

Abstract
Recent evidence has implicated succinate-driven reverse electron transport (RET) through complex I as a major source of damaging reactive oxygen species (ROS) underlying reperfusion injury after prolonged cardiac ischemia. However, this explanation may be incomplete, because RET on reperfusion is self-limiting and therefore transient. RET can only generate ROS when mitochondria are well polarized, and it ceases when permeability transition pores (PTP) open during reperfusion. Because prolonged ischemia/reperfusion also damages electron transport complexes, we investigated whether such damage could lead to ROS production after PTP opening has occurred. Using isolated cardiac mitochondria, we demonstrate a novel mechanism by which antimycin-inhibited complex III generates significant amounts of ROS in the presence of Mg2+ and NAD+ and the absence of exogenous substrates upon inner membrane pore formation by alamethicin or Ca2+-induced PTP opening. We show that H2O2 production under these conditions is related to Mg2+-dependent NADH generation by malic enzyme. H2O2 production is blocked by stigmatellin, indicating its origin from complex III, and by piericidin, demonstrating the importance of NADH-related ubiquinone reduction for ROS production under these conditions. For maximal ROS production, the rate of NADH generation has to be equal or below that of NADH oxidation, as further increases in [NADH] elevate ubiquinol-related complex III reduction beyond the optimal range for ROS generation. These results suggest that if complex III is damaged during ischemia, PTP opening may result in succinate/malate-fueled ROS production from complex III due to activation of malic enzyme by increases in matrix [Mg2+], [NAD+], and [ADP].
Keywords: electron transport system (ETS), mitochondria, nicotinamide adenine dinucleotide (NADH), oxidation-reduction (redox), reactive oxygen species (ROS), complex III
Introduction
When the heart is reperfused after prolonged ischemia, damaging reactive oxygen species (ROS)2 produced by cardiac mitochondria are thought to play a major role in ischemia/reperfusion (I/R) injury. Although at least 10 distinct sites in the electron transport chain and redox-active matrix proteins can be involved in ROS generation (1, 2), the major focus has centered on complexes I and III (3, 4), with recent evidence favoring succinate-driven reverse electron transport (RET) through complex I as the major mechanism (5, 6). However, RET is possible only when membrane potential (ΔΨ) is fully polarized (7–9). This requires mitochondria, which are depolarized during prolonged ischemia, to recover ΔΨ fully to generate succinate-driven ROS via RET. Therefore, when ΔΨ either does not recover (10, 11) or recovers only slowly and partially (12–14) upon reperfusion after prolonged ischemia as a result of permeability transition pore (PTP) opening (15) and increased inner membrane leakiness (16–18), ROS production by succinate-driven RET would be severely limited unless a sufficient subpopulation of mitochondria recovered ΔΨ fully. Given the strong evidence linking succinate accumulation to ROS-induced reperfusion injury (5), we therefore examined whether other mechanisms might come into play to promote additional ROS production by electron transport complexes after PTP opening has occurred and RET-driven ROS production has ceased.
In contrast to succinate-fueled RET-driven ROS production by complex I, ROS production by forward electron transport through complexes I or III requires the flow of electrons out of these complexes to be inhibited. When blocked using specific chemical inhibitors, the general consensus is that maximal ROS production at complex I is about half of that at complex III (19). In the absence of chemical inhibitors, mounting evidence also indicates that complexes I and III are damaged by prolonged I/R in vivo (3, 20), raising the possibility that they also contribute to the damaging burst of ROS in this setting (21–24). Nevertheless, the relative importance of complexes I and III, as well as other potential sources of ROS during reperfusion, is still debated. For forward electron transport into inhibited complex I to generate significant ROS requires a very high NADH/NAD ratio (>10) (25). Although this ratio rises during ischemia, it falls rapidly during reoxygenation (16, 17, 26) as mitochondria attempt to regenerate membrane potential (ΔΨ), leaving only a brief possible time window for significant ROS production. In addition, the complex I inhibitor rotenone has been found to decrease, rather than potentiate, ROS production by I/R (27, 28), suggesting that ROS is produced either by RET at complex I or downstream by complex III. It should be noted that recent experiments in Langendorff-perfused rat hearts subjected to 30 min of global ischemia demonstrated that increased ROS production during reperfusion occurred after, rather than before, PTP opening (29). Also, both ischemic preconditioning and cyclosporin A (CsA), despite inhibiting PTP opening during reperfusion, did not augment ROS production as might be expected if RET were prolonged during reperfusion but instead attenuated ROS production (29).
Forward electron flow into inhibited complex III can generate ROS, but the relationship between respiratory chain redox state and ROS production by complex III is less straightforward than for inhibited complex I. Experiments with isolated mitochondria have shown that ROS production by antimycin-inhibited complex III first increases and then decreases as succinate concentration is gradually increased. Likewise, the relationship between H2O2 production by antimycin-inhibited complex III and the supply of malate and glutamate is also bell-shaped (30). These and other observations (31, 32) suggest that in contrast to the high NADH/NAD+ ratio required for increased ROS production by inhibited complex I (33, 34), a relatively low NADH/NAD+ ratio, corresponding to a low ubiquinol (QH2)/ubiquinone (Q) ratio, is required to maximize ROS production by antimycin-inhibited complex III. Here we report the novel finding that when complex III is inhibited by antimycin in a setting in which succinate and its closely related metabolites are in limited supply, ROS production by complex III is dramatically increased after pore opening in the inner membrane by addition of NAD+/Mg2+ to activate malic enzyme (ME2). These findings may be relevant to the damaging ROS burst during reperfusion after prolonged ischemia when complex III has been damaged, and mitochondrial permeability transition pore opening leads to elevated matrix Mg2+, ADP, and a low NADH/NAD+ ratio.
Results
Oxidation of endogenous substrates and H2O2 production by antimycin-inhibited complex III
When isolated cardiac mitochondria were added to sucrose buffer followed by activation of the respiratory chain with ADP and carbonyl cyanide p-chlorophenylhydrazone, addition of the complex III inhibitor antimycin stimulated modest H2O2 production (0.1–0.15 nmol/min/mg) driven by oxidation of endogenous substrates (Fig. 1A, red trace). To simulate pore opening under these conditions, mitochondria were then exposed to alamethicin. H2O2 production decreased but could be markedly accelerated (6–7-fold) by the addition of Mg2+ and NAD+. The latter effect was inhibited by stigmatellin, indicating that H2O2 production originated from complex III. The dependence on endogenous substrates, despite inner membrane permeabilization, was revealed both by the suppression of H2O2 production with malonate to inhibit succinate oxidation by complex II, and by its relief by adding exogenous succinate in excess (Fig. 1A, blue trace). Fig. 1B presents summary data. The findings suggest that pore opening in the inner membrane, generated in this case by alamethicin, rapidly increased H2O2 production by antimycin-inhibited complex III in the presence of ADP, NAD+, and MgCl2 and in the absence of exogenous substrates. This is further demonstrated in Fig. 1C, where pore opening with alamethicin induced significant acceleration of H2O2 production whether antimycin had been added before (Fig. 1C, red trace) or after ADP, NAD+, and Mg2+ (Fig. 1C, blue trace), indicating that the increase in matrix concentration of these substances was critical for stigmatellin-sensitive ROS production. Note also that H2O2 production was significantly inhibited with piericidin, which blocks Q reduction by NADH, implying that Q reduction was required for ROS production by antimycin-inhibited complex III.
Figure 1.
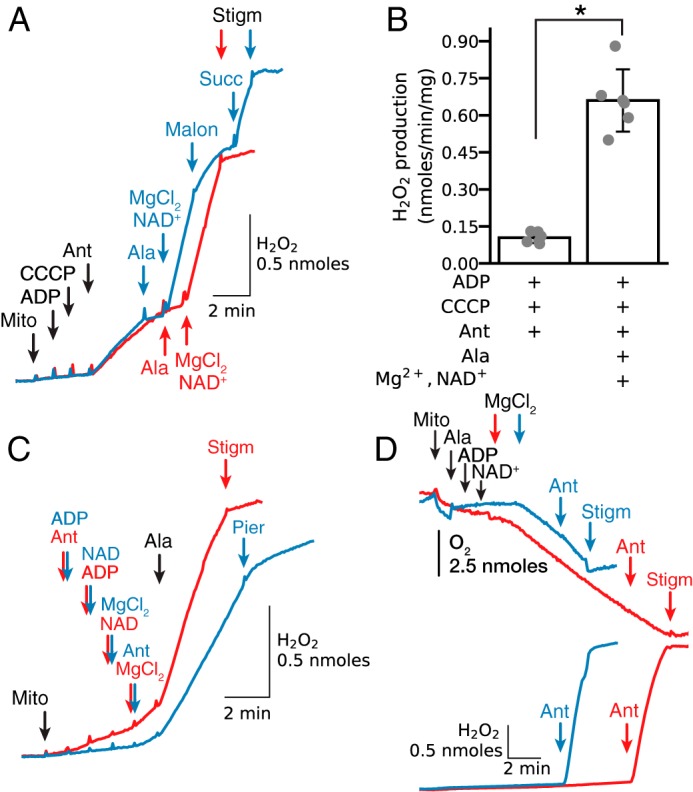
Mg2+ accelerates H2O2 production by antimycin-inhibited complex III in isolated mitochondria respiring on endogenous substrates. A, when isolated mitochondria (Mito) treated with carbonyl cyanide p-chlorophenylhydrazone (CCCP) (1 μm) and ADP (0.25 mm) were exposed to antimycin (Ant, 1 μm), H2O2 production increased. Permeabilization with alamethicin (Ala, 20 μg/ml) reduced H2O2 production, but when MgCl2 (2.5 mm) and NAD+ (0.5 mm) were added, H2O2 production increased markedly and was inhibited by stigmatellin (Stigm, 0.1 μm), indicating the ROS originated from complex III (red trace). During Mg2+/NAD+-stimulated H2O2 production, addition of malonate (2.5 mm) to inhibit endogenous succinate oxidation decreased H2O2 production that was reversed by adding exogenous succinate (Succ) (5 mm) (blue trace). In this and other figures, additions to all traces are indicated by black arrows; additions to one trace only are indicated by the arrows of the same color as the trace. B, summary data (median ± 95% confidence intervals, *, p < 0.05) for antimycin-dependent H2O2 production in intact and permeabilized mitochondria in the absence of exogenous substrates/co-factors. C, complex III was inhibited with antimycin (1 μm) added before (red trace) or after ADP (0.25 mm), NAD+ (0.5 mm), and MgCl2 (2.5 mm) (blue trace). Pore opening with alamethicin rapidly increased H2O2 production that was inhibited by stigmatellin (0.5 μm) or piericidin (pier, 1.5 μm). D, in permeabilized mitochondria, addition of ADP (0.25 mm) and NAD+ (0.5 mm) did not stimulate O2 consumption (upper traces) until Mg2+ (2.5 mm) was added, indicating that Mg2+ activated oxidation of endogenous substrates. Note that the modest increase in O2 consumption was maintained for 4 min (blue trace) and 11 min (red trace), and even after antimycin addition (1 μm) rapidly accelerated H2O2 production (lower traces). Both O2 consumption and H2O2 production were inhibited by stigmatellin (0.1 μm).
The relationship between H2O2 production and O2 consumption in the absence of exogenous substrates is shown in Fig. 1D. When cardiac mitochondria were permeabilized with alamethicin, sequential addition of ADP, NAD+, and MgCl2 caused O2 consumption to increase without significant H2O2 production, consistent with slow oxidation of endogenous substrates. This low oxidation rate was maintained for 4 min (Fig. 1D, blue trace) or 11 min (red trace) before antimycin was added, suggesting that inner membrane permeabilization does not result in rapid depletion of endogenous substrates to a level inhibiting complex III reduction over this time period. Antimycin addition did not affect O2 consumption, but promoted rapid H2O2 production that was not much different for the 4- or 11-min incubation periods (Fig. 1D, lower red and blue traces). Under these conditions, stigmatellin inhibited both mitochondrial O2 consumption and H2O2 production, indicating that (a) H2O2 was produced by complex III and (b) O2 consumption after inhibition of the respiratory chain with antimycin was diverted to H2O2 generation.
Retention of substrates is required for NAD+ reduction during Mg2+-stimulated H2O2 generation in alamethicin-permeabilized mitochondria
A surprising finding in Fig. 1D is that neither NAD+/Mg2+-stimulated O2 consumption nor antimycin-induced ROS production were decreased in alamethicin-permeabilized mitochondria despite the relatively long incubation in the absence of exogenous substrates. However, previous studies have demonstrated that mitochondria isolated from various tissues (35), including rabbit heart (36), contain endogenous substrates in low millimolar concentrations. In this context, the experiments in Fig. 2 were designed to study how quickly efflux of substrates following alamethicin administration begins to limit ADP/NAD+/Mg2+-induced H2O2 production by antimycin-inhibited complex III. Mitochondria were permeabilized with alamethicin and incubated for 1, 3, 6, and 9 min before antimycin was added, followed by ADP, NAD+, and MgCl2 addition. It is evident that stigmatellin-sensitive NAD+/Mg2+-dependent H2O2 production was not significantly reduced even after the longest delay. Thus, even after 9 min, NADH-related reduction of complex III had not yet decreased below the level required for NAD+/Mg2+-induced ROS production by antimycin-inhibited complex III.
Figure 2.
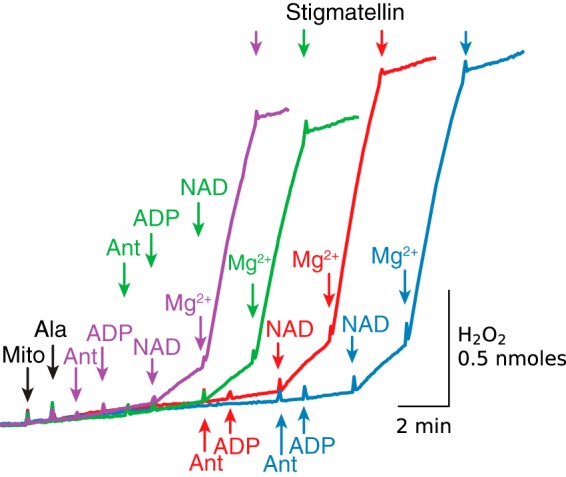
Endogenous substrates supporting NAD+/Mg2+-induced complex III reduction and H2O2 production are retained for >9 min after alamethicin addition. Mitochondria (Mito) were permeabilized with alamethicin (Ala), and antimycin (Ant) was added 1 (purple trace), 3 (green trace), 6 (red trace), and 9 min later (blue trace) as indicated, followed by ADP (0.25 mm), NAD+ (0.5 mm), MgCl2 (2.5 mm), and stigmatellin (0.5 μm). Additions to all traces are indicated by black arrows; additions to one trace only are indicated by the arrows of the same color as the trace.
Mg2+ or Mn2+-stimulated NAD+-dependent malic enzyme (ME2) activity in cardiac mitochondria
Because malic enzyme (ME2), which converts malate + NAD(P)+ to pyruvate + NAD(P)H + CO2, is to our knowledge the only NAD(P)H-generating enzyme in the matrix that has an absolute requirement for Mg2+ or Mn2+ (37), we next examined whether ME2 might be the source of NADH production fueling NAD+/Mg2+-induced ROS production by antimycin-inhibited complex III. ME2 activity in our preparations was assessed as illustrated in Fig. 3A. NAD+ addition to permeabilized mitochondria incubated in the presence of piericidin and malate resulted in a small increase in NADH followed by rapid inhibition. As shown previously (33), the inhibition is due to the increase in [NADH] and [oxaloacetate] that easily reverses the malate dehydrogenase (MDH) reaction in the direction of NADH oxidation and malate generation. Consistent with this mechanism, glutamate addition to promote oxaloacetate removal via aspartate aminotransferase resulted in rapid acceleration of NADH generation (Fig. 3A, green trace). Despite strong inhibition of the MDH reaction in forward direction in the absence of glutamate, however, MgCl2 addition increased NADH generation, indicating Mg2+-induced activation of malate oxidation and NAD+ reduction. This concentration-dependent effect of MgCl2 was inhibited and reversed with EDTA added in excess of MgCl2. Under these conditions, ME2 should be fully inhibited such that only MDH is available for oxidation of the NADH generated by ME2 before EDTA addition. As expected, NADH oxidation by MDH was rapidly reversed into NADH generation by addition of glutamate. Note, that in the presence of 5 mm malate, addition of CoA to accelerate the ME2 reaction by removing pyruvate also accelerated NADH production.
Figure 3.

Stimulation of ME2 activity in cardiac mitochondria by [Mg2+] and [Mn2+]. A, NAD+ (0.5 mm) addition to alamethicin (Ala)- permeabilized mitochondria (Mito), incubated in the presence of piericidin (Pier) (1.5 μm) and malate (Mal) (5 mm), resulted in a limited and transient increase of NADH generation that was rapidly accelerated by glutamate (5 mm) addition (green trace). In the absence of glutamate, MgCl2 addition caused a much slower increase in NADH that was a concentration-dependent effect, slightly accelerated by CoA (0.15 mm) and rapidly inhibited and reversed by adding EDTA > MgCl2 (purple, orange, red, and blue traces). The decrease in [NADH] after EGTA was due to MDH working in the reverse direction as shown by a rapid increase in [NADH] after adding glutamate. B, similar results were obtained using MnCl2 in place of MgCl2 to activate ME2. C, acceleration of H2O2 production by antimycin (Ant)-inhibited complex III after MnCl2 or MgCl2 addition. H2O2 production by alamethicin-permeabilized, antimycin-inhibited mitochondria was stimulated by sequential addition of ADP (0.25 mm), NAD+ (0.5 mm), and MnCl2 in the concentrations indicated at the end of traces. For comparison, an increase in H2O2 production after 2.5 mm MgCl2 is also presented (green trace). In all cases H2O2 production was inhibited with piericidin (1 μm).
The experiments in Fig. 3A were also repeated with MnCl2 addition in place of MgCl2 (Fig. 3B). Compared with Mg2+ addition, the Mn2+-induced increase in NADH production was about 4–5 times faster. In addition, significantly lower [Mn2+] compared with [Mg2+] was required to increase NADH production.
This higher sensitivity of ME2 to Mn2+ was accompanied by greater Mn2+-induced H2O2 production by antimycin-inhibited complex III in the presence of NAD+ and absence of exogenous substrates (Fig. 3C). Under the same conditions in the same preparation, addition of 0.5 mm Mn2+ resulted in 2.8 times higher H2O2 production compared with addition of 2.5 mm Mg2+ (1.99 and 0.70 nmol/min/mg correspondingly). NAD+-dependent Mn2+- or Mg2+-activated H2O2 production by antimycin-inhibited complex III was significantly inhibited with piericidin (Fig. 3C), indicating that ROS production was largely dependent on NADH-related Q reduction.
Collectively, these results demonstrate that cardiac mitochondria contain ME2 which, when activated by Mg2+- or Mn2+, reduces NAD+ in the presence of malate and piericidin. Although this activity is low compared with MDH activity (about 115 versus 25 nmol/min/mg for Mn2+-activated versus Mg2+-activated enzyme), it is more than sufficient to reduce antimycin-inhibited complex III to an extent promoting significant H2O2 production in the presence of only endogenous substrates. In fact, ME2-mediated ROS production by complex III seems to work most efficiently in the presence of endogenous substrates, and it is easily inhibited by increasing [malate] to increase enzyme activity, as described below.
Functional analysis of key endogenous substrates/co-factors supporting Mg2+/NAD+-induced ROS production by antimycin-inhibited complex III, effects of complex I and II inhibitors
The simplest explanation for why small amounts of substrates are retained in alamethicin-permeabilized mitochondria may be binding to matrix structures and constituents, possibly enzymes. There is some evidence that ME2 binds NAD+ first, followed by malate (38). Perhaps this binding can take place before oxidative decarboxylation is stimulated by Mg2+ or Mn2+. Whatever the mechanism for retaining malate and related substrates in the vicinity of ME2, the expected significant efflux of substrates after pore opening lowers their level in the matrix, most likely below the detection limit. Therefore, the functional approach, although indirect, seems to be the most practical method available to clarify the importance of NAD+-related substrates for reduction and ROS production by antimycin-inhibited complex III in the absence of exogenous substrates.
In Fig. 4A, H2O2 production by antimycin-inhibited permeabilized mitochondria was evaluated after sequential addition of ADP, NAD+, and Mg2+ in the presence or absence of respiratory chain inhibitors. Mg2+ addition promoted significant H2O2 production that was inhibited with stigmatellin (orange trace, control). Stigmatellin addition to permeabilized mitochondria before ADP, NAD+, and Mg2+ also inhibited the NAD+- and Mg2+-induced increase in H2O2 production (Fig. 4A, green trace). Importantly, increased H2O2 production was also absent when NADH-related QH2 generation was blocked with piericidin (2 μm, Fig. 4A, red trace). Because piericidin is not expected to inhibit oxidation of endogenous succinate leading to QH2 generation, we conclude that in the absence of exogenous substrates, NADH-related QH2 generation and complex III reduction are mostly responsible for ROS production by antimycin-inhibited complex III. The NAD+/Mg2+-induced increase in H2O2 production was also blocked in the combined presence of piericidin to inhibit complex I and malonate (5 mm) to inhibit succinate oxidation by complex II (Fig. 4A, blue trace). Under those conditions, however, malonate inhibition was overcome by excess succinate (5 mm), which accelerated H2O2 production (Fig. 4A, blue trace). Malonate alone in the absence of piericidin, however, only partially inhibited H2O2 production, which could be rapidly accelerated by addition of malate (150 μm, Fig. 4A, purple trace). Note that H2O2 production rates are similar (about 1.1 nmol/min/mg) for all conditions, suggesting that perhaps the maximum rate was achieved during oxidation of endogenous substrates. These results suggest that cardiac mitochondria after pore opening in the inner membrane still contain NAD+-related substrates that can be utilized (oxidized) for Mg2+-stimulated NAD+ reduction by ME2 to support ROS production by antimycin-inhibited complex III. Fig. 4B summarizes inhibitory effects of stigmatellin, piericidin, malonate, and piericidin plus malonate on Mg2+/NAD+-related H2O2 production by antimycin-inhibited complex III.
Figure 4.
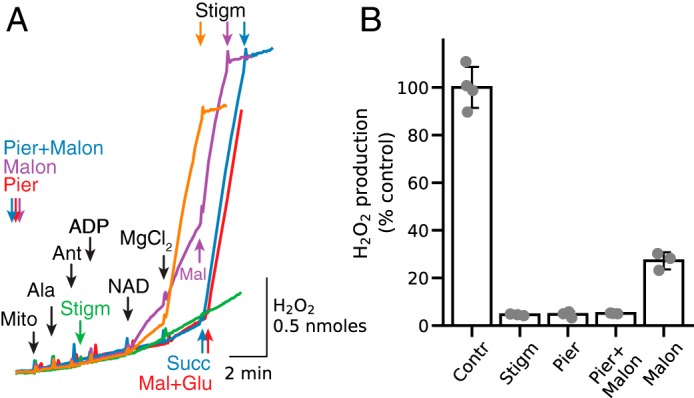
Relative effect of NADH-related and succinate-related Q reduction on NAD+/Mg2+-stimulated H2O2 generation by antimycin-inhibited complex III in the absence of exogenous substrates. A, alamethicin (Ala)-permeabilized, antimycin (Ant)-inhibited mitochondria (Mito) were subjected to sequential addition of ADP (0.25 mm), NAD+ (0.5 mm), and MgCl2 (2.5 mm) that resulted in rapid H2O2 production suppressed by stigmatellin (Stigm, 0.5 μm) (orange trace, control). Stigmatellin (green trace) and piericidin (Pier, 1.5 μm) (red trace) added to permeabilized mitochondria before ADP, NAD+, and MgCl2 inhibited H2O2 production. Piericidin inhibition of H2O2 production was alleviated by addition of malate/glutamate (5 mm each) to enhance the matrix NADH/NAD+ ratio sufficiently to support ROS production by complex I (red trace). Preincubation with malonate (Malon, 5 mm) resulted only in partial inhibition of H2O2 production that was enhanced to the control level by addition of malate (Mal, 0.15 mm) (purple trace). As expected, H2O2 production was inhibited in the presence of both piericidin and malonate and significantly activated after succinate (Succ) (5 mm) addition (blue trace). As in other figures, additions to all traces are indicated by black arrows; additions to one trace only are indicated by the arrows of the same color as the trace. B, summary data for inhibition of NAD+/Mg2+-induced antimycin-dependent H2O2 production by complex III in the absence of exogenous substrates. H2O2 production in the absence of inhibitors other than antimycin averaged 0.99 nmol/min/mg, which was taken as 100%.
Functional analysis: order of substrate/co-factor addition
Although the best way to mimic the effects of pore opening at the start of reperfusion is to apply alamethicin in the presence of ADP, NAD+, and Mg2+ (as in Fig. 1), these conditions are not suitable for studying the regulation of H2O2 production by ADP, NAD+, and Mg2+ individually. Experiments therefore were performed with sequential addition of substrates, cofactors, and inhibitors after mitochondria were permeabilized with alamethicin to permit the relative importance of each factor on ROS production by antimycin-inhibited complex III to be assessed.
In the absence of exogenous substrates, antimycin had no significant effect on H2O2 production when added to permeabilized mitochondria before NAD+ and Mg2+ (Fig. 5A, as well as Figs. 2 and 3C). Addition of NAD+ alone slightly increased H2O2 production (Fig. 5A, red trace), whereas addition of ADP alone had no effect (Fig. 5A, blue trace). In both cases, however, Mg2+ stimulated robust H2O2 production that was rapidly inhibited with stigmatellin. These results show that H2O2 production by antimycin-inhibited complex III in the absence of exogenous substrates depends critically on Mg2+ and NAD+, although for maximum stimulation ADP is also required (Fig. 5B). Fig. 5C summarizes average stigmatellin-sensitive H2O2 production by antimycin-inhibited complex III in permeabilized mitochondria in the presence of ADP (0.25 mm), NAD+ (0.5 mm), and/or MgCl2 (2.5 mm).
Figure 5.
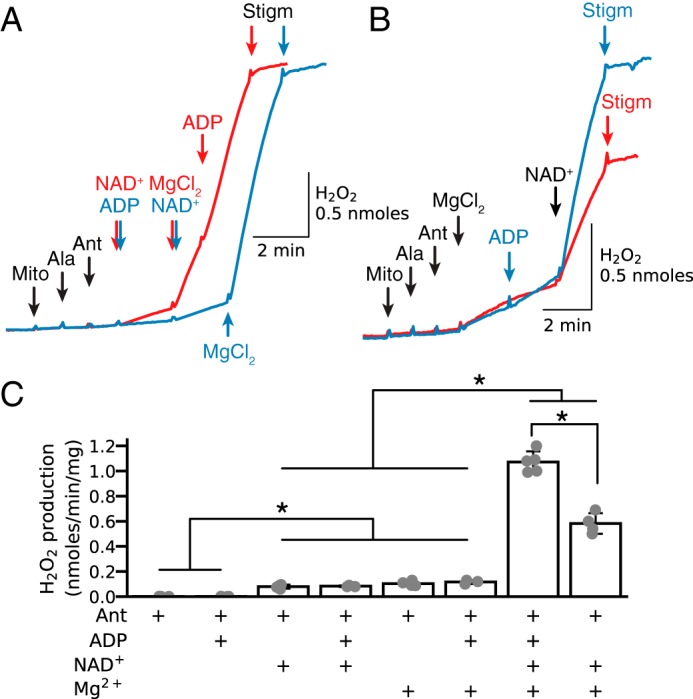
Relative importance of Mg2+, NAD+, and ADP in stimulating H2O2 production by antimycin-inhibited complex III. A, in alamethicin (Ala)-permeabilized mitochondria (Mito) without exogenous substrates, antimycin (Ant, 1 μm) followed by addition of NAD+ (0.5 mm, red trace) and/or ADP (0.25 mm, blue trace) had little effect on H2O2 production until Mg2+ (2.5 mm) was also added. B, similarly, after antimycin, Mg2+ had only a modest effect on H2O2 production until NAD+ was also added (red trace), which was maximized when ADP was also present (blue trace). In all cases, stigmatellin (Stigm, 0.1 μm) was suppressed on H2O2 production. C, summary data (median ± 95% confidence intervals, *, p < 0.05) under the various conditions indicated. As in other figures, additions to all traces are indicated by black arrows; additions to one trace only are indicated by the arrows of the same color as the trace.
With NAD+ present, the rapid stimulation of H2O2 production by Mg2+ was concentration-dependent (Fig. 6A). EDTA almost completely inhibited Mg2+-induced H2O2 production, which was reactivated by adding Mg2+ in excess of EDTA. The small increase in H2O2 production after NAD+ alone may have been due to residual endogenous Mg2+, as it was significantly suppressed in the presence of 250 μm EDTA (Fig. 6A, blue trace). If Mg2+ was added before NAD+, however, then the increase in H2O2 production was modest (Fig. 6B). We attributed this modest H2O2 production to the presence of endogenous NAD+, because significant amounts of matrix NAD(H) are bound to matrix proteins (39, 40), which slow NAD(H) efflux after membrane permeabilization. In the absence of exogenous substrates, H2O2 production was significantly potentiated by exogenous NAD+ (0.5 mm).
Figure 6.
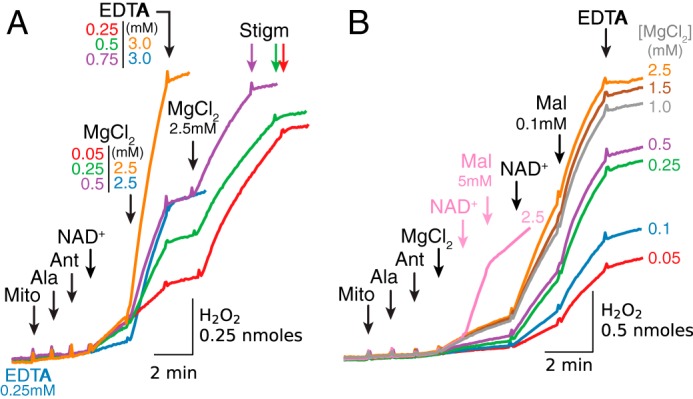
Dependence of H2O2 production by antimycin-inhibited complex III on [Mg2+] and exogenous [malate]. A, when alamethicin (Ala)-permeabilized mitochondria (Mito) without exogenous substrates were exposed to antimycin (Ant) to inhibit complex III, NAD+ (0.5 mm) caused a modest increase in H2O2 production that was progressively accelerated by increasing [Mg2+] from 0.05 to 2.5 mm (traces color-labeled as indicated) and then stopped by adding EDTA in excess, restarted by adding Mg2+ in excess, and terminated by stigmatellin (Stigm, 0.1 μm). Note that with 0.25 mm EDTA present from the start to chelate endogenous Mg2+, NAD+ alone failed to increase H2O2 production until Mg2+ was added in excess (blue trace). B, in the absence of NAD+, Mg2+ added at the various concentrations increased H2O2 production progressively that was markedly accelerated by NAD+ (0.5 mm). Addition of malate (Mal) at low concentration (0.1 mm) further accelerated H2O2 production at submillimolar [Mg2+], which was suppressed by adding EGTA in excess. Addition of malate at a high concentration (5 mm), however, suppressed H2O2 production in the presence of NAD+ (0.5 mm) and Mg2+ (2.5 mm) (pink trace). As in other figures, additions to all traces are indicated by black arrows; additions to one trace only are indicated by the arrows of the same color as the trace.
Addition of exogenous malate significantly influenced ROS production under these conditions. At a concentration of 0.1 mm, malate potentiated H2O2 production, especially at Mg2+ <0.5 mm (Fig. 6B). However, 5 mm malate suppressed H2O2 production (Fig. 6B, pink trace), consistent with the bell-shaped relationship reported previously (30).
These results show that in the absence of exogenous substrates, H2O2 production by antimycin-inhibited complex III depends on matrix [Mg2+] and requires NAD+. In addition, exogenous malate at low concentrations under these conditions further stimulates H2O2 production, whereas high malate concentrations inhibit H2O2 production.
Functional analysis: substrate/co-factor concentration dependence
To account for the ability of Mg2+ and NAD+ to stimulate robust activation of H2O2 production from antimycin-inhibited complex III when only endogenous or low concentrations of exogenous malate were present, we postulated that ME2 activation by Mg2+ and NAD+ generated a small increase in the NADH/NAD+ ratio, which optimized the QH2/Q ratio for maximal H2O2 production by inhibited complex III (30–32). With high concentrations of malate (5 mm), however, the NADH/NAD+ ratio, and consequently the QH2/Q ratio, becomes too high for maximal H2O2 production. We tested this hypothesis by first inhibiting endogenous succinate oxidation to malate by complex II inhibitors (malonate and AA5) and then adding malate at various concentrations to activate ME2. As expected, Mg2+-dependent generation of NADH increased progressively as malate concentration was increased (Fig. 7A). In contrast, H2O2 production rapidly increased after addition of low concentrations of malate but then decreased at concentrations >0.5 mm (Fig. 7B). Under these conditions, Q reduction depends on NADH generation, and the QH2/Q ratio is expected to increase progressively as malate concentration increases to support high NADH levels. Thus, ROS production was greatest when the NADH/NAD+ (and presumably QH2/Q) ratio was low at low malate concentrations, and subsequently decreased as malate concentration increased, with half-maximal inhibition of H2O2 production at about 0.7 mm malate (Fig. 7C). The ability of mitochondria to oxidize NADH was also evaluated by adding NADH after NADH generation/oxidation was close to steady state at different malate concentrations (Fig. 7A). When the rate of NADH oxidation (i.e. the rate of NADH fluorescence decrease after NADH addition) was plotted against H2O2 production, H2O2 production increased as NADH oxidation rate increased (Fig. 7D). Thus, when NADH oxidation was inhibited due to the high NADH/NAD+ ratio, which is also expected to increase the QH2/Q ratio in the presence of antimycin, Mg2+, NAD+ and high [malate], ROS production by complex III was low. In contrast, when NADH oxidation was rapid, as expected to occur at a low QH2/Q ratio, ROS production by antimycin-inhibited complex III was high.
Figure 7.
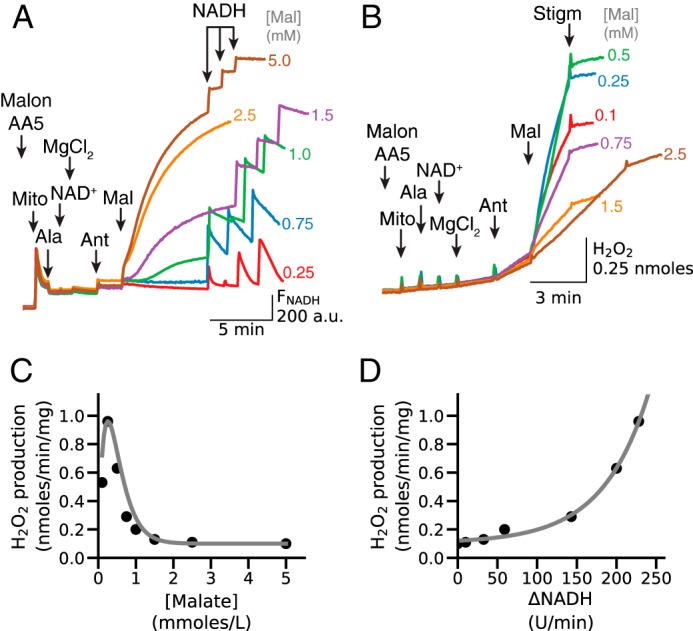
Relationship between [malate]-dependent increases in NADH and H2O2 production by antimycin-inhibited complex III. A, in the presence of AA5 (1 μm) and malonate (Malon, 2.5 mm) to inhibit succinate oxidation by complex II, alamethicin (Ala)-permeabilized mitochondria (Mito) were exposed to NAD+ (0.5 mm), MgCl2 (2.5 mm), and antimycin (Ant, 1 μm), after which malate (Mal) was added in the concentrations indicated at the end of each trace. NADH fluorescence (FNADH) increased progressively with increasing [malate]. After FNADH reached near steady state, NADH boluses (25 μm three times) were then added to assess NADH oxidation rates relative to NADH generation. B, stigmatellin (Stigm, 0.1 μm)-sensitive H2O2 production under the same conditions. C, rate of H2O2 production versus [malate], calculated from the traces in B. H2O2 production peaked at 0.25 mm malate and thereafter rapidly decreased as [malate] increased. D, rate of H2O2 production versus NADH oxidation rates at different [malate], calculated from the traces in A and B, illustrating that decreased ability to lower [NADH] at high [malate] results in inhibition of malate-stimulated H2O2 production in the presence of NAD+ and Mg2+. The rate of NADH oxidation was calculated from the slope of NADH fluorescence decrease over 1 min, measured after last 25 μm NADH bolus. A.U., arbitrary unit.
Endogenous malate can either be present in the matrix from the start or generated by endogenous succinate/fumarate oxidation. Consistent with the latter pathway, Fig. 8 shows that under conditions where respiration was inhibited due to lack of O2, the addition of succinate resulted in NADH generation that was Mg2+-dependent. Because NAD+ reduction by RET is excluded after membrane permeabilization, the most likely mechanism for the Mg2+-dependent increase in NADH is malate generation due to succinate oxidation, with subsequent malate oxidation by ME2 generating NADH, pyruvate, and CO2. This interpretation is supported by the finding that adding CoA to metabolize the pyruvate generated by ME2 further potentiated Mg2+-induced NADH generation. Under those conditions, NADH generation was clearly demonstrated by adding O2 (air) transiently to oxidize the accumulated NADH and observing its regeneration after O2 was again removed. The NADH increase in the presence of 2.5 mm Mg2+ under these conditions was inhibited by 3 mm EDTA and subsequently stimulated by adding Mg2+ in excess of EDTA, demonstrating the Mg2+ dependence of this reaction.
Figure 8.
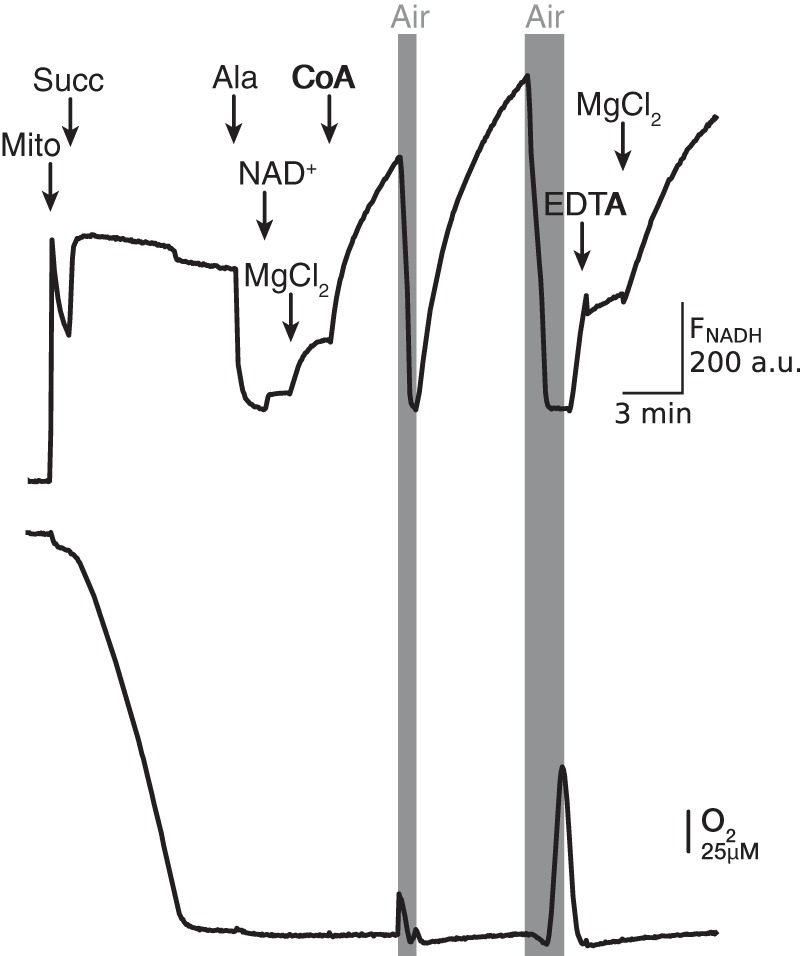
Mg2+-dependent NADH generation in succinate-energized mitochondria. Mitochondria (Mito) were energized with 5 mm succinate (Succ), which maximized NADH fluorescence (FNADH, upper trace) while depleting buffer O2 (lower trace). Mitochondria were then permeabilized with alamethicin (Ala), causing a decrease in FNADH which recovered only slightly when NAD+ (0.5 mm) was added, until Mg2+ (2.5 mm) and CoA (0.15 mm) were also added to stimulate succinate/malate oxidation and NAD+ reduction. The generated NADH was rapidly oxidized by allowing a brief infusion of O2 (air) into the buffer. NADH generation was stopped with excess EDTA (3 mm) and restarted with excess Mg2+ (5 mm). A.U., arbitrary unit.
Similar to malate, succinate's ability to support H2O2 production by antimycin-inhibited complex III was also concentration-dependent (Fig. 9A). In the absence of exogenous malate, H2O2 production increased when succinate concentration was increased from 25 to 100 μm but then decreased as concentration was further elevated to 0.5 or 2 mm. Addition of ADP and NAD+ had little effect in the absence of Mg2+. Most importantly, addition of MgCl2 (2.5 mm) rapidly accelerated H2O2 production in the presence of low concentrations of succinate (25–100 μm), but it had no effect at 0.5 or 2 mm succinate. In the presence of 2 mm succinate, inhibition of succinate oxidation with malonate (2.5 mm) rapidly increased H2O2 production to the level recorded in the presence of low [succinate]. Note that Mg2+ added to mitochondria incubated in the absence of exogenous succinate also induced significant H2O2 production (Fig. 9A, red trace) that was only slightly lower than the Mg2+-stimulated production rates in the presence of 25–100 μm succinate. Changes in O2 consumption recorded simultaneously with ROS production show that O2 consumption slightly increased after acceleration of ROS production induced by MgCl2 or malonate. Both H2O2 production and O2 consumption were inhibited with stigmatellin, indicating that both were related to ROS production by complex III.
Figure 9.

Dependence of H2O2 production by antimycin-inhibited complex III on exogenous [succinate] and [fumarate]. A, alamethicin (Ala)-permeabilized mitochondria were energized with exogenous succinate (Succ) at the concentrations indicated, which induced O2 consumption (lower trace) but not H2O2 production (upper trace) until antimycin (Ant, 1 μm) was added. H2O2 production was not affected by ADP (0.25 mm) and NAD+ (0.25 mm) addition, but was significantly accelerated by Mg2+ (2.5 mm) when [succinate] was 0.1 mm or less but not 0.5 mm or greater (brown and orange traces). With 2 mm [succinate], malonate (Malon, 2.5 mm) administered to inhibit succinate oxidation caused H2O2 production to increase markedly (orange trace). Stigmatellin (Stigm, 0.1 μm) inhibited H2O2 production in all cases. B, ROS production increased after sequential addition of ADP (0.25 mm), NAD+ (0.5 mm), and MgCl2, depending on [MgCl2] indicated at the end of trace. Fumarate (1 mm) accelerated H2O2 production, especially at low [Mg2+]. As in other figures, additions to all traces are indicated by black arrows; additions to one trace only are indicated by the arrows of the same color as the trace.
Fumarate in low millimolar concentrations is known to allosterically activate purified human recombinant ME2 (41). Fig. 9B demonstrates that 1 mm fumarate increased NAD+/Mg2+-activated H2O2 production by antimycin-inhibited complex III. Consistent with the more pronounced effect of fumarate on enzyme activity at lower substrate concentrations (41), the fumarate-induced increase in H2O2 production was more significant at lower [Mg2+]. However, besides allosteric activation of ME2, fumarate is also expected to increase malate production in permeabilized mitochondria, and we cannot exclude the possibility that this effect predominated under our conditions.
In the absence of exogenous substrates, NAD+/Mg2+ addition is expected to activate not only oxidation of malate available in the matrix, but all tricarboxylic acid segments involving succinate → fumarate → malate. Inhibition of succinate oxidation by malonate and AA5 cuts off malate production from succinate, such that subsequent activation of malate oxidation by adding NAD+/Mg2+ before inhibition of complex III should decrease endogenous malate levels available for NADH generation after antimycin is added. In line with these predictions, complex II inhibition depressed NAD+/Mg2+-stimulated H2O2 production by antimycin-inhibited complex III that varied with the timing of malonate + AA5 addition (Fig. 10A). The effect was least expressed when antimycin was added before (Fig. 10A, green trace) or together with NAD+/Mg2+ (orange trace). Adding antimycin 1 min (Fig. 10A, purple trace) or 3 min (Fig. 10A, red trace) after NAD+/Mg2+ resulted in further progressive decreases in H2O2 production. These results contrast with the much greater NAD+/Mg2+-stimulated ROS production by antimycin-inhibited complex III in the absence of complex II inhibitors (Fig. 10A, blue trace). Note that decreased H2O2 production after complex II inhibition was rapidly activated by low concentrations of malate and inhibited with stigmatellin.
Figure 10.

A, complex II inhibition significantly depresses NAD+/Mg2+-induced H2O2 production by antimycin-inhibited complex III in the absence of exogenous substrates. In the presence of AA5 (1 μm) and malonate (Malon, 2.5 mm) to inhibit succinate oxidation by complex II, alamethicin (Ala)-permeabilized mitochondria (Mito) were exposed to NAD+ (0.5 mm), MgCl2 (2.5 mm), and antimycin (Ant, 1 μm) at the times indicated by the same-colored arrows for each trace. Whether antimycin was added before (green trace) or together with NAD+/Mg2+ (orange trace), H2O2 production was greater than when antimycin was added later (lilac and red traces), presumably because less endogenous malate had been oxidized by ME2 before complex III was inhibited. Consistent with this prediction, in all conditions subsequent addition of 0.25 mm malate (Mal) rapidly activated H2O2 production to a similar rate. In the absence of complex II inhibitors, NAD+/Mg2+-stimulated H2O2 production after antimycin was greater because oxidation of endogenous succinate was available to generate malate (blue trace). Stigmatellin (Stigm, 0.1 μm) suppressed H2O2 production in all cases. B, relative effectiveness of MDH versus ME2 at stimulating H2O2 production by antimycin-inhibited complex III. In alamethicin-permeabilized mitochondria exposed to antimycin (1 μm), addition of 1 or 5 mm malate (orange and brown traces, respectively) stimulated robust H2O2 production that was suppressed by addition of NAD+ and Mg2+, presumably by increasing the NADH/NAD+ ratio beyond that optimal for H2O2 production by antimycin-inhibited complex III (see text). Lower [malate] (green, purple, and blue traces) stimulated less H2O2 production initially until NAD+ and Mg2+ were added, after which H2O2 production markedly accelerated. Note that with no exogenous malate (red trace), oxidation of endogenous malate was insufficient to cause any H2O2 production by MDH until NAD+ was added, whereas subsequent activation of ME2 by adding Mg2+ led to a much more robust increase in H2O2 production. Stigmatellin (0.1 μm) suppressed H2O2 production in all cases. C, dependence of H2O2 production by antimycin-inhibited complex III on exogenous [NAD+]. In alamethicin (Ala)-permeabilized mitochondria exposed to antimycin (0.5 μm) in the absence of exogenous substrates, addition of NAD+ at the concentrations indicated stimulated progressive H2O2 production that was markedly accelerated by Mg2+ (2.5 mm) and inhibited by stigmatellin (0.1 μm). As in other figures, additions to all traces are indicated by black arrows; additions to one trace only are indicated by the arrows of the same color as the trace.
As shown above, H2O2 production by antimycin-inhibited complex III was maximal at low succinate or malate concentrations, which limited NADH and QH2 generation, consistent with previous evidence (30, 32, 42) that H2O2 production is maximized by a low QH2/Q ratio and suppressed by a high ratio. In addition to restricting substrate availability, NADH and QH2 generation can also be limited by restricting NAD+ availability. In Fig. 10B, malate was added to antimycin-inhibited permeabilized mitochondria in the absence of exogenous NAD+ and Mg2+. Without Mg2+ to activate ME2, malate can still be oxidized by MDH, but NADH generation is limited because the increase in oxaloacetate and NADH inhibits the reversible MDH reaction, unless oxaloacetate is removed by also adding glutamate (see Fig. 3A) (33). Under these conditions without exogenous NAD+ and Mg2+, malate concentrations of >0.2 mm were required to produce enough NADH by MDH to stimulate H2O2 production by antimycin-inhibited complex III (Fig. 10B). However, if NADH and QH2 generation were then further boosted by adding exogenous NAD+, H2O2 production was rapidly inhibited. In the same experiments, addition of Mg2+ to activate ME2 only stimulated robust H2O2 production at low malate concentrations (<0.2 mm). The rates of Mg2+-stimulated H2O2 production were similar in the absence of malate as for malate concentrations up to 0.2 mm, suggesting that the endogenous malate concentration in the matrix is submillimolar under these conditions.
We also studied other permutations of substrates NAD+ and Mg2+. Addition of 0.1 mm malate to antimycin-inhibited mitochondria slightly increased H2O2 production that was further significantly enhanced by Mg2+. Incubation of mitochondria with 250 μm EDTA suppressed malate-induced H2O2 production, which was then reversed by adding 2.5 mm Mg2+. MgCl2 addition significantly activated H2O2 production at low concentrations of malate/glutamate (5–100 μm) in the absence of exogenous NAD+, but no activation was observed at 1 mm malate/glutamate. Because no NAD+ was added in these experiments, the findings indicate the following: (a) small amount of NAD(H) is still available for Q reduction after inner membrane permeabilization; (b) substrate-dependent activation of NADH generation can compensate for low NAD+ availability; and (c) NADH production above a certain level inhibits H2O2 generation, even when only endogenous NAD(H) is available (results not shown).
However, in the absence of exogenous substrates, Mg2+-stimulated H2O2 production by antimycin-inhibited complex III depended on exogenous [NAD+] in a concentration-dependent manner (Fig. 10C). Note that the NAD+-dependent increase in H2O2 production started at very low concentrations of added NAD+ and was already significant at 50 μm NAD+.
Collectively, our results show that the rate of ROS production by antimycin-inhibited complex III is strongly modulated by electrons derived from NADH and depends on a low NADH/NAD+ and presumably low QH2/Q ratio for significant ROS production, as dually determined by substrate and NAD+ concentrations. Accordingly, the level of H2O2 production can be regulated by increasing NAD+ when substrate availability is limited or by increasing substrate concentrations when NAD+ availability is limited.
Ca2+/Pi-induced PTP opening reproduces alamethicin's effects on Mg2+/NAD+-stimulated ROS production by antimycin-inhibited complex III
The above experiments were performed in mitochondria permeabilized by alamethicin to create nonselective pores in the inner mitochondrial membrane. To determine whether PTPs have similar effects as alamethicin pores, PTP opening was induced by sequential addition of Pi and Ca2+ before MgCl2 and NAD+ were added (Fig. 11A). Similar to alamethicin, addition of NAD+/MgCl2 induced an increase in H2O2 production when complex III was inhibited by antimycin that was suppressed by EGTA or CsA (Fig. 11B). In general, the population of mitochondria with open pores allowing NAD+/MgCl2 access to the matrix induced with 100 μm Ca2+ was about half of that induced with alamethicin (20 μg/ml) (Fig. 11C).
Figure 11.

Ca2+-induced PTP opening accelerates H2O2 production by antimycin-inhibited complex III in the presence of NAD+ and MgCl2. A, mitochondria (Mito) incubated in the presence of ADP (0.5 mm) and antimycin (Ant, 0.2 μm) were challenged with Pi (2.5 mm) and CaCl2 (concentration indicated at the end of tracing). NAD+(0.5 mm) and MgCl2 (2.5 mm) addition resulted in H2O2 production that was dependent on [Ca2+], inhibited in the presence of CsA (1 μm), and rapidly inhibited with stigmatellin (Stigm, 0.2 μm) or EDTA (3 mm). B, mitochondria, incubated under the same condition as in A and challenged with 100 μm Ca2+ in the presence of CsA (1 μm, red trace) or EGTA (250 μm, blue trace), followed by alamethicin and stigmatellin (0.2 μm). Green trace represents control without inhibition of Ca2+/Pi-induced PTP opening. C, summary data (median ± 95% confidence intervals) of NAD+/Mg2+-induced H2O2 production triggered by Ca2+-induced PTP opening as demonstrated in A. Values for H2O2 production in the presence of CsA (1 μm) and EGTA (0.25 mm) were obtained after adding 100 μm Ca2+.
Discussion
The major finding of this study is that Mg2+, NAD+, and ADP induce marked acceleration of ROS production when complex III is inhibited by antimycin in permeabilized cardiac mitochondria. This novel mechanism is able to increase mitochondrial ROS production significantly when formation of alamethicin pores or PTP in the inner membrane allows Mg2+, NAD+, and ADP to equilibrate between the matrix and extramitochondrial space. Under these conditions, Mg2+-dependent activation of ME2 generates reducing equivalents from malate that can markedly stimulate ROS production by inhibited complex III, but only when NADH production/oxidation rates are in an optimal range to maintain a low NADH/NAD+ ratio. A reasonable assumption is that QH2/Q ratio is also low under those conditions (Fig. 12). As discussed later, we hypothesize that if such conditions are present when PTP opening occurs following prolonged I/R, this mechanism could contribute to continued ROS production after succinate-fueled RET-driven ROS production has ceased.
Figure 12.
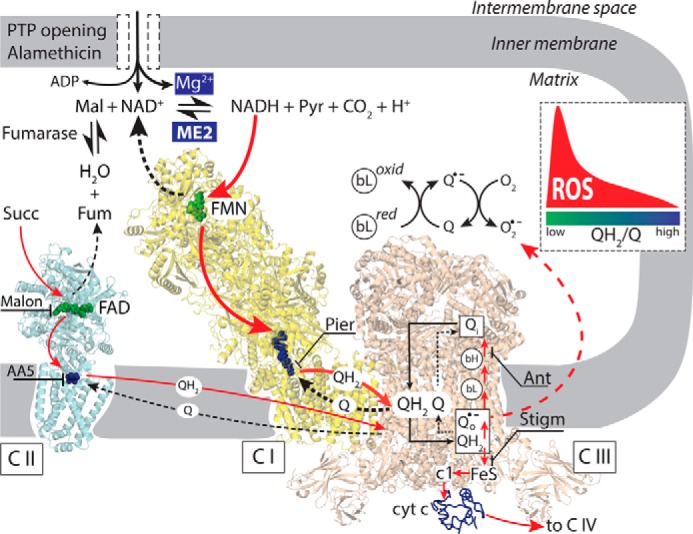
Schematic of ROS production when ME2 is activated by increases in matrix Mg2+, NAD+, and ADP after inner membrane permeabilization by alamethicin or PTP opening. Malate oxidation by ME2 generates NADH that donates electrons (red arrows) to the flavoprotein site (FMN) in complex I (C I, yellow), reducing Q to QH2. Piericidin (Pier) blocks Q reduction. QH2 is oxidized by complex III (C III, tan), donating electrons to cytochrome c (cyt c) and complex IV in the absence of complex III inhibitors. Antimycin (Ant) disrupts electron flow from cytochrome b (bL and bH) in complex III to Q, such that electrons accumulate in FeS clusters and are donated to O2 to form superoxide or H2O2 (dashed red arrow), but only when the QH2/Q ratio is low (inset). Stigmatellin (Stigm) blocks electron transfer to the FeS clusters with which O2 interacts to prevent ROS generation. By oxidizing succinate to fumarate (Fum), complex II (C II, blue) provides fumarate to ME2 to generate malate (Mal). Malonate (Malon) inhibits succinate oxidation by complex II, limiting the supply of malate to ME2. Pyr, pyruvate.
Critical importance of increased matrix [Mg2+] for NADH generation by ME2
In antimycin-inhibited permeabilized mitochondria, Mg2+-induced H2O2 production was rapidly and almost completely inhibited with EDTA > Mg2+ and reactivated with Mg2+ > EDTA, demonstrating that H2O2 production was strongly dependent on Mg2+. To the best of our knowledge, ME2 is the only NADH-generating enzyme in the matrix that has absolute requirement for Mg2+ or Mn2+ (37). In catalyzing oxidative decarboxylation of malate to generate NAD(P)H, pyruvate and CO2, ME2 can reduce either NAD+ and NADP+, but under most conditions prefers NAD+ (37, 43). ME2 is allosterically activated by fumarate and inhibited by ATP (41, 43). Fumarate's effect is mainly to decrease the Km value for malate, Mg2+, and NAD+, such that the greatest activation occurs at lower concentrations of malate, Mg2+, and NAD+ (41). We demonstrated that fumarate activates NAD+/Mg2+-induced ROS production by antimycin-inhibited complex III, the effect being most pronounced at low [Mg2+] (Fig. 9B). Because fumarate addition is expected to increase [malate] in the matrix as well, this effect may dominate in our conditions.
In permeabilized cardiac mitochondria, both Mg2+ and Mn2+ are known to have concentration-dependent effects on NADH generation in the presence of saturating concentrations of malate, NAD+, and complex I inhibitors, which are rapidly inhibited with EDTA (33). Mn2+ activates ME2 more effectively as demonstrated by the increase in NADH production at significantly lower [Mn2+] compared with [Mg2+] (33, 37) In this study we confirmed higher activation of NADH production by ME2 with Mn2+ compared with Mg2+ in permeabilized cardiac mitochondria. In addition, Mn2+ activated NADH production at lower concentrations compared with Mg2+. Although ME2 activity is relatively low in cardiac mitochondria, it is sufficient to generate a small increase in the NADH/NAD+ ratio required for significant ROS production by antimycin-inhibited complex III in the absence of exogenous substrates. In this regard, the higher rates of NADH production stimulated by Mn2+ also resulted in higher H2O2 production rates compared with Mg2+ in the absence of exogenous substrates. We did not study the effect of Mn2+ on H2O2 production in detail, mainly because the effect of I/R on [Mn2+] in cardiac cells is not known. Mg2+ alone had modest effects on H2O2 production that can be attributed to residual NAD(H) bound to matrix proteins (39, 40). Conversely, NAD+ addition alone slightly increased H2O2 production that was suppressed by 250 μm EDTA to chelate residual endogenous matrix Mg2+. Importantly, for low [Mg2+] at which H2O2 production after NAD+ addition was relatively modest, exogenous malate (0.1 mm) significantly accelerated H2O2 production. For higher [Mg2+] at which NAD+ addition stimulated more robust H2O2 production, however, exogenous malate (0.1 mm) had little additive effect on H2O2 production. Adding exogenous malate in relatively high (millimolar) concentrations during NAD+/Mg2+-stimulated H2O2 production, however, rapidly inhibited H2O2 production. These results suggest that the critical level of NAD+-related reduction of complex III, accomplished via Q reduction, is required to maximize stigmatellin-sensitive ROS production (30, 32, 42), and once this level is exceeded, ROS production rapidly decreases (Fig. 6).
Importance of limited NADH generation
Rather surprisingly, alamethicin-permeabilized mitochondria still contained enough endogenous substrates to reduce antimycin-inhibited complex III and markedly increase ROS production when Mg2+ and NAD+ were added to activate ME2. Because piericidin, known to inhibit Q reduction by NADH, effectively inhibited H2O2 production, oxidation of NAD+-related substrates was most likely responsible for NAD+/Mg2+-induced reduction of antimycin-inhibited complex III. However, because complex II inhibition significantly decreased NAD+/Mg2+-stimulated H2O2 production, we conclude that the sequence from succinate to fumarate was involved in malate production, thereby fueling ME2-induced NADH generation. The stimulating effect of ADP on Mg2+/NAD+-induced ROS production (Fig. 5) could be related to succinate generation from succinyl-CoA. In addition to GTP/GDP-specific succinyl-CoA synthetase, cardiac mitochondria also contain an ATP/ADP-specific enzyme (44) that catalyzes the reversible conversion of succinyl-CoA and ADP to CoA, ATP, and succinate. Succinate oxidation can also reduce the Q pool that connects complexes II and III. In intact mitochondria, an increase in added succinate has been shown to increase H2O2 production, reaching a maximum at about 1 mm, followed by a decrease at higher concentrations (30). However, because reported Km values of dicarboxylate transporter for succinate are around 1 mm (45), the rate of succinate influx into the matrix in intact mitochondria may not be sufficient to maintain matrix [succinate] at the buffer level. This could explain why in permeabilized mitochondria, stigmatellin-sensitive H2O2 production reached a maximum at succinate concentration significantly lower than 1 mm. Most importantly, the increased ROS production at low [succinate] was significantly accelerated by adding 2.5 mm Mg2+ in the presence of ADP and NAD+. The ability of Mg2+ to enhance H2O2 production to an almost equal extent in the absence or presence of exogenous succinate (Fig. 9A) suggests that enough malate is available or generated from endogenous succinate under those conditions to optimize ROS production by antimycin-inhibited permeabilized mitochondria. The importance of endogenous substrate oxidation for limited NADH generation and NAD+/Mg2+-dependent ROS production was also demonstrated by the finding that adding exogenous malate only slightly increased H2O2 production when malate was increased from 0 to 0.2 mm, followed by significant inhibition at malate concentrations >1 mm (Fig. 10B). When complex II was inhibited to prevent succinate oxidation, higher exogenous malate concentrations were required to increase NAD+/Mg2+-dependent ROS production by complex III, but ROS production was still significantly inhibited at malate concentrations of >1 mm (Fig. 7B). Here, an important finding is that the decrease in H2O2 production occurred at malate concentrations that stimulated a robust increase in NADH fluorescence in the mitochondrial suspension (Fig. 7A).
At first sight these results may seem confusing because NADH-related reduction of complex III is definitely required for the Mg2+/NAD+-stimulated ROS production by complex III. However, these results might be explained by efficient electron transfer from ME2 to complex I and further from NADH to Q within the I–III supercomplex. It has been proposed that a certain fraction of Q, not utilized for succinate oxidation, is channeled within the I–III supercomplex in a compartmentalized manner. Some complex III units interact with complex I to form a supercomplex exclusively dedicated to NADH oxidation, where other complex III units not as tightly bound to complex I are mainly responsible for oxidation of QH2 generated by SDH using the free Q pool (46). If redox carriers are indeed compartmentalized and can accomplish efficient electron transfer from NADH to complex III by using only Q available within the I–III supercomplex, the corresponding changes in NADH fluorescence that are responsible for ROS production are likely to be small. In contrast, the increase in NADH production beyond the limit required to maximize stigmatellin-sensitive ROS production was readily detected (Fig. 7A). Nevertheless, based on recent kinetic evidence, caution should be exercised regarding substrate channeling between the enzymes in supercomplexes. If metabolic pathways for NADH and succinate interact and catalyze via a universally accessible Q pool as suggested (47), H2O2 production by antimycin-inhibited complex III is still dependent on the QH2/Q ratio.
Implications for ROS production in the setting of ischemia/reperfusion
Based on the work of Chouchani et al. (5, 6), a major source of damaging ROS upon reperfusion after prolonged ischemia is related to RET driven by succinate that has accumulated during ischemia. Because mitochondria become depolarized during ischemia, this mechanism requires that a significant subpopulation of mitochondria is able to fully recover ΔΨ during early reperfusion (7–9). As soon as RET-related ROS production or other factors trigger PTP opening, however, matrix depolarization will immediately terminate ROS production by RET (48). This negative feedback feature tends to limit sustained ROS production by RET. Indeed, after prolonged ischemia, ΔΨ may either recover only briefly before PTP opening occurs, may not recover at all (10, 11), or may recover only slowly and partially (12–14). In this scenario, our findings provide a novel mechanism that could sustain ROS generation from inhibited complex III when PTP opening causes RET to cease, assuming matrix succinate, fumarate, malate, and NAD+ concentrations are in the appropriate range for optimizing complex III reduction for ROS production (30–32) when ME2 is activated by elevated matrix Mg2+ and ADP (33, 37).
PTP opening has been demonstrated to occur at the start of reperfusion after prolonged ischemia (29, 49) and results in a collapse of protonmotive force, reversal of the F0F1-ATPase, and direct access to cytoplasmic ATP. As F0F1-ATPase hydrolyzes MgATP, both ADP/Pi and free Mg2+ increase, because the affinity of ATP for Mg2+ is about 10-fold higher than that of ADP (50). Alternatively, membrane potential depolarization may increase matrix-free Mg2+ and its release (51). Free Mg2+ elevation after ischemia/reperfusion (52–54) or anoxia/reoxygenation has been well documented (52, 53). Thus, if complex III has become damaged during prolonged ischemia, reperfusion sets the stage for robust continued ROS generation when PTP is open and allows the matrix to equilibrate with elevated cytoplasmic Mg2+ and to activate ME2 at the same time that the NADH/NAD+ ratio is low and ADP is elevated. Although increases in matrix [ADP] and [Mg2+] are known to inhibit PTP opening in settings that do not involve ROS production (55, 56), these inhibitors act from the matrix side where they also simultaneously increase ROS generation by the mechanism proposed in this study. Therefore, it seems unlikely that ROS generation will be significantly and rapidly inhibited even if ADP and Mg2+ induce PTP closure. In addition, increased ROS production favors PTP opening that may overpower inhibitory effect of ADP and Mg2+.
Inhibition of complex III has been reported to require ischemia durations of >30 min, whereas complex I inhibition occurs early after 15–20 min of ischemia (3, 57). However, ROS generation by inhibited complex I requires a very high NADH/NAD+ ratio (>10) (25), whereas the NADH/NAD+ ratio declines very rapidly upon reperfusion when O2 again becomes available for ROS production (16, 17, 26, 29), agreeing with other evidence that forward electron flow through damaged complex I is not the major source of ROS during reperfusion (27–29). In contrast, for damaged complex III to produce ROS when ME2 is activated by increases in matrix Mg2+, NAD+, and ADP, the level of endogenous/exogenous substrates generating malate has to be at appropriate concentrations such that the NADH/NAD+ ratio remains low, expected to maintain a low Q/QH2 ratio required to maximize ROS production by antimycin-inhibited complex III (30–32).
Our proposed mechanism is consistent with the observation that inhibition of succinate oxidation by malonate (5, 58, 59) is cardioprotective, because malonate would not only limit RET through complex I, but also inhibit succinate conversion to fumarate and malate, thereby limiting the availability of malate for oxidation by ME2. Likewise, by preventing RET-derived ROS generation, complex I inhibition with rotenone or amobarbital during I/R should also abolish NADH-related reduction of complex III with a resulting decrease in ROS production and (by reducing PTP opening) reduced mitochondrial damage during cardiac ischemia, consistent with the cardioprotective effect of rotenone (27) or amobarbital (60). However, despite existing evidence that complex III plays a central role in ROS production (27, 61), the extent of damage to complex III by ischemia/reperfusion is not well defined. Ischemia/reperfusion may induce changes in the structural integrity of complex III, which in association with decreased electron transfer may result in higher ROS production. It has been demonstrated that proteinase K or heat treatment can induce increases in superoxide production by complex III similar to antimycin treatment. Increased ROS production has been explained by changes in the hydrophobic QH2 oxidation pocket that may also lead to increased accessibility of O2 to the pocket to generate more superoxide or facilitate its release (62). Some useful information may come from comparative studies with the bacterial (Rhodobacter capsulatus) mutant complex III and human complex III mutant Y278C (homolog of R. capsulatus cytochrome b Y302C mutant). In both cases, mutation of a highly conserved tyrosine residue of cytochrome b, in the quinol oxidation site, results in surprisingly similar decreases in catalytic activity of complex III, accompanied by increased ROS production and cellular damage (63). However, the precise mechanism of complex III damage during ischemia/reperfusion remains elusive.
Limitations
Extrapolating our findings in alamethicin-permeabilized mitochondria to I/R in intact hearts is highly speculative, given that the intracellular milieu during I/R is constantly changing and is not precisely defined. Nevertheless, the finding that calcium-induced PTP opening reproduced the effects of alamethicin demonstrates that Mg2+/NAD+-stimulated ROS production by complex III is not an artifact of alamethicin pores, but is a general response to inner membrane permeabilization. Whether complex III is comparably inhibited during genuine ischemia as with antimycin, and whether the relative concentrations of Mg2+, NAD+, succinate, fumarate, malate, ADP, and other metabolites during reperfusion approximate those optimizing ROS production by inhibited complex III when ME2 is activated remain open questions. Although the definitive contribution to cardiac injury of ME2 and complex III, relative to RET via complex I, remains to be established, this is potentially testable by knocking down cardiac ME2 in mice to determine whether they demonstrate a smaller ROS burst and less cardiac injury following ischemia/reperfusion compared with wild-type mice. As shown in the accompanying article (73), complex II can also become a significant source of ROS production after PTP opening when either complex II or III is inhibited, and its quantitative contribution, relative to complex III-derived ROS, remains to be characterized.
Experimental procedures
Ethical approval
This study was approved by the UCLA Chancellor's Animal Research Committee (ARC 2003-063-23B) and was performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (Publication No. 85-23, revised 1996) and with UCLA Policy 990 on the Use of Laboratory Animal Subjects in Research (revised 2010).
Experimental techniques
All measurements were carried out using customized Fiber Optic Spectrofluorometer (Ocean Optics) in a partially open continuously stirred cuvette at room temperature (22–24 °C) (64). Mitochondria were isolated from rabbit hearts as described previously (65). Intact mitochondria were added (0.6 to 1.0 mg/ml) to incubation buffer containing 250 mm sucrose, 10 mm Hepes, pH 7.4, with Tris. Mitochondria were permeabilized with addition of alamethicin (20 μg/ml). Alamethicin creates pores that allow equilibration of low-molecular-weight components across the inner membrane, whereas high-molecular-weight proteins are retained in the matrix and intermembrane space (66). Electron microscopy of alamethicin-treated mitochondria reveals no major disruption of membranes (66), and a consensus exists that enzymes, including those involved in ROS production, remain fully functional and have enhanced access to exogenously delivered substrates (33, 66–69). All chemicals were obtained from Sigma.
H2O2 release from mitochondria was measured using 5 μm Amplex Red and 0.2 units of HRP in the buffer (excitation/emission, 540:590 nm). The increase in resorufin fluorescence was calibrated by adding H2O2 in known concentrations to the buffer containing permeabilized mitochondria. Mitochondrial O2 consumption was measured continuously by monitoring buffer O2 content using a fiber optic oxygen sensor FOXY-AL300 (Ocean Optics) (64).
NADH fluorescence was recorded at 366/460 nm excitation/emission wavelengths. NAD+-dependent ME2 activity was determined in permeabilized mitochondria by measuring the ability of Mg2+ or Mn2+ to increase [NADH] in the presence of piericidin, malate, and NAD+. The increase in NADH fluorescence was calibrated by adding NADH in known concentration to permeabilized mitochondria in the presence of piericidin.
Statistical analysis
For each data set, the mean and accompanying 95% confidence intervals are reported. The conventional percentile bootstrap-resampling approach with 10,000 replications was used for estimating 95% confidence intervals as well as examining the significant difference between groups (effect size statistics) (70–72). A p value <0.05 was considered statistically significant. All analyses were performed by subroutines for bootstrapping developed in the Python programming language (72).
Author contributions
P. K. and J. N. W. conceived and coordinated the study. P. K., G. C., and S. J. A. performed and analyzed the experiments. P. K., J. N. W., G. C., and S. J. A. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by National Institutes of Health Grants R01 HL101228 and R01 HL117385 from the NHLBI, American Heart Association Western States Affiliate Post-doctoral Research Fellowship 11POST6110007, American Heart Association Scientist Development Grant 16SDG31180036, and the Laubisch and Kawata Endowments. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ROS
- reactive oxygen species
- AA5
- atpenin A5
- CoA
- coenzyme A
- CsA
- cyclosporin A
- ME2
- malic enzyme
- MDH
- malate dehydrogenase
- ΔΨ
- mitochondrial membrane potential
- PTP
- permeability transition pore
- RET
- reverse electron transport
- Q
- ubiquinone
- QH2
- ubiquinol
- SDH
- succinate dehydrogenase
- I/R
- ischemia/reperfusion.
References
- 1. Quinlan C. L., Goncalves R. L., Hey-Mogensen M., Yadava N., Bunik V. I., and Brand M. D. (2014) The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 289, 8312–8325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Starkov A. A. (2008) The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N.Y. Acad. Sci. 1147, 37–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen Q., Camara A. K., Stowe D. F., Hoppel C. L., and Lesnefsky E. J. (2007) Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. Am. J. Physiol. Cell Physiol 292, C137–C147 [DOI] [PubMed] [Google Scholar]
- 4. Dröse S., and Brandt U. (2012) Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 748, 145–169 [DOI] [PubMed] [Google Scholar]
- 5. Chouchani E. T., Pell V. R., Gaude E., Aksentijević D., Sundier S. Y., Robb E. L., Logan A., Nadtochiy S. M., Ord E. N., Smith A. C., Eyassu F., Shirley R., Hu C. H., Dare A. J., James A. M., et al. (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chouchani E. T., Pell V. R., James A. M., Work L. M., Saeb-Parsy K., Frezza C., Krieg T., and Murphy M. P. (2016) A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 23, 254–263 [DOI] [PubMed] [Google Scholar]
- 7. Korshunov S. S., Korkina O. V., Ruuge E. K., Skulachev V. P., and Starkov A. A. (1998) Fatty acids as natural uncouplers preventing generation of O2˙̄ and H2O2 by mitochondria in the resting state. FEBS Lett. 435, 215–218 [DOI] [PubMed] [Google Scholar]
- 8. Korshunov S. S., Skulachev V. P., and Starkov A. A. (1997) High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 416, 15–18 [DOI] [PubMed] [Google Scholar]
- 9. Votyakova T. V., and Reynolds I. J. (2001) Δψ(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J. Neurochem. 79, 266–277 [DOI] [PubMed] [Google Scholar]
- 10. Levraut J., Iwase H., Shao Z. H., Vanden Hoek T. L., and Schumacker P. T. (2003) Cell death during ischemia: relationship to mitochondrial depolarization and ROS generation. Am. J. Physiol. Heart Circ. Physiol. 284, H549–H558 [DOI] [PubMed] [Google Scholar]
- 11. Matsumoto-Ida M., Akao M., Takeda T., Kato M., and Kita T. (2006) Real-time 2-photon imaging of mitochondrial function in perfused rat hearts subjected to ischemia/reperfusion. Circulation 114, 1497–1503 [DOI] [PubMed] [Google Scholar]
- 12. Slodzinski M. K., Aon M. A., and O'Rourke B. (2008) Glutathione oxidation as a trigger of mitochondrial depolarization and oscillation in intact hearts. J. Mol. Cell. Cardiol. 45, 650–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berkich D. A., Salama G., and LaNoue K. F. (2003) Mitochondrial membrane potentials in ischemic hearts. Arch. Biochem. Biophys. 420, 279–286 [DOI] [PubMed] [Google Scholar]
- 14. Kato M., Akao M., Matsumoto-Ida M., Makiyama T., Iguchi M., Takeda T., Shimizu S., and Kita T. (2009) The targeting of cyclophilin D by RNAi as a novel cardioprotective therapy: evidence from two-photon imaging. Cardiovasc. Res. 83, 335–344 [DOI] [PubMed] [Google Scholar]
- 15. Halestrap A. P., Clarke S. J., and Javadov S. A. (2004) Mitochondrial permeability transition pore opening during myocardial reperfusion–a target for cardioprotection. Cardiovasc. Res. 61, 372–385 [DOI] [PubMed] [Google Scholar]
- 16. Riess M. L., Camara A. K., Chen Q., Novalija E., Rhodes S. S., and Stowe D. F. (2002) Altered NADH and improved function by anesthetic and ischemic preconditioning in guinea pig intact hearts. Am. J. Physiol. Heart Circ. Physiol. 283, H53–H60 [DOI] [PubMed] [Google Scholar]
- 17. Aldakkak M., Stowe D. F., Chen Q., Lesnefsky E. J., and Camara A. K. (2008) Inhibited mitochondrial respiration by amobarbital during cardiac ischaemia improves redox state and reduces matrix Ca2+ overload and ROS release. Cardiovasc. Res. 77, 406–415 [DOI] [PubMed] [Google Scholar]
- 18. Aldakkak M., Stowe D. F., Heisner J. S., Spence M., and Camara A. K. (2008) Enhanced Na+/H+ exchange during ischemia and reperfusion impairs mitochondrial bioenergetics and myocardial function. J. Cardiovasc. Pharmacol. 52, 236–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lenaz G. (2001) The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life 52, 159–164 [DOI] [PubMed] [Google Scholar]
- 20. Chen Y. R., and Zweier J. L. (2014) Cardiac mitochondria and reactive oxygen species generation. Circ. Res. 114, 524–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bolli R., Jeroudi M. O., Patel B. S., DuBose C. M., Lai E. K., Roberts R., and McCay P. B. (1989) Direct evidence that oxygen-derived free radicals contribute to postischemic myocardial dysfunction in the intact dog. Proc. Natl. Acad. Sci. U.S.A. 86, 4695–4699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zweier J. L., Flaherty J. T., and Weisfeldt M. L. (1987) Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc. Natl. Acad. Sci. U.S.A. 84, 1404–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zweier J. L., Kuppusamy P., Williams R., Rayburn B. K., Smith D., Weisfeldt M. L., and Flaherty J. T. (1989) Measurement and characterization of postischemic free radical generation in the isolated perfused heart. J. Biol. Chem. 264, 18890–18895 [PubMed] [Google Scholar]
- 24. Murphy E., and Steenbergen C. (2008) Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 88, 581–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Korge P., Calmettes G., and Weiss J. N. (2015) Increased reactive oxygen species production during reductive stress: the roles of mitochondrial glutathione and thioredoxin reductases. Biochim. Biophys. Acta 1847, 514–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aldakkak M., Stowe D. F., Lesnefsky E. J., Heisner J. S., Chen Q., and Camara A. K. (2009) Modulation of mitochondrial bioenergetics in the isolated guinea pig beating heart by potassium and lidocaine cardioplegia: implications for cardioprotection. J. Cardiovasc. Pharmacol. 54, 298–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen Q., Vazquez E. J., Moghaddas S., Hoppel C. L., and Lesnefsky E. J. (2003) Production of reactive oxygen species by mitochondria: central role of complex III. J. Biol. Chem. 278, 36027–36031 [DOI] [PubMed] [Google Scholar]
- 28. Becker L. B., vanden Hoek T. L., Shao Z. H., Li C. Q., and Schumacker P. T. (1999) Generation of superoxide in cardiomyocytes during ischemia before reperfusion. Am. J. Physiol. 277, H2240–H2246 [DOI] [PubMed] [Google Scholar]
- 29. Andrienko T., Pasdois P., Rossbach A., and Halestrap A. P. (2016) Real-time fluorescence measurements of ROS and [Ca2+] in ischemic/reperfused rat hearts: detectable increases occur only after mitochondrial pore opening and are attenuated by ischemic preconditioning. PLoS ONE 11, e0167300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Quinlan C. L., Gerencser A. A., Treberg J. R., and Brand M. D. (2011) The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. J. Biol. Chem. 286, 31361–31372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bleier L., and Dröse S. (2013) Superoxide generation by complex III: from mechanistic rationales to functional consequences. Biochim. Biophys. Acta 1827, 1320–1331 [DOI] [PubMed] [Google Scholar]
- 32. Dröse S., Bleier L., and Brandt U. (2011) A common mechanism links differently acting complex II inhibitors to cardioprotection: modulation of mitochondrial reactive oxygen species production. Mol. Pharmacol. 79, 814–822 [DOI] [PubMed] [Google Scholar]
- 33. Korge P., Calmettes G., and Weiss J. N. (2016) Reactive oxygen species production in cardiac mitochondria after complex I inhibition: modulation by substrate-dependent regulation of the NADH/NAD ratio. Free Radic. Biol. Med. 96, 22–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kushnareva Y., Murphy A. N., and Andreyev A. (2002) Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem. J. 368, 545–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bellamy D. (1962) The endogenous citric acid cycle intermediates and amino acids of mitochondria. Biochem. J. 82, 218–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Olson M. S., and Von Korff R. W. (1967) Changes in endogenous substrates of isolated rabbit heart mitochondria during storage. J. Biol. Chem. 242, 325–332 [PubMed] [Google Scholar]
- 37. Lin R. C., and Davis E. J. (1974) Malic enzymes of rabbit heart mitochondria. Separation and comparison of some characteristics of a nicotinamide adenine dinucleotide-preferring and a nicotinamide adenine dinucleotide phosphate-specific enzyme. J. Biol. Chem. 249, 3867–3875 [PubMed] [Google Scholar]
- 38. Davisson V. J., and Schulz A. R. (1985) The purification and steady-state kinetic behaviour of rabbit heart mitochondrial NAD(P)+ malic enzyme. Biochem. J. 225, 335–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blinova K., Carroll S., Bose S., Smirnov A. V., Harvey J. J., Knutson J. R., and Balaban R. S. (2005) Distribution of mitochondrial NADH fluorescence lifetimes: steady-state kinetics of matrix NADH interactions. Biochemistry 44, 2585–2594 [DOI] [PubMed] [Google Scholar]
- 40. Blinova K., Levine R. L., Boja E. S., Griffiths G. L., Shi Z. D., Ruddy B., and Balaban R. S. (2008) Mitochondrial NADH fluorescence is enhanced by complex I binding. Biochemistry 47, 9636–9645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hung H. C., Kuo M. W., Chang G. G., and Liu G. Y. (2005) Characterization of the functional role of allosteric site residue Asp102 in the regulatory mechanism of human mitochondrial NAD(P)+-dependent malate dehydrogenase (malic enzyme). Biochem. J. 392, 39–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dröse S., and Brandt U. (2008) The mechanism of mitochondrial superoxide production by the cytochrome bc1 complex. J. Biol. Chem. 283, 21649–21654 [DOI] [PubMed] [Google Scholar]
- 43. Sauer L. A. (1973) Mitochondrial NAD-dependent malic enzyme: a new regulatory enzyme. FEBS Lett. 33, 251–255 [DOI] [PubMed] [Google Scholar]
- 44. Johnson J. D., Muhonen W. W., and Lambeth D. O. (1998) Characterization of the ATP- and GTP-specific succinyl-CoA synthetases in pigeon. The enzymes incorporate the same α-subunit. J. Biol. Chem. 273, 27573–27579 [DOI] [PubMed] [Google Scholar]
- 45. Siebels I., and Dröse S. (2013) Q-site inhibitor induced ROS production of mitochondrial complex II is attenuated by TCA cycle dicarboxylates. Biochim. Biophys. Acta 1827, 1156–1164 [DOI] [PubMed] [Google Scholar]
- 46. Genova M. L., and Lenaz G. (2014) Functional role of mitochondrial respiratory supercomplexes. Biochim. Biophys. Acta 1837, 427–443 [DOI] [PubMed] [Google Scholar]
- 47. Blaza J. N., Serreli R., Jones A. J., Mohammed K., and Hirst J. (2014) Kinetic evidence against partitioning of the ubiquinone pool and the catalytic relevance of respiratory-chain supercomplexes. Proc. Natl. Acad. Sci. U.S.A. 111, 15735–15740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ross T., Szczepanek K., Bowler E., Hu Y., Larner A., Lesnefsky E. J., and Chen Q. (2013) Reverse electron flow-mediated ROS generation in ischemia-damaged mitochondria: role of complex I inhibition vs. depolarization of inner mitochondrial membrane. Biochim. Biophys. Acta 1830, 4537–4542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Halestrap A. P. (2004) Does the mitochondrial permeability transition have a role in preconditioning? Circulation 110, e303. [DOI] [PubMed] [Google Scholar]
- 50. Leyssens A., Nowicky A. V., Patterson L., Crompton M., and Duchen M. R. (1996) The relationship between mitochondrial state, ATP hydrolysis, (Mg-2+)-i and (Ca-2+)-i studied in isolated rat cardiomyocytes. J. Physiol. 496, 111–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kubota T., Shindo Y., Tokuno K., Komatsu H., Ogawa H., Kudo S., Kitamura Y., Suzuki K., and Oka K. (2005) Mitochondria are intracellular magnesium stores: investigation by simultaneous fluorescent imagings in PC12 cells. Biochim. Biophys. Acta 1744, 19–28 [DOI] [PubMed] [Google Scholar]
- 52. Headrick J. P. (1998) Aging impairs functional, metabolic and ionic recovery from ischemia-reperfusion and hypoxia-reoxygenation. J. Mol. Cell. Cardiol. 30, 1415–1430 [DOI] [PubMed] [Google Scholar]
- 53. Headrick J. P., and Willis R. J. (1991) Cytosolic free magnesium in stimulated, hypoxic, and underperfused rat heart. J. Mol. Cell. Cardiol. 23, 991–999 [DOI] [PubMed] [Google Scholar]
- 54. Murphy E., Steenbergen C., Levy L. A., Raju B., and London R. E. (1989) Cytosolic free magnesium levels in ischemic rat heart. J. Biol. Chem. 264, 5622–5627 [PubMed] [Google Scholar]
- 55. Novgorodov S. A., Gudz T. I., Brierley G. P., and Pfeiffer D. R. (1994) Magnesium ion modulates the sensitivity of the mitochondrial permeability transition pore to cyclosporin A and ADP. Arch. Biochem. Biophys. 311, 219–228 [DOI] [PubMed] [Google Scholar]
- 56. Novgorodov S. A., Gudz T. I., Milgrom Y. M., and Brierley G. P. (1992) The permeability transition in heart mitochondria is regulated synergistically by ADP and cyclosporin A. J. Biol. Chem. 267, 16274–16282 [PubMed] [Google Scholar]
- 57. Rouslin W. (1983) Mitochondrial complexes I, II, III, IV, and V in myocardial ischemia and autolysis. Am. J. Physiol. 244, H743–H748 [DOI] [PubMed] [Google Scholar]
- 58. Valls-Lacalle L., Barba I., Miró-Casas E., Alburquerque-Béjar J. J., Ruiz-Meana M., Fuertes-Agudo M., Rodríguez-Sinovas A., and García-Dorado D. (2016) Succinate dehydrogenase inhibition with malonate during reperfusion reduces infarct size by preventing mitochondrial permeability transition. Cardiovasc. Res. 109, 374–384 [DOI] [PubMed] [Google Scholar]
- 59. Wojtovich A. P., and Brookes P. S. (2008) The endogenous mitochondrial complex II inhibitor malonate regulates mitochondrial ATP-sensitive potassium channels: implications for ischemic preconditioning. Biochim. Biophys. Acta 1777, 882–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen Q., Moghaddas S., Hoppel C. L., and Lesnefsky E. J. (2006) Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. J. Pharmacol. Exp. Ther. 319, 1405–1412 [DOI] [PubMed] [Google Scholar]
- 61. Chen Q., Moghaddas S., Hoppel C. L., and Lesnefsky E. J. (2008) Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am. J. Physiol. Cell Physiol. 294, C460–C466 [DOI] [PubMed] [Google Scholar]
- 62. Yin Y., Yang S., Yu L., and Yu C. A. (2010) Reaction mechanism of superoxide generation during ubiquinol oxidation by the cytochrome bc1 complex. J. Biol. Chem. 285, 17038–17045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lanciano P., Khalfaoui-Hassani B., Selamoglu N., Ghelli A., Rugolo M., and Daldal F. (2013) Molecular mechanisms of superoxide production by complex III: a bacterial versus human mitochondrial comparative case study. Biochim. Biophys. Acta 1827, 1332–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Korge P., Ping P., and Weiss J. N. (2008) Reactive oxygen species production in energized cardiac mitochondria during hypoxia/reoxygenation: modulation by nitric oxide. Circ. Res. 103, 873–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Korge P., Honda H. M., and Weiss J. N. (2001) Regulation of the mitochondrial permeability transition by matrix Ca(2+) and voltage during anoxia/reoxygenation. Am. J. Physiol. Cell Physiol. 280, C517–C526 [DOI] [PubMed] [Google Scholar]
- 66. Gostimskaya I. S., Grivennikova V. G., Zharova T. V., Bakeeva L. E., and Vinogradov A. D. (2003) In situ assay of the intramitochondrial enzymes: use of alamethicin for permeabilization of mitochondria. Anal. Biochem. 313, 46–52 [DOI] [PubMed] [Google Scholar]
- 67. Grivennikova V. G., Kapustin A. N., and Vinogradov A. D. (2001) Catalytic activity of NADH-ubiquinone oxidoreductase (complex I) in intact mitochondria. evidence for the slow active/inactive transition. J. Biol. Chem. 276, 9038–9044 [DOI] [PubMed] [Google Scholar]
- 68. Kareyeva A. V., Grivennikova V. G., and Vinogradov A. D. (2012) Mitochondrial hydrogen peroxide production as determined by the pyridine nucleotide pool and its redox state. Biochim. Biophys. Acta 1817, 1879–1885 [DOI] [PubMed] [Google Scholar]
- 69. Kotlyar A. B., Maklashina E., and Cecchini G. (2004) Absence of NADH channeling in coupled reaction of mitochondrial malate dehydrogenase and complex I in alamethicin-permeabilized rat liver mitochondria. Biochem. Biophys. Res. Commun. 318, 987–991 [DOI] [PubMed] [Google Scholar]
- 70. Nakagawa S., and Cuthill I. C. (2007) Effect size, confidence interval and statistical significance: a practical guide for biologists. Biol. Rev. Camb. Philos. Soc. 82, 591–605 [DOI] [PubMed] [Google Scholar]
- 71. Efron B., and Tibshirani R. (1991) Statistical data analysis in the computer age. Science 253, 390–395 [DOI] [PubMed] [Google Scholar]
- 72. Calmettes G., Drummond G. B., and Vowler S. L. (2012) Making do with what we have: use your bootstraps. J. Physiol. 590, 3403–3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Korge P., John S. A., Calmettes G., and Weiss J. N. (2017) Reactive oxygen species production induced by pore opening in cardiac mitochondria: The role of complex II. J. Biol. Chem. 292, 9896–9905 [DOI] [PMC free article] [PubMed] [Google Scholar]