

Abstract
Hepcidin is a liver-derived hormone that negatively regulates serum iron levels and is mainly regulated at the transcriptional level. Previous studies have clarified that in addition to hepatic iron levels, inflammation also efficiently increases hepatic hepcidin expression. The principle regions responsible for efficient hepcidin transcription are bone morphogenetic protein-responsive elements (BMP-REs) 1 and 2 as well as the signal transducer and activator of transcription 3-binding site (STAT-BS). Here, we show that the proinflammatory cytokine interleukin-1β (IL-1β) efficiently increases hepcidin expression in human HepG2 liver-derived cells and primary mouse hepatocytes. The primary region responsible for IL-1β-mediated hepcidin transcription was the putative CCAAT enhancer-binding protein (C/EBP)-binding site (C/EBP-BS) at the hepcidin promoter spanning nucleotides −329 to −320. IL-1β induces the expression of C/EBPδ but neither C/EBPα nor C/EBPβ in hepatocytes, and C/EBPδ bound to the C/EBP-BS in an IL-1β-dependent manner. Lipopolysaccharide (LPS) induced the expression of IL-1β in Kupffer cells and hepatocytes in the mouse liver; furthermore, the culture supernatants from the macrophage-like cell line RAW264.7 treated with LPS potentiated the stimulation of hepcidin expression in hepatocytes. The present study reveals that: 1) inflammation induces IL-1β production in Kupffer cells and hepatocytes; 2) IL-1β increases C/EBPδ expression in hepatocytes; and 3) induction of C/EBPδ activates hepcidin transcription via the C/EBP-BS that has been uncharacterized yet. In cooperation with the other pathways activated by inflammation, IL-1β pathway stimulation leads to excess production of hepcidin, which could be causative to anemia of inflammation.
Keywords: CCAAT enhancer-binding protein (C/EBP), gene transcription, hepatocyte, inflammation, interleukin 1 (IL-1), hepcidin
Introduction
Inflammation is a symptom of an adaptive response that is triggered by microbial infection or tissue injury (1, 2). Initially, infiltrated neutrophils kill invading microbes by releasing granules containing toxic contents such as reactive oxygen species and proteases; this process is followed by macrophage-mediated resolution and repair (3, 4). The inflammatory process persists when pathogens are insufficiently eliminated during the acute response (4, 5). Chronic inflammation is well known to associate with a wide variety of diseases, including progressive and irreversible damage to the central nervous system, tumorigenesis, and metabolic syndrome (5–7).
One of the various disorders resulting from chronic inflammation is anemia of inflammation (8, 9). The cause of anemia of inflammation is multifactorial, and precise mechanisms underlying its pathogenesis are not fully elucidated (10); however, inflammation-induced hepcidin production has been suggested to be responsible for the onset of anemia of inflammation (8, 9). Hepcidin is a liver-derived peptide hormone that negatively regulates plasma iron levels (11–13). Hepcidin stimulates the internalization and degradation of ferroportin, an iron exporter expressed in macrophages and intestinal epithelial cells. Therefore, overproduction of hepcidin resulting from inflammation inhibits iron release from macrophages for erythropoiesis and the intestinal absorption of iron (11–13), leading to the onset of anemia of inflammation.
Hepcidin expression has been shown to increase with elevated hepatic iron levels; hepatic iron induces bone morphogenetic protein (BMP)2 6 expression, which stimulates hepcidin transcription via BMP-responsive elements (BMP-RE) 1 and 2 on the hepcidin gene (12). However, proinflammatory cytokines such as interleukin (IL)-6 and oncostatin M also up-regulate hepcidin expression; these cytokines transactivate the hepcidin gene via the signal transducer and activator of transcription (STAT) 3-binding site (STAT-BS) on the hepcidin gene (11–13). Furthermore, we recently found that activin B is induced in sinusoidal endothelial cells and Kupffer cells in response to intraperitoneal lipopolysaccharide (LPS) injection, which activates hepcidin transcription via BMP-RE1 and BMP-RE2 (14). In view of the production of diverse cytokines during inflammation, other cytokines may be involved in regulating hepcidin transcription via regions other than known elements. Here, we show that IL-1β, a proinflammatory cytokine, stimulates hepcidin transcription mainly via a CCAAT enhancer-binding protein (C/EBP)-binding site (C/EBP-BS) located in the hepcidin promoter.
Results
IL-1β stimulates hepcidin transcription through region other than BMP-REs and STAT-BS
We first examined the effects of IL-1β on hepcidin expression in primary hepatocytes; consistent with a previous study (15), treatment with IL-1β for 24 h stimulated hepcidin expression in primary mouse hepatocytes (Fig. 1A). In contrast, IL-1β-induced up-regulation of hepcidin expression was not detected in primary rat hepatocytes (Fig. 1A). Considering that in primary hepatocytes from mice and rats, IL-1β induces iNOS, an IL-1β-responsive gene (Fig. 1B) (16), both mouse hepatocytes and rat hepatocytes are defined as IL-1β-responsive cells. IL-1β increased hepcidin expression within 4 h after the treatment in primary mouse hepatocytes, and the increased expression continued after at least 12 h of IL-1β treatment (supplemental Fig. S1A). In contrast, hepcidin expression was slightly higher in primary rat hepatocytes treated with IL-1β within 12 h than in control hepatocytes, but this difference was due to a reduction of hepcidin expression in the control cells (supplemental Fig. S1B). We also examined whether IL-1β stimulates hepcidin expression in HepG2 cells, a human liver-derived cell line. Similar to the primary mouse hepatocytes, HepG2 cells responded to IL-1β by increasing hepcidin expression (Fig. 1C).
Figure 1.

Up-regulation of hepcidin expression by IL-1β in hepatocytes. Primary mouse and rat hepatocytes (A and B) were treated with IL-1β (25 ng/ml) for 24 h. The expression of hepcidin (A) and iNOS (B) was examined by RT-qPCR analysis with the levels in the control cells defined as 1. The data are presented as the mean ± S.E. (n = 4). **, p < 0.01 versus cells treated without IL-1β. C, HepG2 cells were treated with IL-1β (25 ng/ml) for the indicated time. The expression of hepcidin was examined by RT-qPCR analysis with the levels in the control cells prior to IL-1β treatment defined as 1. The data are presented as the mean ± S.E. (n = 3). **, p < 0.01 versus cells treated without IL-1β at the respective time point. D and E, HepG2 cells were either untreated or pretreated with BAY 11-7085 (5 μm, BAY: D) or cycloheximide (1 μg/ml, CHX, E) before treatment with IL-1β (10 or 25 ng/ml) for 12 h. Hepcidin expression was examined by RT-qPCR analysis. The expression levels in control cells treated without either BAY 11-7085 or cycloheximide were defined as 1. The data are presented as the mean ± S.E. (n = 3). *, p < 0.05 and **, p < 0.01 versus cells treated with the respective inhibitor (vehicle, BAY 11-7085, or cycloheximide) in the absence of IL-1β. †, p < 0.05 and ††, p < 0.01 versus cells with corresponding IL-1β treatments in the absence of inhibitor (i.e. BAY 11-7085 (D) or cycloheximide (E)).
IL-1β activates the transcription factor nuclear factor-κB (NF-κB) (17). To evaluate the role of the NF-κB pathway in IL-1β-induced hepcidin expression, HepG2 cells were treated with BAY 11-7085, an inhibitor of NF-κB pathway by blocking phosphorylation of inhibitor κBα (IκBα) (18). IL-1β-induced hepcidin expression was inhibited by pretreatment with BAY 11-7085 in HepG2 cells, suggesting the up-regulation of hepcidin expression through activation of the NF-κB pathway (Fig. 1D).
To examine the necessity of novel protein synthesis for IL-1β-induced hepcidin expression, cycloheximide, an inhibitor of protein synthesis (19), was added to cells, and the data showed that IL-1β-induced hepcidin expression was cycloheximide-sensitive in both HepG2 cells (Fig. 1E) and mouse hepatocytes (supplemental Fig. S2), suggesting that de novo protein synthesis is required for IL-1β-induced hepcidin expression.
A previous study revealed that IL-1β stimulates hepcidin expression by inducing BMP2 expression in Huh7 cells, a human liver-derived cell line (20). In addition, IL-1β up-regulated IL-6 expression in various cell lines, including Huh7 cells (21). Thus, it is possible that BMP2 and/or IL-6 are produced in response to IL-1β treatment in hepatocytes and that these molecules induce hepcidin expression in an autocrine manner.
IL-1β transiently increased BMP2 expression in HepG2 cells within 2 h of treatment initiation (Fig. 2A). In contrast, IL-1β did not increase Bmp2 expression in mouse hepatocytes (supplemental Fig. S3A), whereas IL-1β-induced Bmp2 expression peaked at 8 h in primary rat hepatocytes (supplemental Fig. S3B). After BMP forms a complex with its respective receptors, Smad1/5/8 is phosphorylated and activated, leading to transcriptional activation of hepcidin (11–13). The levels of phosphorylated Smad1/5/8 were slightly increased after IL-1β treatment in HepG2 cells (Fig. 2B), but significant phosphorylation of Smad1/5/8 was not induced in either mouse hepatocytes (supplemental Fig. S3C) or rat hepatocytes (supplemental Fig. S3D).
Figure 2.

IL-1β-induced hepcidin transcription via regions other than BMP-REs. A and B, HepG2 cells were treated with IL-1β (25 ng/ml) for the indicated time. A, expression of BMP2 was examined by RT-qPCR analysis. The expression level in the control cells prior to IL-1β treatment was defined as 1. The data are presented as the mean ± S.E. (n = 3). *, p < 0.05 and **, p < 0.01 versus cells treated without IL-1β at the respective time point. B, levels of phosphorylated Smad1/5/8 and STAT3 were examined by Western blot analysis with β-actin as the loading control. C and D, HepG2 cells were transfected with siRNA targeting the indicated gene. At 48 h after transfection, cells were treated with IL-1β (25 ng/ml) for 4 h. Expression of hepcidin was examined by RT-qPCR analysis. The expression level in cells transfected with siGFP and treated without IL-1β was set at 1. The data are presented as the mean ± S.E. (n = 3). *, p < 0.05 and **, p < 0.01 versus cells transfected with the respective siRNA without IL-1β treatment. † and ††, p < 0.05 and p < 0.01 versus cells with corresponding IL-1β treatments and transfected with siGFP. D, levels of phosphorylated Smad1/5/8 were examined by Western blot analysis with β-actin as the loading control. E and F, HepG2 cells were transfected with tk-Renilla-luc and either the indicated reporters (E) or hepcidin(−2018)-luc (F). At 4 h post-transfection, cells were treated with or without IL-1β (10 ng/ml) for 12 h. Cells were also pretreated with or without LDN-193189 (100 nm, F). Firefly luciferase activity normalized to Renilla luciferase activity was calculated, and the relative luciferase activity in untreated cells transfected with hepcidin(−2018)-luc was defined as 1. The data are presented as the mean ± S.E. (n = 3).
We next examined whether the induction of BMP2 by IL-1β has a role in hepcidin expression in HepG2 cells; siRNA targeting BMP2 was transfected to down-regulate BMP2 expression (supplemental Fig. S4). Although knockdown of the BMP2 gene decreased basal expression of hepcidin, IL-1β still increased expression of hepcidin (Fig. 2C) without an increase in phosphorylation of Smad1/5/8 (Fig. 2D). The role of the induced BMP2 in hepcidin transcription was further evaluated by luciferase-based reporter assays in HepG2 cells. BMP stimulates hepcidin transcription via BMP-REs 1 and 2 in the hepcidin promoter: BMP-RE1 spans nt −155 to −150, and BMP-RE2 spans nt −1678 to −1673 (22–24). IL-1β increased the luciferase activity of wild-type reporter as expected, suggesting that IL-1β-induced hepcidin expression is transcriptionally regulated (Fig. 2E). Mutations in either BMP-RE (mBMP-RE) decreased the basal transcription of hepcidin; however, their responsiveness to IL-1β (i.e. fold-induction of luciferase expression after IL-1β treatment) was not decreased but rather increased (wild-type reporter, 18-fold; reporter with mBMP-RE1, 41-fold; reporter with mBMP-RE2, 58-fold) (Fig. 2E). In addition, mutants of both BMP-RE1 and BMP-RE2 did not decrease the fold-induction in response to IL-1β (Fig. 2E). In contrast, mutations of both BMP-REs in the hepcidin promoter blunted the transcriptional response to either BMP2 or ALK3(QD) expression, the latter of which is a constitutively active ALK3 (25) (supplemental Fig. S5). Furthermore, treatment with LDN-193189, an inhibitor of BMP type I receptor (26), also decreased the basal transcription of hepcidin but did not prevent the responsiveness to IL-1β (Fig. 2F). We concluded that the induction of BMP2 and subsequent transcription of hepcidin via BMP-REs does not contribute to IL-1β-induced hepcidin expression based on the following results: 1) induction of Bmp2 expression was not detected in mouse hepatocytes irrespective of IL-1β-induced hepcidin expression; 2) Bmp2 expression was increased by IL-1β in rat hepatocytes; however, Smad1/5/8 phosphorylation was not detected, and the hepcidin induction was minimal; 3) knockdown of the BMP2 gene blocked IL-1β-induced phosphorylation of Smad1/5/8, but IL-1β still increased expression of hepcidin; and 4) neither BMP-RE mutations within the hepcidin promoter nor LDN-193189 treatment inhibited the transcriptional responsiveness of HepG2 cells to IL-1β.
IL-6 expression was increased by IL-1β treatment in both HepG2 cells and mouse hepatocytes (Fig. 3A and supplemental Fig. S6). In HepG2 cells, up-regulation of IL-6 expression was detected within 2 h after IL-1β treatment and maintained for at least 12 h (Fig. 3A). The rapid induction of IL-6 expression by IL-1β was transcriptionally regulated; IL-1β treatment increased the luciferase expression of the reporter containing the IL-6 promoter (supplemental Fig. S7A). There is a putative NF-κB site within the IL-6 gene located from nt −123 to −111 (supplemental Fig. S7B). Mutations of this potential NF-κB site blunted IL-1β-induced IL-6 transcription (supplemental Fig. 7A), suggesting that IL-1β transcriptionally stimulates IL-6 expression by activating the NF-κB pathway.
Figure 3.

IL-1β-induced hepcidin transcription is mainly mediated via regions other than the STAT-BS. A, HepG2 cells were treated with IL-1β (25 ng/ml) for the indicated time. Expression of IL-6 was examined by RT-qPCR analysis with the level in the control cells prior to IL-1β treatment defined as 1. The data are presented as the mean ± S.E. (n = 3). **, p < 0.01 versus cells treated without IL-1β at the respective time point. B, HepG2 cells were transfected with tk-Renilla-luc and the indicated reporters. At 4 h post-transfection, cells were subjected to the presence or absence of IL-1β (10 ng/ml) for 12 h. Firefly luciferase activity normalized to Renilla luciferase activity was calculated, and the relative luciferase activity in untreated cells transfected with hepcidin(−2018)-luc was defined as 1. The data are presented as the mean ± S.E. (n = 3).
Consistent with the induction of IL-6 by IL-1β, STAT3, a molecule that is phosphorylated in response to IL-6, showed increased phosphorylated levels in HepG2 cells (Fig. 2B); furthermore, IL-1β also increased phosphorylated STAT3 levels in rat hepatocytes but not mouse hepatocytes (supplemental Fig. S3, C and D). Previous studies have shown that IL-6 stimulated hepcidin transcription by activating STAT3 to promote its binding to the STAT-BS spanning nt −143 to −134 of the hepcidin promoter (11–13). Mutations of the STAT-BS slightly decreased the transcriptional responsiveness to IL-1β (Fig. 3B); fold-induction of luciferase expression after IL-1β treatment was 16-fold for the wild-type reporter and 11-fold for the reporter with mSTAT. IL-6-induced hepcidin transcription was expectedly inhibited by mutations of the STAT-BS (supplemental Fig. S8). These results suggest that IL-1β induces IL-6 expression but that this induction does not majorly contribute to hepcidin transcription.
C/EBPδ binding to the C/EBP-BS in the hepcidin promoter is responsible for IL-1β-induced hepcidin transcription
Provided that IL-1β transcriptionally regulates hepcidin expression, the region responsible for gene induction was next explored by using a series of deleted reporters (Fig. 4A). Deletion of a region spanning nt −2018 to −1419 decreased luciferase expression in the absence of IL-1β; this could be explained by the deletion of BMP-RE2 (24). However, the responsiveness to IL-1β was not decreased but rather increased (fold-induction of luciferase expression by IL-1β: hepcidin(−2018)-luc, 13-fold; hepcidin(−1418)-luc, 59-fold). Deletion of a region with the hepcidin promoter from nt −1418 to −356 did not affect IL-1β-induced hepcidin transcription. In contrast, deletion of the region spanning nt −355 to −271 significantly decreased the responsiveness to IL-1β (Fig. 4A). The nucleotide sequence of this region indicates an element closely related to a C/EBP-BS (27) that spans nt −329 to −320 (Fig. 4B). The mutations of this putative C/EBP-BS in the hepcidin promoter blunted the responsiveness to IL-1β (Fig. 4C).
Figure 4.

Involvement of the C/EBP-BS within the hepcidin promoter in IL-1β-induced hepcidin transcription. A, C, and D, HepG2 cells were transfected with tk-Renilla-luc and the indicated reporters and. At 4 h post-transfection, cells were treated with or without IL-1β (10 ng/ml) for 12 h. Firefly luciferase activity normalized to Renilla luciferase activity was calculated, and the relative luciferase activity in untreated cells transfected with hepcidin(−2018)-luc was defined as 1. The data are presented as the mean ± S.E. (n = 3). B, nucleotide sequence of the hepcidin promoter. The putative C/EBP-BS is boxed, and the BMP-REs and STAT-BS are underlined with solid lines and a dotted line, respectively.
We further evaluated the relative importance of C/EBP-BS and interactive relationships among the regulatory elements for hepcidin transcription, C/EBP-BS, BMP-REs, and STAT-BS (Fig. 4D). The mutations of C/EBP-BS greatly decreased responsiveness to IL-1β (fold-induction: wild-type, 20-fold; mC/EBP-BS, 4-fold), whereas that of STAT-BS slightly decreased (13-fold) and that of BMP-RE1,2 did not decrease but rather increased IL-1β responsiveness (38-fold); these results are consistent with those shown in Figs. 2E, 3B, and 4C. Combinational mutations of C/EBP-BS and STAT-BS further decreased responsiveness to IL-1β (2-fold), which was comparable with the results on the reporter with all mutations of C/EBP-BS, STAT-BS, and BMP-RE1,2 (2-fold). These results suggest that C/EBP-BS is the principle region responsible for IL-1β-induced hepcidin transcription and that STAT-BS is also involved in the responsiveness. The present results also suggest the independent role of C/EBP-BS, STAT-BS, and BMP-RE1,2 in IL-1β-induced hepcidin transcription. These results suggest the independent regulation of hepcidin transcription via C/EBP-BS, BMP-REs, and STAT-BS.
There are several C/EBP isoforms: C/EBPα, -β, -γ, -δ, -ϵ, and -ζ (28); the mRNA levels of C/EBPδ were increased within 2 h after IL-1β stimulation and maintained for at least 12 h (Fig. 5A). In contrast, C/EBPα expression was transiently decreased by IL-1β. In addition, IL-1β minimally affected the expression of C/EBPβ and C/EBPζ (>2-fold), and significant expression of neither C/EBPγ nor C/EBPϵ was detected (data not shown). IL-1β-induced C/EBPδ expression was also detected at the protein level (Fig. 5B). Similar to the response in HepG2 cells, clear up-regulation of C/EBPδ expression by IL-1β was detected in mouse and rat hepatocytes (supplemental Fig. S9). Although substantial up-regulation of C/EBPδ expression was detected even at 12 h after IL-1β treatment in HepG2 cells and mouse hepatocytes, the marked increase in C/EBPδ expression by IL-1β was relatively transient in rat primary hepatocytes; the reason of the differential response to IL-1β on C/EBPδ induction is not clear. Moreover, BAY 11-7085 blocked IL-1β-induced C/EBPδ expression at the mRNA level (Fig. 5C) as well as at the protein level (Fig. 5D), suggesting that activation of NFκB by IL-1β is involved in the C/EBPδ gene induction.
Figure 5.

IL-1β induces C/EBPδ. A and B, HepG2 cells were treated with or without IL-1β (25 ng/ml) for the indicated time. Expression of the C/EBP family of proteins was examined by RT-qPCR analysis (A) with the level in the control cells prior to IL-1β treatment defined as 1. The data are presented as the mean ± S.E. (n = 3). *, p < 0.05 and **, p < 0.01 versus cells treated without IL-1β at the respective time point. B, C/EBPδ was examined by Western blot analysis with β-actin as the loading control. C and D, HepG2 cells were pretreated with or without BAY 11-7085 (5 μm, BAY) followed by treatment with or without IL-1β (10 ng/ml). C/EBPδ expression was examined by RT-qPCR analysis (C). The levels in the control cells treated without BAY 11-7085 were defined as 1. The data are presented as the mean ± S.E. (n = 3). **, p < 0.01 versus cells treated with the respective inhibitor (vehicle or BAY 11-7085) without IL-1β treatment. †, p < 0.05 and ††, p < 0.01 versus cells with corresponding IL-1β treatments in the absence of BAY 11-7085. D, C/EBPδ and β-actin expression was examined by Western blot analysis.
To evaluate the involvement of C/EBPδ in IL-1β-induced hepcidin expression, we examined the effect of C/EBPδ gene knockdown. siRNA transfection targeting C/EBPδ decreased the C/EBPδ mRNA levels by ∼80% in HepG2 cells (Fig. 6A). IL-1β increased the expression level of C/EBPδ even in cells transfected with C/EBPδ-siRNA; this could be explained by the imperfect suppression of gene expression by siRNA. Down-regulation of C/EBPδ expression decreased the IL-1β-induced mRNA expression of hepcidin (Fig. 6B). Unlike IL-1β, BMP2 and IL-6 did not increase expression of C/EBPδ in HepG2 cells, irrespective of transfection with siRNA for C/EBPδ (Fig. 6C). In addition, the gene knockdown of C/EBPδ did not modulate responsiveness to BMP2 and IL-6 on hepcidin expression (Fig. 6D), suggesting that C/EBPδ is not involved in BMP2- or IL-6-mediated hepcidin expression. An oligonucleotide pulldown assay indicated that binding of C/EBPδ to the C/EBP-BS on the hepcidin promoter was IL-1β-dependent (Fig. 6E). All these results suggest that IL-1β stimulates hepcidin transcription by activating NF-κB, which in turn induces C/EBPδ production and its subsequent binding to the C/EBP-BS spanning nt −329 to −320 on the hepcidin promoter.
Figure 6.

IL-1β-induced C/EBPδ is responsible for hepcidin induction. A–D, HepG2 cells were transfected with siRNA targeting the indicated gene. At 48 h after transfection, cells were treated with IL-1β (25 ng/ml) (A and B), or BMP2 (100 ng/ml) or IL-6 (10 ng/ml) or both (C and D) for 12 (A and B) or 4 h (C and D). Expression of C/EBPδ (A and C) or hepcidin (B and D) was examined by RT-qPCR analysis. The expression level in cells transfected with siGFP and treated without ligand was set at 1. The data are presented as the mean ± S.E. (n = 3). *, p < 0.05 and **, p < 0.01 versus cells transfected with the respective siRNA without ligand treatment. † and ††, p < 0.05 and p < 0.01 versus cells with corresponding ligand treatments and transfected with siGFP. F, HepG2 cells were treated with or without IL-1β (10 ng/ml) for 4 h. The cell lysates were incubated with biotinylated oligonucleotide probe targeting the putative C/EBP-BS, and the presence of C/EBPδ in the DNA-protein complexes was examined by Western blot analysis.
The nucleotide sequence of the rat C/EBP-BS on the hepcidin promoter contributes to less efficient transcription of hepcidin by IL-1β
As shown above, hepcidin expression was slightly higher in rat hepatocytes treated with IL-1β than in control rat hepatocytes, resulting from reduction of hepcidin expression with time in the control cells; unlike mouse hepatocytes, hepcidin expression was not increased with time after IL-1β treatment in rat hepatocytes (Fig. 1A and supplemental Fig. S1, A and B). In view of the induction of C/EBPδ by IL-1β in primary rat hepatocytes (supplemental Fig. 9B), we hypothesized that the C/EBP-BS in the rat hepcidin gene cannot mediate efficient transcription in response to IL-1β. A comparison of the nucleotide sequence among human, mouse, and rat hepcidin promoters indicates that one nucleotide difference was detected between mouse C/EBP-BS and rat C/EBP-BS: the guanylic acid at nt −323 in the mouse hepcidin promoter is a thymidylic acid at nt −319 of the rat hepcidin promoter (Fig. 7A). Mutating the reporter construct containing the mouse hepcidin promoter to mimic that of the rat C/EBP-BS (i.e. mutation of the guanylic acid at nt −323 to a thymidylic acid) decreased IL-1β-induced hepcidin transcription (Fig. 7B). In contrast, mutating the rat hepcidin promoter in the luciferase reporter to mimic the sequence of the mouse-type C/EBP-BS increased IL-1β-induced luciferase expression (Fig. 7C). These changes in transcriptional activity of the swapped reporters are not nonspecific events; stimulating the BMP pathway by ALK3(QD) expression did not affect the transcription of these reporters (supplemental Fig. S10). Furthermore, mutations of the mouse and rat C/EBP-BSs to TTAtGGGcAA and TTAtGGTcAA, respectively (small characters indicate the mutated nucleic acids), decreased the responsiveness of both the mouse and rat hepcidin promoters to IL-1β (Fig. 7, B and C). All these results indicate that the reduced activity of IL-1β in up-regulating hepcidin expression in rat hepatocytes at least partly results from the nucleotide sequence of the rat C/EBP-BS.
Figure 7.

Inability of IL-1β to efficiently stimulate transcription of rat hepcidin. A, comparison of regulatory elements for transcription on human, mouse, or rat hepcidin promoter. Asterisk indicates common nucleotide among human, mouse, and rat hepcidin promoters. BMP-REs and STAT-BS are underlined with solid and dotted lines, respectively, and C/EBP-BS is boxed. B and C, HepG2 cells were transfected with tk-Renilla-luc and the indicated reporters. At 4 h post-transfection, cells were treated with or without IL-1β (10 ng/ml) for 12 h. Firefly luciferase activity was normalized to Renilla luciferase activity, and the relative luciferase activity in cells transfected with mouse hepcidin(−2018)-luc (B) or rat hepcidin(−1861)-luc (C) without IL-1β treatment was defined as 1. The data are presented as the mean ± S.E. (n = 3).
Induction of IL-1β in hepatocytes and Kupffer cells in response to LPS stimulates hepcidin transcription via C/EBPδ production
Previous studies have shown that IL-1β expression is up-regulated in response to inflammation in the liver (14, 29–31). To determine the source of IL-1β during inflammation, immunohistochemical analyses were performed in the livers of mice injected with either phosphate-buffered saline (PBS) or LPS (Fig. 8A, Table 1). Immunoreactive IL-1β was strongly detected in the cytoplasm of Kupffer cells from LPS-treated livers. Additionally, a large number of hepatocytes was positively stained by an anti-IL-1β antibody. IL-1β-positive hepatocytes were also slightly detected in control livers (Fig. 8A).
Figure 8.

Localization of IL-1β and C/EBPδ in LPS-treated livers and hepatic expression of genes in response to LPS. A–D, C57BL/6 mice were intraperitoneally injected with PBS or LPS (5 mg/kg). At 6 h post-injection, the livers were recovered. Localization of IL-1β (A) and C/ebpδ (B) was examined by immunohistochemistry. A representative result of the livers from LPS-treated mice is shown. Scale bar: 20 μm. A, upper: localization of IL-1β-positive cells (green, IL-1β antibody). Arrows, Kupffer cells (red, F4/80 antibody). B, upper: localization of C/ebpδ-positive cells (red). Area surrounded by a square in the left panel of each treatment was enlarged and shown in the right panel. Arrowheads: C/ebpδ-positive sinusoidal endothelial cells. C and D, expression of C/ebpδ (C) and hepcidin (D) was examined by RT-qPCR analysis. The levels in the control mice were defined as 1. The data are presented as the mean ± S.E. (n = 4). * and **, p < 0.05 and p < 0.01, respectively, versus PBS-treated liver. E and F, hepatocytes (HC) and non-parenchymal cells (NPC) were isolated from livers of ICR mice injected with PBS or LPS (5 mg/kg) intraperitoneally 6 h prior to sacrifice. Expression of IL-1β (E) and C/ebpδ (F) was examined by RT-qPCR analysis. The levels in the hepatocytes from PBS-treated liver were defined as 1. The data are presented as the mean ± S.E. (n = 4). * and **, p < 0.05 and p < 0.01, respectively, versus corresponding cells from PBS-treated liver. † and ††, p < 0.05 and p < 0.01 versus hepatocytes from liver with corresponding treatment.
Table 1.
Immunolocalization of IL-1β and C/EBPδ in livers treated with LPS
| IL-1β |
C/EBPδ |
|||
|---|---|---|---|---|
| Control | LPS | Control | LPS | |
| Hepatocytes | ± | ++ | ± | + |
| Kupffer cells | + | ++ | − | − |
| Hepatic stellate cells | − | ± | − | − |
| Endothelial cells | ||||
| Central vein | − | − | ± | ± |
| Interlobular arteriovenous | − | − | − | − |
| Sinusoid | − | − | ± | + |
| Vessel lumen | ||||
| Central vein | − | + | − | − |
| Interlobular arteriovenous | − | ± | − | − |
| Sinusoid | − | + | − | − |
Symbols represent: −, negative; ±, faint staining; +; moderate staining; ++, intense staining.
C/EBPδ was localized in hepatocytes that resided in a limited area around the central vein in control mice (Fig. 8B, Table 1). LPS increased the number of C/EBPδ-positive hepatocytes, and immunoreactive C/EBPδ was also detected in some but not all sinusoidal endothelial cells in LPS-treated mice. Consistent with the results of immunohistochemical analyses, the expression level of C/EBPδ was higher in LPS-treated livers than in control livers (Fig. 8C). Concurrently, hepcidin expression in the liver was significantly increased by LPS (Fig. 8D).
We also isolated hepatocytes and non-parenchymal cells from livers treated with or without LPS; cells of the hepatocyte fraction exclusively expressed albumin, a gene predominantly expressed in hepatocytes, but not stabilin-1 (endothelial cell marker) and Nramp-1 (Kupffer cell marker), and those of non-parenchymal fraction expressed vice versa (supplemental Fig. S11). LPS greatly increased the expression level of IL-1β in non-parenchymal cells; LPS-induced up-regulation of IL-1β was also detected in hepatocytes (Fig. 8E). In addition, expression of C/ebpδ was increased by LPS in hepatocytes as well as non-parenchymal cells (Fig. 8F).
In RAW264.7 cells, a mouse macrophage-like cell line, LPS increased the expression of IL-1β within 2 h after treatment, but the expression levels began to gradually decrease after 4 h (Fig. 9A). In fact, IL-1β protein was detected in culture supernatant from LPS-treated RAW264.7 cells but not from control RAW264.7 cells (Fig. 9B). Expression of IL-6 but not inhibin βB, a molecule consisting of activin B, was also increased by LPS in RAW264.7 cells (supplemental Fig. S12). In contrast, significant IL-1β induction was not detected in HepG2 cells in response to LPS treatment (data not shown). However, treatment with conditioned medium from LPS-treated RAW264.7 cells increased expression of IL-1β in primary mouse hepatocytes (Fig. 9C).
Figure 9.

IL-1β expression in macrophages and hepatocytes. A, RAW264. 7 cells were treated with or without LPS (100 ng/ml) for the indicated time. B and C, conditioned media from RAW264.7 cells treated with (CM-LPS) or without (CM-C) LPS (100 ng/ml) for 30 h were prepared as described under “Experimental procedures.” B, an equal volume of the conditioned media was subjected to Western blot analysis to detect IL-1β. HepG2 cells were treated with the conditioned medium from RAW264.7 cells for 24 h, and the expression of IL-1β was examined by RT-qPCR analysis. The levels in the control cells prior to IL-1β treatment (A) or in cells treated with control conditioned medium from RAW264.7 cells (CM-C) (C) were defined as 1. The data are presented as the mean ± S.E. (n = 3). **, p < 0.05 versus cells treated without IL-1β at the respective time points (A) or cells treated with control conditioned medium from RAW264.7 cells (C).
Conditioned medium from LPS-treated RAW264.7 cells potentiated the induction of C/EBPδ (Fig. 10A) and hepcidin (Fig. 10B) gene expression; this activity was inhibited by BAY 11-7085 in HepG2 cells. Primary mouse hepatocytes also showed an increase in C/EBPδ and hepcidin expression after treatment with conditioned medium from LPS-treated RAW264.7 cells (supplemental Fig. S13). Furthermore, either mutations of the C/EBP-BS on the hepcidin promoter or down-regulation of C/EBPδ expression by siRNA targeting C/EBPδ decreased the ability of conditioned medium from LPS-treated RAW264.7 cells to induce efficient hepcidin transcription and expression (Fig. 10, C and D). Based on these data and the results of IL-1β induction during hepatic inflammation, the induced IL-1β expression in Kupffer cells and hepatocytes stimulated hepcidin transcription via C/EBPδ production in an autocrine/paracrine manner.
Figure 10.

Induction of hepcidin gene transcription in HepG2 cells by activated macrophages. A and B, conditioned media from RAW264. 7 cells treated with (CM-LPS) or without (CM-C) LPS (100 ng/ml) for 30 h were prepared as described under “Experimental procedures.” A and B, HepG2 cells were pre-treated with or without BAY 11-7085 (5 μm, BAY) followed by treatment with or without conditioned media from RAW264.7 cells for 24 h. The expression of C/EBPδ (A) and hepcidin (B) was examined by RT-qPCR analysis with the level in cells treated with control conditioned medium for 12 h defined as 1. The data are presented as the mean ± S.E. (n = 3). **, p < 0.01 versus cells treated with the respective inhibitor (vehicle or BAY 11-7085) and control conditioned medium. †, p < 0.05 and ††, p < 0.01 versus cells treated with the corresponding conditioned medium from RAW264.7 cells in the absence of BAY 11-7085. C, HepG2 cells were transfected with the indicated reporters and CMV-βGal. At 4 h post-transfection, cells were treated with the indicated conditioned medium from RAW264.7 cells for 24 h. Firefly luciferase activity was normalized to β-galactosidase, and the relative luciferase activity in cells transfected with hepcidin(−2018)-luc and CM-C was defined as 1. The data are presented as the mean ± S.E. (n = 3). D, HepG2 cells were transfected with siRNA targeting the indicated gene. At 48 h after transfection, cells were treated with either CM-C or CM-LPS for 24 h. *, p < 0.05 and **, p < 0.01 versus cells transfected with the corresponding siRNA (GFP or C/EBPδ) and treated with control conditioned medium. ††, p < 0.01 versus cells treated with respective conditioned media and transfected with siRNA targeting GFP.
IL-1β enhances hepcidin expression induced by activin B and IL-6
Various molecules are produced during inflammation; among them, activin B and IL-6 stimulate hepcidin transcription via BMP-REs and STAT-BS, respectively (14, 32). We explored whether IL-1β enhances activin B- or IL-6-induced hepcidin transcription and expression (Fig. 11). IL-1β increased activin B- or IL-6-induced hepcidin expression and further enhanced hepcidin expression induced by co-treatment with activin B and IL-6 (Fig. 11A). Similar results were also obtained by hepcidin transcription assays (Fig. 11B). These results indicate that molecules produced during hepatic inflammation could independently activate hepcidin transcription, leading to excessive hepcidin production.
Figure 11.

Enhancement of hepcidin expression and transcription by IL-1β, activin B, and IL-6. A, HepG2 cells were treated with the indicated combination of IL-1β (10 ng/ml), activin B (ActB, 50 ng/ml), and IL-6 (10 ng/ml) for 12 h. Expression of hepcidin was examined by RT-qPCR analysis with the level in the control cells treated without ligand defined as 1. The data are presented as the mean ± S.E. (n = 3). **, p < 0.01 versus cells treated without IL-1β, activin B, or IL-6. B, HepG2 cells were transfected with tk-Renilla-luc and the indicated reporters. At 4 h post-transfection, cells were treated with the indicated combination of IL-1β (10 ng/ml), activin B (ActB, 50 ng/ml), and IL-6 (10 ng/ml) for 12 h. The firefly luciferase activity normalized to Renilla luciferase activity was calculated, and the relative luciferase activity in cells transfected with hepcidin(−2018)-luc in the absence of activin B, IL-6, and IL-1β was defined as 1. The data are presented as the mean ± S.E. (n = 3).
Discussion
Hepcidin is a liver-derived hormone that regulates plasma iron levels, and aberrant hepcidin expression leads to a severe disturbance of the intestinal absorption of iron and iron release from macrophages, these events indicate the central role of hepcidin in homeostatic regulation of iron metabolism (11–13). Previous studies have extensively revealed that hepcidin expression is transcriptionally regulated via BMP-REs and STAT-BS on the hepcidin promoter (11–13). The present study reveals that: 1) IL-1β up-regulates hepcidin expression by stimulating transcription; 2) BMP-REs on the hepcidin promoter are involved in IL-1β-induced hepcidin transcription, whereas the STAT-BS slightly participate in the transcriptional regulation resulting from stimulation of IL-6 production; 3) a C/EBP-BS spanning from nt −329 to −320 is essential for transcriptional activation; 4) IL-1β induces expression of C/EBPδ, which binds to the C/EBP-BS on the hepcidin promoter to activate transcription; 5) IL-1β is localized in Kupffer cells in basal murine livers, and LPS stimulation increased IL-1β expression in Kupffer cells as well as in hepatocytes; and 6) molecules produced during hepatic inflammation such as IL-1β, activin B, and IL-6 cooperatively stimulate hepcidin expression through distinct transcriptional mechanisms. Our results shown here indicate that Kupffer cells sense a proinflammatory stimulus to accelerate IL-1β production, leading to hepcidin production through up-regulation of C/EBPδ expression in hepatocytes. In addition, IL-1β-induced IL-6 production slightly contributes to hepcidin transcription via STAT-BS (Fig. 12). The relay of the proinflammatory signal from Kupffer cells to hepatocytes via IL-1β leads to the excessive production of hepcidin.
Figure 12.
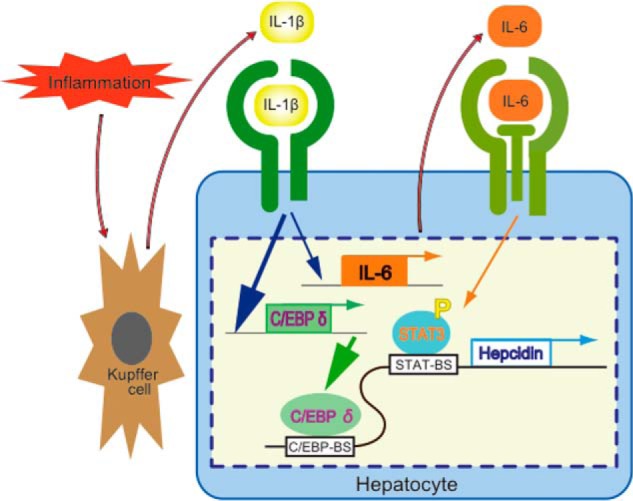
Schematic model of IL-1β function in the liver. IL-1β is expressed in Kupffer cells and hepatocytes in response to hepatic inflammation. This induced IL-1β stimulates the expression of C/EBPδ and IL-6; the induced C/EBPδ enhances hepcidin transcription via the C/EBP-BS on the hepcidin promoter spanning nt −329 to −320, and the induced IL-6 stimulates STAT3 phosphorylation and hepcidin transcription via the STAT-BS spanning nt −143 to −134 slightly. Note the species difference on IL-1β-induced hepcidin expression between mouse hepatocytes and rat hepatocytes, resulting from difference of the nucleotide sequence of C/EBP-BS.
Previously, it was shown that interferon (IFN) γ and Mycobacterium tuberculosis could increase hepcidin transcription in RAW264.7 cells via the putative NF-κB-binding site spanning nt −556 to −547 on the hepcidin promoter (33). However, this region is not involved in IL-1β-induced hepcidin transcription in hepatocytes, as deletion of this region did not affect hepcidin transcription induced by IL-1β (Fig. 4A). Although IL-6-stimulated hepcidin transcription has been well established (supplemental Figs. 8 and 11–13), hepcidin expression was not increased by IL-6 in macrophages (34, 35). These results reveal the distinct regulatory mechanisms of hepcidin transcription between hepatocytes and macrophages, implying cell type-dependent regulation of hepcidin transcription; the relative importance of which region is most responsible for hepcidin transcription may be different between cell types. In fact, in alveolar macrophages, IL-1 did not induce hepcidin expression (34). Considering that hepcidin is predominantly expressed in hepatocytes (12, 36), the results of this study clarify the primary regulatory system of hepcidin expression during inflammation.
A previous study has shown the involvement of C/EBPα in hepcidin expression: overexpression of C/EBPα-stimulated hepcidin transcription in U-2 OS osteosarcoma cells (37). Binding of C/EBPα to the C/EBP-BS was verified in rat liver nuclear extracts, but the functional role of this region in hepcidin transcription was not determined. Furthermore, how this C/EBPα activity is regulated was unclear (37). Considering that C/EBPα expression was decreased in response to IL-1β (Fig. 5A), IL-1β-induced hepcidin transcription is unlikely to be mediated by C/EBPα in hepatocytes during inflammation.
Our results revealed that C/EBPδ expression is up-regulated in response to LPS-induced IL-1β expression, which led to efficient binding of C/EBPδ to the C/EBP-BS on the hepcidin promoter. C/EBPδ has an intrinsic ability to bind to the C/EBP-BS (38). However, the activity of C/EBPδ as a transcription factor is enhanced through post-translational modifications (39, 40). As compared with the increase in C/EBPδ expression in response to IL-1β treatment, more C/EBPδ bound to the C/EBP-BS (Fig. 6E). Thus, IL-1β may exert activities not only to increase C/EBPδ expression but also to promote C/EBPδ activity.
Etiological studies have shown that increased concentrations of serum IL-1β were detected in patients with coronary artery disease, schizophrenia, insulin-dependent diabetes, and Alzheimer disease (41–44). Patients with Alzheimer disease suffer from anemia with decreased plasma iron levels (45–46). In addition, hepcidin has been hypothesized to be involved in dysfunctional iron metabolism in patients with Alzheimer disease (47). In various pathological conditions with increased IL-1β levels, IL-1β-mediated hepcidin expression may partially contribute to aberrant iron metabolism.
IL-1β induction was detected not only in Kupffer cells but also in hepatocytes from LPS-treated mice. In fact, IL-1β expression was up-regulated in RAW264.7 cells treated with LPS (Fig. 9A). However, LPS did not induce IL-1β expression in HepG2 cells (data not shown). These results suggest that IL-1β induction in hepatocytes but not Kupffer cells during hepatic inflammation is indirect. Considering that LPS stimulates Kupffer cells as well as sinusoidal endothelial cells (14, 48–50), various molecules secreted from the non-parenchymal cells are possibly responsible for IL-1β induction in hepatocytes. Transcription of IL-1β is stimulated by activation of NF-κB (51); in fact, IL-1β transcription was stimulated by IL-1 in U937 myeloid cells in an autoregulatory manner (52). However, we could not detect significant induction of IL-1β in response to IL-1β in HepG2 cells (data not shown). Future studies should clarify the regulation of IL-1β expression in hepatocytes at the molecular level.
Previous studies revealed that activin B and IL-6 production are increased in the liver during inflammation and that these cytokines stimulate hepcidin transcription via BMP-REs and STAT-BS, respectively, in hepatocytes (14, 24, 32). Activin B and IL-6 independently increased hepcidin expression, and co-treatment with activin B and IL-6 further enhanced hepcidin expression (53). The present study expands the available information on regulating hepcidin expression in hepatocytes during inflammation, as inflammation-induced IL-1β production leads to the stimulation of hepcidin transcription in hepatocytes via the C/EBP-BS on the hepcidin promoter. The concurrent stimulation of the three cis-elements cooperatively enhanced hepcidin transcription and expression in hepatocytes compared with the stimulation of each individual element (Fig. 11); the molecules induced during inflammation possibly resulted in increased hepcidin expression in hepatocytes. Considering that hepcidin was originally identified as an antimicrobial peptide (54–56), enhanced hepcidin production via these three elements may be helpful to exclude pathogens but could potentially promote anemia of inflammation through overproduction of hepcidin.
Experimental procedures
Materials and methods
The following reagents were purchased: recombinant human IL-1β was from RayBiotech, Inc. (Norcross, GA); recombinant mouse IL-1β and rat IL-1β were from Bioworld Technology (Louis Park, MN); recombinant human IL-6, recombinant activin B, and goat polyclonal antibody against IL-1β (AF-401-NA) was from R&D Systems (Minneapolis, MN); recombinant human BMP2 was from PeproTech (Rocky Hill, NJ); cycloheximide and control mouse IgG were from Sigma; LDN-193189 was from Stemgent (San Diego, CA); BAY 11-7085 was from Cayman Chemical (Ann Arbor, MI); rabbit polyclonal antibody against phospho-Smad1 (Ser463/Ser465)/Smad5 (Ser463/Ser465)/Smad8 (Ser426/Ser428), and mouse monoclonal antibody against phospho-STAT3 (Tyr705) (3E2) were from Cell Signaling Technology (Danvers, MA); rabbit polyclonal antibody against human C/EBPδ that cross-reacts with mouse C/EBPδ and was used in immunohistochemical analysis, mouse monoclonal antibody against β-actin (AC-15), and rat monoclonal antibody against F4/80 (CI:A3-1) were from Abcam (Cambridge, MA); rabbit polyclonal antibody against C/EBPδ (M-17) that was used in Western blot analysis was from Santa Cruz Biotechnology (Santa Cruz, CA); Alexa 488 donkey anti-goat IgG antibody and Alexa 594 donkey anti-rat IgG antibody were from Thermo Fisher Scientific (Waltham, MA).
Cell isolation and cell culture
All procedures for animal use were approved by the Kyoto University Animal Experiment Committee. Primary hepatocytes from the livers of 4-week-old male Sprague-Dawley rats were collected as previously described (57). Primary hepatocytes were also recovered from 5–8-week-old male ICR mice by a similar procedure to isolate primary rat hepatocytes. Isolated hepatocytes were plated in 12-well collagen-coated plates at 1.5 × 105 cells per well and cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum (FBS), insulin, dexamethasone, and antibiotics. Adherent cells were immediately used. Non-parenchymal cells were isolated as supernatant fraction to recover hepatocyte fraction as cell pellet of liver digested with collagenase after 50 × g for 3 min. Subsequently, non-parenchymal cells were washed with HBSS and pelleted at 800 × g for 10 min at 4 °C. Furthermore, non-parenchymal cells resuspended in HBSS were layered onto a 2-step Percoll gradient (25% Percoll layer and 50% Percoll layer, respectively), followed by centrifugation at 800 × g for 30 min to purify further. Non-parenchymal cells reside in the 25% Percoll layer as well as the 50% Percoll layer; interface of HBSS and 25% Percoll contains cell debris, red blood cells are at the bottom of the 50% Percoll layer. After recovery of non-parenchymal cells, the cells were pelleted at 800 × g for 10 min at 4 °C. HepG2 human hepatoma cells and RAW264.7 mouse macrophage-like cells were cultured in DMEM supplemented with 10% heat-inactivated FBS and antibiotics.
Preparation of conditioned medium from RAW264.7 cells
RAW264.7 cells were treated with or without LPS (100 ng/ml) for 30 h in serum-free DMEM. The conditioned medium of LPS-treated cells (CM-LPS) and control cells (CM-C) were concentrated by Centriprep-10 (Merck, Darmstadt, Germany), and the solvent was replaced with HEPES buffer (21 mm HEPES, pH 7.5, 0.7 mm Na2HPO4, 137 mm NaCl, 5 mm KCl, 6 mm dextrose). HepG2 cells were treated with the conditioned medium; concentrations of CM-C and CM-LPS were equivalent to the conditioned medium of RAW264.7 cells.
siRNA transfection
HepG2 cells (3 × 104 cells per well) were seeded onto 24-well plates. Cells were reverse-transfected with 2 μl of Lipofectamine RNAi Max (Invitrogen) and 50 pmol of siRNA. The nucleotide sequence of the double-stranded siRNA is shown in supplemental Table S1. At 48 h after seeding, cells were serum-starved with medium containing 0.2% FBS for 4 h followed by treatment with IL-1β (25 ng/ml) for 12 h.
RNA isolation and RT quantitative PCR
Total RNA isolation, cDNA synthesis, and real-time quantitative PCR (qPCR) were performed as previously described (57). The sequences of the oligonucleotide primers are shown in supplemental Table S2. The ΔΔCt method was used to normalize the levels of the target transcripts to the TBP levels (58).
Western blot analyses
Western blot analyses were performed as previously described (59). The immunoreactive proteins were visualized using the ECL Select Western blotting detection system (GE Healthcare, Buckinghamshire, UK) according to the manufacturer's protocol.
Plasmids and luciferase reporter assay
Constitutively active ALK3 (ALK3(QD)) (25) was kindly provided by Dr. K. Miyazono. A mouse hepcidin promoter fragment (nt −2018 to −35) or rat hepcidin promoter fragment (nt −1861 to −35) was inserted into the luciferase reporter vector pGL4 (mhepcidin-luc or rhepcidin-luc, respectively). In addition, a mouse IL-6 promoter fragment (nt −300 to −79) was inserted into pGL4. The translation initiation site is numbered as +1. Mutations were prepared by PCR-based methods. The nucleotide sequences of the reporter constructs were verified by DNA sequencing. HepG2 cells (6 × 104 cells per well) were seeded onto 24-well plates. The next day, 0.5 μg of a pGL4-based hepcidin reporter and either 0.5 μg of Renilla luciferase expression vector under the control of a thymidine kinase promoter (tk-Renilla-luc) or 0.1 μg of a β-galactosidase expression plasmid under control of a cytomegalovirus-derived promoter (CMV-βGal) were transfected into cells in 0.2% FBS medium using polyethylenimine Max reagent (Polysciences, Warrington, PA). After 4 h, cells were stimulated with ligands or the culture supernatant from RAW264.7 cells. Firefly luciferase activity was normalized to either Renilla luciferase activity or β-galactosidase activity as appropriate.
Oligo DNA pulldown assay
HepG2 cells were scraped from the plates and centrifuged at 1500 rpm. The cell pellets were resuspended in lysis buffer (20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% (w/v) Triton X-100, 1 mm PMSF, 1% (v/v) aprotinin, 1 mm Na3VO4), vortexed, incubated on ice for 15 min, and centrifuged to remove cell debris. The supernatants were treated with 50 pmol of 5′-biotinylated probe with or without 500 pmol of unlabeled probe for 12 h at 4 °C followed by an incubation with 50 μl of 25% (v/v) streptavidin-agarose beads for 1 h at 4 °C. Subsequently, the beads were washed with lysis buffer three times, and the proteins were eluted into 6× SDS-PAGE sample buffer. C/EBPδ binding was analyzed by Western blotting. The probe was prepared from the following oligonucleotides: 5′-catcgtgatggggaaagggctcccc-3′ (forward, 5′-biotinylated) and 5′-atctggggagccctttccccatcac-3′ (reverse). The probe included the C/EBP-BS from the human hepcidin promoter.
Immunohistochemistry
To identify the localization of IL-1β in the liver, male C57BL/6 mice (aged 9 weeks) were intraperitoneally injected with LPS (5 mg/kg) or PBS (n = 4 per group). At 6 h after injection, livers were fixed with Bouin's solution, embedded in paraffin, and sliced into 4-μm sections (14). After the sections were deparaffinized, they were treated with 3% normal bovine serum for 1 h at room temperature followed by incubation with primary antibodies (anti-mouse IL-1β antibody (5 μg/ml), anti-mouse F4/80 antibody (5 μg/ml), and anti-C/EBPδ antibody (5 μg/ml)) for 17 h at 4 °C in a humidified atmosphere. Subsequently, the sections were reacted with secondary antibodies (Alexa 488 anti-goat IgG (2 μg/ml) and Alexa 594 anti-rat IgG (2 μg/ml). Sections were observed by confocal microscopy (LSM710, Carl Zeiss, Oberkochen, Germany).
Statistical analysis
The data are expressed as the mean ± S.E. The data regarding gene expression were log-transformed to provide an approximation of a normal distribution before analysis. Differences in the gene expression among the cells were examined using unpaired t-tests. Differences of p < 0.05 were considered significant.
Author contributions
Y. K. and M. F. designed the experiments. Y. K., M. M., M. S., and O. H. conducted the experiments. Y. K., M. M., M. S., O. H., T. M., and M. F. analyzed the data. Y. K. and M. F. wrote the main manuscript text. All authors reviewed the manuscript.
Supplementary Material
Acknowledgment
We thank Dr. Kohei Miyazono for providing the ALK3(QD) expression vector.
This work was supported in part by Japan Society for the Promotion of Science KAKENHI Grant 15J02394 (to Y. K.), research project Grant 2014-K-6 from Azabu University (to M. M.), and a grant from the Tokyo Institute of Technology (to Y. K.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Tables S1 and S2 and Figs. S1–S13.
- BMP
- bone morphogenetic protein
- BMP-RE
- BMP-responsive element
- C/EBP
- CCAAT enhancer-binding protein
- C/EBP-BS
- CCAAT enhancer-binding protein-binding site
- HBSS
- Hanks' balanced salt solution
- qPCR
- quantitative PCR
- nt
- nucleotide(s).
References
- 1. Beutler B. (2004) Inferences, questions and possibilities in Toll-like receptor signalling. Nature 430, 257–263 [DOI] [PubMed] [Google Scholar]
- 2. Chen G. Y., and Nuñez G. (2010) Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol. 10, 826–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barton G. M. (2008) A calculated response: control of inflammation by the innate immune system. J. Clin. Invest. 118, 413–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Medzhitov R. (2008) Origin and physiological roles of inflammation. Nature 454, 428–435 [DOI] [PubMed] [Google Scholar]
- 5. Hunter P. (2012) The inflammation theory of disease. The growing realization that chronic inflammation is crucial in many diseases opens new avenues for treatment. EMBO Rep. 13, 968–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mantovani A., Allavena P., Sica A., and Balkwill F. (2008) Cancer-related inflammation. Nature 454, 436–444 [DOI] [PubMed] [Google Scholar]
- 7. Marques-Rocha J. L., Samblas M., Milagro F. I., Bressan J., Martínez J. A., and Marti A. (2015) Noncoding RNAs, cytokines, and inflammation-related diseases. FASEB J. 29, 3595–3611 [DOI] [PubMed] [Google Scholar]
- 8. Nemeth E., and Ganz T. (2014) Anemia of inflammation. Hematol. Oncol. Clin. North Am. 28, 671–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang C. Y., and Babitt J. L. (2016) Hepcidin regulation in the anemia of inflammation. Curr. Opin. Hematol. 23, 189–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weiss G. (2015) Anemia of chronic disorders: new diagnostic tools and new treatment strategies. Semin. Hematol. 52, 313–320 [DOI] [PubMed] [Google Scholar]
- 11. Hentze M. W., Muckenthaler M. U., Galy B., and Camaschella C. (2010) Two to tango: regulation of mammalian iron metabolism. Cell 142, 24–38 [DOI] [PubMed] [Google Scholar]
- 12. Meynard D., Babitt J. L., and Lin H. Y. (2014) The liver: conductor of systemic iron balance. Blood 123, 168–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Michels K., Nemeth E., Ganz T., and Mehrad B. (2015) Hepcidin and host defense against infectious diseases. PLoS Pathog. 11, e1004998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kanamori Y., Sugiyama M., Hashimoto O., Murakami M., Matsui T., and Funaba M. (2016) Regulation of hepcidin expression by inflammation-induced activin B. Sci. Rep. 6, 38702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee P., Peng H., Gelbart T., Wang L., and Beutler E. (2005) Regulation of hepcidin transcription by interleukin-1 and interleukin-6. Proc. Natl. Acad. Sci. U.S.A. 102, 1906–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yamada M., Nishizawa M., Nakatake R., Habara K., Yoshida H., Ozaki T., Matsui K., Hamada Y., Kamiyama Y., Ito S., and Okumura T. (2007) Characterization of alternatively spliced isoforms of the type I interleukin-1 receptor on iNOS induction in rat hepatocytes. Nitric Oxide 17, 98–105 [DOI] [PubMed] [Google Scholar]
- 17. Speaker K. J., and Fleshner M. (2012) Interleukin-1β: a potential link between stress and the development of visceral obesity. BMC Physiol. 12, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pierce J. W., Schoenleber R., Jesmok G., Best J., Moore S. A., Collins T., and Gerritsen M. E. (1997) Novel inhibitors of cytokine-induced IκBα phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J. Biol. Chem. 272, 21096–21103 [DOI] [PubMed] [Google Scholar]
- 19. Baliga B. S., Pronczuk A. W., and Munro H. N. (1969) Mechanism of cycloheximide inhibition of protein synthesis in a cell-free system prepared from rat liver. J. Biol. Chem. 244, 4480–4489 [PubMed] [Google Scholar]
- 20. Shanmugam N. K., Chen K., and Cherayil B. J. (2015) Commensal bacteria-induced interleukin 1β (IL-1β) secreted by macrophages up-regulates hepcidin expression in hepatocytes by activating the bone morphogenetic protein signaling pathway. J. Biol. Chem. 290, 30637–30647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hu Y., Xue J., Yang Y., Zhou X., Qin C., Zheng M., Zhu H., Liu Y., Liu W., Lou G., Wang J., Wu S., Chen Z., and Chen F. (2015) Lipocalin 2 upregulation protects hepatocytes from IL1-β-induced stress. Cell Physiol. Biochem. 36, 753–762 [DOI] [PubMed] [Google Scholar]
- 22. Casanovas G., Mleczko-Sanecka K., Altamura S., Hentze M. W., and Muckenthaler M. U. (2009) Bone morphogenetic protein (BMP)-responsive elements located in the proximal and distal hepcidin promoter are critical for its response to HJV/BMP/SMAD. J. Mol. Med. 87, 471–480 [DOI] [PubMed] [Google Scholar]
- 23. Truksa J., Lee P., and Beutler E. (2009) Two BMP responsive elements, STAT, and bZIP/HNF4/COUP motifs of the hepcidin promoter are critical for BMP, SMAD1, and HJV responsiveness. Blood 113, 688–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kanamori Y., Murakami M., Matsui T., and Funaba M. (2015) Role of a TPA-responsive element in hepcidin transcription induced by the bone morphogenetic protein pathway. Biochem. Biophys. Res. Commun. 466, 162–166 [DOI] [PubMed] [Google Scholar]
- 25. Imamura T., Takase M., Nishihara A., Oeda E., Hanai J., Kawabata M., and Miyazono K. (1997) Smad6 inhibits signalling by the TGF-β superfamily. Nature 389, 622–626 [DOI] [PubMed] [Google Scholar]
- 26. Yu P. B., Deng D. Y., Lai C. S., Hong C. C., Cuny G. D., Bouxsein M. L., Hong D. W., McManus P. M., Katagiri T., Sachidanandan C., Kamiya N., Fukuda T., Mishina Y., Peterson R. T., and Bloch K. D. (2008) BMP type I receptor inhibition reduces heterotopic ossification. Nat. Med. 14, 1363–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Akira S., Isshiki H., Sugita T., Tanabe O., Kinoshita S., Nishio Y., Nakajima T., Hirano T., and Kishimoto T. (1990) A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. EMBO J. 9, 1897–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nerlov C. (2007) The C/EBP family of transcription factors: a paradigm for interaction between gene expression and proliferation control. Trends Cell Biol. 17, 318–324 [DOI] [PubMed] [Google Scholar]
- 29. DeCicco L. A., Rikans L. E., Tutor C. G., and Hornbrook K. R. (1998) Serum and liver concentrations of tumor necrosis factor α and interleukin-1β following administration of carbon tetrachloride to male rats. Toxicol. Lett. 98, 115–121 [DOI] [PubMed] [Google Scholar]
- 30. O'Bryan M. K., Gerdprasert O., Nikolic-Paterson D. J., Meinhardt A., Muir J. A., Foulds L. M., Phillips D. J., de Kretser D. M., and Hedger M. P. (2005) Cytokine profiles in the testes of rats treated with lipopolysaccharide reveal localized suppression of inflammatory responses. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R1744–R1755 [DOI] [PubMed] [Google Scholar]
- 31. Csak T., Ganz M., Pespisa J., Kodys K., Dolganiuc A., and Szabo G. (2011) Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 54, 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wrighting D. M., and Andrews N. C. (2006) Interleukin-6 induces hepcidin expression through STAT3. Blood 108, 3204–3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sow F. B., Alvarez G. R., Gross R. P., Satoskar A. R., Schlesinger L. S., Zwilling B. S., and Lafuse W. P. (2009) Role of STAT1, NF-κB, and C/EBPβ in the macrophage transcriptional regulation of hepcidin by mycobacterial infection and IFN-γ. J. Leukoc. Biol. 86, 1247–1258 [DOI] [PubMed] [Google Scholar]
- 34. Nguyen N. B., Callaghan K. D., Ghio A. J., Haile D. J., and Yang F. (2006) Hepcidin expression and iron transport in alveolar macrophages. Am. J. Physiol. Lung Cell Mol. Physiol. 291, L417–L425 [DOI] [PubMed] [Google Scholar]
- 35. Sow F. B., Florence W. C., Satoskar A. R., Schlesinger L. S., Zwilling B. S., and Lafuse W. P. (2007) Expression and localization of hepcidin in macrophages: a role in host defense against tuberculosis. J. Leukoc. Biol. 82, 934–945 [DOI] [PubMed] [Google Scholar]
- 36. Zhang A. S., Xiong S., Tsukamoto H., and Enns C. A. (2004) Localization of iron metabolism-related mRNAs in rat liver indicate that HFE is expressed predominantly in hepatocytes. Blood 103, 1509–1514 [DOI] [PubMed] [Google Scholar]
- 37. Courselaud B., Pigeon C., Inoue Y., Inoue J., Gonzalez F. J., Leroyer P., Gilot D., Boudjema K., Guguen-Guillouzo C., Brissot P., Loréal O., and Ilyin G. (2002) C/EBPα regulates hepatic transcription of hepcidin, an antimicrobial peptide and regulator of iron metabolism. J. Biol. Chem. 277, 41163–41170 [DOI] [PubMed] [Google Scholar]
- 38. Shi X. M., Blair H. C., Yang X., McDonald J. M., and Cao X. (2000) Tandem repeat of C/EBP binding sites mediates PPARγ2 gene transcription in glucocorticoid-induced adipocyte differentiation. J. Cell Biochem. 76, 518–527 [DOI] [PubMed] [Google Scholar]
- 39. Ko C. Y., Chang L. H., Lee Y. C., Sterneck E., Cheng C. P., Chen S. H., Huang A. M., Tseng J. T., and Wang J. M. (2012) CCAAT/enhancer binding protein δ (CEBPD) elevating PTX3 expression inhibits macrophage-mediated phagocytosis of dying neuron cells. Neurobiol. Aging 33, 422.e11–e25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ko C. Y., Wang W. L., Wang S. M., Chu Y. Y., Chang W. C., and Wang J. M. (2014) Glycogen synthase kinase-3β-mediated CCAAT/enhancer-binding protein δ phosphorylation in astrocytes promotes migration and activation of microglia/macrophages. Neurobiol. Aging 35, 24–34 [DOI] [PubMed] [Google Scholar]
- 41. Hasdai D., Scheinowitz M., Leibovitz E., Sclarovsky S., Eldar M., and Barak V. (1996) Increased serum concentrations of interleukin-1β in patients with coronary artery disease. Heart 76, 24–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schmitt A., Bertsch T., Tost H., Bergmann A., Henning U., Klimke A., and Falkai P. (2005) Increased serum interleukin-1β and interleukin-6 in elderly, chronic schizophrenic patients on stable antipsychotic medication. Neuropsychiatr. Dis. Treat. 1, 171–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dogan Y., Akarsu S., Ustundag B., Yilmaz E., and Gurgoze M. K. (2006) Serum IL-1β, IL-2, and IL-6 in insulin-dependent diabetic children. Mediators Inflamm. 2006, 59206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Forlenza O. V., Diniz B. S., Talib L. L., Mendonça V. A., Ojopi E. B., Gattaz W. F., and Teixeira A. L. (2009) Increased serum IL-1β level in Alzheimer's disease and mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 28, 507–512 [DOI] [PubMed] [Google Scholar]
- 45. Faux N. G., Rembach A., Wiley J., Ellis K. A., Ames D., Fowler C. J., Martins R. N., Pertile K. K., Rumble R. L., Trounson B., Masters C. L., AIBL Research Group, and Bush A. I. (2014) An anemia of Alzheimer's disease. Mol. Psychiatry 19, 1227–1234 [DOI] [PubMed] [Google Scholar]
- 46. Hare D. J., Doecke J. D., Faux N. G., Rembach A., Volitakis I., Fowler C. J., Grimm R., Doble P. A., Cherny R. A., Masters C. L., Bush A. I., and Roberts B. R. (2015) Decreased plasma iron in Alzheimer's disease is due to transferrin desaturation. ACS Chem. Neurosci. 6, 398–402 [DOI] [PubMed] [Google Scholar]
- 47. Myhre O., Utkilen H., Duale N., Brunborg G., and Hofer T. (2013) Metal dyshomeostasis and inflammation in Alzheimer's and Parkinson's diseases: possible impact of environmental exposures. Oxid. Med. Cell Longev. 2013, 726954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Knolle P. A., and Gerken G. (2000) Local control of the immune response in the liver. Immunol. Rev. 174, 21–34 [DOI] [PubMed] [Google Scholar]
- 49. Brenner C., Galluzzi L., Kepp O., and Kroemer G. (2013) Decoding cell death signals in liver inflammation. J. Hepatol. 59, 583–594 [DOI] [PubMed] [Google Scholar]
- 50. Tsutsui H., and Nishiguchi S. (2014) Importance of Kupffer cells in the development of acute liver injuries in mice. Int. J. Mol. Sci. 15, 7711–7730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cogswell J. P., Godlevski M. M., Wisely G. B., Clay W. C., Leesnitzer L. M., Ways J. P., and Gray J. G. (1994) NF-κB regulates IL-1β transcription through a consensus NF-κB binding site and a nonconsensus CRE-like site. J. Immunol. 153, 712–723 [PubMed] [Google Scholar]
- 52. Hiscott J., Marois J., Garoufalis J., D'Addario M., Roulston A., Kwan I., Pepin N., Lacoste J., Nguyen H., and Bensi G. (1993) Characterization of a functional NF-κB site in the human interleukin 1β promoter: evidence for a positive autoregulatory loop. Mol. Cell Biol. 13, 6231–6240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Besson-Fournier C., Latour C., Kautz L., Bertrand J., Ganz T., Roth M. P., and Coppin H. (2012) Induction of activin B by inflammatory stimuli up-regulates expression of the iron-regulatory peptide hepcidin through Smad1/5/8 signaling. Blood 120, 431–439 [DOI] [PubMed] [Google Scholar]
- 54. Krause A., Neitz S., Mägert H. J., Schulz A., Forssmann W. G., Schulz-Knappe P., and Adermann K. (2000) LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 480, 147–150 [DOI] [PubMed] [Google Scholar]
- 55. Park C. H., Valore E. V., Waring A. J., and Ganz T. (2001) Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 276, 7806–7810 [DOI] [PubMed] [Google Scholar]
- 56. Pigeon C., Ilyin G., Courselaud B., Leroyer P., Turlin B., Brissot P., and Loréal O. (2001) A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem. 276, 7811–7819 [DOI] [PubMed] [Google Scholar]
- 57. Kanamori Y., Murakami M., Matsui T., and Funaba M. (2014) The regulation of hepcidin expression by serum treatment: requirements of the BMP response element and STAT- and AP-1-binding sites. Gene 551, 119–126 [DOI] [PubMed] [Google Scholar]
- 58. Duran E. M., Shapshak P., Worley J., Minagar A., Ziegler F., Haliko S., Moleon-Borodowsky I., and Haslett P. A. (2005) Presenilin-1 detection in brain neurons and FOXP3 in peripheral blood mononuclear cells: normalizer gene selection for real time reverse transcriptase PCR using the ΔΔCt method. Front. Biosci. 10, 2955–2965 [DOI] [PubMed] [Google Scholar]
- 59. Funaba M., and Murakami M. (2008) A sensitive detection of phospho-Smad1/5/8 and Smad2 in Western blot analyses. J. Biochem. Biophys. Methods 70, 816–819 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.