, indicating the existence of other V. fischeri glutamine transporters in the genome. We tested whether VF_1172 (annotated as a tyrosine-specific transporter) can also transport leucine, by evaluating growth of E. coli BW25113 with a plasmid overexpressing VF_1172 in minimal medium supplemented with glucose and leucine (supplemental Fig. S2). Overexpression of VF_1172 slowed growth in media supplemented with leucine, a phenotype that can be attributed to leucine toxicity that was previously observed in an E. coli K-12 strain overexpressing branched-chain amino acid transporters (32). Like VF_1172, other amino acid transporters of V. fischeri might have broad substrate specificity.
Figure 5.

Plasmids expressing V. fischeri genes complemented growth of V. fischeri knock-out mutants. Each panel shows the growth curves for a V. fischeri mutant complemented with an empty vector or vector expressing the deleted gene. The gene listed at the top of each panel indicates the gene deleted in the mutant and expressed in the complementation plasmid. Each solid line represents A600 over time, and the associated colored transparent shade indicates the range of standard deviations across biological replicates (n = 3). The medium used in each panel was the DMM minimal medium supplemented with (a) 20 mm glucose, (b) 20 mm mannitol, or (c) 20 mm N-acetylneuraminate. Blue lines represent growth of V. fischeri ES114 with an empty pVSV105 vector. Green lines represent growth of the V. fischeri knock-out mutants (ΔVF_0892, ΔVF_A0062, and ΔVF_0668) with an empty pVSV105 vector. Yellow lines represent growth of V. fischeri knock-out mutants carrying a pVSV105 plasmid expressing the corresponding V. fischeri gene (e.g. ΔVF_0892 + pVF0892).
Detection of β-alanine using in vitro enzyme assays
To further confirm the enzymatic function of VF_0892, we purified a His6-tagged VF_0892 protein and tested its functionality as an aspartate 1-decarboxylase in vitro. Only the reaction condition that contained both aspartate and the VF_0892 enzyme was able to produce β-alanine from aspartate (12.7 ± 0.8 nmol, where reported error is the standard deviation across three biological replicates). In contrast, conditions containing aspartate alone, VF_0892 enzyme alone, aspartate and heat-inactivated VF_0892 enzyme, or aspartate and proteins purified from cells containing the empty vector did not produce any detectable β-alanine (less than 0.125 nmol). Proteins purified from cells containing the empty vector were used as a control to provide information of contaminant proteins (supplemental Fig. S3a). These in vitro enzyme assays further confirmed that VF_0892 (1644 bp) can catalyze the conversion of aspartate to β-alanine and thus is functionally equivalent to panD (381 bp) despite their sequence dissimilarity (i.e. BLAST found no significant similarity between the two proteins).
Kinetic characterization of PanD, VF_0892, and VF_1064 enzymes
VF_0892 is currently annotated in NCBI as a glutamate decarboxylase (EC 4.1.1.15). To compare the activity and substrate specificity of VF_0892 and E. coli PanD toward the two potential substrates (aspartate and glutamate), decarboxylase activities (supplemental Fig. S3b) were evaluated using the DC-PEPC-MDH-linked assays at pH 8.05. Here, the kinetic parameters were determined from three technical replicates and are reported as an average ± S.E. The kcat and Km values of the VF_0892 enzyme were 0.075 ± 0.04 μm CO2/μm enzyme-sec and 1.44 ± 0.35 mm, respectively, at 28 °C, and 0.008 ± 0.001 μm CO2/μm enzyme-sec and 1.70 ± 0.56 mm, respectively, at 37 °C (Fig. 6a). The VF_0892 enzyme was around 10-fold more active at 28 °C compared with 37 °C. Precipitation of purified VF_0892 enzyme was observed over time at 37 °C. Greater activity at 28 °C is consistent with the optimal growth of V. fischeri at 28 °C and its intolerance to higher temperatures. E. coli PanD showed a kcat of 0.008 ± 0.001 μm CO2/μm enzyme-sec and Km of 1.44 ± 0.33 mm at 37 °C (Fig. 6b). However, it is possible that not all of the purified PanD was post-translationally processed into its active form, resulting in a low kcat. Both PanD and the VF_0892 enzyme showed a much higher reaction rate when aspartate, rather than glutamate, was used as the substrate (PanD, 10-fold; VF_0892, 5-fold at 37 °C, and 26-fold at 28 °C, Fig. 6c).
Figure 6.
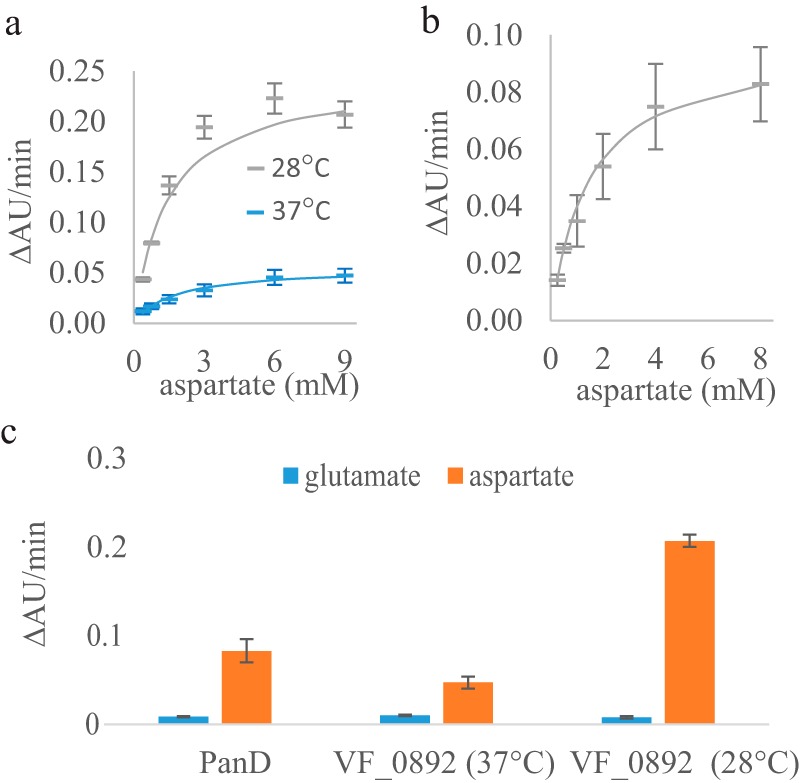
Kinetic characterization and substrate specificity of the E. coli PanD and the VF_0892 (PanP) enzymes. Average reaction rates (ΔAU units/min) are shown for three technical replicates of the DC-PEPC-MDH-linked assays performed at the specified aspartate or glutamate concentration with standard deviations shown as the error bars. a, reactions rates with the VF_0892 enzyme at various concentrations of aspartate at either 28 °C (gray) or 37 °C (blue). Solid lines show the nonlinear fitting to the Michaelis-Menten equation using KaleidaGraph. b, reaction rates of PanD at various concentrations of aspartate at 37 °C. c, reaction rates of PanD with a concentration of 8 mm of either glutamate or aspartate and reaction rates of the VF_0892 enzyme with a concentration of 9 mm of either glutamate or aspartate at either 28 or 37 °C.
In addition to VF_0892, another V. fischeri gene (VF_1064) is currently annotated as a glutamate decarboxylase. Based on BLASTP, VF_1064 has 21% identity to VF_0892 with an E-value of 0.38 but 58% identity and an E-value of 0.0 to the E. coli glutamate decarboxylase gadB. We evaluated experimentally whether VF_1064 decarboxylates glutamate and/or aspartate. In the DC-PEPC-MDH-linked assays, the purified VF_1064 enzyme (supplemental Fig. S3b) showed about a 20-fold higher reaction rate with 10 mm glutamate than with 10 mm aspartate (supplemental Fig. S4a). With glutamate at pH 8.05 and 37 °C, VF_1064 exhibited a kcat of 0.055 ± 0.037 μm CO2/μm enzyme-sec and Km of 61.7 ± 46.4 mm (supplemental Fig. S4b). Because of the solubility of glutamate, we were not able to test the enzyme kinetics of VF_1064 at glutamate concentrations at or greater than its Km value, resulting in large standard errors for kcat and Km. A pH of 8.05 was used to keep CO2 produced by the decarboxylase primarily as HCO3−, but this pH might not be optimal for VF_1064 because E. coli GadB is most active at pH 3.8 (33). Additional experimental evidence suggests that VF_1064 cannot function as an aspartate 1-decarboxylase; specifically, only VF_0892, and not VF_1064, is essential in LBS medium (28). Also, the single gene plasmid expressing VF_1064 did not complement the ΔpanD E. coli mutant in the selective medium, whereas the plasmid expressing VF_0892 did (supplemental Fig. S5 and Fig. 4a).
Based on the measured substrate preferences for the VF_0892 and VF_1064 enzymes, we conclude that VF_0892 is associated with the aspartate 1-decarboxylase reaction, whereas VF_1064 is only associated with the glutamate decarboxylase reaction in the V. fischeri model.
Growth and VF_A0062 expression in wild-type V. fischeri using sole carbon sources
Using MEGS, we identified VF_A0062, a gene currently annotated as dehydrogenase, as a mannitol 1-phosphate 5-dehydrogenase. Here, we provide more evidence that VF_A0062 is involved in mannitol and not l-sorbose utilization. VF_A0062 has significant similarity to genes annotated as l-sorbose 1-phosphate reductases in other Vibrio species, including Vibrio rotiferianus and Vibrio owensii (BLASTP E-value = 0, amino acid identity >88%). In addition to the high-throughput growth-phenotyping assays, we confirmed in shake flasks that l-sorbose does not support growth of wild-type V. fischeri as a sole carbon source, although mannitol and glucose do (supplemental Fig. S6). qPCR analysis showed a 6.5-fold increase in expression of VF_A0062 in the mannitol minimal medium compared with the glucose minimal medium. (A relative expression ratio of 6.5 ± 2.3 was calculated from three biological replicates. The reported error is the standard deviation). In some other Vibrio species, e.g. Vibrio cholerae and Vibrio metoecus, which do not contain a gene similar to VF_A0062, an ortholog of mtlD is present in their genomes instead. Therefore, Vibrio species apparently contain a mannitol 1-phosphate 5-dehydrogenase that is similar to either mtlD in E. coli or VF_A0062 in V. fischeri.
Squid light-organ colonization phenotypes
We further tested whether V. fischeri knock-out mutants (ΔVF_0892, ΔVF_A0062, ΔVF_0668, and ΔVF_0924ΔVF_1172) could successfully colonize the squid light organ in competition with a wild-type strain (we excluded VF_0892 due to its inability to grow in LBS medium without supplementation of β-alanine or pantothenate). We observed no significant colonization phenotypes in our knock-out mutants during the initial stage of colonization (24 h after inoculation) (supplemental Fig. S7). However, VF_A0062 and VF_0668 might play a role in persistence after colonization because transposon insertions in either VF_A0062 or VF_0668 failed to persist in the squid when competing with a pool containing other transposon library mutants and the wild type (after 48 h of colonization) (28).
Discussion
In this work, we developed MEGS to improve gene annotations by combining metabolic modeling with genomic functional selection. Computational models, built from an existing genome annotation, were used to identify missing or incorrect annotations in the current genome annotation and to design selections (using other organisms) for genes responsible for these missing functions. We successfully identified five genes responsible for four metabolic functions that were missing from our draft V. fischeri metabolic model. Using MEGS, we provided the first experimental evidence that (i) VF_0892 functions as an aspartate 1-decarboxylase; (ii) VF_A0062 functions as a mannitol 1-phosphate 5-dehydrogenase; (iii) VF_0668 functions as an N-acetylneuraminate transporter, and (iv) VF_0924 and VF_1172 function as glutamine transporters. Importantly, none of these genes are orthologous to the E. coli genes with the same functions. These discoveries improved the quality of the V. fischeri genome-scale metabolic model, iVF846, which has been used in studying V. fischeri metabolism and its symbiotic relationship in the squid light organ, especially during the habitat transition between seawater and the symbiotic niche (34).
MEGS leverages both computational and experimental techniques to provide functional annotations for genes encoding enzymes and transporters. Metabolic genes are responsible for the physiological and biochemical states of a cell, and knowledge of their functions is critical for understanding and controlling cell behavior. By taking advantage of metabolic modeling, MEGS identifies errors and omissions in existing genome annotations due to either a lack of experimental evidence or prior knowledge in databases and designs experiments to correct these errors. Because MEGS uses genomic functional selections to find genes instead of sequence similarity or genomic context, it can discover genes with unique sequences and/or genes that have not been studied in the laboratory. MEGS is experimentally and computationally inexpensive and efficient. A draft genome-scale metabolic model can be prepared automatically using available software platforms in only a few hours (17, 35–37). Metabolic reactions missing from such a draft model or associated with the wrong genes, as well as genomic functional-selection strategies, can also be identified within a few hours (23). The genomic library used in our method (which takes 2 days to construct) contains information from across the entire genome, so that all the genes in the library go through the selection simultaneously. Once a library is created, MEGS cycles can be repeated to search for additional genes associated with different missing reactions. Similarly, once a recipient strain is constructed, it can be used to select for genes responsible for a particular metabolic reaction from multiple genomes (using separate genomic libraries). The time required to build the recipient strains depends on the number of gene additions and deletions needed; however, this time can be significantly reduced by using existing mutant strain collections (29, 38, 39). Functional selection from the genomic library via growth complementation of the recipient strain in a selective medium is fast (1–3 days) and also provides direct experimental evidence of gene functions. One cycle of the entire MEGS process can be completed within a week using existing recipient strain collections.
MEGS offers an alternative and complementary approach to bioinformatics-based methods for predicting gene functions. In retrospect, some of the genes we identified using MEGS are also top-scoring candidates that have been or could be derived bioinformatically using various genomic context-based methods. VF_0892 was already tentatively suggested to be a member of the pyridoxal-dependent aspartate 1-decarboxylases (TIGR03799) by partial phylogenetic profiling (29). This protein family was given a suggested name of PanP, to distinguish it from a non-orthologous family of aspartate 1-decarboxylase (PanD TIGR00223), which is pyruvoyl-dependent and more widely distributed than PanP. PanP is present in a number of marine bacteria (a list of all proteins that belong to TIGR03799 are listed in supplemental Table S3); however, no direct experimental evidence was available previously to support its annotation as an aspartate 1-decarboxylase. We found that PanP was not properly incorporated in six manually curated genome-scale metabolic models. Five of the models included an aspartate 1-decarboxylase reaction without any associated genes (40, 41) and one associated PanP with l-cysteate, 3-sulfino-l-alanine, glutamate, and aspartate decarboxylase reactions (42). Similarly, VF_A0062 could have been predicted as a mannitol 1-phosphate 5-dehydrogenase candidate using bioinformatics methods because it is located in the same operon as yggD (VF_A0063), and YggD has been characterized as a mannitol operon repressor in Shigella flexneri (34). VF_0668 is a predicted member of SiaR regulon (controlling sialic acid degradation) according to the RegPrecise database (43), and it was tentatively annotated as a possible sialic acid transporter (permease), NanT. The SiaR regulon in Haemophilus influenzae includes a different tripartite ATP-independent periplasmic transporter (44). However, the function of the V. fischeri NanT has not yet been experimentally characterized.
Similar to operon- and regulon-based bioinformatics methods, MEGS does not depend on sequence similarity to well-characterized genes. Annotations based solely on sequence similarity may lack detailed functions when the unknown sequence is not similar to a characterized gene (e.g. conserved hypothetical proteins) or may be incorrect if the gene is similar to a gene that is incorrectly annotated in sequence databases. In the case of VF_A0062, we demonstrated that this gene actually encodes a mannitol 1-phosphate 5-dehydrogenase even though it shares high sequence similarity with other annotated l-sorbose 1-phosphate reductase genes.
MEGS and bioinformatics-based approaches have different strengths and limitations and, as a result, can complement each other. For example, MEGS can complement bioinformatics-based approaches when top-scoring candidates do not exist. Sometimes correlations between genes with unknown function and genes with known functions may not exist, and such correlations can still lead to erroneous and/or nonspecific function assignments using bioinformatics methods alone (8). Another strength of MEGS compared with the bioinformatics-based approach is that genes identified from MEGS already have direct experimental evidence for their functions from the genomic-library selection experiments, while bioinformatics-derived gene functions must be tested in subsequent experiments. MEGS can be applied to organisms for which there are currently no genetic tools, opening up ways to evaluate their gene functions experimentally in another host. Some limitations of MEGS include the following: (i) it requires heterologous expression in the recipient strain, which might not be optimal; (ii) the recipient strains might be difficult to construct (e.g. essential genes cannot be deleted unless growth can be complemented by nutritional supplementation); and (iii) if multiple genes are responsible for a missing metabolic function, they must be co-localized on the chromosome. Additionally, MEGS might identify genes with low promiscuous activities that, when overexpressed, can complement growth defects associated with essential metabolic functions. Recent approaches for improving heterologous gene expression (45) and conditional mutation systems (46) are likely to help overcome some of these limitations. Ultimately, more detailed biochemical characterization of enzymes identified using bioinformatics or MEGS might be needed to confirm the physiological functions of gene products. However, these computational and experimental approaches are useful for identifying which genes to evaluate biochemically.
As more genome sequences become available, we speculate that MEGS will be successfully applied to further discover the roles of uncharacterized genes and improve our understanding of metabolism in a variety of both familiar and uncharacterized microorganisms. Newly discovered gene functions will propagate through genome databases as they are used by other existing approaches to improve annotations of genes in additional organisms.
Experimental procedures
In silico modeling
A newly constructed genome-scale metabolic model of V. fischeri strain ES114, iVF846, was used in this work (supplemental Excel file S1). Reactions and metabolites from an E. coli model, iJO1366 (26), were transferred into the draft model of iVF846 when orthologs to E. coli metabolic genes were found in V. fischeri. By our definition, the orthologous genes shared the KEGG ortholog identifier and were best reciprocal hits in the KEGG Sequence Similarity Database. The model contains 846 genes and 1583 reactions. FBA (27) was used to calculate the growth rate of V. fischeri by maximizing flux through a defined biomass objective function. In FBA, the minimal or rich medium was simulated by giving negative values to the lower limits for the exchange fluxes of the medium components in the model (supplemental Table S4 lists negative exchanges used for different simulations). A carbon source was predicted to be a sole carbon source if the FBA-predicted growth rate was greater than zero in minimal medium supplemented with this carbon source. To simulate a knock-out strain, the fluxes of metabolic reactions associated with the gene were fixed at zero, unless isozymes were present. A gene was predicted as essential if the FBA-predicted growth rate of the strain containing a single gene deletion was zero. A modified version of the mixed integer linear programming algorithm, SMILEY (14), was used to predict missing metabolic genes (and their associated reactions) when the model incorrectly predicted wild-type or mutant V. fischeri strains cannot grow in a particular medium. The algorithm was modified from its original implementation by minimizing the total number of metabolic genes (instead of reactions) that need to be added from iJO1366 (26) (instead of KEGG) to the V. fischeri model to enable growth and reconcile false predictions.
Strain construction
Wild-type E. coli BW25113 and wild-type V. fischeri ES114 were used in this work. E. coli knock-out strains (derived from BW25113) containing kanamycin resistance genes were obtained from the Keio collection (Open Biosystems) (29, 30). The temperature-sensitive plasmid pCP20 was used to remove the kan gene from the mutants as described previously (47). The resulting kanamycin-sensitive E. coli knock-out strains were used as recipient strains for the V. fischeri genomic library. Knock-out mutants of V. fischeri ES114 were constructed using conjugation and homologous recombination as described previously (48–51). To construct ΔVF_0892, 10 mm pantothenate and 10 mm β-alanine were supplemented in the LBS growth medium. All strains used in this study are reported in supplemental Table S5.
Plasmid construction for single-gene complementation
For E. coli complementation experiments, a single V. fischeri gene was cloned into the multiple cloning site of pZE21MCS (EXPRESSYS) using Gibson cloning. The construct was transformed into the corresponding E. coli knock-out strains, and colonies were selected on LB agar containing 50 μg of kanamycin per ml. For V. fischeri complementation experiments, a single V. fischeri gene was cloned into the multiple cloning site of pVSV105 (52) using Gibson cloning. These plasmids containing a V. fischeri gene were introduced into V. fischeri ES114 knock-out strains by conjugation. All plasmids used in this work are listed in supplemental Table S5.
Growth conditions and complementation experiments
Unless otherwise noted, E. coli strains were grown at 37 °C, and V. fischeri strains were grown at 28 °C, both with shaking at 225 rpm. E. coli strains were grown in Luria-Bertani (LB) or a MOPS-buffered minimal medium (53). Because E. coli grows poorly on glutamine as a sole carbon source, vitamin supplements (0.05 mm thiamine, 0.05 mm niacinamide, and 20 nm biotin) were added to the minimal medium to shorten the experiments in which the glutamine transporter is complemented. V. fischeri strains were grown in Luria-Bertani salt (LBS) (24) or V. fischeri DMM (supplemental Table S6). Overnight cultures of ΔVF_0892 were grown in LBS supplemented with 10 mm pantothenate and 10 mm β-alanine. When appropriate, 50 μg of kanamycin or 5 μg of chloramphenicol per ml was added to the media. For complementation experiments, an overnight LB (or LBS) culture of each strain was subcultured by ∼1:100 dilution into fresh minimal medium with a starting absorbance of 0.02 at 600 nm (A600). The A600 of the culture in a 96-well plate was measured by an Infinite M200 plate reader (Tecan) every 15 min with 3-mm orbital shaking. In complementation experiments with a VF_0892 plasmid in ΔpanD or ΔVF_0892, an overnight LB (or LBS) culture of each strain was washed twice in minimal medium and subcultured by 1:100 dilution into fresh minimal medium. Once the subculture grew to about mid-exponential phase, it was washed twice and subcultured again into fresh minimal medium for the growth curve measurements. All experiments testing a VF_A0062 plasmid were performed at 28 °C, because pZEVFA0062 did not complement ΔmtlD well when grown at 37 °C.
Growth-phenotyping experiments
To test sole carbon sources of V. fischeri, V. fischeri strain ES114 was grown in inoculating fluid supplemented with 255 mm sodium chloride, on PM1 or PM2A plates containing single carbon sources (Biolog) according to the manufacturer's protocol. The plates were incubated at 28 °C in an OmniLog incubator reader (Biolog), and turbidity was measured every 15 min for 48 h. The increase in turbidity was compared with a negative control with no added carbon source to determine whether cells grew. Additional experiments were performed in 17 × 100-mm test tubes to confirm growth of strains on a carbon source of interest or to detect strains with an intermediate increase in turbidity.
Genomic library construction
The genomic DNA of V. fischeri ES114 strain was extracted from LBS culture using the DNeasy Blood and Tissue kit (Qiagen). The extracted DNA was then fragmented at 10% amplitude for 5 s using the Sonic Dismembrator Model 500 (Thermo Fisher Scientific). DNA fragments between ∼2 and ∼5 kbp were size-selected from a 1% agarose gel in Tris acetate/EDTA buffer and purified with the Zymoclean Gel DNA Recovery kit (Zymo). The DNA fragments were ligated into the HinCII site of the multiple cloning site of the pZE21MCS1 vector and transformed into 50 μl of E. coli MegaX suspension (Invitrogen) following the protocol of Forsberg et al. (22). The average insert size of the library was ∼2.3 kilobase pairs, and the titer of the library was 22 million colony forming units (CFUs). Transformed cells were transferred to 10 ml of LB containing 50 μg of kanamycin per ml and grown overnight. The overnight culture was used to extract the library of plasmids using the QIAprep Spin Miniprep kit (Qiagen).
Gene selection from a V. fischeri genomic library
The V. fischeri genomic library was transformed into competent cells of an E. coli recipient strain and recovered in 1 ml of Super Optimal broth with Catabolite repression (SOC) medium for an hour. The recovered cells were then pelleted at 6000 rpm for 3 min and transferred to 50 ml of selective medium (listed in supplemental Table S2) with 50 μg of kanamycin per ml to grow at 37 °C. After the A600 reached 1, the cells were subcultured into 50 ml of fresh selection medium. After the A600 of the cells reached 1 again, they were plated on LB plus kanamycin plates. Single colonies were picked to confirm growth in selective medium, and the first and last 700 bp of the plasmid inserts were subsequently sequenced to identify the V. fischeri gene(s) included in the plasmid.
Expression and purification of decarboxylases
E. coli panD (b0131), V. fischeri panP (VF_0892), and V. fischeri gadA (VF_1064) were amplified from the genomic DNA using Phusion High-fidelity DNA Polymerase (New England Biolabs). The PCR fragments containing VF_0892 were digested with NcoI and XhoI and cloned into pET28 (Novagen). The resulting plasmid pETVF0892 was transformed into E. coli X90(DE3) competent cells (54). The PCR fragments containing b0131 or VF_1064 were inserted into the pET28 vector with an N-terminal His-tag sequence followed by a tobacco etch virus site. The resulting plasmids pETb0131 and pETVF1064 were transformed into E. coli BL21 (DE3) competent cells. For expression, an inoculum was started in LB medium plus 50 μg of kanamycin per ml from an overnight culture. The cells were grown at 28 °C until reaching an A600 of 0.6. Then cells were induced by 1 mm isopropyl β-d-1-thiogalactopyranoside overnight at 18 °C. The cell lysate was extracted from the collected cells by BugBuster Master Mix (EMD Millipore) following the manufacturer's protocol. The lysate was incubated with HisPur nickel-nitrilotriacetic acid resin (Thermo Fisher Scientific) for 1 h at 4 °C and passed through a Pierce Centrifuge Column (Thermo Fisher Scientific). The column was washed in 50 mm NaH2PO4, 300 mm NaCl, and 20 mm imidazole (pH 8), and the VF_0892 protein was eluted in 50 mm NaH2PO4, 300 mm NaCl, and 250 mm imidazole (pH 8). The eluted protein was dialyzed against 100 mm Tris-HCl (pH 7.5) and concentrated with an Amicon Ultra Centrifugal Filter Unit (EMD Millipore). The final products were analyzed with SDS-polyacrylamide gels, and their concentrations were determined with a bicinchoninic acid kit (Sigma). The eluted protein was dialyzed in 20 mm Tris-HCl to reduce the amount of solutes that went through the GC-MS column. E. coli X90(DE3) containing the empty pET28b(+) vector was subjected to the same expression and purification procedures, and the product was used as a control in detecting V. fischeri PanP activity using GC-MS.
Detection of β-alanine from in vitro enzyme assays
The V. fischeri PanP activity was detected by measuring the formation of β-alanine in five different conditions, each with a 200-μl reaction volume. All reaction mixtures contained 20 mm Tris-HCl, 5 mm MgSO4, and 750 μm pyridoxal 5′-phosphate (pH 7.5). In addition to the buffer components, the first condition also contained 36 μg of purified V. fischeri PanP and 50 nmol of aspartate as the substrate. The second condition contained 36 μg of purified V. fischeri PanP but no substrate. The third condition contained 50 nmol of aspartate but no protein. The fourth condition contained 36 μg of purified V. fischeri PanP that had been heat-inactivated at 90 °C for 30 min and 50 nmol of aspartate. The last condition contained 20 μg of protein obtained from the empty vector lysate and 50 nmol of aspartate. The reaction mixtures were incubated at 37 °C for 15 h and then stopped by heat inactivation at 90 °C for 30 min. The second and the fourth conditions were tested in biological duplicates, and the other conditions were tested in biological triplicates. The heat-inactivated reaction mixtures were spun down at 15,000 rpm for 3 min, and their supernatants were taken for quantitation. A uniformly labeled, β-[U-13C]alanine internal standard (Cambridge Isotope Laboratories) was used for quantification via an isotope-ratio method using GC-MS (55). Prior to sample quantification, a suitable β-alanine fragment formula was identified. This process includes the following: (i) analyzing the mass spectrum of an unlabeled β-alanine sample to propose a feasible structure for the molecular ion of interest; (ii) comparing the theoretical and measured isotopic distributions of said structure; and (iii) verifying that the number of carbon backbone atoms present on the derivatized structure matches the base peak mass shift predicted to be seen in the β-[U-13C]alanine mass spectrum relative to the unlabeled spectrum (56). Once a fragment for quantification was identified, the purity (i.e. extent of labeling) of labeled β-alanine was measured and subsequently used to correct for the unlabeled portion of the labeled standard in downstream calculations. Following this step, aliquots of labeled internal standard were quantified using a known amount of unlabeled standard and calculating the ratio of 12C/13C after correcting both for natural abundances of other isotopes present in the derivatized fragment, and for the unlabeled portion of the labeled standard, using the freely available software, IsoCor (57). Once characterized, known amounts of labeled standard were mixed with the supernatant of the enzymatic assay and dried at 90 °C. This step was followed by derivatization in a volumetric 1:1 ratio of pyridine with N-tert-butyl-dimethylsilyl-N-methyltrifluoroacetamide plus 1% tert-butyl-dimethylchlorosilane at 90 °C for 30 min to confer thermal stability and increased volatility amenable for analysis on the GC-MS instrument. Derivatized samples were centrifuged at 15,000 rpm for 3 min to sediment insoluble material, producing a cleaner supernatant for injection onto the GC-MS. Samples were run on a single quadrupole GC-MS QP2010S (Shimadzu) in electron ionization mode with an Rtx of 5 ms (Restek) low-bleed, fused-silica column for separation with helium as a carrier gas operating under a linear velocity control mode with a split ratio of 0.50, and a column flow of 1.50 ml/min. The temperature program for separation of β-alanine began with holding the column oven temperature at 35 °C for 10 min, ramping up at 25 °C/min to 300 °C, and holding for 19.4 min. Operational parameters included an injection temperature of 240 °C, ion source temperature of 260 °C, interface temperature of 240 °C, and a mass scan range of 100–450 m/z. To test the detection limit of our β-alanine quantification method, the method was used to measure a known amount of unlabeled β-alanine. Each sample (n = 3) contained 0.125, 0.25, 0.5, 1, or 2 nmol of unlabeled β-alanine. The method was able to detect the presence of unlabeled β-alanine in all these samples. Therefore, the detection limit of the method is below 0.125 nmol.
DC-PEPC-MDH-linked assays
Decarboxylase (DC), phosphoenolpyruvate carboxylase (PEPC), and malate dehydrogenase (MDH)-linked assays were performed by mixing 80 μl of freshly prepared mixture A and 120 μl of mixture B. Mixture A contained 100 mm Tris-HCl, 10 mm MgSO4, 1 mm pyridoxal 5′-phosphate (PLP), and various concentrations of aspartate and glutamate (pH 8.05). Mixture B contained purified decarboxylase and Infinity Carbon Dioxide Liquid Stable Reagent (Infinity, Thermo Fisher Scientific). The final decarboxylase concentrations used were 54 μm PanD, 35 μm VF_1064 enzyme, 28 μm V. fischeri PanP for assays at 37 °C, and 14 μm V. fischeri PanP for assays at 28 °C. Like similar DC-PEPC-MDH-linked assays (58–62), our assays were carried out in air. The interference of exogenous CO2 from air and buffer solution was accounted for using a negative control, in which no substrate was added. The signal produced by the negative control was linear during the measurement period and was subtracted from all signals produced by other samples containing substrates. Values of Km and kcat were determined from nonlinear fitting into the Michaelis-Menten equation using KaleidaGraph (Synergy Software). Averages across the three technical replicates were used as data points, whereas standard deviations for each data point were used as weights during the curve fitting. The Km and kcat values are reported with standard errors.
qPCR analysis
Wild-type V. fischeri cells were harvested at an A600 of ∼0.6 in DMM supplemented with glucose or mannitol as sole carbon source. Total RNA from the cell pellets was harvested using the Quick-RNA MicroPrep Kit (Zymo Research) with a 15-min on-column DNase treatment. RNA concentration and quality were assessed on a NanoDrop (Thermo Fisher Scientific) spectrophotometer. Reverse transcription for synthesizing cDNA and real-time qPCR were performed using the GoTaq Two-step RT-qPCR System (Promega) and an AriaMx real-time PCR machine (Agilent). The differential expression of VF_A0062 (forward primer, TGGATATTCCGGGTGGTAAA, and reverse primer, ACGGGTCTTGTTCTGCAAGT) in the mannitol minimal medium and the glucose minimal medium was normalized to the control gene (V. fischeri polA, VF_0074, forward primer, CGACAGCAGCAGAAGTGAAG, and reverse primer, AGCAAGACCAAACGCACTC) using the GED formula (63).
Growth inhibition test with leucine
An overnight LB culture of each E. coli strain was washed twice in minimal medium and subcultured by 1:100 dilution into fresh MOPS minimal medium with 20 mm glucose. Once the subculture grew to about mid-exponential phase, it was washed twice again and subcultured into fresh minimal medium with or without 4 mm leucine, and its A600 was measured on a 96-well plate by an Infinite M200 plate reader (Tecan).
Squid colonization competitions
Freshly hatched juvenile squid were collected from the rearing facility at the University of Hawaii and placed in filter-sterilized seawater. Squid were exposed to an ∼1:1 mixed population of two strains consisting of V. fischeri ES114 carrying a chromosomally inserted erythromycin-GFP marker (64) and the indicated mutant strain, at a total of 3000–5000 CFU/ml. Squid were incubated with bacteria for 3 h, then transferred to individual vials of V. fischeri-free seawater for an additional 18–21 h. Colonized squid were subsequently anesthetized on ice and placed at −80 °C for surface sterilization. Individual squid were then homogenized and plated on LBS and LBS + erythromycin agar plates as described previously (65), and the ratio of strains present in the light organ was determined by counting the unmarked and erythromycin marked colonies. The relative competitive index (RCI) of the co-colonizing strains was calculated as follows: RCI = log(CFU mutant/CFU wild type)/(inoculum CFU mutant/inoculum CFU wild type).
Author contributions
S. P., E. G. R., and J. L. R. conceived and designed the experiments. S. P., K. N., P. A. A., and M. P. performed the experiments. S. P., K. N., and P. A. A. performed statistical analysis. S. P., K. N., P. A. A., and M. P. analyzed the data. S. P., K. N., P. A. A., M. P., J. L. R., and E. G. R. wrote the paper. All authors read and approved the final manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Kevin J. Forsberg for discussions on preparing genomic libraries, members from the Brian Pfleger group for help with the protein purification, Audrey Haines for help with the leucine toxicity experiments, and Caroline Mitchell for help with editing the manuscript.
This work was supported by the Gordon and Betty Moore Foundation Grant 3396, National Institutes of Health Ruth L. Kirschstein National Research Service Award F32GM112214 from the NIGMS (to K. N.), National Science Foundation Grant CBET 1053712 (to J. L. R.), National Institutes of Health Grant AI50661 from NIAID, and Office of the Director Grant OD011024 (to E. G. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Excel files S1 and S2, Figs. S1–S7, Tables S1–S6, and Refs. 1–11.
- MEGS
- model-enabled gene search
- qPCR
- quantitative PCR
- DMM
- defined minimal medium
- FBA
- flux-balance analysis
- DC
- decarboxylase
- PEPC
- phosphoenolpyruvate carboxylase
- MDH
- malate dehydrogenase
- KEGG
- Kyoto Encyclopedia of Genes and Genomes.
References
- 1. Ye Y., and Godzik A. (2004) FATCAT: a web server for flexible structure comparison and structure similarity searching. Nucleic Acids Res. 32, W582–W585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guda C., Lu S., Scheeff E. D., Bourne P. E., and Shindyalov I. N. (2004) CE-MC: a multiple protein structure alignment server. Nucleic Acids Res. 32, W100–W103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Overbeek R., Fonstein M., D'Souza M., Pusch G. D., and Maltsev N. (1999) The use of gene clusters to infer functional coupling. Proc. Natl. Acad. Sci. U.S.A. 96, 2896–2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Enright A. J., Iliopoulos I., Kyrpides N. C., and Ouzounis C. A. (1999) Protein interaction maps for complete genomes based on gene fusion events. Nature 402, 86–90 [DOI] [PubMed] [Google Scholar]
- 5. Pellegrini M., Marcotte E. M., Thompson M. J., Eisenberg D., and Yeates T. O. (1999) Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc. Natl. Acad. Sci. U.S.A. 96, 4285–4288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Deutschbauer A., Price M. N., Wetmore K. M., Tarjan D. R., Xu Z., Shao W., Leon D., Arkin A. P., and Skerker J. M. (2014) Towards an informative mutant phenotype for every bacterial gene. J. Bacteriol. 196, 3643–3655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Green M. L., and Karp P. D. (2004) A Bayesian method for identifying missing enzymes in predicted metabolic pathway databases. BMC Bioinformatics 5, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Galperin M. Y., and Koonin E. V. (2010) From complete genome sequence to ‘complete’ understanding? Trends Biotechnol. 28, 398–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Golyshev M. A., and Korotkov E. V. (2015) Developing of the computer method for annotation of bacterial genes. Adv. Bioinformatics 2015, 635437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. UniProt Consortium. (2015) UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–D212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schnoes A. M., Brown S. D., Dodevski I., and Babbitt P. C. (2009) Annotation error in public databases: Misannotation of molecular function in enzyme superfamilies. PLoS Comput. Biol. 5, e1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Radivojac P., Clark W. T., Oron T. R., Schnoes A. M., Wittkop T., Sokolov A., Graim K., Funk C., Verspoor K., Ben-Hur A., Pandey G., Yunes J. M., Talwalkar A. S., Repo S., Souza M. L., et al. (2013) A large-scale evaluation of computational protein function prediction. Nat. Methods 10, 221–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Satish Kumar V., Dasika M. S., and Maranas C. D. (2007) Optimization based automated curation of metabolic reconstructions. BMC Bioinformatics 8, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reed J. L., Patel T. R., Chen K. H., Joyce A. R., Applebee M. K., Herring C. D., Bui O. T., Knight E. M., Fong S. S., and Palsson B. O. (2006) Systems approach to refining genome annotation. Proc. Natl. Acad. Sci. U.S.A. 103, 17480–17484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kumar V. S., and Maranas C. D. (2009) GrowMatch: An automated method for reconciling in silico/in vivo growth predictions. PLoS Comput. Biol. 5, e1000308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Green M. L., and Karp P. D. (2007) Using genome-context data to identify specific types of functional associations in pathway/genome databases. Bioinformatics 23, i205–i211 [DOI] [PubMed] [Google Scholar]
- 17. Henry C. S., DeJongh M., Best A. A., Frybarger P. M., Linsay B., and Stevens R. L. (2010) High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotechnol. 28, 977–982 [DOI] [PubMed] [Google Scholar]
- 18. Chen L., and Vitkup D. (2006) Predicting genes for orphan metabolic activities using phylogenetic profiles. Genome Biol. 7, R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vitkin E., and Shlomi T. (2012) MIRAGE: a functional genomics-based approach for metabolic network model reconstruction and its application to cyanobacteria networks. Genome Biol. 13, R111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Varaljay V. A., Satagopan S., North J. A., Witte B., Dourado M. N., Anantharaman K., Arbing M. A., Hoeft McCann S., Oremland R. S., Banfield J. F., Wrighton K. C., and Tabita F. R. (2016) Functional metagenomic selection of ribulose 1, 5-bisphosphate carboxylase/oxygenase from uncultivated bacteria. Environ. Microbiol. 18, 1187–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simon C., Herath J., Rockstroh S., and Daniel R. (2009) Rapid identification of genes encoding DNA polymerases by function-based screening of metagenomic libraries derived from glacial ice. Appl. Environ. Microbiol. 75, 2964–2968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Forsberg K. J., Patel S., Gibson M. K., Lauber C. L., Knight R., Fierer N., and Dantas G. (2014) Bacterial phylogeny structures soil resistomes across habitats. Nature 509, 612–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tervo C. J., and Reed J. L. (2012) FOCAL: an experimental design tool for systematizing metabolic discoveries and model development. Genome Biol. 13, R116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee K. H., and Ruby E. G. (1994) Effect of the squid host on the abundance and distribution of symbiotic Vibrio fischeri in nature. Appl. Environ. Microbiol. 60, 1565–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dunn A. K. (2012) Vibrio fischeri metabolism: symbiosis and beyond. Adv. Microb. Physiol. 61, 37–68 [DOI] [PubMed] [Google Scholar]
- 26. Orth J. D., Conrad T. M., Na J., Lerman J. A., Nam H., Feist A. M., and Palsson B. Ø. (2011) A comprehensive genome-scale reconstruction of Escherichia coli metabolism–2011. Mol. Syst. Biol. 7, 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Orth J. D., Thiele I., and Palsson B. Ø. (2010) What is flux balance analysis? Nat. Biotechnol. 28, 245–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brooks J. F. 2nd., Gyllborg M. C., Cronin D. C., Quillin S. J., Mallama C. A., Foxall R., Whistler C., Goodman A. L., and Mandel M. J. (2014) Global discovery of colonization determinants in the squid symbiont Vibrio fischeri. Proc. Natl. Acad. Sci. U.S.A. 111, 17284–17289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K. A., Tomita M., Wanner B. L., and Mori H. (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006.0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yamamoto N., Nakahigashi K., Nakamichi T., Yoshino M., Takai Y., Touda Y., Furubayashi A., Kinjyo S., Dose H., Hasegawa M., Datsenko K. A., Nakayashiki T., Tomita M., Wanner B. L., and Mori H. (2009) Update on the Keio collection of Escherichia coli single-gene deletion mutants. Mol. Syst. Biol. 5, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Neidhardt F. C. (ed) (1996) Escherichia coli and Salmonella: Cellular and Molecular Biology, 2nd Ed., p. 359, American Society for Microbiology, Washington, D. C. [Google Scholar]
- 32. Quay S. C., Dick T. E., and Oxender D. L. (1977) Role of transport systems in amino acid metabolism: leucine toxicity and the branched-chain amino acid transport systems. J. Bacteriol. 129, 1257–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Capitani G., De Biase D., Aurizi C., Gut H., Bossa F., and Grütter M. G. (2003) Crystal structure and functional analysis of Escherichia coli glutamate decarboxylase. EMBO J. 22, 4027–4037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thompson L. R., Nikolakakis K., Pan S., Reed J., Knight R., and Ruby E. G. (2017) Transcriptional characterization of Vibrio fischeri during colonization of juvenile Euprymna scolopes. Environ. Microbiol. 19, 1845–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Swainston N., Smallbone K., Mendes P., Kell D., and Paton N. (2011) The SuBliMinaL Toolbox: automating steps in the reconstruction of metabolic networks. J. Integr. Bioinform. 8, 186. [DOI] [PubMed] [Google Scholar]
- 36. Agren R., Liu L., Shoaie S., Vongsangnak W., Nookaew I., and Nielsen J. (2013) The RAVEN toolbox and its use for generating a genome-scale metabolic model for Penicillium chrysogenum. PLoS Comput. Biol. 9, e1002980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Karp P. D., Paley S. M., Krummenacker M., Latendresse M., Dale J. M., Lee T. J., Kaipa P., Gilham F., Spaulding A., Popescu L., Altman T., Paulsen I., Keseler I. M., and Caspi R. (2010) Pathway Tools version 13.0: integrated software for pathway/genome informatics and systems biology. Brief. Bioinform. 11, 40–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kobayashi K., Ehrlich S. D., Albertini A., Amati G., Andersen K. K., Arnaud M., Asai K., Ashikaga S., Aymerich S., Bessieres P., Boland F., Brignell S. C., Bron S., Bunai K., Chapuis J., et al. (2003) Essential Bacillus subtilis genes. Proc. Natl. Acad. Sci. U.S.A. 100, 4678–4683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Giaever G., and Nislow C. (2014) The yeast deletion collection: a decade of functional genomics. Genetics 197, 451–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ong W. K., Vu T. T., Lovendahl K. N., Llull J. M., Serres M. H., Romine M. F., and Reed J. L. (2014) Comparisons of Shewanella strains based on genome annotations, modeling, and experiments. BMC Syst. Biol. 8, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fondi M., Maida I., Perrin E., Mellera A., Mocali S., Parrilli E., Tutino M. L., Liò P., and Fani R. (2015) Genome-scale metabolic reconstruction and constraint-based modelling of the Antarctic bacterium Pseudoalteromonas haloplanktis TAC125. Environ. Microbiol. 17, 751–766 [DOI] [PubMed] [Google Scholar]
- 42. Kim H. U., Kim S. Y., Jeong H., Kim T. Y., Kim J. J., Choy H. E., Yi K. Y., Rhee J. H., and Lee S. Y. (2011) Integrative genome-scale metabolic analysis of Vibrio vulnificus for drug targeting and discovery. Mol. Syst. Biol. 7, 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Novichkov P. S., Kazakov A. E., Ravcheev D. A., Leyn S. A., Kovaleva G. Y., Sutormin R. A., Kazanov M. D., Riehl W., Arkin A. P., Dubchak I., and Rodionov D. A. (2013) RegPrecise 3.0–a resource for genome-scale exploration of transcriptional regulation in bacteria. BMC Genomics 14, 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Johnston J. W., Zaleski A., Allen S., Mootz J. M., Armbruster D., Gibson B. W., Apicella M. A., and Munson R. S. (2007) Regulation of sialic acid transport and catabolism in Haemophilus influenzae. Mol. Microbiol. 66, 26–39 [DOI] [PubMed] [Google Scholar]
- 45. Gaida S. M., Sandoval N. R., Nicolaou S. A., Chen Y., Venkataramanan K. P., and Papoutsakis E. T. (2015) Expression of heterologous σ factors enables functional screening of metagenomic and heterologous genomic libraries. Nat. Commun. 6, 7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Herring C. D., and Blattner F. R. (2004) Conditional lethal amber mutations in essential Escherichia coli genes. J. Bacteriol. 186, 2673–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Datsenko K. A., and Wanner B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stabb E. V., and Ruby E. G. (2002) RP4-based plasmids for conjugation between Escherichia coli and members of the Vibrionaceae. Methods Enzymol. 358, 413–426 [DOI] [PubMed] [Google Scholar]
- 49. Le Roux F., Binesse J., Saulnier D., and Mazel D. (2007) Construction of a Vibrio splendidus mutant lacking the metalloprotease gene vsm by use of a novel counterselectable suicide vector. Appl. Environ. Microbiol. 73, 777–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shibata S., and Visick K. L. (2012) Sensor kinase RscS induces the production of antigenically distinct outer membrane vesicles that depend on the symbiosis polysaccharide locus in Vibrio fischeri. J. Bacteriol. 194, 185–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pan M., Schwartzman J. A., Dunn A. K., Lu Z., and Ruby E. G. (2015) A single host-derived glycan impacts key regulatory nodes of symbiont metabolism in a coevolved mutualism. MBio. 6, e00811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dunn A. K., Millikan D. S., Adin D. M., Bose J. L., and Stabb E. V. (2006) New rfp- and pES213-derived tools for analyzing symbiotic Vibrio fischeri reveal patterns of infection and lux expression in situ. Appl. Environ. Microbiol. 72, 802–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Neidhardt F. C., Bloch P. L., and Smith D. F. (1974) Culture medium for enterobacteria. J. Bacteriol. 119, 736–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hayes C. S., Bose B., and Sauer R. T. (2002) Proline residues at the C terminus of nascent chains induce SsrA tagging during translation termination. J. Biol. Chem. 277, 33825–33832 [DOI] [PubMed] [Google Scholar]
- 55. Long C. P., and Antoniewicz M. R. (2014) Quantifying biomass composition by gas chromatography/mass spectrometry. Anal. Chem. 86, 9423–9427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Miranda-Santos I., Gramacho S., Pineiro M., Martinez-Gomez K., Fritz M., Hollemeyer K., Salvador A., and Heinzle E. (2015) Mass isotopomer analysis of nucleosides isolated from RNA and DNA using GC-MS. Anal. Chem. 87, 617–623 [DOI] [PubMed] [Google Scholar]
- 57. Millard P., Letisse F., Sokol S., and Portais J. C. (2012) IsoCor: Correcting MS data in isotope labeling experiments. Bioinformatics 28, 1294–1296 [DOI] [PubMed] [Google Scholar]
- 58. Liu Y. C., Hsu D. H., Huang C. L., Liu Y. L., Liu G. Y., and Hung H. C. (2011) Determinants of the differential antizyme-binding affinity of ornithine decarboxylase. PLoS ONE 6, e26835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hsieh J. Y., Yang J. Y., Lin C. L., Liu G. Y., and Hung H. C. (2011) Minimal antizyme peptide fully functioning in the binding and inhibition of ornithine decarboxylase and antizyme inhibitor. PLoS ONE 6, e24366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Su K. L., Liao Y. F., Hung H. C., and Liu G. Y. (2009) Critical factors determining dimerization of human antizyme inhibitor. J. Biol. Chem. 284, 26768–26777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jackson L. K., Goldsmith E. J., and Phillips M. A. (2003) X-ray structure determination of Trypanosoma brucei ornithine decarboxylase bound to d-ornithine and to G418 insights into substrate binding and odc conformational flexibility. J. Biol. Chem. 278, 22037–22043 [DOI] [PubMed] [Google Scholar]
- 62. Liao C., Wang Y., Tan X., Sun L., and Liu S. (2015) Discovery of novel inhibitors of human S-adenosylmethionine decarboxylase based on in silico high-throughput screening and a non-radioactive enzymatic assay. Sci. Rep. 5, 10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schefe J. H., Lehmann K. E., Buschmann I. R., Unger T., and Funke-Kaiser H. (2006) Quantitative real-time RT-PCR data analysis: current concepts and the novel 'gene expression's CT difference' formula. J. Mol. Med. 84, 901–910 [DOI] [PubMed] [Google Scholar]
- 64. Bongrand C., Koch E. J., Moriano-Gutierrez S., Cordero O. X., McFall-Ngai M., Polz M. F., and Ruby E. G. (2016) A genomic comparison of 13 symbiotic Vibrio fischeri isolates from the perspective of their host source and colonization behavior. ISME J. 10, 2907–2917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mandel M. J., Schaefer A. L., Brennan C. A., Heath-Heckman E. A., Deloney-Marino C. R., McFall-Ngai M. J., and Ruby E. G. (2012) Squid-derived chitin oligosaccharides are a chemotactic signal during colonization by Vibrio fischeri. Appl. Environ. Microbiol. 78, 4620–4626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Derst C., Henseling J., and Röhm K. H. (2000) Engineering the substrate specificity of Escherichia coli asparaginase. II. Selective reduction of glutaminase activity by amino acid replacements at position 248. Protein Sci. 9, 2009–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Masters P. S., and Hong J. S. (1981) Genetics of the glutamine transport system in Escherichia coli. J. Bacteriol. 147, 805–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nohno T., Saito T., and Hong J. (1986) Cloning and complete nucleotide sequence of the Escherichia coli glutamine permease operon (glnHPQ). MGG Mol. Gen. Genet. 205, 260–269 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.