

Abstract
Enterovirus 71 (EV71) has emerged as one of the most important enteroviruses since the eradication of poliovirus, and it causes severe neurological symptoms for which no effective antiviral drugs are available. Type I interferons (IFN) α/β have been used clinically as antiviral therapy as the first line of defense against virus infections successfully for decades. However, treatment with type I interferons has not been effective in patients with EV71 infection. In this study, we found that in cells pretreated with IFN-β, EV71 infection could still lead to a cytopathic effect, and the viral replication was not affected. The mechanism by which EV71 antagonizes interferon signaling, however, has been controversial. Our study indicated that EV71 infection did not inhibit phosphorylation of STAT1/2 induced by IFN-β stimulation, but p-STAT1/2 transport into the nucleus was significantly blocked. We showed that EV71 infection reduced the formation of STAT/karyopherin-α1 (KPNA1) complex upon interferon stimulation and that the virus down-regulated the expression of KPNA1, a nuclear localization signal receptor for p-STAT1. Using specific caspase inhibitors and siRNA for caspase-3, we demonstrated that EV71 infection induced degradation of cellular KPNA1 in a caspase-3-dependent manner, which led to decreased induction of interferon-inducible genes and IFN response. Viral 2A and 3C proteases did not degrade KPNA1, inhibit the activity of ISRE or suppress the transcription of interferon-inducible genes induced by IFN-β. Our study demonstrates a novel mechanism by which antiviral signaling is suppressed through degradation of KPNA1 by activated caspase-3 induced in an enteroviral infection.
Keywords: interferon, Janus kinase (JAK), protein expression, RNA virus, STAT transcription factor
Introduction
Enterovirus 71 (EV71)3 is a single positive-stranded RNA virus belonging to the genus Enterovirus in the family Picornaviridae (1). First isolated in California in 1969, the virus causes largely self-limiting disease in patients, but infection can lead to potentially life-threatening neurological complications ranging from aseptic meningitis to acute flaccid paralysis or brainstem encephalitis, similar to those caused by poliovirus. EV71 circulates endemically each year from summer through fall, causing high morbidity with significant casualties among children. Despite concentrated efforts, specific antiviral therapy is still not available against EV71, which has emerged to be one of the most important current neurotropic pathogens (2, 3).
Type I interferon response is the first line of defense against viral infection through suppression of viral replication and by blocking viral spread, mediated by hundreds of interferon-inducible genes (ISGs). Type I IFNs induce the ISGs through the JAK/STAT signal pathway (4, 5). First, type I interferons (IFN-α/β) bind to heterodimeric receptors consisting of interferon-α receptors 1 (IFNAR1) and 2 (IFNAR2), causing transphosphorylation and activation of the “Janus” tyrosine kinases Tyk2 and Jak1. Then the STATs are recruited to the receptor-bound Jaks and phosphorylated at tyrosine. Phosphorylated STAT1/2 subsequently associates with IFN-regulatory factor 9 (IRF9) to form a heterotrimeric complex, named interferon-stimulated gene factor 3 (ISGF3), which is translocated to the nucleus and binds the IFN-stimulated response elements (ISREs) to activate transcription of ISGs.
Accumulated evidence shows that viral infections can circumvent the IFN response by either suppressing IFN induction or blocking IFN stimulation to induce ISGs (6–9). IFNs have been clinically used in treatment of many viral infections because of their powerful antiviral activities (4), but surprisingly, they have had very limited effect on patients infected with EV71 (10). It has been reported experimentally that only early administration of high dose of IFN-α protected mice against EV71 infection (11). In vitro analysis in Vero cells also showed that conventional type I IFNs had an inhibitory effect on EV71 replication only at very high concentrations (12), which would cause serious side effects if comparable doses of IFNs were applied clinically.
The mechanisms underlying EV71 resistance to IFN treatment are not fully understood, and reports so far have been inconsistent. A previous study reported that EV71 blocked type I IFN signaling by reducing cellular expression of IFNAR1 (10), whereas another study showed that EV71 inhibited the IFN response by down-regulating JAK1 and that the expression of IFNAR1 was not significantly altered in EV71-infected cells. We aimed to elucidate the mechanism by which EV71 resists the antiviral effect of IFNs and reconcile previously reported conflicting data. In this study, we report that in HeLa cells, IFNAR1 or JAK1 was not significantly down-regulated in expression or degraded during EV71 infection in cell cultures with an inoculum of the virus at an m.o.i. of either 1 or 10. EV71 infection did not affect the phosphorylation of STAT1/2, indicating that IFNs binds to the receptor and that Jaks function well. Our data indicated that EV71 disrupted the interaction of STAT1 and KPNA1 by inducing degradation of KPNA1, resulting in the blockage of the translocation of p-STAT1/2 into the nucleus. Moreover, we provide evidence to show that KPNA1, a nuclear localization signal receptor for p-STAT1, was degraded through a caspase-3-dependent process induced during EV71 infection. Our data demonstrate a novel mechanism for EV71 resist IFN-mediated host antiviral response.
Results
IFN-β failed to inhibit EV71 replication
We started by evaluating antiviral effect of IFN-β on EV71 replication in an in vitro culture. Vero cells were pretreated with IFN-β at varying concentrations 2 h prior to EV71 infection at an m.o.i. of 0.2 and observed under a light microscope for cytopathic effect (CPE). Apparent CPE occurred in both pretreated and untreated cells at 48 h postinfection (p.i.), and no difference was exhibited in cells treated with IFN-β ranging from 100 to 1,000 ng/ml (Fig. 1A). Culture supernatants were collected at 48 h p.i. for infectious viral titration. The results indicated that there were no significant differences in viral titers (TCID50) between IFN-β untreated and treated cells. IFN-β had no evident inhibitory effect on EV71 replication even at a high concentration of 1,000 ng/ml (Fig. 1B). We also extracted total RNA or prepared cell lysates from infected cells and examined the viral VP1 gene transcript levels by real-time PCR and VP1 at protein levels by Western blotting, respectively. No significant changes between the cells treated with IFN-β or left untreated were detected in VP1 both at RNA transcript (Fig. 1C) and protein (Fig. 1D) levels. Taken together, these findings showed that IFN-β failed to inhibit EV71 replication in cell cultures pretreated at high concentrations of up to 1,000 ng/ml.
Figure 1.

IFN-β failed to inhibit EV71 replication. Vero cells were pretreated with or without IFN-β at concentrations as indicated for 2 h and then infected with EV71 at an m.o.i. of 0.2. A, cytopathic effects of infected cells pretreated with IFN-β. The infected or uninfected cells were fixed at 72 h p.i. with 3% formaldehyde and stained with 0.5% crystal violet. After washes the cells were subjected to light microscopy. The assay was performed in duplicate (top and bottom rows). B, the culture medium supernatants from the cells were harvested at 48 h p.i., and infectious viral titers (TCID50) were determined for the cells pretreated with IFN-β at various concentrations. C, total RNA was isolated from the cells pretreated with IFN-β for real-time RT-PCR to measure viral VP1 gene copy numbers. D, cell lysates were prepared from infected or uninfected cells and then pretreated with IFN-β for Western blot analyses with anti-VP1 antibodies. The experiments were performed at least twice, and representative results are presented. NS, p > 0.05 (Student's t test); MW, molecular mass.
EV71 inhibited induction of ISGs stimulated by IFN-β
Type I IFN signaling leads to induction of a variety of ISGs. To validate the inhibitory effect of EV71 on IFN-β signaling, we examined the expression of ISGs induced by IFN-β in cells either infected or uninfected with EV71. HeLa cells were infected with EV71 for 24 h and then further treated with IFN-β at 20 ng/ml or left untreated for another 15 h. Total RNA was prepared from the cells for real-time RT-PCR analyses to measure the induction of ISGs. As shown in Fig. 2, the transcript levels of MX1, MX2, OAS1, IFI27, ISG54, and ISG56 increased up to 128.5-, 700.4-, 225.7-, 443.7-, 84.2-, and 94.1-fold, respectively, in IFN-β-treated cells compared with those in untreated cells. However, in IFN-β-treated cells infected with EV71, the induction of MX1, MX2, OAS1, IFI27, ISG54, and ISG56 transcripts was present up only to 29.9-, 117.1-, 37.5-, 73.2-, 20.3-, and 21.5-fold, respectively, significantly lower than IFN-β-treated cells without EV71 infection (Fig. 2A). The induction of ISGs in EV7-infected cells without IFN-β stimulation was barely detectable, similar to that in mock-treated cells (Fig. 2A). These data suggest that IFN-β signaling is strongly suppressed in EV71-infected Vero cells.
Figure 2.

EV71 inhibited induction of ISGs stimulated by IFN-β. HeLa cells were infected with EV71 or inoculated with UV-inactivated EV71 for 24 h, followed by treatment with or without IFN-β at 20 ng/ml for another 15 h. A, EV71 infection inhibited induction of ISGs stimulated by IFN-β. Total RNA was prepared from the cells for cDNA synthesis, and fold changes of the ISG gene transcripts were measured by real-time PCR assay. B, inactivated EV71 failed to inhibit induction of ISGs stimulated by IFN-β. Total RNA was prepared from the cells inoculated with inactivated EV71 and subjected to measurement of fold change of ISGs stimulated by IFN-β. C, EV71 inhibited the induction of MX1 and MX2. The cell lysates were prepared from infected or uninfected cells, stimulated with or without IFN-β, and subjected to Western blot analyses with anti-MX1 or anti-MX2 antibodies. The experiments were repeated at least three times. *, p < 0.05 (Student's t test); MW, molecular mass.
We were able to demonstrate that suppression of IFN-β signaling was dependent on EV71 replication, because the inhibitory effect on the induction of ISGs disappeared when the cells were infected with inactivated EV71 without effective viral replication. EV71 virions inactivated by UV illumination were used for infection in Vero cells, and viral inactivation was verified by our failure to detect viral replication at 72 h p.i. The induction of ISG transcripts were comparable in IFN-β-treated cells, which were later inoculated with inactivated viruses (Fig. 2B).
In parallel we collected cell lysates from infected or uninfected cells, stimulated with or without IFN-β, and subjected them to Western blot analysis. As shown in Fig. 2C, the expression of MX1 and MX2 was clearly induced by IFN-β stimulation, which was, however, significantly down-regulated by EV71 infection, confirming that the expression of ISGs, induced by IFN-β stimulation, was effectively suppressed by EV71 replication.
EV71 infection did not alter the expression of IFNAR1 or JAK1
A previous study showed that EV71 infection disrupted IFN signaling by down-regulating the expression of IFNAR1 (10). However, a recent report found that EV71 down-regulated JAK1, whereas IFNAR1 expression was not significantly altered. To evaluate the EV71 mechanism for antagonizing interferon signaling, we examined the expression of IFNAR1 and JAK1 through EV71 infection. As shown in Fig. 3, our data indicated that no evident changes of IFNAR1 or JAK1 expression occurred during EV71 infection at an m.o.i. of 1 in HeLa or RD cells (Fig. 3, A and B). To validate our finding, we infected the cells with EV71 at an m.o.i. of 10 for 24 h, followed by stimulation of IFN-β for 2 h. The cell lysates were collected and examined by Western blot analysis. Our results showed that with or without IFN-β stimulation, EV71 infection at an m.o.i. of 10 did not alter IFNAR1 or JAK1 expression, even though both IFNAR1 and JAK were up-regulated slightly upon IFN-β stimulation (Fig. 3C). Therefore, we believe that down-regulation of IFNAR1 or JAK1 may not be the mechanism by which EV71 resists IFN signaling, and other mechanisms need to be explored for explaining how EV71 targets IFN-β signaling.
Figure 3.

EV71 infection did not alter the expression or phosphorylation status of IFNAR1 or JAK1. A and B, expression of IFNAR1 or JAK1 was not altered. HeLa (A) and RD (B) cells were infected with EV71 at an m.o.i. of 1. The cell lysates were prepared at 12, 24, and 36 h p.i. and subjected to Western blot analyses with specific antibodies for IFNAR1, JAK1, or VP1. C, expression of IFNAR1 or JAK1, stimulated by IFN-β, was not altered. HeLa cells were infected with EV71 at an m.o.i. of 10 for 24 h, followed by stimulation with IFN-β for another 2 h. The cell lysates were prepared for Western blot analysis with specific antibodies. D and E, phosphorylation of STAT1/2 was not affected by EV71 infection. HeLa cells were infected with EV71 at an m.o.i. of 1 (D) or 10 (E) for 24 h, followed by stimulation with IFN-β for another 30 min. The cell lysates were prepared and analyzed by Western blot analyses with antibodies specific for STAT1, STAT2, p-STAT1, or p-STAT2. The faint VP-like band in EV71 (−) samples, which is supposed to be negative, was due to sample overloading of the neighboring well. ctl, control; MW, molecular mass.
EV71 infection had no effect on the protein or phosphorylation level of STAT1/2
To determine the site where IFN-β signaling was targeted by EV71, we first examined STAT1 and STAT2 expression after IFN-β stimulation. HeLa cells were infected with EV71 for 24 h, followed by stimulation with IFN-β at 20 ng/ml for another 30 min. The whole cell lysates were analyzed by SDS-PAGE and Western blot analysis with relevant antibodies. As shown in Fig. 3 (D and E), without IFN-β stimulation, STATs were not phosphorylated in EV71-infected cells, probably because of the suppression of IFN signaling by EV71. Our data also showed that after IFN-β stimulation, both STAT1 and STAT2 are significantly up-regulated within a short period. However, EV71 infection at an m.o.i. of either 1 or 10 had no effect on the induced protein levels of either STAT1 or STAT2 (Fig. 3, D and E), indicating that EV71 infection did not target STAT1 or STAT2 for degradation.
STAT1/2 are phosphorylated and then associated with IRF9 to form the complex ISGF3, which is subsequently translocated into the nucleus for ISG induction (13). Our results, however, did not detect any difference in phosphorylation status of STAT1 or STAT2 between uninfected and infected cells, although both STAT1 and STAT2 were significantly phosphorylated upon IFN-β stimulation (Fig. 3, D and E). This result suggests that EV71 infection did not target the phosphorylation of STAT1 or STAT2 for suppressing IFN-β signaling.
EV71 infection prevented STAT1/STAT2 from translocation into the nucleus
Once STAT1 and STAT2 are phosphorylated upon IFN-β stimulation, the two are associated with IRF9 to assemble and form the ISGF3 complex, which is translocated into the nucleus to initiate gene transcription by binding to ISREs. To examine whether EV71 infection affects the translocation of ISGF3 in JAK/STAT signaling, we infected HeLa cells with EV71 for 24 h, followed by stimulation of IFN-β for 30 min. The cells were fixed and permeabilized before incubation with anti-EV71 VP1 and anti-STAT1 antibodies. Our data indicated that in EV71-infected cells (VP1-positive), STAT1 was almost invisible because of its low expression (Fig. 4B), whereas in IFN-β-stimulated cells without EV71 infection, STAT1 was induced visibly significantly and translocated into the nucleus (Fig. 4C). However, in EV71-infected cells, STAT1 remained mainly in the cytoplasm even though stimulated with IFN-β (Fig. 4D), indicating clearly that EV71 infection blocked STAT1 from translocation into the nucleus.
Figure 4.

EV71 infection blocked STAT1 from translocation into the nucleus. HeLa cells were infected with EV71 for 24 h, followed by stimulation with IFN-β at 20 ng/ml for another 30 min. The cells were fixed and incubated with anti-VP1 or anti-STAT1 antibodies, followed by incubation with FITC- or TRITC-conjugated secondary antibodies, before the cells were stained with DAPI to visualize the nuclei. After washes, the coverslips were analyzed under an Olympus confocal microscope.
To confirm this observation, we fractionated the cell lysates prepared from uninfected or infected cells, stimulated with IFN-β or not, and collected the nuclear and cytoplasmic fractions for SDS-PAGE and Western blot analysis. As shown in Fig. 5, both STAT1 and STAT2 were phosphorylated upon IFN-β stimulation. In EV71-infected cells, however, p-STAT1 was mostly retained in the cytosol (Fig. 5A), whereas it was present significantly less in the nucleus (Fig. 5B), in contrast to its distribution in uninfected cells. On the other hand, although p-STAT2 was distributed in the cytosol comparably between uninfected and infected cells (Fig. 5A), much less p-STAT2 was present in the nucleus in infected cells (Fig. 5B). These data strongly demonstrate that EV71 infection blocked the translocation of p-STAT1/2 from the cytosol to the nucleus upon IFN-β stimulation.
Figure 5.

EV71 infection inhibited the translocation of p-STAT1/2. HeLa cells were infected with EV71 at an m.o.i. of 1 for 24 h, followed by stimulation with IFN-β for 30 min. The cytosolic (A) and nuclear (B) fractions were prepared and subjected to SDS-PAGE and Western blot analyses with antibodies specific for p-STAT1 and p-STAT2. Tubulin and histone H1 were used as loading controls for cytosolic and nuclear fractions, respectively. MW, molecular mass.
EV71 infection disrupted the formation of STAT/KPNA1 complex after IFN-β stimulation and induced KPNA1 degradation
Accumulation of activated p-STAT1 in the nucleus requires its interaction with a specific nuclear localization signal receptor, KPNA1 (9, 14). Our finding that EV71 infection blocked the IFN-β-stimulated nuclear translocation of p-STAT1/2 raised the possibility that EV71 infection may target a nuclear localization signal receptor. Because KPNA1 is mainly involved in translocation of p-STAT1, we examined whether the interaction of STAT1 and KPNA1 was affected by EV71 infection after IFN-β stimulation. We immunoprecipitated KPNA1 by incubation of cell lysates prepared from infected or control cells with or without IFN-β stimulation, with an anti-KPNA1 antibody. The immunoprecipitates were analyzed by SDS-PAGE and Western blot analysis with anti-p-STAT1 antibodies. Our results indicated that KPNA1 was associated with p-STAT1 only in IFN-β-stimulated cells, and EV71 infection significantly decreased the association of KPNA1 and p-STAT1, even though STAT1 was phosphorylated comparably in infected and uninfected cells (Fig. 6A).
Figure 6.

EV71 infection disrupted the interaction of STAT1 and KPNA1 complex and induced KPNA1 degradation. A, the IFN-β-induced complex of STAT/KPNA1 was disrupted. HeLa cells were infected EV71 for 24 h and afterward were treated with IFN-β for 30 min. The cell lysates were prepared and immunoprecipitated with an anti-KPNA1 antibody. The immunoprecipitates were analyzed by SDS-PAGE and Western blot analyses with anti-p-STAT1 antibodies. The inputs of p-STAT1, KPNA1, and β-actin were shown by Western blot analyses with specific antibodies. B, KPNA1 was degraded in EV71 infected RD cells. RD cells were infected with EV71 at an m.o.i. of 2. The cell lysates were prepared at indicated time points for Western blot analysis with antibodies specific for KPNA1. C and D, KPNA1 was degraded in Vero but not in HT-29 cells. Vero (C) and HT-29 (D) cells were infected with EV71, and cell lysates were prepared for Western blot analysis with antibodies specific for KPNA1, TRIF, IRNAR1, and JAK1. ctl, control; IB, immunoblot; IP, immunoprecipitation; MW, molecular mass.
Importantly, our data suggested that the expression level of KPNA1 was significantly lower in EV71-infected cells, with or without IFN-β stimulation, whereas IFN-β had no impact on KPNA1 expression (Fig. 6A). To confirm this observation in different cell types, we tested KPNA1 expression in RD or Vero cells following EV71 infection by performing Western blot analyses with the anti KPNA1 antibody. The results showed that EV71 infection decreased the levels of KPNA1 in either RD or Vero cells (Fig. 6, B and C), which may account for less formation of the STAT/KPNA1 complex and blocked translocation of p-STAT1 into the nucleus.
KPNA1 was degraded in RD and Vero cells but not in HT-29 cells
We further compared the status of IFNAR1, JAK1, KPNA1, and TRIF in Vero and human intestinal epithelial HT-29 cells. We previously showed a robust induction of IFN-β during EV71 infection in HT-29 cells (15, 16). Our results indicated that, on the one hand, the levels of IFNAR1 and JAK1 remained unchanged in EV71-infected Vero and HT-29 cells; on the other hand, the levels of both KPNA1 and TRIF decreased in Vero cells (Fig. 6C), but in contrast, those two proteins remained not affected in infected HT-29 cells during the infection (Fig. 6D). These data further confirmed the results that IFNAR1 and JAK1 may not be targeted and degraded during EV71 infection; the targeted proteins would mostly likely be KPNA1 as well as TRIF in Vero cells (and in RD cells). However, none of KPNA1 or TRIF was affected or degraded in EV71-infected HT-29 cells (Fig. 6, C and D).
2Apro and 3Cpro proteases did not cleave KPAN1 or inhibit IFN-β signaling
We next aimed to determine what caused the lower level of KPNA1 in EV71-infected cells. It was reported that 3Cpro of foot-and-mouth disease virus (FMDV) induced proteasome- and caspase-independent protein degradation of KPNA1 (17). Thus we examined whether 2Apro and 3Cpro of EV71, the two proteases responsible for viral polyprotein processing, had any effect on the degradation of KPNA1 and subsequent IFN-β signaling.
We cloned cDNA of EV71 2A and 3C into an expression vector, pRK5, with either an HA or Flag tag. HEK293 cells were transfected with plasmids expressing tagged 2A protein (2Apro) or 3C protein (3Cpro); their expression is shown in Fig. 7A. The cell lysates were further analyzed with Western blot for KPNA1 expression. Our data showed that ectopic expression of 3Cpro at various doses for transfection could cleave TRIF but had no effect on KPNA1, indicating that KPNA1 was not the cellular target of 3Cpro (Fig. 7A). On the other hand, ectopic expression of 2Apro has almost no effect on degrading KPNA1 as well (Fig. 7B). These data showed that KPNA1 was not the target of both viral proteases 3C and 2A.
Figure 7.

2Apro and 3Cpro proteases did not cleave KPNA1 or inhibit IFN-β response. A and B, HEK293 cells were transfected with pRK5-Flag-3C (A) or pRK-HA-2A (B), respectively, and the cell lysates were prepared for Western blot analyses with antibodies specific for KPNA1, TRIF, or eIF4G. C, HEK293 cells were co-transfected with ISRE reporter plasmids, along with either pRK5-HA-2A, pRK5-Flag-3C, or a control plasmid at the indicated doses, respectively. At 24 h post-transfection, the cells were stimulated with 20 ng/ml IFN-β. The cell lysates were collected 16 h later and analyzed for luciferase activities. The experiments were repeated at least three times. *, p < 0.05; **, p < 0.01 (Student's t test). MW, molecular mass.
Our observation was confirmed with a dual-luciferase reporter assay, in which we tested whether 3Cpro or 2Apro had an effect on ISRE promoter activity stimulated by IFN-β. HEK293 cells were co-transfected with reporter plasmid, pISRE-Luc, along with pRK5-HA-2A, pRK5-Flag-3C, or a control plasmid at the indicated doses, respectively. At 24 h post-transfection, the cells were stimulated with 20 ng/ml of IFN-β. The cell lysates were prepared after 16 h and analyzed for luciferase activities. Our results showed that ectopically 2A- and 3C-expressing cells induced luciferase activity at levels comparable with those of the controls after IFN-β stimulation (Fig. 7C).
We also examined the effect of the two viral proteins on the mRNA levels of ISGs induced by IFN-β stimulation with real-time RT-PCR. Our data indicated that overexpression of 2Apro or 3Cpro had little inhibitory effect on the induction of MX1, MX2, IFIH1, IFI27, OAS2, or IFIT1 (Fig. 8). Thus we conclude that either 2Apro or 3Cpro had no role in the degradation of KPNA1 and disruption of KPNA/STAT1 complex and had no role in suppressing ISGs induced by IFN-β signaling.
Figure 8.

2Apro and 3Cpro proteases had no inhibitory effect on the induction of ISGs. HEK293 cells were transfected with pRK5-HA-2A or pRK5-Flag-3C at the indicated doses or with a control plasmid and stimulated with IFN-β for 16 h. Total RNA was prepared for real-time RT-PCR analyses to measure changes of ISG gene transcripts in response to IFN-β stimulation. Specific primers for MX1 (A), MX2 (B), IFIHI (C), IFI27 (D), OAS2 (E), and IFIT1 (F) were used for RT-PCR. The experiments were repeated at least three times. *, p < 0.05; **, p < 0.01 (Student's t test).
EV71-induced KPNA1 degradation is caspase-3-dependent
To exclude whether the lowered level of KPNA1 was due to down-regulation or a decrease of mRNA transcription, we measured mRNA levels of KPNA1 during EV71 infection by real-time RT-PCR with a control from uninfected cells and detected no significant changes of the KPNA1 gene transcripts between uninfected and infected cells during various time points, suggesting that KPNA1 was not regulated transcriptionally (Fig. 9A).
Figure 9.
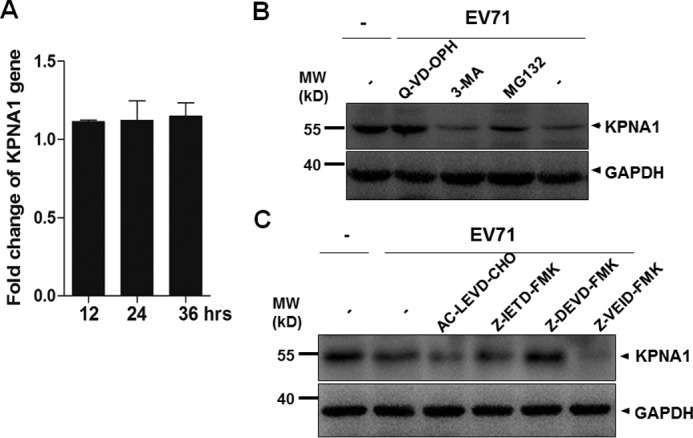
EV71-induced KPNA1 degradation is caspase-3-dependent. A, HeLa cells were infected with EV71 at an m.o.i. of 2. Total RNA was prepared at 12, 24, and 36 h p.i. by the TRIzol reagent. 500 ng of total RNA were reverse-transcribed and used for real-time PCR with specific primers to KPNA1 for quantitative comparison of its gene transcripts without and during infection. B, caspase inhibitor prevented KPNA1 from degradation. The cells were pretreated with 5 μm of a proteasome inhibitor MG132, 10 mm of an autophagy inhibitor 3-MA, or 20 nm of a broad-spectrum caspase inhibitor Q-VD-OPH for 12 h, followed by infection with EV71 at an m.o.i. of 10 for another 12 h. The cell lysates were prepared for Western blot analyses with anti-KPNA1 antibodies. C, HeLa cells were pretreated with inhibitors specific for caspase-4 (AC-LEVD-CHO, 30 μm), caspase-8 (Z-IETD-FMK, 30 μm), caspase-3 (Z-DEVD-FMK, 100 μm), and caspase-6 (Z-VEID-FMK, 50 μm) for 6 h prior to infection with EV71. The cell lysates were prepared and analyzed by Western blot with anti-KPNA1 antibody. MW, molecular mass.
Cellular proteins are generally degraded by ubiquitin-proteasome-dependent degradation, the autophagy-lysosomal pathway (36), or caspase-mediated proteolysis. To assess which pathway was involved in the degradation of KPNA1, the cells were pretreated with proteasome inhibitor MG132 at 5 μm, autophagy inhibitor 3-methyladenine (3-MA) at 10 mm, or a broad-spectrum caspase inhibitor quinoline-Val-Asp-difluorophenoxymethylketone (Q-VD-OPH) at 20 nm for 12 h, subsequently followed by infection with EV71 at an m.o.i. of 10 for another 12 h. The cell lysates were harvested for measuring the levels of KPNA1. As shown in Fig. 9, pretreatment with Q-VD-OPH significantly blocked the degradation of KPNA1 in a process induced by EV71 infection, whereas pretreatment with MG132 or 3-MA had no effect on KPNA1 expression, suggesting that EV71-induced degradation of KPNA1 was caspase-dependent (Fig. 9B).
To further investigate exactly which caspase was involved in this process, inhibitors for caspase-4 (AC-LEVD-CHO), caspase-8 (Z-IETD-FMK), caspase-3 (Z-DEVD-FMK), and caspase-6 (Z-VEID-FMK) were added to the culture medium for 6 h prior to infection with EV71. The cell lysates were analyzed by SDS-PAGE and Western blot analysis. As shown in Fig. 9C, only pretreatment of the cells with the caspase-3-specific inhibitor blocked the degradation of KPNA1 in EV71 infection, thus suggesting that the degradation of KPNA1 during the infection was caspase-3-dependent and that the process of caspase activation was induced by EV71 infection.
Inhibition of caspase-3 decreased EV71 replication through KPNA1
We also examined the effect of the inhibitors for caspases, proteasome, and autophagy on viral replication in infected cells. HeLa cells were pretreated with MG132, 3-MA, Q-VD-OPH, AC-LEVD-CHO, Z-IETD-FMK, Z-DEVD-FMK, or Z-VEID-FMK, followed by infection with EV71 for another 24 h. Infectious viral titers in the cultural medium were determined, which showed that both Q-VD-OPH and Z-DEVD-FMK significantly decreased virus replication, unlike other caspase inhibitors (Fig. 10, A and B). Pretreatment with MG132 or 3-MA could marginally reduce virus titers (Fig. 10A). Neither of them, however, affected the expression of KPNA1 (Fig. 9B), indicating that the inhibition of proteasome and autophagy affected EV71 replication independent of KPNA1.
Figure 10.

Caspase-3 knockdown reversed degradation of KPNA1 induced by EV71 infection. A, a broad-spectrum caspase inhibitor Q-VD-OPH inhibited virus replication. The cells were pretreated with 5 μm MG132, 10 mm 3-MA, or 20 nm Q-VD-OPH for 12 h, followed by infection with EV71 at an m.o.i. of 10 for another 24 h. Infectious viral titers in the cultural medium were determined by TCID50 assays in Vero cells. B, a specific inhibitor for caspase-3 inhibited virus replication. HeLa cells were pretreated with inhibitors specific for caspase-4 (AC-LEVD-CHO, 30 μm), caspase-8 (Z-IETD-FMK, 30 μm), caspase-3 (Z-DEVD-FMK, 100 μm), and caspase-6 (Z-VEID-FMK, 50 μm) for 6 h prior to infection with EV71. Infectious viral titers were determined by TCID50 assays. C, caspase-3-specific inhibitor increased the induction of MX2 in IFN-treated cells during EV71 infection. HeLa cells were pretreated with or without Z-DEVD-FMK for 6 h. Then the cells were infected with EV71, followed by IFN-β treatment for another 24 h. Total RNA was isolated and used for real-time PCR with specific primers to MX2. D, caspase-3 knockdown reversed the degradation of KPNA1 induced by EV71 infection. HeLa cells were transfected with caspase-3-specific siRNA-1, siRNA-2, or control siRNA (final siRNA concentration, 50 nm). At 24 h after transfection, cells were infected with EV71 at an m.o.i. of 10. 24 h later, the cell lysates were collected and examined by SDS-PAGE and Western blot analysis with specific antibodies to determine the levels of KPNA1 and cleaved caspase-3. E and F, caspase-3 knockdown promoted translocation of STAT2 into the nucleus in IFN-β-treated cells infected with EV71. HeLa cells were transfected with siRNA-1 or control siRNA (final concentration, 50 nm). At 24 h after transfection, the cells were infected with EV71 at an m.o.i. of 10 for 24 h, followed by stimulation with IFN-β for 30 min. The cytosolic (E) and nuclear (F) fractions were prepared and subjected to SDS-PAGE. Actin and histone H1 were used as loading controls for cytosolic and nuclear fractions, respectively. MW, molecular mass. *, p < 0.05; **, p < 0.01.
We further pretreated HeLa cells with or without Z-DEVD-FMK for 6 h, followed by EV71 infection together with IFN-β treatment for another 24 h. The results showed that the inhibitor for caspase-3 increased the level of MX2 in IFN-treated cells during the infection (Fig. 10C), indicating that EV71-suppresed induction of MX2 by IFN-b could be reversed by inhibiting caspase-3 and protecting KPNA1.
To confirm the effect of caspase-3 on KPNA1, we transfected the cells with caspase-3 siRNA to knock down caspase-3 expression and then infected cells with EV71 at an m.o.i. of 10 for 24 h. The cell lysates were examined with Western blot analyses, and the results showed that although the levels of caspase-3 decreased in the knockdown cells with siRNA1 and siRNA2, KPNA1 degradation during the infection was inhibited, and the levels of KPNA1 returned to the normal level (Fig. 10D), demonstrating that caspase-3 is involved in the degradation of KPNA1 during EV71 infection.
To further understand whether caspase-3 affects retention of STATs in the cytoplasm, we analyzed cytosolic and nuclear fractions of the cells treated with caspase-3-specific siRNA1 in a Western blot analysis. As shown in Fig. 10 (E and F), in EV71-infected cells, IFN-β-induced STAT2 was predominately retained in the cytoplasm, and in uninfected cells, IFN-β-induced STAT2 was mainly translocated into the nucleus (Fig. 10, E and F). However, when caspase-3 was knocked down by siRNA-1, even though IFN-β-induced STAT2 was still mainly retained in the cytoplasm because of EV71 infection, a significant portion of STAT2 was translocated into the nucleus (Fig. 10F), indicating that knockdown of caspase-3 increased the translocation of STATs into the nucleus. These data support that caspase-3 plays a critical role in preventing STAT2 from entering the nucleus for IFN response in infected cells treated with IFN-β.
Discussion
Since the eradication of poliovirus, enterovirus 71 has emerged as a clinically important neurotropic virus in Asian countries in the past decade. It has become endemic in some regions, and the patients are almost exclusively children. Although IFNs serve as effective antiviral therapeutics for many infections of viruses including HIV and hepatitis C and B virus, IFN treatment had little effect on patients infected with EV71. High doses of IFN administration can raise survival rates in EV71-infected mice but are not clinically plausible or acceptable in humans because of predictable side effects. Importantly, reports thus far on how EV71 resists antiviral effect of IFNs remain controversial, which impedes further application of IFNs clinically for severe cases associated with higher mortality rates.
Viruses have evolved various strategies to counteract IFN-mediated host antiviral immunity. Generally viruses circumvent the IFN responses by inhibiting the induction of IFNs, blocking IFN stimulation to induce ISGs, or both. Previous studies have demonstrated a variety of strategies used by EV71 to block the induction of type I IFN (18–21). In addition, EV71 is capable of antagonizing IFN for downstream stimulation of induction of ISGs, which is mediated by the JAK/STAT signaling pathway. A previous study showed that EV71 disrupted type I IFN signaling by down-regulating IFNAR1 and that 2A was responsible for the inhibitory effect on IFN response (10). However, a recent study reported that EV71 infection did not alter either the total expression level or surface expression of IFNAR1; instead, the infection down-regulated the protein level of JAK1 (22). In this report, we present evidence to show that neither IFNAR1 nor JAK1 levels were significantly affected during EV71 infection and were unchanged even in an infection with an inoculum as high as 10 m.o.i. We were unable to detect the change of the expression of STAT1 and STAT2, and tyrosine phosphorylation of STAT1 and STAT2 was also not affected, indicating that the stimulation on IFNAR1 by IFN-β and the activation of Jaks were barely affected during EV71 infection.
Previous studies have demonstrated that IFN induction was significantly inhibited, and the expression of IFNs was barely detectable in a variety of tissues or cell types such as RD, HeLa, or Vero cells (19, 20). An exception is probably intestinal epithelial cells as shown in our earlier report, in which a robust IFN-β production was still induced (15). Induction of IFNs in the gastrointestinal tract may explain asymptomatic or mild symptoms, because the virus may have been restrained in the gastrointestinal system, as observed in most children infected with EV71. However, severe neurological complications still occur in some cases, suggesting that EV71 is somehow capable of resisting antiviral responses if IFN is induced in the gastrointestinal system, where it can evade innate immunity and spread systemically.
In this study our data clearly showed that following EV71 infection, p-STAT1 and p-STAT2, in response to IFN-β stimulation, were retained in the cytosol and unable to be translocated into the nucleus even though STAT1 and STAT2 were phosphorylated. We explored the mechanism by which activated STAT1/2 was blocked from entering the nucleus and identified a disruptive interaction by STAT1 and KPNA1. KPNA1 is a member of the KPNA family, which includes KPNA-1, -2, -3, -4, and -5, for karyopherin-α1, -α2, -α3, -α4, and -α5, respectively. We further characterized the interaction of STAT1 and KPNA1 and observed that KPNA1 was targeted for degradation during EV71 infection.
Extensive studies have shown how KPNA1 was involved in IFN signaling (14, 23). Primarily, KPNA1 is involved in nucleocytoplasmic trafficking and directly interacts with tyrosine phosphorylated STAT1, facilitating the transport of STAT1 into the nucleus (14, 23). Many viruses have been found to be able to dampen the trafficking of STAT1 by interference with KPNA1. It was recently reported that infection by FMDV, which belongs to the genus Aphthovirus in the family Picornaviridae, induced the degradation of KPNA1 and thus blocked the nuclear translocation of p-STAT1 and STAT2 (17).
Viral proteins have been shown to play critical roles in targeting members of the KPNA family. VP24 of Ebola virus binds KPNA1 and blocks STAT1 nuclear accumulation (9). VP24 also recognizes a unique nonclassical nuclear location signal-binding site on KPNA5, which is necessary for efficient p-STAT1 nuclear transport. By binding to KPNA5 with high affinity, VP24 is able to compete with p-STAT1 effectively and inhibit the interaction of p-STAT1 and KPNA1, resulting in the translocational blockage of p-STAT1 in the complex of ISGF3 into the nucleus (24). Nsp1β of porcine reproductive and respiratory syndrome virus, or PRRSV, inhibits IFN-stimulated JAK/STAT signaling by inducing KPNA1 degradation (25), as does 3Cpro of FMDV (17). The ORF6 product of severe acute respiratory syndrome coronavirus, or SARS CoV, disrupts nuclear import of p-STAT1 by tethering KPNA2 to the endoplasmic reticulum/Golgi membrane (26). We sought to elucidate how KPNA1 was degraded in EV71 infection; our data showed that two viral proteases, 3Cpro and 2Apro, both effectively degraded numerous host proteins including MAVS, MDA5, TRIF, and IRF7 (18, 19, 21, 27), leading to lower IFN response as also observed in our study, but failed to cleave and degrade KPNA1. Therefore, KPNA1 may be degraded by an alternative process induced in EV71 infection.
We demonstrated that the degradation of KPNA1 occurred in RD, HeLa, and Vero cells, but not in human intestinal epithelial cells during EV71 infection (Fig. 6) in this study. Our previous studies have already shown that a robust IFN-β induction remained intact, and IRF3 could be activated, and p-IRF3 be translocated into the nucleus in EV71-infected HT-29 cells, unlike in other cell types (15, 16). We have not observed the degradation of either IFNAR1 or JAK1 in infected HT-29 cells, which is in accordance with our observation that the IFN response remains intact as well in human intestinal epithelial cells.
Proteins are generally degraded by ubiquitin-proteasome dependent degradation, the autophagy-lysosomal pathway, or caspase-mediated proteolysis. In this study, we observed that blocking the caspase-mediated proteolysis pathway, but not the ubiquitin-proteasome or autophagy-lysosomal signal pathway, resulted in the restoration of KPNA1 levels in EV71-infected cells, suggesting that the degradation of KPNA1 could be apoptosis- or caspase-dependent. We selected a group of caspases for further studies, including caspase-3, -4, -6, -7, and -8, among which caspase-3 and -6 or -7 are executioner caspases, whereas caspase-4 and -8 are initiator caspases. By using specific synthetic inhibitors for these caspases, we found that only the inhibitor specific for caspase-3 could block the degradation of KPNA1, suggesting that the reduction of KPNA1 in protein level during EV71 infection was caspase-3-dependent. This was further confirmed in the cells treated with siRNA specific for caspase-3. When the levels of caspase-3 decreased, the degradation of KPNA1 was reversed in EV71-infected cells (Fig. 10D).
Numerous reports have shown that caspases are activated during an apoptotic process, which can be either beneficial or detrimental to viral infection (29, 30). We have shown that apoptosis was triggered in EV71 infection, and caspases, including caspase-3, were activated (31). Caspase-3 activation is crucial in apoptosis and mediates the proteolysis of many host proteins at the final stage of apoptosis (15). The host proteins can be structural ones, resulting in disintegration of cellular structures, or functional ones, playing important roles in signaling or host defense. A recent study showed that the cleavage of MDA5 was caspase-dependent during EV71 infection (32, 33), and in poliovirus infection MDA-5 was degraded in a proteasome- and caspase-dependent manner (21). Our data indicate that caspase cascade may play a unique role in EV71 viral replication, demonstrating that caspase-3-mediated degradation of KPNA1 could serve as a novel mechanism for viral evasion of IFN signaling responses by an enterovirus.
Experimental procedures
Cells and reagents
African green monkey kidney cells (Vero), human cervical carcinoma cells (HeLa), and human kidney carcinoma cells (293T) were grown in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (HyClone, Logan, UT), 1 mm sodium pyruvate (HyClone), and 1% antibiotic-antimycotic solution (Invitrogen). The cells were cultured at 37 °C in a humidified atmosphere with 5% CO2.
Anti-EV71 VP1 antibody was purchased from Millipore (Billerica, MA). Antibodies for STAT1 (catalog no. 9172), STAT2 (catalog no. 4594), phosphorylated STAT1 (catalog no. 9171), phosphorylated STAT2 (catalog no. 4441), and JAK1 (catalog no. 3344) were obtained from Cell Signaling Technology (Danvers, MA). An anti-IFNAR1 antibody (ab45172; Abcam, Cambridge, MA) was used for detecting IFNAR1 protein. HRP-conjugated anti-rabbit and anti-mouse IgG antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The Super Signal ECL reagent kit was purchased from Thermo Fisher (Rockford, IL). Caspase inhibitors including Z-DEVD-FMK (caspase-3), Z-VEID-FMK (caspase-6), AC-LEVD-CHO (caspase-4), Z-IETD-FMK (caspase-8), and Q-VD-OPH, a broad-spectrum caspase inhibitor, were purchased from CalBiochem (San Diego, CA). The proteasome inhibitors MG132 and 3-MA were purchased from Sigma-Aldrich.
Virus and virus infection
EV71 Fuyang strain belonging to the C4a cluster of the C4 subgenotype as verified through sequence analysis of the VP1 region (34) was kindly provided by Dr. Wu Bin at Jiangsu Provincial Centers for Disease Control and Prevention. The virus was propagated in Vero cells, and its infectious titers were titrated by a routine TCID50 assay. For the TCID50 assay, serially diluted viruses from 10−2 to 10−9 in DMEM were inoculated into Vero cells in 96-well plates. The cells were incubated for 7 days at 37 °C before observation under a light microscope, and titers were calculated by counting the wells with CPE using the formula: logTCID50 = L − d (s − 0.5), where L is the log of the lowest dilution, d is the difference between dilution steps, and s is the sum of the proportion of positive wells (35).
Quantitative real-time RT-PCR
Total RNA was prepared from cells by using TRIzol (Invitrogen) following the manufacturer's protocol. After quantification by a Nanodrop Reader (Thermo), RNA was used for reverse transcription for cDNA using reverse transcriptase Moloney murine leukemia virus (RNase H−) (Invitrogen). Real-time PCR was performed as previously described (15). The 2−ΔΔCt method (28) was used to quantify and normalize the relative quantification data. The data were calculated as fold change (2−ΔΔCt), which was the copy numbers of corresponding gene transcripts normalized to an internal control, GAPDH.
Immunoprecipitation and Western blot analysis
The cell lysates were prepared with 1% Nonidet P-40 and after quantification were used for incubation with either preimmune or specific antibodies at 4 °C for 2 h and then precipitated with protein A/G beads. After incubation, the beads were washed four times with lysis buffer, and the immunoprecipitates were subjected to SDS-PAGE. The proteins were then transferred onto an immunoblot PVDF membrane (Millipore) for primary antibody incubation overnight. HRP-conjugated secondary antibodies were used for further incubation with the membranes for 90 min. After thorough washes, the membranes were developed by ECL reagents (Invitrogen).
Confocal fluorescence microscopy
HeLa grown on glass coverslips were infected with EV71 for 24 h, followed by IFN-β treatment for 30 min, washed twice with PBS, and fixed with 4% paraformaldehyde in PBS. The cells were premeabilized with 0.1% Triton X-100 in PBS and blocked with 5% bovine serum albumin for 1 h before incubation with anti-EV71 VP1 and anti-STAT1 antibodies in PBS containing 1% BSA for 1.5 h at room temperature, followed by three washes. The cells were subsequently incubated with secondary antibodies at dilutions of 1:100 at 37 °C for 30 min. After three washes the coverslips were mounted on glass slides for confocal microscopy.
Plasmids and transfection of viral protease genes
cDNA of 2Apro and 3Cpro were subcloned into expression plasmids pRK5-HA and pRK5-Flag, respectively, for ectopic expression. The ISRE reporter plasmid pISRE-TA-luc was purchased from Beyotime (Jiangsu, China). HeLa or HEK293T cells were transfected with plasmids in Lipofectamine 2000 reagent (Invitrogen) for expression of respective proteins.
siRNA and knockdown of caspase-3
siRNA molecules targeting caspase-3 were synthesized with sequences as follows: siRNA-1, 5′-UGGAUUAUCCUGAGAUGGGTT-3′, 5′-CCCAUCTCAGGAUAAUCCATT-3′; and siRNA-2, 5′-AGUGAAGCAAAUCAGAAACTT-3′, 5′GUUUCUGAUUUGCUUCACUTT-3′). We also synthesized a scramble control siRNA with its sequences: 5′-UUCUCCGAACGUGUCACGUTT-3′, 5′-ACGUGACACGUUCGGAGAATT-3′. siRNA were synthesized by GenePharma Inc. (Shanghai, China). HeLa cells were transfected with siRNA-1, siRNA-2, or control siRNA with final concentration of 50 nm. At 24 h after transfection, the cells were infected with EV71 at an m.o.i. of 10. 24 h later, and the cell lysates were collected and examined by SDS-PAGE and Western blot analyses with specific antibodies to ensure the efficacy of caspase-3 knockdown.
Luciferase reporter assay
HeLa cells were seeded in 12-well plates and grown to 70–80% confluence before use. The cells were transfected with 0.25, 0.5, or 1 μg of pRK5-Flag-3C or pRK5-HA-2A, together with 0.5 μg of pISRE-TA-Luc and 0.05 μg of pRL-TK using Lipofectamine 2000. After 24 h post-transfection, the cells were incubated with 20 ng/ml of IFN-β (300-02BC; PeproTech, Rocky Hill, NJ) for 16 h, then washed in PBS, and lysed in 0.1 ml of reporter lysis buffer (Promega, Madison, MI). Firefly luciferase activities were measured with the luciferase assay system (Promega) following the manufacturer's protocol. The results were expressed as fold change over the nontransfected controls.
Statistical analysis
Two-tailed Student's t test was used to evaluate the data. The data shown are the means of three independent experiments ± S.E. A p value ≤ 0.05 level was considered statistically significant.
Author contributions
C. W. and Z. X. conceived and coordinated the study. C. W., M. S., X. Y., L. J., and Z. X. designed, performed, and analyzed the experiments shown in Figs. 1–10. C. J. C. and Y. J. provided reagents and technical assistance and contributed to completion of the studies. C. W. and Z. X. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We thank Sandy Shank for dedicated and excellent work in editing the manuscript.
This work was supported by National Natural Science Foundation of China Grant 81571993 (to Z. X.) and Natural Science Foundation of Jiangsu Province Grant BK20141078 (to Y. J.). The authors declare that they have no conflicts of interest with the contents of this article.
- EV71
- enterovirus 71
- ISG
- interferon-inducible gene
- IFNAR
- interferon-α receptor
- IRF
- IFN-regulatory factor
- ISGF3
- interferon-stimulated gene factor 3
- ISRE
- IFN-stimulated response elements
- m.o.i.
- multiplicity of infection
- KPNA1
- karyopherin-α1
- CPE
- cytopathic effect
- p.i.
- postinfection
- FMDV
- foot-and-mouth disease virus
- 3-MA
- 3-methyladenine
- Z
- benzyloxycarbonyl
- FMK
- fluoromethyl ketone
- TRITC
- tetramethylrhodamine isothiocyanate.
References
- 1. Wong S. S., Yip C. C., Lau S. K., and Yuen K. Y. (2010) Human enterovirus 71 and hand, foot and mouth disease. Epidemiol. Infect. 138, 1071–1089 [DOI] [PubMed] [Google Scholar]
- 2. Yi L., Lu J., Kung H.-F., He M.-L. (2011) The virology and developments toward control of human enterovirus 71. Crit. Rev. Microbiol. 37, 313–327 [DOI] [PubMed] [Google Scholar]
- 3. Solomon T., Lewthwaite P., Perera D., Cardosa M. J., McMinn P., and Ooi M. H. (2010) Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet 10, 778–790 [DOI] [PubMed] [Google Scholar]
- 4. Platanias L. C. (2005) Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386 [DOI] [PubMed] [Google Scholar]
- 5. Darnell J. E. Jr., Kerr I. M., and Stark G. R. (1994) Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264, 1415–1421 [DOI] [PubMed] [Google Scholar]
- 6. Weber F., Kochs G., and Haller O. (2004) Inverse interference: how viruses fight the interferon system. Viral Immunol. 17, 498–515 [DOI] [PubMed] [Google Scholar]
- 7. Johnson C. L., and Gale M. Jr. (2006) CARD games between virus and host get a new player. Trends Immunol. 27, 1–4 [DOI] [PubMed] [Google Scholar]
- 8. Katze M. G., He Y., and Gale M. Jr. (2002) Viruses and interferon: a fight for supremacy. Nat. Rev. Immunol. 2, 675–687 [DOI] [PubMed] [Google Scholar]
- 9. Reid S. P., Leung L. W., Hartman A. L., Martinez O., Shaw M. L., Carbonnelle C., Volchkov V. E., Nichol S. T., and Basler C. F. (2006) Ebola virus VP24 binds karyopherin α1 and blocks STAT1 nuclear accumulation. J. Virol. 80, 5156–5167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu J., Yi L., Zhao J., Yu J., Chen Y., Lin M. C., Kung H. F., and He M. L. (2012) Enterovirus 71 disrupts interferon signaling by reducing the level of interferon receptor 1. J. Virol. 86, 3767–3776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu M. L., Lee Y. P., Wang Y. F., Lei H. Y., Liu C. C., Wang S. M., Su I. J., Wang J. R., Yeh T. M., Chen S. H., and Yu C. K. (2005) Type I interferons protect mice against enterovirus 71 infection. J. Gen. Virol. 86, 3263–3269 [DOI] [PubMed] [Google Scholar]
- 12. Yi L., He Y., Chen Y., Kung H. F., and He M. L. (2011) Potent inhibition of human enterovirus 71 replication by type I interferon subtypes. Antivir. Ther. 16, 51–58 [DOI] [PubMed] [Google Scholar]
- 13. Dupuis S., Jouanguy E., Al-Hajjar S., Fieschi C., Al-Mohsen I. Z., Al-Jumaah S., Yang K., Chapgier A., Eidenschenk C., Eid P., Al Ghonaium A., Tufenkeji H., Frayha H., Al-Gazlan S., Al-Rayes H., et al. (2003) Impaired response to interferon-α/β and lethal viral disease in human STAT1 deficiency. Nat. Genet. 33, 388–391 [DOI] [PubMed] [Google Scholar]
- 14. McBride K. M., Banninger G., McDonald C., and Reich N. C. (2002) Regulated nuclear import of the STAT1 transcription factor by direct binding of importin-α. EMBO J. 21, 1754–1763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chi C., Sun Q., Wang S., Zhang Z., Li X., Cardona C. J., Jin Y., and Xing Z. (2013) Robust antiviral responses to enterovirus 71 infection in human intestinal epithelial cells. Virus Res. 176, 53–60 [DOI] [PubMed] [Google Scholar]
- 16. Wang C., Ji L., Yuan X., Jin Y., Cardona C. J., and Xing Z. (2016) Differential regulation of TLR signaling on the induction of antiviral interferons in human intestinal epithelial cells infected with enterovirus 71. PLoS One 11, e0152177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Du Y., Bi J., Liu J., Liu X., Wu X., Jiang P., Yoo D., Zhang Y., Wu J., Wan R., Zhao X., Guo L., Sun W., Cong X., Chen L., et al. (2014) 3Cpro of foot-and-mouth disease virus antagonizes the interferon signaling pathway by blocking STAT1/STAT2 nuclear translocation. J. Virol. 88, 4908–4920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lei X., Xiao X., Xue Q., Jin Q., He B., and Wang J. (2013) Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J. Virol. 87, 1690–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lei X., Sun Z., Liu X., Jin Q., He B., and Wang J. (2011) Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J. Virol. 85, 8811–8818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lei X., Liu X., Ma Y., Sun Z., Yang Y., Jin Q., He B., and Wang J. (2010) The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J. Virol. 84, 8051–8061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kuo R. L., Kao L. T., Lin S. J., Wang R. Y., and Shih S. R. (2013) MDA5 plays a crucial role in enterovirus 71 RNA-mediated IRF3 activation. PLoS One 8, e63431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu Y., Zhang Z., Zhao X., Yu R., Zhang X., Wu S., Liu J., Chi X., Song X., Fu L., Yu Y., Hou L., and Chen W. (2014) Enterovirus 71 inhibits cellular type I interferon signaling by downregulating JAK1 protein expression. Viral Immunol. 27, 267–276 [DOI] [PubMed] [Google Scholar]
- 23. Chook Y. M., and Blobel G. (2001) Karyopherins and nuclear import. Curr. Opin. Struct. Biol. 11, 703–715 [DOI] [PubMed] [Google Scholar]
- 24. Xu W., Edwards M. R., Borek D. M., Feagins A. R., Mittal A., Alinger J. B., Berry K. N., Yen B., Hamilton J., Brett T. J., Pappu R. V., Leung D. W., Basler C. F., and Amarasinghe G. K. (2014) Ebola virus VP24 targets a unique NLS binding site on karyopherin α5 to selectively compete with nuclear import of phosphorylated STAT1. Cell Host Microbe 16, 187–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang R., Nan Y., Yu Y., and Zhang Y. J. (2013) Porcine reproductive and respiratory syndrome virus Nsp1β inhibits interferon-activated JAK/STAT signal transduction by inducing karyopherin-α1 degradation. J. Virol. 87, 5219–5228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Frieman M., Yount B., Heise M., Kopecky-Bromberg S. A., Palese P., and Baric R. S. (2007) Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 81, 9812–9824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang B., Xi X., Lei X., Zhang X., Cui S., Wang J., Jin Q., and Zhao Z. (2013) Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathogens 9, e1003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 29. Liang C., Oh B. H., and Jung J. U. (2015) Novel functions of viral anti-apoptotic factors. Nat. Rev. Microbiol. 13, 7–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ghosh Roy S., Sadigh B., Datan E., Lockshin R. A., and Zakeri Z. (2014) Regulation of cell survival and death during Flavivirus infections. World J. Biol. Chem. 5, 93–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Best S. M., Wolfinbarger J. B., and Bloom M. E. (2002) Caspase activation is required for permissive replication of Aleutian mink disease parvovirus in vitro. Virology 292, 224–234 [DOI] [PubMed] [Google Scholar]
- 32. Wang K. K., Posmantur R., Nath R., McGinnis K., Whitton M., Talanian R. V., Glantz S. B., and Morrow J. S. (1998) Simultaneous degradation of αII- and βII-spectrin by caspase 3 (CPP32) in apoptotic cells. J. Biol. Chem. 273, 22490–22497 [DOI] [PubMed] [Google Scholar]
- 33. Prévôt D., Darlix J. L., and Ohlmann T. (2003) Conducting the initiation of protein synthesis: the role of eIF4G. Biol. Cell 95, 141–156 [DOI] [PubMed] [Google Scholar]
- 34. Zhang Y., Zhu Z., Yang W., Ren J., Tan X., Wang Y., Mao N., Xu S., Zhu S., Cui A., Zhang Y., Yan D., Li Q., Dong X., Zhang J., et al. (2010) An emerging recombinant human enterovirus 71 responsible for the 2008 outbreak of hand foot and mouth disease in Fuyang city of China. Virol. J. 7, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kärber G. (1931) Contribution to the collective treatment of pharmacological series tests. Naunyn-Schmiedebergs Arch. Exp. Pathol. Pharmakol 162, 480–483 [Google Scholar]
- 36. Rubinsztein D. C. (2006) The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443, 780–786 [DOI] [PubMed] [Google Scholar]