

Abstract
Succinate-driven reverse electron transport (RET) through complex I is hypothesized to be a major source of reactive oxygen species (ROS) that induces permeability transition pore (PTP) opening and damages the heart during ischemia/reperfusion. Because RET can only generate ROS when mitochondria are fully polarized, this mechanism is self-limiting once PTP opens during reperfusion. In the accompanying article (Korge, P., Calmettes, G., John, S. A., and Weiss, J. N. (2017) J. Biol. Chem. 292, 9882–9895), we showed that ROS production after PTP opening can be sustained when complex III is damaged (simulated by antimycin). Here we show that complex II can also contribute to sustained ROS production in isolated rabbit cardiac mitochondria following inner membrane pore formation induced by either alamethicin or calcium-induced PTP opening. Two conditions are required to maximize malonate-sensitive ROS production by complex II in isolated mitochondria: (a) complex II inhibition by atpenin A5 or complex III inhibition by stigmatellin that results in succinate-dependent reduction of the dicarboxylate-binding site of complex II (site IIf); (b) pore opening in the inner membrane resulting in rapid efflux of succinate/fumarate and other dicarboxylates capable of competitively binding to site IIf. The decrease in matrix [dicarboxylate] allows O2 access to reduced site IIf, thereby making electron donation to O2 possible, explaining the rapid increase in ROS production provided that site IIf is reduced. Because ischemia is known to inhibit complexes II and III and increase matrix succinate/fumarate levels, we hypothesize that by allowing dicarboxylate efflux from the matrix, PTP opening during reperfusion may activate sustained ROS production by this mechanism after RET-driven ROS production has ceased.
Keywords: electron transfer complex, electron transport system, mitochondria, mitochondrial membrane potential, mitochondrial respiratory chain complex, nicotinamide adenine dinucleotide (NADH), reactive oxygen species (ROS), complex II
Introduction
Following prolonged myocardial ischemia, reperfusion results in the generation of excessive reactive oxygen species (ROS)2 from mitochondria that contribute to reperfusion injury. A recent metabolomics study demonstrated that succinate accumulation during ischemia correlated strongly with ROS production and injury during reperfusion (1). The authors proposed that the major source of damaging ROS under these conditions was succinate-driven reverse electron transport (RET) through complex I (2). Although RET is known to generate copious ROS, this is possible only when ΔΨ is fully polarized (3–5). This raises the issue that mitochondria, which are depolarized during prolonged ischemia, would have to recover ΔΨ fully during early reperfusion to generate ROS by this mechanism, which would then cease once ROS production induced PTP opening. Under conditions in which ΔΨ either does not recover (6, 7) or recovers only slowly and partially (8–10) upon reperfusion as a result of PTP opening (11) and increased inner membrane leakiness (12–14), ROS production by RET would be self-limited. We therefore examined whether additional mechanisms might come into play to sustain succinate-driven ROS production after PTP opening has occurred, consistent with experimental evidence that ROS production increases simultaneously with PTP opening (15). In the accompanying paper (52), we showed that when ischemic injury to complex III is mimicked with antimycin, inner membrane permeabilization using either alamethicin or calcium-induced PTP opening stimulated robust ROS production by complex III by allowing Mg2+ influx into the matrix to activate malic enzyme 2 (ME2). Using malate derived from endogenous succinate and fumarate, ME2 reduced NAD+ to provide the reducing equivalents for ROS production by antimycin-inhibited complex III. In this study, we report that when the inner membrane is permeabilized, complex II can also serve as a robust source of ROS production after either inhibition of ubiquinone (Q) reduction in complex II or ubiquinol (QH2) oxidation in complex III, providing another potential mechanism for succinate-driven ROS production during and after PTP opening.
Mitochondrial complex II (succinate dehydrogenase (SDH)) is responsible for succinate oxidation into fumarate in the TCA cycle, coupled to Q reduction to QH2 and respiratory chain activity. Besides this main function, complex II is known to produce ROS directly from the reduced flavin site called site IIf (16) in the presence of low levels of dicarboxylates, as shown first by Imlay (17) in inverted respiratory vesicles from Escherichia coli. Subsequent studies by several groups have demonstrated ROS production by SDH-containing membranes prepared from E. coli strain KM5 (18), submitochondrial particles (SMP) from beef heart mitochondria (16), or mitochondria isolated from skeletal muscle (19). With electrons supplied by succinate, ROS production by complex II can be stimulated by the complex II inhibitor atpenin A5 (AA5) or by the complex III inhibitor stigmatellin, which blocks QH2 oxidation by complex III. Conversely, ROS production by complex II is inhibited by malonate, consistent with ROS arising from the fully reduced flavin site IIf (16), although alternative ROS-generating sites have also been proposed (20). Brand and co-workers (19) have shown that H2O2 production increases with [succinate] up to 400 μm, but thereafter it significantly decreases at the millimolar [succinate] commonly used to energize mitochondria. This is because complex II produces ROS only when site IIf is unoccupied by dicarboxylates (16, 19). Succinate and other TCA intermediates (fumarate in particular) that bind to the dicarboxylate-binding site can restrict O2 access to this site and thereby suppress ROS production by complex II. Because ischemia/hypoxia are known to significantly increase [succinate] and [fumarate] in the matrix (1, 21), this may prevent significant ROS production from complex II simply by preventing contact of reduced site IIf with O2. Given that ROS production by inhibited complex II has been established by several groups, this study was designed to evaluate whether pore opening could create conditions promoting ROS production by complex II and, if so, determine what factors regulate it. We demonstrate that by accelerating succinate/fumarate efflux from the matrix and decreasing [dicarboxylate] in the vicinity of site IIf, pore formation in the inner mitochondrial membrane induced by the alamethicin or calcium-induced PTP opening allows O2 access to reduced IIf sites with a resulting increase in ROS production (Fig. 1). This mechanism of ROS production also requires either complex II inhibition at the level of Q reduction or inhibition of QH2 oxidation downstream in complex III. These conditions may be met during I/R because the catalytic activities of complexes II and III are inhibited in mitochondria isolated from ischemic hearts after reperfusion (22–26). If so, then during reperfusion this mechanism may also contribute to continued damaging ROS production after PTP opening has occurred and succinate-fueled RET-driven ROS production has ceased.
Figure 1.

Proposed mechanism of increased ROS production by inhibited complex II following PTP opening. Left panel, under physiological conditions succinate (S) oxidation by complex II (succinate dehydrogenase composed of four subunits SDHA–D) results in flavin site reduction (FADH2 generation) and further tunneling of electrons via Fe-S clusters to ubiquinone (Q) in the Q-binding site that generates ubiquinol (QH2) by two sequential single electron transfers, requiring also two protons. QH2 is further oxidized by complex III. No ROS are generated under those conditions because the flavin site is rapidly oxidized if there is no downstream inhibition, and dicarboxylates such as succinate and fumarate (F) shield access of O2 to their binding site. Right panel, when electron transport is inhibited at the level of Q reduction by AA5 or further downstream, FADH2 accumulates. After PTP opening in the inner membrane, matrix succinate/fumarate levels decrease, giving O2 access to reduced flavin site to generate ROS.
Results
To examine conditions under which complex II can generate significant ROS during pore opening in the inner membrane, we exposed isolated cardiac mitochondria to the complex II inhibitor AA5 (0.5 μm) while measuring H2O2 in the buffer. AA5 binds to complex II with high affinity, and experiments with isolated cardiac mitochondria and submitochondrial particles have shown complete inhibition of complex II activity by 0.1 μm AA5 (27). As shown in Fig. 2A, H2O2 production was very low until AA5-inhibited mitochondria were permeabilized with alamethicin, at which point H2O2 production accelerated markedly and then leveled off. Addition of exogenous succinyl-CoA then restored H2O2 production to the initial level. The increase in ROS production after succinyl-CoA addition indicates that despite permeabilization, the matrix contained sufficient ADP/GDP and Pi to generate succinate from the added succinyl-CoA. The concentration dependence of the ability of AA5 to stimulate H2O2 production after alamethicin was practically unchanged between 0.1 and 1 μm (results not shown), and therefore we used 0.5 or 1 μm in all our experiments. Importantly, H2O2 production required pore formation by alamethicin and was proportional to the concentration of mitochondria.
Figure 2.

Alamethicin permeabilization promotes a rapid increase in H2O2 production when complex II is inhibited with atpenin A5 or complex III is inhibited with stigmatellin. A, isolated cardiac mitochondria (Mito) were added in concentrations of 1.0, 0.75, or 0.5 mg/ml (traces a–c, respectively) to buffer containing atpenin A5 (AA5, 0.5 μm). After permeabilization with alamethicin (Ala, 20 μg/ml), H2O2 production increased and plateaued as endogenous substrates were depleted and then reaccelerated after addition of succinyl-CoA (Suc-CoA) (0.15 mm). B, mitochondria were added at concentrations of 1.0 (trace a) and 0.5 (trace b) mg/ml to buffer containing stigmatellin (Stigm, 1 μm), followed by alamethicin and succinate (50 μm) as indicated. Decreased H2O2 production was reactivated with 50 μm succinate and inhibited in trace a with 0.5 and 5 mm succinate (Succ) and in trace b with 1 mm malonate (Malon). In this and other figures, additions to all traces are indicated by black arrows; additions to one trace only are indicated by the arrows of the same color as the trace. C, summary data (median ±95% confidence intervals, *, p < 0.05) for H2O2 production in AA5- and stigmatellin-inhibited mitochondria after alamethicin permeabilization and inhibition by malonate.
Fig. 2B demonstrates that in the absence of AA5, downstream electron transport inhibition at complex III with stigmatellin also resulted in increased ROS production after alamethicin. Stigmatellin effectively inhibits QH2 oxidation by complex III and also prevents antimycin-induced ROS production by complex III. Because alamethicin eliminates Δψ and thereby stimulates NADH oxidation, endogenous substrates alone are unlikely to be able to increase the NADH/NAD ratio to the level required for ROS production during forward electron transport through complex I or ketoglutarate dehydrogenase. Moreover, without polarized Δψ, RET is not possible. By exclusion, complex II is the only plausible source of H2O2 production under these conditions. After the rapid initial increase in H2O2 production declined due to oxidation of IIf sites and depletion of endogenous succinate, addition of 50 μm succinate reactivated H2O2 production. Increasing [succinate] to 500 μm, however, suppressed H2O2 production, consistent with the previously mentioned data (16, 19) that higher [succinate] masks reduced IIf sites and prevents access to O2. H2O2 production was also inhibited by malonate, which prevents succinate oxidation by complex II (Fig. 2B, blue trace b). Although complex II activity, determined in submitochondrial particles from cardiac mitochondria in the presence of 2.5 mm succinate was already 50% inhibited in the presence of 42 μm malonate (28), we used 1 mm malonate routinely and 5 mm for experiments with higher [succinate]. Summary findings are shown in Fig. 2C.
Because alamethicin dissipates Δψ, which increases respiratory activity and accelerates consumption of endogenous substrates, including succinate, and also promotes efflux of endogenous substrates through alamethicin pores, it was important to add AA5 before the inner membrane was permeabilized. In Fig. 3A, H2O2 production was highest (0.157 nmol/min/mg) when AA5 was added 2 min before alamethicin (purple trace), but it decreased to 22, 18, and 12% when AA5 was added 1, 2, or 3 min after alamethicin (green, red and blue traces, respectively). Decreased H2O2 production under these conditions could be rapidly reversed by adding exogenous succinate in low concentrations (50 μm). However, succinate in higher concentrations (500 μm) rapidly inhibited H2O2 production.
Figure 3.
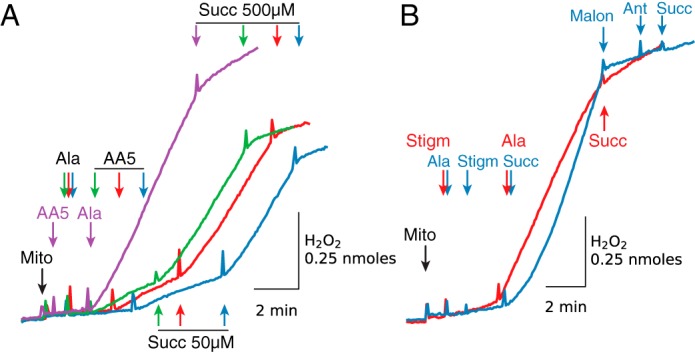
H2O2 production by inhibited complex II is maximized when mitochondria are permeabilized after AA5 (A) or stigmatellin (B) addition. A, compared with AA5 addition before alamethicin (Ala, purple trace), AA5 (0.5 μm) added 1 min (green trace), 2 min (red trace), and 3 min (blue trace) after alamethicin resulted in reduced H2O2 production. Low H2O2 production was enhanced by adding 50 μm succinate (Succ) but suppressed by 500 μm. B, alamethicin added after stigmatellin (Stigm) (2 μm) induced a rapid increase in H2O2 production that was inhibited with 5 mm succinate (red trace). Stigmatellin added after alamethicin (blue trace) had little effect, but H2O2 production was increased with a low concentration of succinate (75 μm) and inhibited with malonate (Malon) (5 mm). Note that ROS production by antimycin (Ant)-inhibited complex III after addition of 5 mm succinate was inhibited due to the presence of stigmatellin (blue trace). As in other figures, additions to all traces are indicated by black arrows; additions to one trace only are indicated by the arrows of the same color as the trace. Mito, mitochondria.
Similarly to increased H2O2 production induced by alamethicin after (but not before) AA5 (Fig. 3A), downstream inhibition of QH2 oxidation with stigmatellin resulted in increased ROS production only when added before alamethicin (Fig. 3B, red trace). Stigmatellin added after alamethicin (Fib. 3B, blue trace) did not increase H2O2 production until exogenous succinate (75 μm) was added. ROS production induced by 75 μm succinate in the presence of stigmatellin was inhibited by malonate (5 mm) and not reactivated after sequential addition of antimycin and 5 mm succinate (Fig. 3B, blue trace). Note that under these conditions, stigmatellin inhibits ROS production by antimycin-inhibited complex III by blocking QH2 oxidation.
The ability of high [succinate] to suppress H2O2 production by AA5-inhibited complex II (16, 19) is further illustrated in Fig. 4A. Compared with the case with only endogenous substrates present, addition of exogenous 10 μm succinate allowed AA5-inhibited mitochondria to maintain H2O2 production after permeabilization for a longer duration (Fig. 4A, compare purple and red traces). However, raising [succinate] to 25 μm did not further increase H2O2 production (blue trace), indicating that succinate-driven H2O2 production was already optimized at 10 μm succinate. Further increases in [succinate] suppressed H2O2 production, with half-maximal suppression at 297 μm succinate (Fig. 4C), consistent with succinate binding causing a progressive decrease in the concentration of reduced site IIf accessible to O2. The suppressant effect of succinate on H2O2 production was also evident when succinate was added after H2O2 production had been initiated by alamethicin (Fig. 4B).
Figure 4.

Concentration dependence of succinate-induced inhibition of H2O2 production by complex II-inhibited mitochondria. A, mitochondria (Mito) were incubated with AA5 (1 μm) and succinate (Succ) at the concentrations indicated at the end of trace. After permeabilization with alamethicin (Ala), H2O2 production was lower at higher [succinate]. The initial rate of H2O2 production with 10 μm succinate present (red trace) was similar to that generated by endogenous substrates in the absence of added succinate (purple trace). As in other figures, additions to all traces are indicated by black arrows; additions to one trace only are indicated by the arrows of the same color as the trace. B, H2O2 production in AA5 (0.5 μm)-inhibited mitochondria initiated by alamethicin was progressively suppressed by adding exogenous succinate in increasing concentrations, as indicated at the end of traces. At the end H2O2 production was inhibited with malonate (Malon) (5 mm). C, summary data for inhibition of H2O2 production as a function the [succinate], showing half-maximal inhibition at 297 μm. Shaded areas indicate 95% confidence intervals. Ant, antimycin; Stigm, stigmatellin.
SDH is a reversible enzyme and its dicarboxylate-binding site can bind a range of substrates and inhibitors, including succinate, fumarate, citrate, malate, malonate, and oxaloacetate (OAA). OAA is an intermediate of the TCA cycle that inhibits SDH by binding to the enzyme with high affinity (Kd = 10−8 m), so that isolated SDH is obtained largely in deactivated form (29). Because of that tight binding, only a fraction of complex II in mitochondrial membranes isolated from cardiac tissue is found to be in the free active form (30), suggesting that the concentration of reduced IIf sites accessible to O2 after inhibition of Q reduction or QH2 oxidation is decreased in our permeabilized mitochondria. To evaluate the importance of OAA binding on the ability of complex II to produce ROS, we followed a previously reported protocol (30) to remove bound OAA by incubating the preparation with malonate (see “Experimental procedures”). Fig. 5 shows that (AA5 + succinate)-induced H2O2 production by malonate-treated permeabilized mitochondria was about 65% higher compared with mitochondria incubated in the absence of malonate. Assuming that malonate treatment removed all bound OAA, the difference between H2O2 production with and without malonate treatment is due to OAA bound to SDH. OAA binding to complex II apparently lowers ROS production values in our experiments but is unlikely to change our main conclusion that PTP opening can increase ROS production by complex II under certain conditions. It is interesting that short cardiac ischemia resulted in a dramatic decrease in OAA in mouse heart, but it had no effect on the amount of OAA-free complex II (30).
Figure 5.
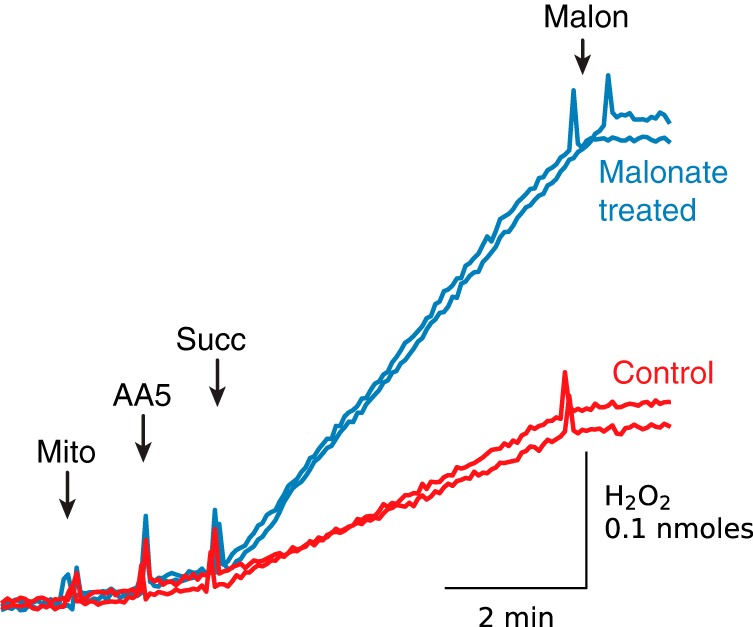
Malonate-induced removal of oxaloacetate bound to complex II enhances succinate-driven H2O2 production by AA5-inhibited permeabilized mitochondria. Permeabilized mitochondria were incubated with (blue traces) and without 1 mm malonate (Malon) (red traces), and the inhibitor was then removed by washing and centrifugation. The resuspended pellet was used for stimulation of ROS production by adding succinate (Succ) (50 μm) to AA5 (1 μm)-inhibited mitochondria (Mito). Dual traces are superimposed for each condition.
In addition to strong inhibition by malonate (Fig. 4B) and high [succinate] (Fig. 4A), H2O2 production by AA5 inhibited complex II after permeabilization was even more sensitive to increases in [fumarate] (Fig. 6A), with half-maximal inhibition achieved at 11 μm fumarate (Fig. 6B).
Figure 6.
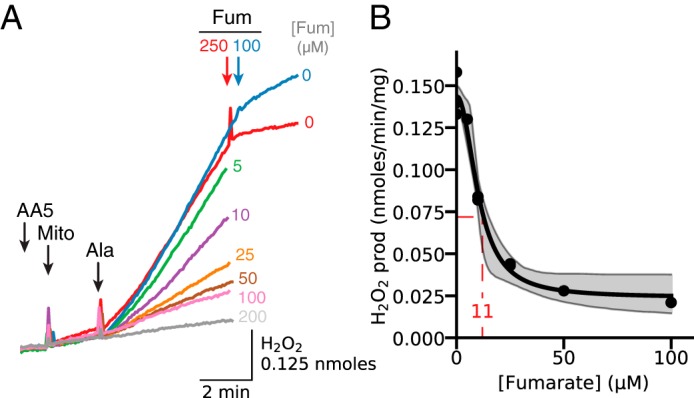
Concentration dependence of fumarate-induced inhibition of H2O2 production by complex II-inhibited mitochondria. A, mitochondria (Mito) were incubated with AA5 (1 μm) and fumarate (Fum) at the concentrations indicated at the end of each trace. After permeabilization with alamethicin (Ala), H2O2 production progressively decreased with higher [fumarate]. As in other figures, additions to all traces are indicated by black arrows; additions to one trace only are indicated by the arrows of the same color as the trace. B, summary data for inhibition of H2O2 production as a function [fumarate], showing half-maximal inhibition at 11 μm. Shaded areas indicate 95% confidence intervals.
As shown earlier in Figs. 2B and 3B, dicarboxylate- and malonate-sensitive H2O2 production could also be demonstrated in stigmatellin-inhibited mitochondria as a result of inhibition of QH2 oxidation by complex III. Fig. 7 illustrates the effects of stigmatellin in greater detail. When applied to intact isolated mitochondria energized with succinate, stigmatellin caused immediate almost full depolarization relative to alamethicin (Fig. 7A). The effects on H2O2 production are shown in Fig. 7B. When isolated normally polarized mitochondria were energized with 5 mm succinate (Fig. 7B, purple trace), H2O2 production due to RET increased significantly but was then suppressed by stigmatellin-induced depolarization. Addition of alamethicin at this point had no significant further effect on H2O2 production. In contrast, before alamethicin, 0.1 mm succinate (Fig. 7B, red trace) had no initial effect on H2O2 production either before or after stigmatellin-induced depolarization. At this point, however, alamethicin addition accelerated H2O2 production to a much higher rate than RET-induced H2O2 production. At 0.5 mm (Fig. 7B, blue trace), succinate had intermediate effects. These findings demonstrate that the rate of H2O2 production from complex II when [succinate] is optimal (<0.5 mm) exceeds that induced from complex I via RET at high [succinate] in fully polarized mitochondria. Here, we have to underline that ROS produced by complex I in the matrix is actively neutralized by antioxidant enzymes, such that only a fraction of total H2O2 generated diffuses into the buffer. Also, we have shown previously that H2O2 efflux generated by complex I is more complete in alamethicin-permeabilized mitochondria (31) because of a significant decrease in matrix antioxidant activity. In addition, the inner membrane may have more limited permeability to H2O2 before alamethicin (32). These factors may underestimate the H2O2 production rate by RET.
Figure 7.

Comparison of succinate-driven H2O2 production by RET in intact fully polarized mitochondria with succinate-driven H2O2 production by complex II after permeabilization with alamethicin. A, demonstration that stigmatellin (Stigm, 1 μm) dissipates ΔΨ required for H2O2 production by RET in intact mitochondria (Mito) energized with succinate (Succ, 0.5 mm). Pi 2.5 mm. B, as expected, stigmatellin (1 μm) inhibited succinate-induced H2O2 production by RET. The [succinate] is indicated at the end of each trace. Subsequent addition of alamethicin (Ala) robustly stimulated H2O2 production in the presence of endogenous or low exogenous [succinate], but it had no effect at 5 mm [succinate] (purple trace). Malonate (Malon, 1 mm) suppressed the increase H2O2 production induced by alamethicin. Pi 2.5 mm. As in other figures, additions to all traces are indicated by black arrows; additions to one trace only are indicated by the arrows of the same color as the trace. C, summary data (median ± 95% confidence intervals, *, p < 0.05) of the effect of [succinate] on H2O2 production in stigmatellin-inhibited mitochondria after alamethicin permeabilization and inhibition by malonate.
The above experiments were performed in mitochondria permeabilized by alamethicin to create non-selective pores in the inner mitochondrial membrane. To determine whether PTPs have equivalent effects as alamethicin pores, mitochondria incubated with AA5 for 2 min were exposed to sequential addition of Pi (2.5 mm) and Ca2+ (50 μm) to induce PTP opening (Fig. 8A, blue trace). H2O2 production increased significantly and was only mildly potentiated by addition of alamethicin but was suppressed by high [succinate] (1.5 mm). In contrast, if mitochondria were incubated in 250 μm EGTA to prevent PTP opening, Pi and calcium addition had no significant effect on H2O2 production until alamethicin was also added (Fig. 8A, red trace). Similarly, incubation of mitochondria in the presence of CsA (1.5 μm) significantly inhibited the Ca2+/Pi-induced increase in H2O2 production, although less effectively than EGTA (Fig. 8B). Thus, membrane permeabilization by alamethicin and calcium/Pi-induced PTP opening had nearly equivalent effects on stimulating ROS production by complex II.
Figure 8.
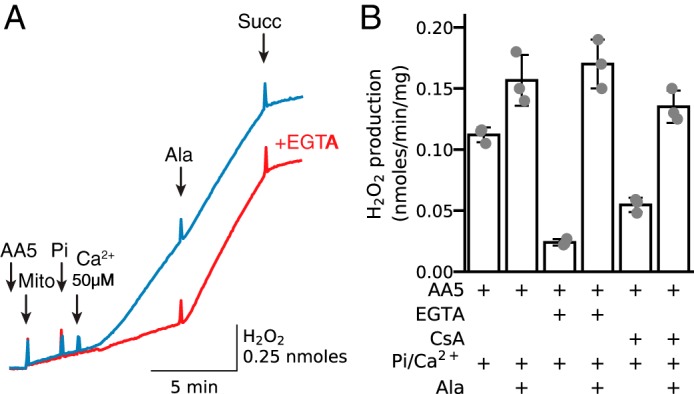
Calcium-induced PTP opening induces H2O2 production from complex II in AA5-inhibited mitochondria. A, in mitochondria (Mito) incubated with AA5 (0.5 μm), addition of Pi (2.5 mm) and Ca2+ (50 μm) to induce transient PTP opening caused a robust increase in H2O2 production (blue trace). Subsequent addition of alamethicin (Ala) had little additional effect. Pre-incubating with EGTA (0.25 mm) (red trace) abolished the increase in H2O2 production after Ca2+/Pi addition, but alamethicin remained effective. B, summary data (median ± 95% confidence intervals) of H2O2 production in AA5-inhibited mitochondria when PTP opening was triggered by Ca2+/Pi, including the effects of the PTP inhibitor CsA (1.5 μm). Succ, succinate.
Discussion
Summary of major findings
The major finding of this study is that in the absence of exogenous substrates, inner membrane permeabilization for low-molecular-weight solutes achieved by exposing isolated cardiac mitochondria to either alamethicin or Ca2+/Pi to induce PTP opening rapidly increased ROS production when electron flow through complex II was inhibited by AA5 prior to pore opening. Inner membrane permeabilization also increased ROS production by complex II in the absence of AA5 when downstream electron transport in complex III was inhibited with stigmatellin to prevent QH2 oxidation (or antimycin-stimulated ROS production by complex III). In these settings, ROS production declined gradually due to oxidation of reduced flavin sites and depletion of endogenous substrates, but it could be reactivated by adding succinate in low concentrations. Under these conditions, ROS could not originate from complex I or α-ketoglutarate dehydrogenase without a high NADH/NAD+ ratio (33–35), and RET was not possible due to Δψ dissipation. ROS production by complex III was also excluded by the experiments with stigmatellin, leaving complex II as the only plausible source of ROS under these conditions.
Most importantly, whereas low [succinate] maintained ROS production, higher [succinate] (>100 μm) inhibited ROS production, as did fumarate and malonate. These results demonstrate that although reduction of the dicarboxylate-binding site is required for ROS production by complex II, elevated dicarboxylate concentrations prevent O2 access to reduced site IIf and thereby suppress ROS generation (Fig. 1), as described previously (16, 17, 19). Upon inner membrane permeabilization with either alamethicin or Ca2+-induced PTP opening, dicarboxylate efflux from the matrix lowers their concentrations and exposes reduced site IIf, creating optimal conditions for ROS generation by complex II. The implication is that if either complex II or III is damaged during prolonged ischemia (22–26), then PTP opening during reperfusion could create similar conditions, promoting continued succinate-driven ROS production from reduced complex II after RET-driven ROS production has ceased.
Alamethicin pores versus PTP
PTP opening is known to increase inner membrane permeability at the start of cardiac reperfusion (11, 36). PTP opening can be also promoted in vitro by exposing isolated mitochondria to various inducers, including high [Ca2+], [Pi], and/or oxidant chemicals. However, the presence of oxidant chemicals significantly complicates the evaluation of the role of PTP opening in ROS production. To avoid this complication, we used either alamethicin or PTP opening induced by Ca2+/Pi to mimic reperfusion-induced PTP opening. At low concentrations, exogenous pore-forming peptides like alamethicin and mastoparan have been shown to form regulated pores whose conductance properties are similar to PTP induced by low concentrations of oxidant chemicals (37). At higher concentrations, such as used in this study, alamethicin pores become unregulated, similar to PTP openings in the high conductance mode induced by chemicals (37). We emphasize that no direct comparison between PTP and alamethicin-generated pores is intended, except that both allow rapid equilibration of small molecules between matrix and extramitochondrial space. In this regard, any regulation of PTP open/closed states, including spontaneous reclosure of PTP such as observed in rat heart after ischemia/reperfusion (38), does not occur with alamethicin-generated pores, but it could modify distribution of solutes during ischemia/reperfusion. In addition, higher [Ca2+], often used to induce PTP opening, may promote H2O2 generation via activation of α-glycerophosphate dehydrogenase located in the outer surface of the inner membrane (39). Although ROS production by this enzyme is slow, it could modify ROS production by complex II in permeabilized mitochondria. With these caveats, however, alamethicin-generated pores appear to be a reasonable tool to study the importance of dicarboxylate redistribution on ROS production by complex II.
Dicarboxylate concentration dependence of ROS production by complex II
We found that H2O2 production by AA5-inhibited complex II was already optimized at 10–50 μm exogenous succinate and suppressed by higher concentrations. Brand and co-workers (19) reported peak H2O2 production at 400 μm succinate, with significant inhibition at higher [succinate] commonly used to energize mitochondria (5 mm). In these experiments using intact skeletal muscle mitochondria, ROS production by complexes I and III was inhibited with rotenone and myxothiazol, respectively, with the latter also providing downstream block for electron transport. Using SMP from beef heart mitochondria, maximal ROS production by AA5-inhibited complex II was observed at around 100 μm, closer to our findings. AA5-inhibited SMP also demonstrated progressive inhibition of ROS production in response to increased [succinate], similar to our results (16). Because the dicarboxylate carrier Km for succinate is around 1 mm (16, 19), matrix [succinate] in non-permeabilized mitochondria may be significantly lower than the exogenous [succinate] in the extramitochondrial buffer (16) when succinate is being actively metabolized in the matrix. This may account for the requirement for higher [succinate] to reach maximum ROS production compared with the permeabilized mitochondria in our experiments. Indeed, more recent experiments with rotenone/myxothiazol-inhibited intact or permeabilized cardiac mitochondria have shown that H2O2 production is maximal at about 50 μm [succinate], followed by a gradual decrease at higher [succinate] (20). Furthermore, superoxide-generating activity of AA5-inhibited purified complex II is already high at very low [succinate] (2–5 μm) and decreases as the fumarate/succinate ratio increases (20).
A critically important finding from previous studies (16, 19) is that complex II can generate ROS only when site IIf is unoccupied by substrate. All TCA intermediates that bind to the dicarboxylate-binding site can therefore affect ROS production by complex II. We found that fumarate, a product of succinate oxidation that binds with comparable affinity to the dicarboxylate-binding site (40), is even more effective than succinate at inhibiting ROS production by AA5-inhibited complex II when added to permeabilized mitochondria, with only 11 μm fumarate required for 50% inhibition (Fig. 6). Attenuation of ROS generation by fumarate has been explained by two mechanisms as follows: (a) occupying the dicarboxylate-binding site, and (b) increasing the fumarate/succinate ratio which decreases complex II reduction (16). In alamethicin-permeabilized cardiac mitochondria incubated with myxothiazol, an increase in [fumarate] to modestly increase the fumarate/succinate ratio resulted in rapid inhibition of H2O2 production (41). However, antimycin-induced ROS production by cardiac submitochondrial particles was maximal at a [succinate]/[fumarate] ratio of 1:5 and was suppressed by either decreased or increased redox potential (42). Subsequent studies have confirmed a partially oxidized Q pool requirement for maximal ROS production by antimycin-inhibited complex III and also demonstrated that complex II can be a source as well as an inhibitor or enhancer of ROS production by complexes I and III (43). Together, these findings indicate that high matrix dicarboxylate concentrations inhibit ROS production by complex II, even when complex II or III become damaged during ischemia. Inner membrane permeabilization due to alamethicin or calcium-induced PTP opening allows dicarboxylate efflux from the matrix, depleting succinate, fumarate, and other dicarboxylates to levels that unmask reduced site IIf, allowing O2 to interact and generate ROS. A somewhat different mechanism was recently proposed by Vinogradov and co-workers (20), who suggested that iron-sulfur clusters are involved in ROS production by complex II when Q reduction is inhibited. However, they also observed maximum ROS production at low [succinate] (50 μm). To explain the importance of the dicarboxylate-free IIf site for ROS production, the authors postulated long distance interaction between IIf and Q-reactive sites (20).
Relevance to I/R injury
Succinate accumulation during ischemia has been linked to excessive ROS production and enhanced cardiac injury during reperfusion (1). The hypothesized mechanism is that when ischemic depolarized mitochondria are reoxygenated, a subpopulation may recover Δψ sufficiently to generate ROS by RET through complex I using succinate as the electron donor. The ROS generated by RET may then trigger PTP openings promoting cardiac injury. However, once the repolarized mitochondria have been depolarized by PTP opening, further ROS production via succinate-driven RET ceases, introducing a negative feedback factor to self-limit ROS production. This purely RET-based mechanism, however, does not account for observations from Halestrap and co-workers (36), who found that in Langendorff-perfused rat hearts subjected to 30 min of global ischemia, increased ROS production during reperfusion primarily occurred after, rather than before, PTP opening. Both ischemic preconditioning and CsA inhibited PTP opening during reperfusion, which might be expected to increase ROS production if succinate-driven RET were the primary mechanism. However, both interventions attenuated ROS production during reperfusion (36).
In contrast, if PTP opening induced by RET-derived ROS were to induce a secondary mechanism to sustain ROS production after RET ceased, this positive feedback mechanism would promote ROS-induced ROS release amplifying the levels of damaging ROS causing reperfusion injury. This study focusing on complex II and the accompanying article (52) focusing on complex III provide possible mechanisms for this positive feedback scenario in ROS production after PTP opening has occurred. In this case, PTP opening would promote succinate and fumarate efflux from the matrix, lowering their concentrations sufficiently to activate ROS production by complex II (Fig. 8). Here it is relevant to note that increases in cardiac fumarate levels due to knock-out of fumarate hydratase or administration of dimethyl fumarate for 5 days resulted in a robust cardioprotective effect against ischemia/reperfusion injury (44). Attenuation of reperfusion injury by increased [fumarate] was attributed to fumarate-induced activation of the Nrf2 pathway that confers cellular protection against oxidative stress by up-regulating antioxidant enzymes. Our results offer an additional explanation for fumarate's cardioprotective effect, namely that fumarate binding to site IIf suppresses amplification of damaging ROS production by complex II following PTP opening. In addition, as shown in the accompanying article (52), the entry of Mg2+ into the matrix would also activate malic enzyme, such that oxidation of low concentrations of malate arising from succinate and fumarate would provide electrons for ROS generation by damaged complex III. Thus, it is intriguing to speculate that when a population of mitochondria initially regenerates Δψ during reperfusion, succinate-driven ROS production by RET (1) triggers PTP opening, which then amplifies ROS production from complexes II and III by the mechanisms shown in these studies (Fig. 9). This scenario is consistent with recent experimental findings that SDH inhibition with malonate at the start of reperfusion inhibits both ROS production and PTP opening/cell damage (45). It is also consistent with the observation that the complex I inhibitor rotenone protects against I/R injury by suppressing RET and inhibiting the initial PTP opening that activates succinate-driven ROS production by complexes II and III. In addition rotenone blocks Q reduction by NADH, which, as we demonstrate in accompanying article (52), also inhibits NAD+/Mg2+-induced ROS production by antimycin-inhibited complex III. However, given the complexity of the rapidly changing cellular environment during I/R, it is unclear whether the conditions for robust ROS production by complex II and III would be met, and whether one pathway is more important quantitatively than the other. However, because PTP opening is necessary to achieve the appropriate conditions for activation of ROS production by these pathways, preventing succinate accumulation during ischemia seems a promising approach to both suppress initial PTP opening by RET-derived ROS and to inhibit further ROS production by inhibited complexes II and III. It is also intriguing to speculate that RET-derived ROS during brief ischemic episodes that are too short to damage electron transport complexes may play a role in ROS-dependent activation of cardioprotective signaling through the RISK and/or SAFE pathways, because succinate accumulation was significant after only 5 min of ischemia (1). Further studies will be necessary to investigate these possibilities.
Figure 9.
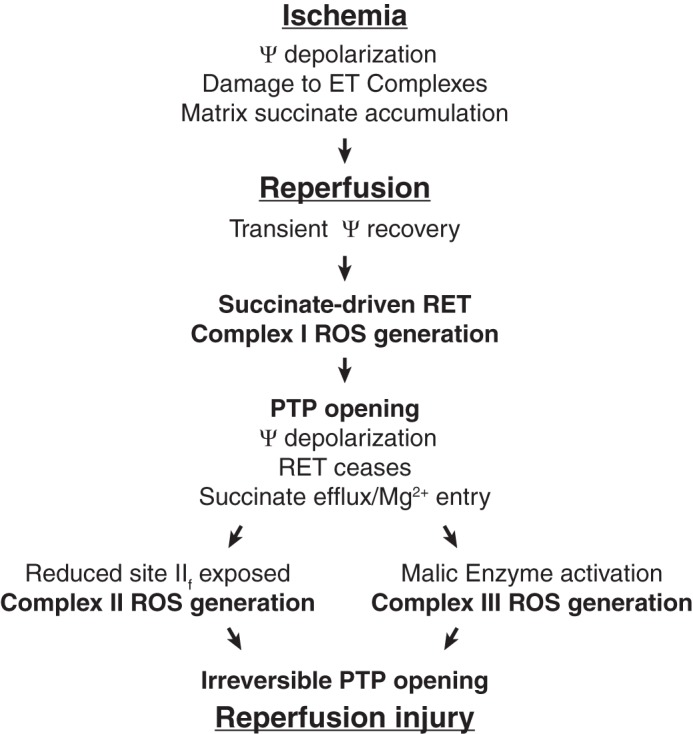
Proposed scenario of ROS generation during reperfusion after prolonged ischemia sufficient to damage complexes II and/or III. See text for description. ET, electron transport; RET, reverse electron transport through complex I; ΔΨ, membrane potential.
Experimental procedures
Ethical approval
This study was approved by the UCLA Chancellor's Animal Research Committee (ARC 2003-063-23B) and performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85-23, revised 1996) and with UCLA Policy 990 on the Use of Laboratory Animal Subjects in Research (revised 2010).
Experimental techniques
All measurements were carried out using customized Fiber Optic Spectrofluorometer (Ocean Optics) in a partially open continuously stirred cuvette at room temperature (22–24 °C) (46). Mitochondria were isolated from rabbit hearts as described previously (47). Intact mitochondria were added (0.6 to 1.0 mg/ml) to the incubation buffer containing 250 mm sucrose, 10 mm Hepes, pH 7.4, with Tris.
Mitochondria were permeabilized with addition of alamethicin (20 μg/ml). Alamethicin creates pores allowing equilibration of low-molecular-weight components across the inner membrane, whereas high-molecular-weight proteins are retained in the matrix and intermembrane space (48). For stimulation of ROS production, it was important to add alamethicin or Ca2+ after complex II inhibition with AA5 or downstream inhibition of QH2 oxidation in complex III with stigmatellin.
PTP opening was induced by adding 2.5 mm Pi and 50 μm Ca2+ to AA5-inhibited mitochondria. Selectivity of membrane permeabilization due to PTP opening was confirmed by adding EGTA (0.25 mm) or cyclosporin A (1.5 μm) to prevent PTP opening.
Isolated permeabilized mitochondria are known to contain OAA that is rather tightly bound to complex II (29) and therefore is expected to decrease free reduced flavin site concentration available for ROS generation. To remove OAA bound to complex II, permeabilized mitochondria were incubated with 1 mm malonate for 30 min at room temperature, a treatment demonstrated to reduce OAA bound to the enzyme (30). After dilution of the preparation with sucrose buffer, malonate is expected to rapidly dissociate from the enzyme, after which the essentially OAA/malonate-free preparation is separated by centrifugation. Control mitochondria were treated similarly in the absence of malonate.
H2O2 release from mitochondria was measured using 5 μm Amplex Red and 0.2 units of HRP in the buffer (excitation/emission, 540:590 nm). The increase in resorufin fluorescence was calibrated by adding H2O2 in known concentrations to the buffer containing permeabilized mitochondria.
Mitochondrial O2 consumption was measured continuously by monitoring buffer O2 content using a fiber optic oxygen sensor FOXY-AL300 (Ocean Optics) (46). Membrane potential (Δψ) was determined by using tetramethylrhodamine as described previously (47). All chemicals were obtained from Sigma.
Statistical analysis
For each data set, the mean and accompanying 95% confidence intervals are reported. The conventional percentile bootstrap-resampling approach with 10,000 replications was used for estimating 95% confidence intervals as well as examining the significant difference between groups (effect size statistics) (49–51). A p value <0.05 was considered statistically significant. All analyses were performed by subroutines for bootstrapping developed in the Python programming language (51).
Author contributions
P. K. and J. N. W. conceived and coordinated the study. P. K., G. C., and S. J. A. performed and analyzed the experiments. P. K., J. N. W., G. C., and S. J. A. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by National Institutes of Health Grants R01 HL101228 and R01 HL117385 from the NHLBI, American Heart Association Western States Affiliate Post-doctoral Research Fellowship 11POST6110007, American Heart Association Scientist Development Grant 16SDG31180036, and the Laubisch and Kawata Endowments. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ROS
- reactive oxygen species
- AA5
- atpenin A5
- CoA
- coenzyme A
- CsA
- cyclosporin A
- ME2
- malic enzyme
- ΔΨ
- mitochondrial membrane potential
- PTP
- permeability transition pore
- RET
- reverse electron transport
- Q
- ubiquinone
- QH2
- ubiquinol
- SDH
- succinate dehydrogenase
- Suc
- succinate
- TCA
- tricarboxylic acid
- OAA
- oxaloacetate
- SMP
- submitochondrial particle
- I/R
- ischemia/reperfusion.
References
- 1. Chouchani E. T., Pell V. R., Gaude E., Aksentijević D., Sundier S. Y., Robb E. L., Logan A., Nadtochiy S. M., Ord E. N., Smith A. C., Eyassu F., Shirley R., Hu C. H., Dare A. J., James A. M., et al. (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chouchani E. T., Pell V. R., James A. M., Work L. M., Saeb-Parsy K., Frezza C., Krieg T., and Murphy M. P. (2016) A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 23, 254–263 [DOI] [PubMed] [Google Scholar]
- 3. Korshunov S. S., Korkina O. V., Ruuge E. K., Skulachev V. P., and Starkov A. A. (1998) Fatty acids as natural uncouplers preventing generation of O2˙̄ and H2O2 by mitochondria in the resting state. FEBS Lett. 435, 215–218 [DOI] [PubMed] [Google Scholar]
- 4. Korshunov S. S., Skulachev V. P., and Starkov A. A. (1997) High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 416, 15–18 [DOI] [PubMed] [Google Scholar]
- 5. Votyakova T. V., and Reynolds I. J. (2001) Δψ(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J. Neurochem. 79, 266–277 [DOI] [PubMed] [Google Scholar]
- 6. Levraut J., Iwase H., Shao Z. H., Vanden Hoek T. L., and Schumacker P. T. (2003) Cell death during ischemia: relationship to mitochondrial depolarization and ROS generation. Am. J. Physiol. Heart Circ. Physiol. 284, H549–H558 [DOI] [PubMed] [Google Scholar]
- 7. Matsumoto-Ida M., Akao M., Takeda T., Kato M., and Kita T. (2006) Real-time 2-photon imaging of mitochondrial function in perfused rat hearts subjected to ischemia/reperfusion. Circulation 114, 1497–1503 [DOI] [PubMed] [Google Scholar]
- 8. Slodzinski M. K., Aon M. A., and O'Rourke B. (2008) Glutathione oxidation as a trigger of mitochondrial depolarization and oscillation in intact hearts. J. Mol. Cell. Cardiol. 45, 650–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Berkich D. A., Salama G., and LaNoue K. F. (2003) Mitochondrial membrane potentials in ischemic hearts. Arch. Biochem. Biophys. 420, 279–286 [DOI] [PubMed] [Google Scholar]
- 10. Kato M., Akao M., Matsumoto-Ida M., Makiyama T., Iguchi M., Takeda T., Shimizu S., and Kita T. (2009) The targeting of cyclophilin D by RNAi as a novel cardioprotective therapy: evidence from two-photon imaging. Cardiovasc. Res. 83, 335–344 [DOI] [PubMed] [Google Scholar]
- 11. Halestrap A. P., Clarke S. J., and Javadov S. A. (2004) Mitochondrial permeability transition pore opening during myocardial reperfusion–a target for cardioprotection. Cardiovasc. Res. 61, 372–385 [DOI] [PubMed] [Google Scholar]
- 12. Riess M. L., Camara A. K., Chen Q., Novalija E., Rhodes S. S., and Stowe D. F. (2002) Altered NADH and improved function by anesthetic and ischemic preconditioning in guinea pig intact hearts. Am. J. Physiol. Heart Circ. Physiol. 283, H53–H60 [DOI] [PubMed] [Google Scholar]
- 13. Aldakkak M., Stowe D. F., Chen Q., Lesnefsky E. J., and Camara A. K. (2008) Inhibited mitochondrial respiration by amobarbital during cardiac ischaemia improves redox state and reduces matrix Ca2+ overload and ROS release. Cardiovasc. Res. 77, 406–415 [DOI] [PubMed] [Google Scholar]
- 14. Aldakkak M., Stowe D. F., Heisner J. S., Spence M., and Camara A. K. (2008) Enhanced Na+/H+ exchange during ischemia and reperfusion impairs mitochondrial bioenergetics and myocardial function. J. Cardiovasc. Pharmacol. 52, 236–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zorov D. B., Juhaszova M., and Sollott S. J. (2014) Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 94, 909–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Siebels I., and Dröse S. (2013) Q-site inhibitor induced ROS production of mitochondrial complex II is attenuated by TCA cycle dicarboxylates. Biochim. Biophys. Acta 1827, 1156–1164 [DOI] [PubMed] [Google Scholar]
- 17. Imlay J. A. (1995) A metabolic enzyme that rapidly produces superoxide, fumarate reductase of Escherichia coli. J. Biol. Chem. 270, 19767–19777 [PubMed] [Google Scholar]
- 18. Messner K. R., and Imlay J. A. (2002) Mechanism of superoxide and hydrogen peroxide formation by fumarate reductase, succinate dehydrogenase, and aspartate oxidase. J. Biol. Chem. 277, 42563–42571 [DOI] [PubMed] [Google Scholar]
- 19. Quinlan C. L., Orr A. L., Perevoshchikova I. V., Treberg J. R., Ackrell B. A., and Brand M. D. (2012) Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 287, 27255–27264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grivennikova V. G., Kozlovsky V. S., and Vinogradov A. D. (2017) Respiratory complex II: ROS production and the kinetics of ubiquinone reduction. Biochim. Biophys. Acta 1858, 109–117 [DOI] [PubMed] [Google Scholar]
- 21. Lukyanova L. D., and Kirova Y. I. (2015) Mitochondria-controlled signaling mechanisms of brain protection in hypoxia. Front. Neurosci. 9, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Veitch K., Hombroeckx A., Caucheteux D., Pouleur H., and Hue L. (1992) Global ischaemia induces a biphasic response of the mitochondrial respiratory chain. Anoxic pre-perfusion protects against ischaemic damage. Biochem. J. 281, 709–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Petrosillo G., Di Venosa N., Ruggiero F. M., Pistolese M., D'Agostino D., Tiravanti E., Fiore T., and Paradies G. (2005) Mitochondrial dysfunction associated with cardiac ischemia/reperfusion can be attenuated by oxygen tension control. Role of oxygen-free radicals and cardiolipin. Biochim. Biophys. Acta 1710, 78–86 [DOI] [PubMed] [Google Scholar]
- 24. Lesnefsky E. J., Gudz T. I., Migita C. T., Ikeda-Saito M., Hassan M. O., Turkaly P. J., and Hoppel C. L. (2001) Ischemic injury to mitochondrial electron transport in the aging heart: damage to the iron-sulfur protein subunit of electron transport complex III. Arch. Biochem. Biophys. 385, 117–128 [DOI] [PubMed] [Google Scholar]
- 25. Chen C. L., Chen J., Rawale S., Varadharaj S., Kaumaya P. P., Zweier J. L., and Chen Y. R. (2008) Protein tyrosine nitration of the flavin subunit is associated with oxidative modification of mitochondrial complex II in the post-ischemic myocardium. J. Biol. Chem. 283, 27991–28003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen Y. R., Chen C. L., Pfeiffer D. R., and Zweier J. L. (2007) Mitochondrial complex II in the post-ischemic heart: oxidative injury and the role of protein S-glutathionylation. J. Biol. Chem. 282, 32640–32654 [DOI] [PubMed] [Google Scholar]
- 27. Wojtovich A. P., and Brookes P. S. (2009) The complex II inhibitor atpenin A5 protects against cardiac ischemia-reperfusion injury via activation of mitochondrial KATP channels. Basic Res. Cardiol. 104, 121–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wojtovich A. P., and Brookes P. S. (2008) The endogenous mitochondrial complex II inhibitor malonate regulates mitochondrial ATP-sensitive potassium channels: implications for ischemic preconditioning. Biochim. Biophys. Acta 1777, 882–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ackrell B. A., Kearney E. B., and Mayr M. (1974) Role 3f oxalacetate in the regulation of mammalian succinate dehydrogenase. J. Biol. Chem. 249, 2021–2027 [PubMed] [Google Scholar]
- 30. Stepanova A., Shurubor Y., Valsecchi F., Manfredi G., and Galkin A. (2016) Differential susceptibility of mitochondrial complex II to inhibition by oxaloacetate in brain and heart. Biochim. Biophys. Acta 1857, 1561–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Korge P., Calmettes G., and Weiss J. N. (2016) Reactive oxygen species production in cardiac mitochondria after complex I inhibition: modulation by substrate-dependent regulation of the NADH/NAD ratio. Free Radic. Biol. Med. 96, 22–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grivennikova V. G., Kareyeva A. V., and Vinogradov A. D. (2010) What are the sources of hydrogen peroxide production by heart mitochondria? Biochim. Biophys. Acta 1797, 939–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hirst J., King M. S., and Pryde K. R. (2008) The production of reactive oxygen species by complex I. Biochem. Soc. Trans. 36, 976–980 [DOI] [PubMed] [Google Scholar]
- 34. Kussmaul L., and Hirst J. (2006) The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. U.S.A. 103, 7607–7612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tretter L., and Adam-Vizi V. (2004) Generation of reactive oxygen species in the reaction catalyzed by α-ketoglutarate dehydrogenase. J. Neurosci. 24, 7771–7778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Andrienko T., Pasdois P., Rossbach A., and Halestrap A. P. (2016) Real-time fluorescence measurements of ROS and [Ca2+] in ischemic/reperfused rat hearts: detectable increases occur only after mitochondrial pore opening and are attenuated by ischemic preconditioning. PLoS ONE 11, e0167300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. He L., and Lemasters J. J. (2002) Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Lett. 512, 1–7 [DOI] [PubMed] [Google Scholar]
- 38. Griffiths E. J., and Halestrap A. P. (1995) Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem. J. 307, 93–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tretter L., and Adam-Vizi V. (2012) High Ca2+ load promotes hydrogen peroxide generation via activation of α-glycerophosphate dehydrogenase in brain mitochondria. Free Radic. Biol. Med. 53, 2119–2130 [DOI] [PubMed] [Google Scholar]
- 40. Iverson T. M. (2013) Catalytic mechanisms of complex II enzymes: a structural perspective. Biochim. Biophys. Acta 1827, 648–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Starkov A. A., and Fiskum G. (2001) Myxothiazol induces H(2)O(2) production from mitochondrial respiratory chain. Biochem. Biophys. Res. Commun. 281, 645–650 [DOI] [PubMed] [Google Scholar]
- 42. Ksenzenko M., Konstantinov A. A., Khomutov G. B., Tikhonov A. N., and Ruuge E. K. (1984) Relationships between the effects of redox potential, α-thenoyltrifluoroacetone and malonate on O(2) and H2O2 generation by submitochondrial particles in the presence of succinate and antimycin. FEBS Lett. 175, 105–108 [DOI] [PubMed] [Google Scholar]
- 43. Dröse S. (2013) Differential effects of complex II on mitochondrial ROS production and their relation to cardioprotective pre- and postconditioning. Biochim. Biophys. Acta 1827, 578–587 [DOI] [PubMed] [Google Scholar]
- 44. Ashrafian H., Czibik G., Bellahcene M., Aksentijević D., Smith A. C., Mitchell S. J., Dodd M. S., Kirwan J., Byrne J. J., Ludwig C., Isackson H., Yavari A., Støttrup N. B., Contractor H., Cahill T. J., et al. (2012) Fumarate is cardioprotective via activation of the Nrf2 antioxidant pathway. Cell Metab. 15, 361–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Valls-Lacalle L., Barba I., Miró-Casas E., Alburquerque-Béjar J. J., Ruiz-Meana M., Fuertes-Agudo M., Rodríguez-Sinovas A., and García-Dorado D. (2016) Succinate dehydrogenase inhibition with malonate during reperfusion reduces infarct size by preventing mitochondrial permeability transition. Cardiovasc. Res. 109, 374–384 [DOI] [PubMed] [Google Scholar]
- 46. Korge P., Ping P., and Weiss J. N. (2008) Reactive oxygen species production in energized cardiac mitochondria during hypoxia/reoxygenation: modulation by nitric oxide. Circ. Res. 103, 873–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Korge P., Honda H. M., and Weiss J. N. (2001) Regulation of the mitochondrial permeability transition by matrix Ca(2+) and voltage during anoxia/reoxygenation. Am. J. Physiol. Cell Physiol. 280, C517–C526 [DOI] [PubMed] [Google Scholar]
- 48. Gostimskaya I. S., Grivennikova V. G., Zharova T. V., Bakeeva L. E., and Vinogradov A. D. (2003) In situ assay of the intramitochondrial enzymes: use of alamethicin for permeabilization of mitochondria. Anal. Biochem. 313, 46–52 [DOI] [PubMed] [Google Scholar]
- 49. Nakagawa S., and Cuthill I. C. (2007) Effect size, confidence interval and statistical significance: a practical guide for biologists. Biol. Rev. Camb. Philos. Soc. 82, 591–605 [DOI] [PubMed] [Google Scholar]
- 50. Efron B., and Tibshirani R. (1991) Statistical data analysis in the computer age. Science 253, 390–395 [DOI] [PubMed] [Google Scholar]
- 51. Calmettes G., Drummond G. B., and Vowler S. L. (2012) Making do with what we have: use your bootstraps. J. Physiol. 590, 3403–3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Korge P., Calmettes G., John S. A., and Weiss J. N. (2017) Reactive oxygen species production induced by pore opening in cardiac mitochondria: The role of complex III. J. Biol. Chem. 292, 9882–9895 [DOI] [PMC free article] [PubMed] [Google Scholar]