

Abstract
The R7 regulator of G protein signaling family (R7-RGS) critically regulates nervous system development and function. Mice lacking all R7-RGS subtypes exhibit diverse neurological phenotypes, and humans bearing mutations in the retinal R7-RGS isoform RGS9-1 have vision deficits. Although each R7-RGS subtype forms heterotrimeric complexes with Gβ5 and R7-RGS-binding protein (R7BP) that regulate G protein-coupled receptor signaling by accelerating deactivation of Gi/o α-subunits, several neurological phenotypes of R7-RGS knock-out mice are not readily explained by dysregulated Gi/o signaling. Accordingly, we used tandem affinity purification and LC-MS/MS to search for novel proteins that interact with R7-RGS heterotrimers in the mouse brain. Among several proteins detected, we focused on Gα13 because it had not been linked to R7-RGS complexes before. Split-luciferase complementation assays indicated that Gα13 in its active or inactive state interacts with R7-RGS heterotrimers containing any R7-RGS isoform. LARG (leukemia-associated Rho guanine nucleotide exchange factor (GEF)), PDZ-RhoGEF, and p115RhoGEF augmented interaction between activated Gα13 and R7-RGS heterotrimers, indicating that these effector RhoGEFs can engage Gα13·R7-RGS complexes. Because Gα13/R7-RGS interaction required R7BP, we analyzed phenotypes of neuronal cell lines expressing RGS7 and Gβ5 with or without R7BP. We found that neurite retraction evoked by Gα12/13-dependent lysophosphatidic acid receptors was augmented in R7BP-expressing cells. R7BP expression blunted neurite formation evoked by serum starvation by signaling mechanisms involving Gα12/13 but not Gαi/o. These findings provide the first evidence that R7-RGS heterotrimers interact with Gα13 to augment signaling pathways that regulate neurite morphogenesis. This mechanism expands the diversity of functions whereby R7-RGS complexes regulate critical aspects of nervous system development and function.
Keywords: G protein, heterotrimeric G protein, neurite outgrowth, proteomics, regulator of G protein signaling (RGS)
Introduction
Regulator of G protein signaling (RGS)2 proteins regulate the amplitude and kinetics of G protein-coupled receptor signaling by functioning in part as GTPase-activating proteins (GAPs) that accelerate the rate that G protein α-subunits hydrolyze GTP to GDP (1). Among ∼30 mammalian proteins containing an RGS domain, the R7 regulator of G protein signaling family (R7-RGS), consisting of RGS6, RGS7, RGS9, and RGS11, is particularly important. R7-RGS proteins are highly expressed in the nervous system and exhibit GAP activity in vitro only for Gαi/o (2–7). Humans bearing mutations in the retinal RGS9-1 isoform exhibit a vision deficit termed bradyopsia (8), and mice lacking selected or all R7-RGS proteins exhibit various neurological phenotypes manifested by impairment of perinatal viability, weight gain, retina structure and function, neurobehavioral development, motor coordination, cerebellar and hippocampal development, and analgesic response to opioids (9–12), thereby establishing these regulators as crucial players in neurological development and function.
Evidence suggests that R7-RGS proteins have diverse mechanistic functions beyond serving as Gαi/o-specific GAPs. First, in contrast to several other classes of RGS proteins that are GAPs for Gi/o α-subunits (13), R7-RGS proteins are structurally complex. Each R7-RGS isoform possesses N-terminal disheveled, Egl-10, and pleckstrin (DEP), DEP helical extension (DHEX), and G protein γ-like (GGL) domains followed by a C-terminal RGS domain that is necessary and sufficient for GAP activity. The GGL domain binds the most diverged member of the Gβ family, Gβ5 (4, 14), to form obligate heterodimeric complexes structurally similar to classical Gβγ dimers (15). The DEP domain interacts with either of two SNARE-like membrane anchor proteins (16–21), R7-RGS-binding protein (R7BP) and RGS9 anchor protein (R9AP), to form R7-RGS heterotrimers. Whereas R9AP is a transmembrane protein localized to photoreceptor disk membranes, R7BP is reversibly and dynamically palmitoylated to regulate plasma membrane localization of R7-RGS heterotrimers throughout much of the nervous system (17, 22–24). Second, as shown in Caenorhabditis elegans, the N-terminal DEP/DHEX domain rather than the RGS domain of R7-RGS proteins determines which neurological processes mediated by distinct G proteins are regulated (25). Third, RGS7·Gβ5 and RGS9-2·Gβ5 complexes, respectively, can attenuate Gq-coupled type 3 muscarinic or Gi/o-coupled type 3 dopamine receptors by GAP-independent mechanisms (19, 21), similar to other structurally complex RGS proteins that serve as multifunctional regulators, integrators, and/or effectors in signaling networks (26, 27). Lastly, RGS6 can scaffold DNA methyltransferase 1 to the acyltransferase Tip60 to promote DNA methyltransferase 1 degradation in response to Ras activation in mouse embryonic fibroblasts (28, 29).
Despite such evidence, whether the membrane anchor R7BP is required for functions of R7-RGS complexes other than Gαi/o-specific GAP activity has not been investigated. Here we have addressed this question by using a proteomics approach to identify proteins that interact with R7BP-bound R7-RGS complexes in mouse brain. Our results expand understanding of R7BP-bound R7-RGS complexes by indicating that they facilitate Gα13 signaling to regulate neurite morphogenesis.
Results
RGS7, Gβ5, and R7BP form macromolecular complexes larger than simple heterotrimers in neuronal cells
If R7BP-bound R7-RGS proteins have novel functions, they would be expected to participate in novel protein-protein interactions. Accordingly, we investigated whether RGS7, Gβ5, and R7BP exist in macromolecular complexes larger than simple heterotrimers in living cells. We used Neuro2a (N2a) cells endogenously expressing RGS7 and Gβ5 and stably transfected with N-terminally FLAG-tagged R7BP at physiologically normal levels (5) that were treated with vehicle or disuccinimidyl suberate (DSS), a cell-permeable, amine-reactive cross-linker. Cell lysates were immunoprecipitated with FLAG antibody, resolved via SDS-PAGE, and immunoblotted to detect Gβ5, the obligate core subunit of all R7-RGS complexes. As expected if additional proteins associate with R7-RGS heterotrimers, this approach detected several distinct species with relative molecular weights larger than R7-RGS heterotrimers (Fig. 1A). These experiments also detected non-cross-linked Gβ5 and cross-linked R7-RGS·Gβ5·FLAG-R7BP heterotrimers (Fig. 1A). These results are consistent with an independent study in which complexes containing R7BP were detected in lysates of transfected HEK293T cells and mouse brain lysates cross-linked with paraformaldehyde (30).
Figure 1.
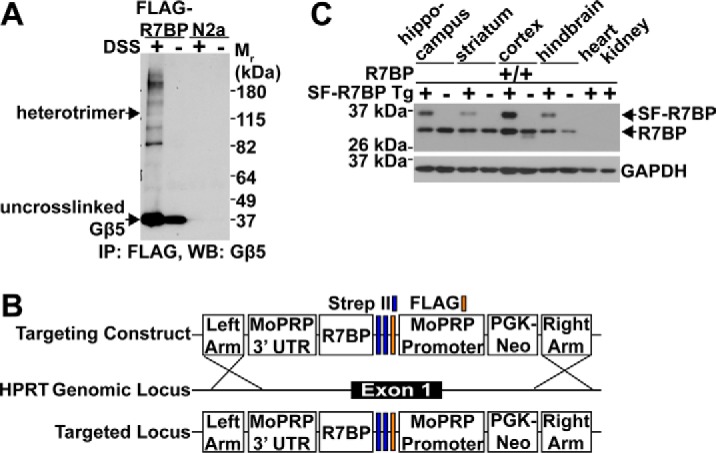
Generation of SF-R7BP transgenic mice to identify novel proteins that interact with R7-RGS heterotrimers. A, live-cell chemical cross-linking. N2a cells lacking or stably expressing 3xFLAG-R7BP were treated with DSS followed by immunoprecipitation (IP) of R7BP-containing cross-linked complexes. Immunoprecipitation eluates were separated by SDS-PAGE and immunoblotted for Gβ5 (shown), RGS7, and R7BP (not shown). The band corresponding to R7-RGS heterotrimers was indicated by detection with Gβ5, RGS7, and R7BP antibodies. Data shown are representative of three independent experiments. B, SF-R7BP transgenic mice were generated by targeting SF-R7BP in reverse orientation to the Hprt locus on the X chromosome as described under “Experimental procedures.” SF-R7BP expression was by the neuron-specific MoPRP. C, Western blot (WB) analysis of lysates from the indicated brain regions and other organs from non-transgenic control and SF-R7BP transgene-positive male mice. Wild-type untagged R7BP was expressed from its endogenous locus. Data are representative of three independent SF-R7BP transgenic lines.
Generation of transgenic mice expressing tandem affinity purification (TAP)-tagged R7BP
To identify potentially novel proteins that associate with R7BP-containing R7-RGS heterotrimers, we isolated these complexes from brain tissue by affinity purification and identified proteins recovered with these complexes by liquid chromatography-coupled tandem mass spectrometry (LC-MS/MS). This approach was motivated by prior studies showing that the membrane anchor R9AP and transducin α-subunits co-purify with RGS9-1·Gβ5 heterodimers in photoreceptor membranes (20) and that an orphan G protein-coupled receptor (GPR158) associates with RGS7·Gβ5 heterodimers in brain lysates (18, 31). However, our approach was novel because it identified proteins that associate with heterotrimers containing any R7-RGS isoform, Gβ5, and R7BP.
We developed a TAP strategy in which the FLAG-StrepII-StrepII tag (32) was appended to the N terminus of R7BP (SF-R7BP; Fig. 1B) and generated transgenic mice expressing this protein specifically in neurons. This was accomplished by driving SF-R7BP expression from the mouse prion promoter upon integration of the transgene in single copy at the X-linked Hprt locus (33, 34) (Fig. 1B). As indicated by immunoblotting with anti-R7BP antibodies, SF-R7BP and endogenously expressed R7BP were expressed similarly in various brain regions and were undetectable in heart and kidney (Fig. 1C). Importantly, SF-R7BP was not overexpressed relative to endogenous R7BP, reducing concern of identifying biologically irrelevant protein-protein interactions due to overexpression. SF-R7BP protein expression profiles were similar in three independently derived transgenic lines, all of which were used for TAP experiments. In all subsequent experiments, SF-R7BP-expressing adult male mice were used to avoid potentially mosaic expression of SF-R7BP due to X chromosome inactivation in females.
Tandem affinity purification and LC-MS/MS analysis of R7BP-containing protein complexes
R7BP-containing protein complexes were isolated from the combined lysates of three SF-R7BP adult male mouse brains as detailed under “Experimental procedures.” The purification procedure was optimized to achieve low nonspecific protein binding as indicated by SYPRO Ruby staining of TAP eluates from control non-transgenic mice relative to SF-R7BP transgenic mice (Fig. 2A). Moreover, the purification conditions preserved R7-RGS heterotrimers as indicated by immunoblotting showing that Gβ5 and RGS7 co-purified with SF-R7BP (Fig. 2B).
Figure 2.
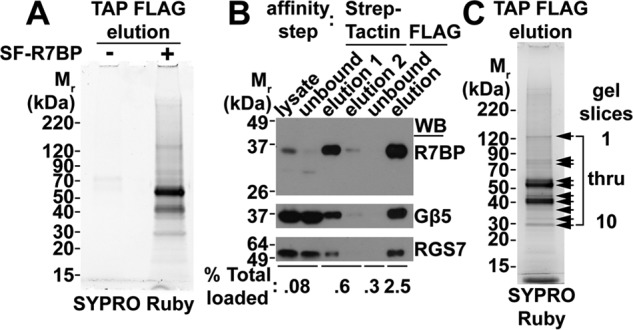
TAP of SF-R7BP and interacting proteins from mouse brain. A, assessing nonspecific background in TAP experiments. TAP was performed as described under “Experimental procedures” using detergent-solubilized brain homogenates from either three SF-R7BP-positive or three SF-R7BP-negative male mice as indicated. The final eluates were separated by SDS-PAGE, and proteins were visualized by SYPRO Ruby staining. B, Western blot (WB) analysis of SF-R7BP, Gβ5, and RGS7 for each step of the TAP protocol. C, SYPRO Ruby-stained gel of the TAP eluate from three SF-R7BP-positive mouse brains. Arrows indicate regions of the gel that were excised and analyzed by LC-MS/MS. Mass spectrometry data summarized in Table 1 and supplemental Table 1 are organized by gel slice numbers indicated in this panel.
Proteins that co-purified with R7-RGS heterotrimers were identified by resolving TAP FLAG eluates on SDS-PAGE, excising and extracting SYPRO Ruby-stained gel bands, and digesting with Glu-C and trypsin (Fig. 2C). The resulting peptides were sequenced by LC-MS/MS, and the corresponding proteins were identified as described under “Experimental procedures.” Detailed peptide and protein identification data are summarized in Table 1 and supplemental Table 1. As expected, SF-R7BP (denoted in Table 1 as Rgs7bp protein), Gβ5, and three of the four R7-RGS isoforms (RGS11 was not detected, presumably due to limited expression in brain) were identified (Table 1 and supplemental Table 1). Gαo, which is a substrate for the GAP function of R7-RGS heterotrimers, also was identified with high confidence.
Table 1.
Subset of proteins identified by LC-MS/MS analysis of TAP fractions
Protein bands marked with an arrow in Fig. 2C were analyzed by LC-MS/MS to identify proteins that co-purified with SF-R7BP from transgenic mouse brain. Peptide identifications were accepted if they could be established at greater than 80% probability by the Scaffold local false discovery rate algorithm. All proteins shown here have at least a 99% protein identification (ID) probability as determined using the Protein Prophet algorithm and at least two exclusive unique peptides assigned. Tabulated are protein identification information for R7BP (Rgs7bp protein); R7-RGS family members; and Gβ5, Gαo, and a novel interacting protein, Gα13. See supplemental Table 1 for a complete list of all proteins identified and peptide sequence information.
| Gel slice | Protein name | Accession number | Protein ID probability | Exclusive unique peptides | Exclusive unique spectra | Total spectra | Sequence coverage |
|---|---|---|---|---|---|---|---|
| % | % | ||||||
| 6 | Rgs7bp protein (Mus musculus) | gi 111600355 | 100.00 | 3 | 4 | 12 | 19.50 |
| 7 | Rgs7bp protein (M. musculus) | gi 111600355 | 100.00 | 3 | 4 | 87 | 13.60 |
| 8 | Rgs7bp protein (M. musculus) | gi 111600355 | 100.00 | 2 | 3 | 73 | 22.60 |
| 2 | Regulator of G-protein signaling 9 isoform 1 (M. musculus) | gi 146134422 | 100.00 | 5 | 7 | 21 | 24.30 |
| 3 | Regulator of G-protein signaling 9 isoform 1 (M. musculus) | gi 146134422 | 100.00 | 4 | 4 | 14 | 18.70 |
| 4 | Regulator of G-protein signaling 6 (M. musculus) | gi 29789076 | 100.00 | 6 | 8 | 15 | 20.30 |
| 5 | Regulator of G-protein signaling 6 (M. musculus) | gi 29789076 | 100.00 | 7 | 9 | 11 | 18.60 |
| 4 | Regulator of G-protein signaling 7 (M. musculus) | gi 312222673 | 100.00 | 9 | 11 | 26 | 21.70 |
| 5 | Regulator of G-protein signaling 7 (M. musculus) | gi 312222673 | 99.90 | 5 | 6 | 12 | 12.30 |
| 6 | Guanine nucleotide-binding protein subunit β5 isoform 2 (M. musculus) | gi 41281679 | 100.00 | 4 | 5 | 5 | 5.67 |
| 7 | Guanine nucleotide-binding protein subunit β5 isoform 2 (M. musculus) | gi 41281679 | 100.00 | 17 | 20 | 25 | 26.10 |
| 8 | Guanine nucleotide-binding protein subunit β5 isoform 2 (M. musculus) | gi 41281679 | 100.00 | 5 | 6 | 6 | 8.22 |
| 9 | Guanine nucleotide-binding protein subunit β5 isoform 2 (M. musculus) | gi 41281679 | 100.00 | 3 | 4 | 4 | 5.67 |
| 10 | Guanine nucleotide-binding protein subunit β5 isoform 2 (M. musculus) | gi 41281679 | 100.00 | 2 | 3 | 3 | 5.10 |
| 7 | Guanine nucleotide-binding protein subunit α13 (M. musculus) | gi 89001109 | 100.00 | 2 | 2 | 3 | 10.10 |
| 7 | Guanine nucleotide-binding protein Go subunit α isoform B (M. musculus) | gi 164607137 | 99.00 | 2 | 2 | 3 | 7.63 |
Gα13 is a novel binding partner of R7-RGS heterotrimers
Gα13 was identified with high confidence in our LC-MS/MS analysis of proteins that co-purified with SF-R7BP-bound R7-RGS complexes (Table 1 and supplemental Table 1). Similarly, Gα13 was identified in TAP-LC-MS/MS experiments using N2a cells expressing SF-R7BP (data not shown). Although unique peptides diagnostic of other Gα isoforms or classical Gβγ subunits were not detected, we cannot exclude that these proteins were present in R7-RGS complexes bound to SF-R7BP.
Recovery of Gα13 was intriguing for two reasons. First, Gα13 is not a substrate for the GAP function of R7-RGS heterotrimers that is specific in vitro for Gαi/o subunits (2–4). Therefore, co-purification of Gα13 with R7-RGS complexes suggested that R7-RGS heterotrimers potentially influence the function of this Gα subunit by GAP-independent mechanisms. Second, mice deficient in all R7-RGS heterotrimers due to knock-out of the shared obligate subunit Gβ5 have abnormal dendritic morphology as seen in retinal ON-bipolar and Purkinje neurons (10, 11). Because Gα13 is a well established regulator of the actin cytoskeleton, which regulates dendritic morphogenesis, a functional relationship between R7-RGS heterotrimers and Gα13 might account in part for the dendritic morphology phenotypes of Gβ5−/− mice. Accordingly, the remainder of the present study focused on the interaction between R7-RGS heterotrimers and Gα13.
To provide independent evidence whether Gα13 can associate with R7-RGS heterotrimers, we adopted split-luciferase complementation assays to assess protein-protein interactions in living cells (35, 36). Because split-luciferase complementation apparently had not been used before with Gα subunits, we first determined whether this technique is appropriate for our purposes. We inserted the N-terminal fragment of click beetle green luciferase (CBGN; Ref. 36) into the αB-αC loop of the helical domain in wild-type and constitutively active (i.e. GTPase-defective Gln → Leu (Q/L) mutants) forms of Gαq and Gα13 because insertion of GFP at this site preserves Gα function (37). The C-terminal fragment of click beetle green luciferase (CBGC) was fused to the N termini of the following proteins: RGS2 (CBGC-RGS2), a GAP for Gαq but not Gα13; the RH-RGS domain of LARG (CBGC-LARG·RGS), a GAP for Gα13 but not Gαq; and Gβ1 (CBGC-Gβ1). These constructs were used to determine whether split-luciferase complementation detects specific and activity-dependent interaction between Gαq or Gα13 and their cognate binding partners in intact cells.
This expectation was confirmed by results of luciferase assays performed with HEK293 cells co-transfected with various combinations of split luciferase-tagged proteins (Fig. 3A). Wild-type CBGN-tagged Gαq or Gα13 complemented strongly with CBGC-Gβ1 co-transfected with Gγ2. Constitutively active Gα13(Q/L)-CBGN complimented strongly with CBGC-LARG·RGS but not CBGC-RGS2. Likewise, constitutively Gαq(Q/L)-CBGN complemented strongly with CBGC-RGS2 but not CBGC-LARG·RGS. Wild-type Gα13-CBGN and Gαq-CBGN complemented insignificantly, respectively, with CBGC-LARG·RGS and CBGC-RGS2. Thus, split-luciferase complementation detected specific protein-protein interactions determined by Gα subunit identity and activation state.
Figure 3.

R7BP is required to detect interaction between Gα13 and R7-RGS complexes containing any R7-RGS family member. A, proof of concept that split-luciferase complementation detects protein-protein interactions specific for Gα subunit identity and activity state. Data are represented as the -fold difference in normalized split-luciferase signal relative to the positive control for each Gα-CBGN construct. Data shown are the average of three independent experiments. B, split-luciferase complementation between the indicated Gα-CBGN proteins and CBGC-Gβ5·RGS7 with or without R7BP. Each box represents a data point from one of three independent experiments. C, split-luciferase complementation between Gα13(Q/L)-CBGN and CBGC-3xFLAG-Gβ5 co-expressed with each R7-RGS isoform with or without R7BP. Luciferase signals were normalized to the level of CBGC-3xFLAG-Gβ5 expression determined by immunoblotting. Each box represents a data point from one of five independent experiments. D, representative FLAG Western blot (WB) showing CBGC-3xFLAG-Gβ5 and 3xFLAG-R7BP expression for each condition in C. The band marked with an asterisk is a degradation product of CBGC-3xFLAG-Gβ5. E, Gα13(Q/L)-CBGN-EE expression does not change with co-expression of R7-RGS heterotrimer proteins. Shown are representative immunoblots for C. Error bars indicate standard deviation of the mean. Statistical significance was determined using two-tailed, two-sample Student's t test assuming equal or unequal variance as deemed appropriate by F-test analysis. *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., p > 0.05.
Split-luciferase complementation then was used to study interaction between Gα13 and R7-RGS heterotrimers and to determine whether this interaction is affected by the activity state of Gα13. In these experiments, HEK293 cells were co-transfected with CBGC-FLAG-Gβ5, HA-RGS7 with or without FLAG-R7BP, various Gα-CBGN constructs, and Renilla luciferase as a transfection control. When co-expressed with HA-RGS7 and FLAG-R7BP, CBGC-FLAG-Gβ5 strongly complemented with either wild-type or constitutively active Gα13-CBGN but not with wild-type or constitutively active Gαq-CBGN (Fig. 3B). These results confirmed that R7-RGS heterotrimers and Gα13 interact in a specific manner rather than by nonspecific collisional interaction within the plane of the plasma membrane and indicated that this interaction is independent of Gα13 activation. These interactions required the presence of R7BP (Fig. 3B), presumably because palmitoylated R7BP targets R7-RGS·Gβ5 complexes to the plasma membrane where Gα13 localizes.
Next we used split-luciferase complementation to determine whether complexes containing any R7-RGS isoform can interact with Gα13 or whether particular R7-RGS family members are preferred. For these experiments, HEK293 cells were transfected with Gα13(Q/L)-CBGN and CBGC-FLAG-Gβ5 with various R7-RGS isoforms in the presence or absence of R7BP. Because the proteolytic stability of Gβ5 varies depending on which R7-RGS isoform is expressed in the presence or absence of R7BP and because luciferase signals depend on the expression level of CBGC-FLAG-Gβ5, we normalized split-luciferase signals in these experiments to the expression of CBGC-FLAG-Gβ5 determined by immunoblotting (Fig. 3D). Normalization for Gα13(Q/L)-CBGN expression was unnecessary because this protein was well expressed with or without RGS7 or R7BP as indicated by immunoblotting of glutamate-glutamate (EE)-tagged Gα13(Q/L)-CBGN (Fig. 3E). Data analyzed in this way showed efficient luciferase complementation when Gα13(Q/L)-CBGN was co-expressed with CBGC-FLAG-Gβ5, R7BP, and any R7-RGS family member, whereas complementation was low in the absence of either an R7-RGS subunit or R7BP (Fig. 3C). Therefore, Gα13 interacted in living cells with heterotrimers bearing R7BP, Gβ5, and any R7-RGS isoform.
R7-RGS heterotrimers augment neurite retraction evoked by Gα13-coupled lysophosphatidic acid (LPA) receptors
To determine whether R7-RGS heterotrimers affect biological processes driven by Gα13 and its close paralog Gα12, we took advantage of studies showing that Gα12/13 signaling causes growth cone collapse and neurite retraction in cultured neurons and neuronal cell lines (38–44). Accordingly, we determined whether R7-RGS heterotrimers affect neurite retraction in differentiated N2a cells. N2a cells were chosen as a model because they elaborate neurites when differentiated by serum starvation, and expression of constitutively active Gα13 or RhoA in these cells antagonizes neurite elaboration (40, 44). Moreover, N2a cells endogenously express RGS7 and Gβ5 but lack R7BP (5), which allowed us to compare Gα12/13-driven processes in the absence or presence of R7BP stably transfected at physiological levels to promote interaction of R7-RGS complexes with Gα13.
LPA evokes neurite retraction in primary neurons by activating receptors coupled to Gα12/13 (41, 42). Whether LPA triggers neurite retraction in N2a cells had not been established, although this seemed likely because expression of constitutively active Gα13 in these cells antagonizes neurite formation. To address this question, we differentiated N2a cells by serum starvation and treated them with LPA for 15 min. After fixation and permeabilization, cells were stained with rhodamine-phalloidin to visualize neurites and actin organization. We found that LPA treatment robustly evoked neurite retraction (Fig. 4, A–C). Because LPA receptors can couple to Gi/o, Gq/11, Gs, and G12/13 (45), we determined whether LPA-induced neurite retraction in differentiated N2a cells is mediated by G12/13 signaling. To address this question, we inhibited G12/13 signaling by overexpressing a truncated HA-tagged version of p115-RhoGEF (HA-p115·RGS) that is targeted to the plasma membrane by a prenylation motif and retains GAP activity for Gα12/13 but cannot activate RhoA (46). Expression of HA-p115·RGS blocked LPA-induced neurite retraction in differentiated N2a cells (Fig. 4, A–C), indicating that G12/13 signaling is indeed required for this process.
Figure 4.

R7BP augments neurite retraction evoked by activation of Gα12/13-coupled LPA receptors or serum starvation. Rhodamine-phalloidin staining and fluorescence microscopy were used to visualize neurites and cell morphology. Cells were scored by counting the number of neurites elaborated from the cell body that were greater than half the cell body diameter. At least 50 cells were scored per condition per experiment. A, representative fluorescence images of parental N2a cells transfected with either GFP (control) or HA-p115·RGS and treated with vehicle (veh) or 10 μm LPA. Control experiments confirmed antibody staining specificity (data not shown). B, quantification of neurites formed per cell under conditions shown in A. Data shown are the average of three independent experiments. C, number of cells with neurites under conditions shown in A. Each box represents a data point from one of three independent experiments. D, representative fluorescence images of parental N2a or N2a cells stably expressing FLAG-R7BP treated with the indicated concentrations of LPA. E, quantification of neurite formation under conditions shown in D. Each box represents a data point from one of four independent experiments. Scale bars, 50 μm. Error bars indicate standard deviation of the mean. Statistical significance was determined using two-tailed, two-sample Student's t test assuming equal or unequal variance as deemed appropriate by F-test analysis. *, p < 0.05; **, p < 0.01. Proj., projection.
Based on the preceding results, we investigated whether R7-RGS heterotrimers can regulate LPA-induced neurite retraction. Because R7BP facilitates interaction between R7-RGS complexes and Gα13, we compared the effects of LPA on N2a cells, which express RGS7 and Gβ5 but lack R7BP (5), with N2a cells stably expressing FLAG-R7BP at physiological levels (5). Results indicated that LPA-evoked neurite retraction was augmented in cells expressing FLAG-R7BP (Fig. 4, D and E), suggesting that R7BP-bound R7-RGS complexes augment signaling by G12/13-coupled LPA receptors.
In the preceding experiments, we also noted that, prior to LPA stimulation, R7BP expression antagonized neurite formation evoked by serum starvation alone (Fig. 4, D and E). To explore the mechanisms involved, we tested two models. In the first model, R7BP expression enables R7-RGS complexes to augment serum starvation-evoked G12/13 signaling, which attenuates neurite formation. If so, inhibiting G12/13 activity would be expected to rescue neurite formation in N2a cells stably expressing FLAG-R7BP. To test this prediction, FLAG-R7BP-expressing N2a cells were transiently transfected with HA-p115·RGS or a GFP-expressing control plasmid, differentiated, and analyzed for neurite formation. Results indicated that neurite formation trended higher but was incompletely rescued in cells transiently expressing HA-p115·RGS as compared with controls (Fig. 5, A and B; p = 0.24). Thus, R7BP-bound R7-RGS heterotrimers appeared to regulate neurite morphogenesis under basal conditions in serum-starved cells by mechanisms that depend partly on G12/13 signaling.
Figure 5.
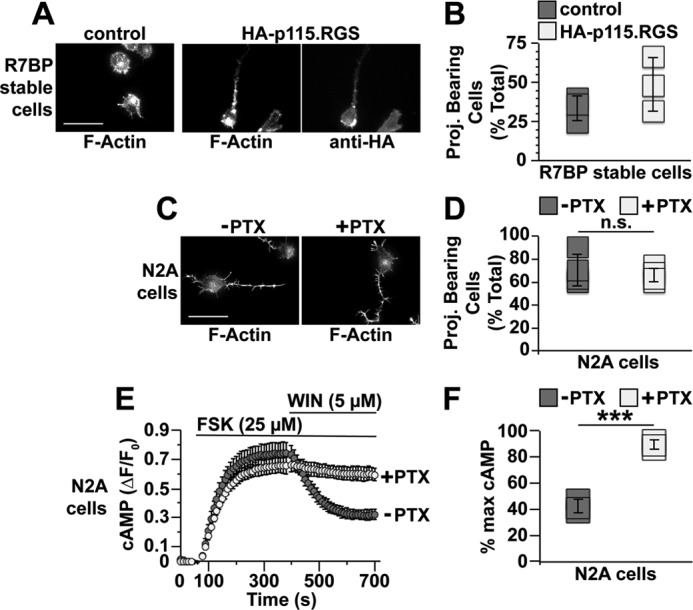
Gα12/13 activity rather than Gαi/o activity regulates neurite formation by serum-starved N2a cells. N2a cells lacking or stably expressing 3xFLAG-R7BP were plated, differentiated, fixed, stained, and scored as described in Fig. 4 and under “Experimental procedures.” A, representative fluorescence images of cells stably expressing FLAG-R7BP and transfected with either GFP (control) or HA-p115·RGS. B, quantification of neurite formation under conditions shown in A. In these experiments, all cells quantified were transfected, whereas those in Fig. 4E were not transfected, which likely explains why the percentage of R7BP-expressing cells bearing neurites in response to serum starvation was different between the two types of experiments. Each box represents a data point from one of three independent experiments. C, representative fluorescence images of parental N2a cells treated with PTX. D, quantification of the neurites formed by cells under conditions described in C. Each box represents a data point from one of four independent experiments. E, inhibition of Gi/o signaling by PTX in N2a cells. N2a cells were transfected with the cAMP FRET sensor Epac-SH187 and treated for 16–24 h with or without 100 ng/ml PTX. Baseline cAMP levels were recorded for 1 min, and then cells were treated with 25 μm forskolin (FSK) to stimulate cAMP production. After 5 min of forskolin treatment, endogenous Gi/o-coupled cannabinoid type 1 receptors were activated by addition of 5 μm WIN 55,212-2 (WIN), and cAMP levels were recorded for an additional 5 min. Data shown are the averages of three independent experiments. F, quantification of percent maximum cAMP levels after WIN 55,212-2 stimulation as shown in E. Percent maximum (% max) cAMP = [(minimum ΔF/F0)/(maximum ΔF/F0)] × 100. Each box represents a data point from one of three independent experiments. Scale bars, 50 μm. Error bars indicate standard deviation of the mean. Statistical significance was determined using two-tailed, two-sample Student's t test assuming equal variance as deemed appropriate by F-test analysis. ***, p < 0.001; n.s., p > 0.05. Proj., projection.
One potential explanation for the preceding result is provided by a second model in which R7BP-bound R7-RGS complexes regulate neurite morphogenesis in serum-starved cells in part by attenuating Gi/o signaling. Indeed, Gi/o-coupled cannabinoid type 1 receptor signaling promotes neurite outgrowth in N2a cells (47, 48), which potentially could be negatively regulated by the GAP activity of R7BP-bound R7-RGS complexes (17, 23, 49–51). However, whether Gi/o activity in N2a cells promotes neurite formation in response to serum starvation had not been tested. We found that this appeared not to be the case because blocking Gi/o activation with pertussis toxin (PTX) did not inhibit neurite formation evoked by serum starvation (Fig. 5, C and D) in contrast to control experiments demonstrating that PTX treatment strongly inhibited the ability of a cannabinoid type 1 agonist (WIN 55,212-2) to blunt forskolin-evoked cAMP accumulation detected with a FRET reporter (Fig. 5, E and F). Therefore, these results argued against models in which R7BP-bound R7-RGS complexes promote neurite retraction by attenuating Gi/o signaling.
Lastly, we explored how R7BP augments Gα13-dependent neurite retraction by studying Gα13-activated RhoGEFs (LARG, p115RhoGEF, and PDZ-RhoGEF) that mediate RhoA activation and consequent neurite retraction. Our initial approach relied on prior investigations showing that these RhoGEFs can be recruited to the plasma membrane by activated Gα13 (46). Thus, if R7BP-bound R7-RGS complexes promote Gα13 signaling, then expression of R7BP in N2a cells might be expected to augment plasma membrane recruitment of these RhoGEFs. However, by confocal imaging, we were unable to detect differences in plasma membrane recruitment of GFP-tagged LARG, p115RhoGEF, or PDZ-RhoGEF in response to LPA and/or R7BP expression (data not shown).
As an alternative approach that might provide greater sensitivity, we determined whether RhoGEFs affect interaction between constitutively active Gα13 and R7BP-bound R7-RGS complexes in split-luciferase complementation assays. Rather than assessing Gα13 activity, this approach addressed the key question whether complexes containing activated Gα13 and R7-RGS heterotrimers are capable of physically engaging effector RhoGEFs that trigger RhoA activation and neurite retraction. Accordingly, HEK293 cells were transfected with Gα13(Q/L)-CBGN, CBGC-FLAG-Gβ5, RGS7, and R7BP in the absence or presence of Myc-tagged forms of LARG, p115RhoGEF, or PDZ-RhoGEF. As shown in Fig. 6, split-luciferase complementation resulting from interaction between activated Gα13 and R7BP-bound RGS7-Gβ5 complexes was increased ∼7-fold by LARG, 4-fold by PDZ-RhoGEF, and 2-fold by p115RhoGEF. Accordingly, the results of these and preceding experiments support models in which R7BP-bound R7-RGS complexes interact with Gα13 in neuronal cells to augment signaling mechanisms that promote neurite retraction and blunt neurite formation.
Figure 6.
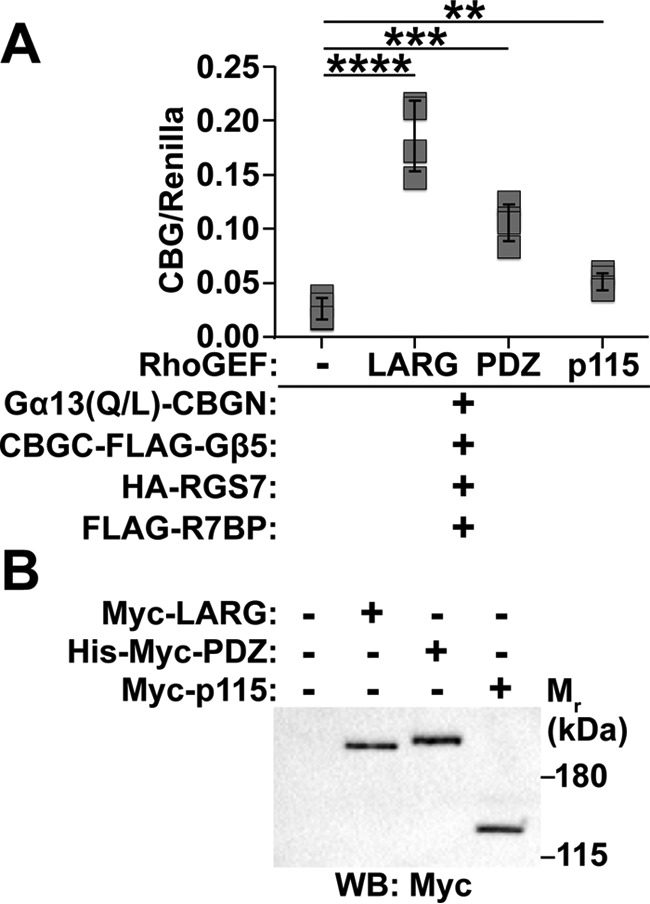
RH-RhoGEFs enhance interaction between Gα13 and R7-RGS heterotrimers. A, split-luciferase complementation between Gα13(Q/L)-CBGN and CBGC-FLAG-Gβ5 co-expressed with HA-RGS7 and FLAG-R7BP with or without Myc-LARG, His-Myc-PDZ-RhoGEF, or Myc-p115RhoGEF. Each box represents a data point from one of four independent experiments. B, representative Myc Western blot (WB) showing Myc-LARG, His-Myc-PDZ-RhoGEF, and Myc-p115RhoGEF protein expression levels for conditions used in A. Error bars indicate standard deviation of the mean. Statistical significance was determined using two-tailed, two-sample Student's t test assuming equal variance as deemed appropriate by F-test analysis. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Discussion
Our findings are the first to indicate that R7-RGS heterotrimers form macromolecular complexes with Gα13 and control neurite morphogenesis regulated in part by G12/13-dependent signaling. Accordingly, they expand the functional diversity of R7-RGS heterotrimers beyond their canonical roles as GAPs for Gαi/o. Because genetic ablation of R7-RGS complexes affects diverse aspects of neurological development and function (9–12), our findings suggest that R7-RGS heterotrimers potentially regulate certain processes in part by augmenting Gα13 signaling rather than solely by attenuating Gi/o signaling. Indeed, certain phenotypes of mice lacking R7-RGS heterotrimers are not recapitulated in mice expressing RGS-insensitive mutant forms of Gαo or Gαi (52).
Several prior lines of evidence are consistent with the hypothesis that R7-RGS heterotrimers regulate G12/13 signaling to control nervous system development. R7-RGS heterotrimers are dramatically up-regulated during postnatal brain development as synapses mature (5, 53). Mice lacking all R7-RGS complexes exhibit abnormal Purkinje cell maturation and dendritic arborization, delayed morphological development of the hippocampus, and increased ectopic granule cells in the hippocampus (10). These postnatal neurological phenotypes potentially could be caused in part by diminished G12/13 signaling, which controls similar processes during embryonic brain development. For example, conditional ablation of Gα13 and its paralog Gα12 in the nervous system causes overmigration of cortical plate and Purkinje neurons in embryonic mice (41). Accordingly, increased ectopic granule cells in the hippocampus of mice lacking all R7-RGS complexes might arise from overmigration of these cells because Gα13 activity is reduced and fails to provide sufficiently strong migratory “stop” signals, similar to what occurs for cortical plate neurons and cerebellar Purkinje cells in embryonic Gα13−/− mice. Further evidence linking Gα13 and R7-RGS complexes is provided by the observation that Gα13 mRNA is up-regulated in cerebellum of 2-week-old postnatal mice lacking all R7-RGS complexes (11) potentially to compensate for diminished Gα13 signaling activity caused by the absence of R7-RGS complexes.
Establishing the biological functions of Gα13 signaling regulated by R7-RGS heterotrimers will require understanding the molecular mechanisms involved. Although much remains to be learned, we suggest two mechanistic hypotheses (Fig. 7), which are not mutually exclusive; other models also can be imagined. Both models are consistent with our findings that R7-RGS heterotrimers can interact with Gα13 regardless of its activity state and that this interaction is augmented by Gα13-regulated RhoGEFs. In the first model, R7BP-bound R7-RGS heterotrimers interact with Gα13 to facilitate RhoGEF signaling that promotes neurite retraction (46). Consistent with this mechanism, targeting p115RhoGEF to the plasma membrane by appending a prenylation motif triggers neurite retraction in PC12 cells (46), which express R7BP and R7-RGS complexes (55). In a second model, R7-RGS heterotrimers are proposed to augment neurite retraction by inhibiting deactivation of Gα13 via the GAP activity of RGS domain-containing RhoGEFs. This model is suggested by evidence that certain RGS domains possess allosteric inhibitory sites distal to the surface that binds Gα subunits to stimulate GTP hydrolysis (56, 57). Whether such inhibitory sites exist in the RGS domains of Gα12/13-activated RhoGEFs and are targeted by R7-RGS complexes remains to be investigated. Distinguishing between these two models would require determining whether R7BP-bound R7-RGS complexes regulate the activation or deactivation of Gα13 and RhoGEFs in vitro.
Figure 7.
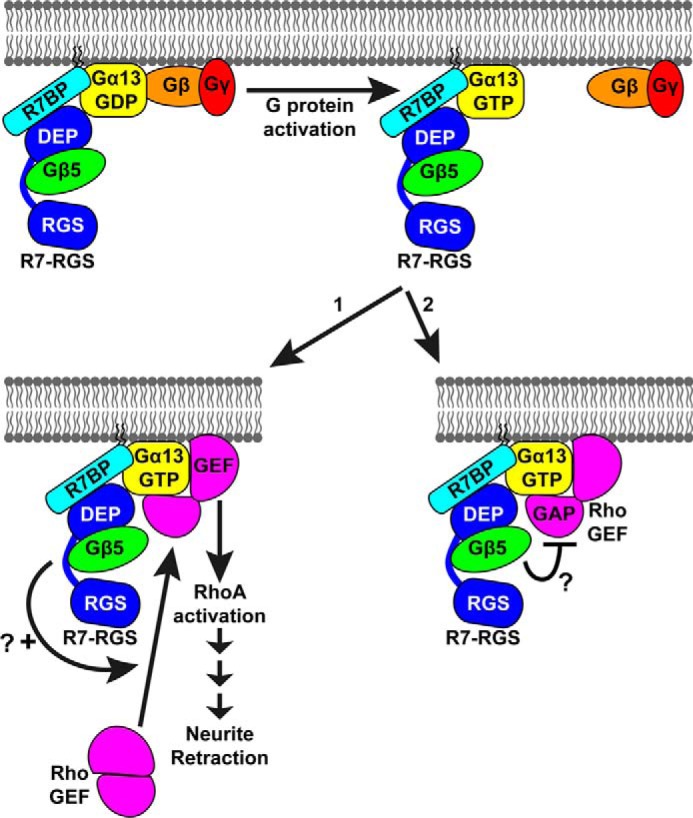
Models for enhancement of Gα13-evoked neurite retraction by R7-RGS heterotrimers. R7-RGS heterotrimers form macromolecular complexes with G13 heterotrimers and remain bound to Gα13 upon activation of the G protein. Activated Gα13 recruits a RhoGEF to the plasma membrane where it activates RhoA and downstream signaling events that ultimately result in neurite retraction. RhoGEFs also negatively regulate this process because they are GAPs for Gα13. R7-RGS heterotrimers could facilitate Gα13-evoked neurite retraction by two non-mutually exclusive mechanisms: 1) enhancing recruitment and activation of RhoGEFs, thereby enhancing RhoA signaling, or 2) inhibiting RhoGEF GAP activity for Gα13, thereby prolonging Gα13 activity.
Whether G12/13 signaling is facilitated similarly by complexes containing any R7-RGS isoform is unclear. Although we found that Gα13 can interact with heterotrimers containing any R7-RGS subtype, our functional studies were limited to N2a cells in which RGS7 appears to be the principal R7-RGS isoform expressed.3 Indeed, biological processes mediated by distinct G protein subtypes can be regulated by distinct R7-RGS isoforms as indicated by studies of the R7-RGS proteins Egl-10 and Eat-16 in C. elegans (25). Thus, it would be important to determine whether G12/13 signaling also can be augmented by R7-RGS heterotrimers bearing RGS6, -9, or -11.
R7-RGS heterotrimers also may control neurite morphogenesis by regulating signaling mechanisms independent of Gα13. This hypothesis is suggested by our finding that the neurite morphogenesis deficit of serum-starved R7BP-expressing N2a cells was rescued incompletely by expression of the G12/13 inhibitor HA-p115·RGS. In contrast, LPA-evoked neurite retraction absolutely required G12/13 signaling as indicated by complete blockade of this process by HA-p115·RGS. Further studies will be required to determine precisely how R7-RGS heterotrimers regulate neurite morphogenesis by Gα13-dependent and -independent mechanisms.
In conclusion, we have identified a novel interaction between R7-RGS heterotrimers and Gα13 and provided evidence that R7-RGS heterotrimers augment the function of Gα13 as a regulator of neurite morphogenesis. Our findings expand understanding of R7-RGS heterotrimers as multifunctional complexes critical for diverse aspects of neuronal development and function.
Experimental procedures
Reagents and antibodies
The following commercially produced antibodies were used: mouse anti-GAPDH (GeneTex, catalog number GTX627408, lot 40953), mouse anti-FLAG M2 (Sigma, catalog number F1804, various lots), mouse anti-actin C4 (Millipore, catalog number MAB1501, lot NG1812617), mouse anti-EE (Covance, catalog number MMS-115P, lot E12BF00285), mouse anti-HA.11 (Covance, catalog number MMS-101R), mouse anti-Myc 9E10 (Covance, catalog number MMS-150R, lot 139023001), HRP-conjugated goat anti-mouse IgG (Thermo Scientific, catalog number 31430, various lots), HRP-conjugated goat anti-rabbit IgG (Thermo Scientific, catalog number 31460, various lots), and Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen, catalog number A11001, lot 481679). Our rabbit anti-R7BP polyclonal antibody has been described (5). Rabbit anti-Gβ5 (ATDG) and rabbit anti-RGS7 (R4613) antibodies were generous gifts from Drs. William Simonds and Theodore Wensel, respectively (58, 59). Other reagents were as follows: Complete protease inhibitor mixture tablets (Roche Applied Science, catalog number 11697498001), mouse anti-FLAG M2-agarose (Sigma, catalog number A-2220), DSS (Thermo Scientific, catalog number 21555), StrepTactin Superflow (IBA, catalog number 2-1206-002), d-desthiobiotin solution (IBA, catalog number 2-1000-025), 3xFLAG peptide (Sigma, catalog number F4799), SYPRO Ruby stain (Sigma, catalog number S4942), Dual-Luciferase Reporter Assay System (Promega, catalog number E1960), LPA (Avanti Polar Lipids, catalog number 857130P), rhodamine-conjugated phalloidin (Life Technologies, catalog number R415), poly-d-lysine (Sigma, catalog number P0899), pertussis toxin (Calbiochem, catalog number 516561), forskolin (Sigma, catalog number F6886), and WIN 55,212-2 (Sigma, catalog number W102). All remaining reagents were from Sigma unless noted otherwise.
Generation of SF-R7BP transgenic mice
All procedures involving mice were conducted under protocols approved by the Animal Studies Committee of Washington University School of Medicine. To generate mice expressing SF-R7BP in neurons, a vector was generated to target SF-R7BP under control of the mouse prion promoter to the Hprt locus on the X chromosome. The backbone of the targeting vector was a modified version of pHPRT-targeting-Tet-EGFP-protein-entry (generously provided by Dr. Andrey Shaw) in which everything between the HPRT-targeting arms (including the Tet promoter and GFP) was replaced with a multiple cloning site and a PGK-Neo cassette. A transgene consisting of the mouse prion protein (MoPRP) promoter, MoPRP 5′-intronic sequence, SF-R7BP, and MoPRP 3′-untranslated region was inserted in reverse orientation in the multiple cloning site of the parent targeting vector. The MoPRP sequences were originally from the MoPrP·XhoI plasmid (34), and the SF tag sequence was originally from N-SF-TAP-pcDNA3 (a gift from Dr. Christian Johannes Gloeckner; Ref. 60). Standard methods were used to transfect ES cells with the targeting construct by electroporation (Washington University Murine Embryonic Stem Cell Core). Homologous recombination of the transgene causes deletion of exon 1 of Hprt, resulting in loss of HPRT protein expression and resistance to 6-thioguanine. Homologous recombinants were selected using both G418 and 6-thioguanine. ES cells that survived the selection process were confirmed as homologous recombinants by PCR using a forward primer specific for the PGK-Neo cassette on the transgene and a reverse primer specific for an HPRT genomic sequence outside of the right targeting arm. ES cells that were positive in the PCR test and had a normal karyotype were used for injection into C57BL/6 mouse blastocysts (Washington University School of Medicine, Department of Pathology Microinjection Core Facility) to generate chimeric mice. Chimeric male mice were bred with female C57BL/6 mice (Charles River) to produce offspring that were screened for germ line transmission by PCR. Mice positive for the SF-R7BP transgene were crossbred with R7BP−/− mice (50) to generate R7BP+/− offspring that also contained the SF-R7BP transgene. These offspring were subsequently crossed to generate R7BP−/− mice that were positive for the SF-R7BP transgene. Because endogenous R7BP is not expected to affect the makeup of the protein interaction profile, SF-R7BP male mice with both R7BP+/− and R7BP−/− backgrounds were used for TAP experiments based on availability.
Cell culture and transfection
All cells were maintained in DMEM/F-12 (Gibco, catalog number 11330-032) supplemented with 10% (v/v) FBS (Atlanta Biologicals, catalog number S11150) and penicillin/streptomycin at 37 °C with 5% CO2 in a humidified incubator. Growth medium for 3xFLAG-R7BP stable cells also contained 200 μg/ml Geneticin (Gibco, catalog number 10131). Chemical cross-linking experiments used a previously described clonal derivative of N2a cells stably expressing 3xFLAG-R7BP (5). Microscopy experiments used N2a cells transfected with 3xFLAG-R7BP (17) and treated with 400 μg/ml Geneticin for 16 days to select cells that stably express 3xFLAG-R7BP. Transfections were performed using TransIT-LT1 transfection reagent (Mirus, catalog number MIR 2305) according to the manufacturer's protocol.
Plasmids
pcDNA3.1+ was the backbone for all constructs unless stated otherwise. CBGN-Gα fusion sequences were made using overlap extension PCR to insert a glycine/serine linker (GGGSSGGG) followed by CBGN and another glycine/serine linker (GGGSSGGG) into Gα13 and Gαq between residues 135 and 136 and between residues 124 and 125, respectively. Split-luciferase fragments were amplified from plasmids containing fragments of click beetle green 99 luciferase (CBGN, amino acids 2–412; CBGC, amino acids 396–542; generous gifts from Dr. David Piwnica-Worms; Ref. 36). Human Gα13 and Gαq were amplified from plasmids purchased from Missouri University of Science and Technology cDNA Resource Center. Constitutively active point mutants (Q226L) of Gα13-CBGN and Gαq-CBGN were generated using site-directed mutagenesis. Gα13-CBGN-EE was generated using overlap extension PCR to mutate Gα13 amino acids 188–193 from DYIPSQ to EYMPTE. CBGC-LARG·RGS was created using overlap extension PCR to splice CBGC followed by a glycine/serine linker and BglII restriction site to sequences encoding the RGS domain of human LARG (amino acids 348–558). CBGC-RGS2 was constructed by splicing CBGC followed by a glycine/serine linker and 3xFLAG sequence to sequences encoding the N terminus of human RGS2. CBGC-3xFLAG-Gβ5 was generated by first constructing a plasmid containing CBGC followed by a glycine/serine linker and a 3xFLAG sequence immediately upstream of the multiple cloning site of pcDNA3.1+. The coding sequence of the short isoform of human Gβ5 (Gβ5s) was inserted in-frame into this plasmid. A plasmid that expresses plasma membrane-localized HA-p115·RGS was constructed by using overlap extension PCR to fuse sequences encoding a glycine/serine linker followed by the last 20 amino acids of human H-Ras to the C terminus of HA-p115·RGS (amino acids 1–252 of human p115RhoGEF). The plasmid expressing 3xFLAG-R7BP has been described (17). All newly described constructs were verified by DNA sequencing. Published plasmids expressing Renilla luciferase and HA-tagged R7-RGS proteins were generous gifts from Drs. David Piwnica-Worms and T. Kendall Harden, respectively. Plasmids expressing Myc-LARG, His-Myc-PDZ-RhoGEF, and Myc-p115RhoGEF were generous gifts from Dr. Tohru Kozasa. The plasmid expressing Epac-SH187 cAMP FRET sensor was generously provided by Dr. Kees Jalink (61).
In-cell chemical cross-linking
Wild-type N2 cells or N2a cells stably expressing 3xFLAG-R7BP were trypsinized, pelleted, and washed twice with PBS. Cross-linking reagents were prepared just before use by making a 200 mm stock of DSS in DMSO and then diluting in 100 mm HEPES (pH 8) for a final working solution of 1 mm DSS in 5% (v/v) DMSO. Cells were suspended in 1 mm DSS solution at 2.2 × 106 cells/ml and incubated for 30 min at room temperature. Cross-linking reactions were quenched by adding 20 mm Tris (pH 7.4) and incubating for 15 min at room temperature. Cells were then pelleted and lysed for immunoprecipitation.
Immunoblotting and immunoprecipitation
The following immunoprecipitation protocol was used for the data shown in Fig. 1A. Cells were lysed in radioimmune precipitation assay buffer (150 mm NaCl, 10 mm sodium phosphate, 1% (w/v) sodium deoxycholate, 1% (v/v) IGEPAL CA-630, 0.5% (w/v) SDS, 1 mm DTT, Complete protease inhibitor mixture) by agitating for 30 min at 4 °C and then cleared by ultracentrifugation at 100,000 × g for 10 min at 4 °C. Cross-linked species containing 3xFLAG-R7BP were immunoprecipitated by incubating with mouse anti-FLAG M2-agarose for 2 h at 4 °C followed by three washes with radioimmune precipitation assay buffer and eluted by boiling with 2× SDS-PAGE sample buffer (50 mm Tris (pH 6.8), 2% (w/v) SDS, 10% (v/v) glycerol, 1% (v/v) 2-mercaptoethanol, 12.5 mm EDTA, 0.02% (w/v) bromphenol blue) for 10 min.
Tissue lysates for experiments shown in Fig. 1C were generated by the following protocol. Mice were euthanized by asphyxiation with CO2 followed by cervical dislocation. The indicated tissues were isolated, and brains were further dissected in Hanks' balanced salt solution (Gibco, catalog number 14175) followed by homogenization in TAP lysis buffer (30 mm Tris (pH 7.4), 150 mm NaCl, 0.5% (v/v) IGEPAL CA-630, 0.5% (w/v) deoxycholate, Complete protease inhibitor mixture) using a Dounce homogenizer and then transferred to tubes and rotated end-over-end for 30 min at 4 °C. Lysates were cleared by ultracentrifugation at 100,000 × g for 15 min at 4 °C. A Bradford assay was performed to determine total protein content in the cleared lysates to equalize protein loading for SDS-PAGE.
All Western blot analyses were performed according to the following protocol. Samples were separated by SDS-PAGE, transferred to PVDF (Millipore, catalog number IPVH00010), blocked for at least 1 h using 5% (w/v) milk in TBST (25 mm Tris (pH 7.2), 150 mm NaCl, 2.7 mm KCl, 0.1% (v/v) Tween 20), incubated with the indicated primary antibody in blocking buffer for at least 2 h, washed three times with TBST, incubated for 1 h with appropriate HRP-conjugated secondary antibodies diluted in blocking buffer, and washed three times with TBST. Signals were detected using ECL solution (GE Healthcare, catalog number RPN2106, or Bio-Rad, catalog number 170-5060) and either film or a ChemiDoc Imaging System (Bio-Rad). In some cases after image acquisition, membranes were rinsed, reblocked, and probed for another protein. All quantified Western blot signals were within the linear range of the detection system as determined by an independent standard curve.
Tandem affinity purification
TAP was performed as described previously with following modifications (32). Three adult male mice expressing SF-R7BP (one R7BP−/− and two R7BP+/−) were euthanized by asphyxiation with CO2 followed by cervical dislocation. All subsequent steps were performed on ice or at 4 °C. Whole brains were dissected, homogenized with a Dounce homogenizer in lysis buffer (30 mm Tris (pH 7.4), 150 mm NaCl, 0.5% (v/v) IGEPAL CA-630, 0.5% (w/v) deoxycholate, Complete protease inhibitor mixture), rotated end-over-end for 30 min, cleared by ultracentrifugation at 100,000 × g for 15 min, and cleared further by passing through a 0.22-μm polyethersulfone filter. Cleared lysates were incubated with StrepTactin resin overnight with end-over-end rotation and washed five times in batch with a 10× column volume of wash buffer (30 mm Tris (pH 7.4), 150 mm NaCl, 0.1% (v/v) IGEPAL CA-630, Complete protease inhibitor mixture). Protein complexes were eluted two consecutive times by incubating with 3× column volumes of elution buffer (1× desthiobiotin buffer E (IBA), 0.1% (v/v) IGEPAL CA-630, Complete protease inhibitor mixture) for 30 min in batch. StrepTactin elution fractions were combined and incubated with anti-FLAG M2-agarose in batch for 1 h and washed five times in batch with 10× column volumes of wash buffer. Protein complexes were eluted from FLAG-agarose by incubating with 4× column volumes of FLAG elution buffer (200 μg/ml 3xFLAG peptide in wash buffer) for 30 min in batch. The final eluate was concentrated in preparation for gel electrophoresis using an Amicon Ultra-0.5 10,000 centrifugal filter device (Millipore, catalog number UFC501008).
LC-MS/MS
Mass spectrometry analysis was performed by the Proteomics and Mass Spectrometry Facility at the Donald Danforth Plant Science Center (St. Louis, MO). The final TAP eluate described above was resolved by SDS-PAGE, and protein bands were visualized with SYPRO Ruby (Fig. 2C). Prominent bands were excised, and proteins were digested using trypsin and Glu-C and analyzed by LC-MS/MS using a 1-h gradient and an LTQ-Orbitrap mass spectrometer. Peak lists were generated using Mascot Distiller version 2.4. Mass spectrometric data were searched using Mascot version 2.4.1 search engine and NCBInr Mammalia database (April 2013 version; 1,392,029 entries). The following parameters were used: trypsin and Glu-C allowed proteases, two missed cleavages allowed, no fixed modifications allowed, +16 on Met (oxidation) allowed as a variable modification, 15-ppm mass tolerance for precursor ions, and 0.80-Da mass tolerance for fragment ions. The database cRAP_20110301 (118 entries) was used to exclude known contaminants. Scaffold (version 4.0.5) was used to validate peptide and protein identities. Peptide identifications were accepted if they could be established at greater than 80% probability by the Scaffold local false discovery rate algorithm. All reported proteins have at least 99% protein identification probabilities as determined using the Protein Prophet algorithm and at least two exclusive unique peptides assigned. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Proteins sharing significant peptide evidence were grouped into clusters. Glu-C, trypsin, and keratin proteins were removed manually from the results. Reported are the proteins identified from a single TAP experiment starting with three SF-R7BP mouse brains.
Split-luciferase complementation
HEK293 cells were plated in 24-well dishes and transfected the next day with the indicated split-luciferase plasmids, Renilla luciferase for signal normalization, GFP to assess transfection efficiency, and pcDNA to normalize total DNA amounts across samples. At least three replicate wells of cells were transfected per condition in each independent experiment. Luciferase assays were performed 24–26 h after transfection using the Dual-Luciferase Reporter Assay System according to the manufacturer's protocol. A GloMax96 luminometer equipped with two injection pumps was used to measure luciferase activity on a per-well basis by first injecting the substrate solution for CBG and then injecting a quencher for click beetle green as well as the substrate for Renilla luciferase. Data collection started 0.4 s after substrate addition with a 10-s integration time.
Fluorescence microscopy
For microscopy experiments using HA-p115·RGS, cells were transfected and then replated the next day at low density on poly-d-lysine (PDL)-coated glass coverslips. For experiments not requiring transfection, cells were simply plated at low density on PDL-coated glass coverslips. Three hours after plating, the medium was exchanged for differentiation medium (DMEM/F-12 plus penicillin/streptomycin). For experiments using PTX, cells were plated at low density on PDL-coated glass coverslips and treated with 100 ng/ml PTX overnight. The next morning, the medium was exchanged for differentiation medium containing 100 ng/ml PTX, and this was repeated again 24 h later. In all cases, cells were allowed to differentiate for 48 h. If indicated, cells were treated with various concentrations of LPA (in 5 mg/ml fatty acid-free BSA in DPBS) for 15 min prior to fixing. Cells were fixed by incubation with 4% paraformaldehyde in DPBS (pH 7.2) for 10 min at room temperature followed by washes with DPBS. Fixed cells were permeabilized with 0.5% (v/v) Triton X-100 in DPBS for 10 min at room temperature, washed with DPBS, and blocked with blocking buffer (1% (w/v) BSA, 0.1% (v/v) Tween 20 in DPBS) for at least 30 min at room temperature. If immunostaining was required, coverslips were incubated with primary antibody (diluted in blocking buffer) in a humidified chamber overnight at 4 °C, washed three times with wash buffer (0.1% (v/v) Tween 20 in DPBS), and incubated with appropriate Alexa Fluor-conjugated secondary antibody and rhodamine-conjugated phalloidin in a humidified chamber for 1 h at room temperature. For experiments that did not require immunostaining, rhodamine-phalloidin was incubated with the coverslips for 30 min in a humidified chamber at room temperature immediately following the permeabilization step. All coverslips were washed three times with wash buffer, rinsed briefly in water, and mounted on glass slides using Vectashield mounting medium. Microscopy images were acquired with an Olympus IX81 inverted fluorescence microscope equipped with a UPlanSApo 60× oil objective (numerical aperture, 1.35) and an EXi BLUE digital camera (Q Imaging). Images were acquired using MetaMorph for Olympus Advanced and processed using Fiji software. Brightness and contrast were adjusted on an entire-image basis. Images of rhodamine-phalloidin fluorescence were used to score neurite projections, defined as any process extending from the cell body that was longer than half the cell body length. Cell body lengths were measured using the line tool, and neurite lengths were measured using the segmented line tool in Fiji.
cAMP FRET sensor assay
N2a cells were transfected with the Epac-SH187 cAMP FRET sensor. After 24 h, cells were trypsinized and replated in a black-wall clear-bottom 96-well plate (Costar, catalog number 3603) coated with poly-d-lysine. Cells were incubated in serum-free medium with or without 100 ng/ml pertussis toxin for 16–24 h. Prior to imaging, growth medium was replaced with prewarmed (37 °C) imaging buffer (125 nm NaCl, 5 mm KCl, 1.5 mm MgCl2, 1.5 mm CaCl2, 10 mm d-glucose, 20 mm HEPES (pH 7.4)) and assayed immediately using a Synergy H4 Hybrid Reader (BioTek) equipped with injection pumps for adding forskolin and WIN 55,212-2. Cells were maintained at 37 °C throughout the recording period. FRET donor was excited using a xenon flash lamp and a 420/20 bandpass filter. Donor emission and acceptor emission were detected every 10 s using 480/20 and 540/20 bandpass filters, respectively. A dichroic mirror with a 455-nm cutoff was used to separate the excitation and emission light paths. Changes in cAMP levels were expressed as follows.
| (Eq. 1) |
where FRET ratio = (480 nmemission/540 nmemission) at a given time point and the baseline FRET ratio = average (480 nmemission/540 nmemission) prior to forskolin stimulation. Triplicate wells were assayed per condition in each independent experiment.
Author contributions
S. L. S. designed and performed experiments, analyzed data, and wrote the paper. M. D. C. developed the split-luciferase system used in Figs. 3 and 6. K. M. K. designed, generated, and maintained SF-R7BP transgenic mice and performed experiments shown in Fig. 6. S. M. K. designed and performed experiments shown in Fig. 5, E and F. K. J. B. conceived and directed the study and wrote the paper. All authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Drs. Johannes Gloeckner, David Harris, David Piwnica-Worms, T. Kendall Harden, Andrey Shaw, Tohru Kozasa, Kees Jalink, and Philip Wedegaertner for providing plasmids; Drs. William Simonds and Theodore Wensel for providing antibodies; Drs. Leslie Hicks and Sophie Alvarez at the Danforth Center Proteomics facility for conducting proteomics experiments; and Joel Rurik for conducting confocal microscopy experiments.
This work was supported by National Institutes of Health Grants GM044592 and HL075632 (to K. J. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Table 1.
S. L. Scherer, M. D. Cain, S. M. Kanai, K. M. Kaltenbronn, and K. J. Blumer, unpublished data.
- RGS
- regulator of G protein signaling
- R7-RGS
- R7 regulator of G protein signaling family
- R7BP
- R7-RGS-binding protein
- Gβ5
- G protein β-subunit isoform 5
- DSS
- disuccinimidyl suberate
- CBG
- click beetle green luciferase
- CBGN
- N-terminal fragment of click beetle green luciferase
- CBGC
- C-terminal fragment of click beetle green luciferase
- TAP
- tandem affinity purification
- PTX
- pertussis toxin
- LPA
- lysophosphatidic acid
- GAP
- GTPase-activating protein
- N2a
- Neuro2a
- GEF
- guanine nucleotide exchange factor
- LARG
- leukemia-associated Rho guanine nucleotide exchange factor
- DEP
- disheveled, Egl-10, and pleckstrin
- DHEX
- DEP helical extension
- GGL
- G protein γ-like
- R9AP
- RGS9 anchor protein
- RH
- RGS homology
- HPRT
- hypoxanthine-guanine phosphoribosyltransferase
- PGK-Neo
- phosphoglycerate kinase-neomycin resistance
- MoPRP
- mouse prion protein
- PDL
- poly-d-lysine
- DPBS
- Dulbecco's PBS
- SF
- Strep/FLAG.
References
- 1. Ross E. M., and Wilkie T. M. (2000) GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu. Rev. Biochem. 69, 795–827 [DOI] [PubMed] [Google Scholar]
- 2. Hooks S. B., Waldo G. L., Corbitt J., Bodor E. T., Krumins A. M., and Harden T. K. (2003) RGS6, RGS7, RGS9, and RGS11 stimulate GTPase activity of Gi family G-proteins with differential selectivity and maximal activity. J. Biol. Chem. 278, 10087–10093 [DOI] [PubMed] [Google Scholar]
- 3. Posner B. A., Gilman A. G., and Harris B. A. (1999) Regulators of G protein signaling 6 and 7. Purification of complexes with Gβ5 and assessment of their effects on G protein-mediated signaling pathways. J. Biol. Chem. 274, 31087–31093 [DOI] [PubMed] [Google Scholar]
- 4. Snow B. E., Krumins A. M., Brothers G. M., Lee S.-F., Wall M. A., Chung S., Mangion J., Arya S., Gilman A. G., and Siderovski D. P. (1998) A G protein γ subunit-like domain shared between RGS11 and other RGS proteins specifies binding to Gβ5 subunits. Proc. Natl. Acad. Sci. U.S.A. 95, 13307–13312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grabowska D., Jayaraman M., Kaltenbronn K. M., Sandiford S. L., Wang Q., Jenkins S., Slepak V. Z., Smith Y., and Blumer K. J. (2008) Postnatal induction and localization of R7BP, a membrane-anchoring protein for regulator of G protein signaling 7 family-Gβ5 complexes in brain. Neuroscience 151, 969–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang J. H., Lai Z., and Simonds W. F. (2000) Differential expression of the G protein β5 gene: analysis of mouse brain, peripheral tissues, and cultured cell lines. J. Neurochem. 75, 393–403 [DOI] [PubMed] [Google Scholar]
- 7. Gold S. J., Ni Y. G., Dohlman H. G., and Nestler E. J. (1997) Regulators of G-protein signaling (RGS) proteins: region-specific expression of nine subtypes in rat brain. J. Neurosci. 17, 8024–8037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nishiguchi K. M., Sandberg M. A., Kooijman A. C., Martemyanov K. A., Pott J. W., Hagstrom S. A., Arshavsky V. Y., Berson E. L., and Dryja T. P. (2004) Defects in RGS9 or its anchor protein R9AP in patients with slow photoreceptor deactivation. Nature 427, 75–78 [DOI] [PubMed] [Google Scholar]
- 9. Krispel C. M., Chen C.-K., Simon M. I., and Burns M. E. (2003) Prolonged photoresponses and defective adaptation in rods of Gβ5−/− mice. J. Neurosci. 23, 6965–6971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rao A., Dallman R., Henderson S., and Chen C.-K. (2007) Gβ5 is required for normal light responses and morphology of retinal ON-bipolar cells. J. Neurosci. 27, 14199–14204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang J.-H., Pandey M., Seigneur E. M., Panicker L. M., Koo L., Schwartz O. M., Chen W., Chen C.-K., and Simonds W. F. (2011) Knockout of G protein β5 impairs brain development and causes multiple neurologic abnormalities in mice. J. Neurochem. 119, 544–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zachariou V., Georgescu D., Sanchez N., Rahman Z., DiLeone R., Berton O., Neve R. L., Sim-Selley L. J., Selley D. E., Gold S. J., and Nestler E. J. (2003) Essential role for RGS9 in opiate action. Proc. Natl. Acad. Sci. U.S.A. 100, 13656–13661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Siderovski D. P., and Willard F. S. (2005) The GAPs, GEFs, and GDIs of heterotrimeric G-protein α subunits. Int. J. Biol. Sci. 1, 51–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen C.-K., Eversole-Cire P., Zhang H., Mancino V., Chen Y.-J., He W., Wensel T. G., and Simon M. I. (2003) Instability of GGL domain-containing RGS proteins in mice lacking the G protein β-subunit Gβ5. Proc. Natl. Acad. Sci. U.S.A. 100, 6604–6609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cheever M. L., Snyder J. T., Gershburg S., Siderovski D. P., Harden T. K., and Sondek J. (2008) Crystal structure of the multifunctional Gβ5-RGS9 complex. Nat. Struct. Mol. Biol. 15, 155–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martemyanov K. A., Yoo P. J., Skiba N. P., and Arshavsky V. Y. (2005) R7BP, a novel neuronal protein interacting with RGS proteins of the R7 family. J. Biol. Chem. 280, 5133–5136 [DOI] [PubMed] [Google Scholar]
- 17. Drenan R. M., Doupnik C. A., Boyle M. P., Muglia L. J., Huettner J. E., Linder M. E., and Blumer K. J. (2005) Palmitoylation regulates plasma membrane-nuclear shuttling of R7BP, a novel membrane anchor for the RGS7 family. J. Cell Biol. 169, 623–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Orlandi C., Posokhova E., Masuho I., Ray T. A., Hasan N., Gregg R. G., and Martemyanov K. A. (2012) GPR158/179 regulate G protein signaling by controlling localization and activity of the RGS7 complexes. J. Cell Biol. 197, 711–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sandiford S. L., and Slepak V. Z. (2009) The Gβ5-RGS7 complex selectively inhibits muscarinic M3 receptor signaling via the interaction between the third intracellular loop of the receptor and the DEP domain of RGS7. Biochemistry 48, 2282–2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu G., and Wensel T. G. (2002) R9AP, a membrane anchor for the photoreceptor GTPase accelerating protein, RGS9-1. Proc. Natl. Acad. Sci. U.S.A. 99, 9755–9760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zheng M., Cheong S.-Y., Min C., Jin M., Cho D.-I., and Kim K.-M. (2011) β-Arrestin2 plays permissive roles in the inhibitory activities of RGS9-2 on G protein-coupled receptors by maintaining RGS9-2 in the open conformation. Mol. Cell. Biol. 31, 4887–4901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jia L., Linder M. E., and Blumer K. J. (2011) Gi/o signaling and the palmitoyltransferase DHHC2 regulate palmitate cycling and shuttling of RGS7 family-binding protein. J. Biol. Chem. 286, 13695–13703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jia L., Chisari M., Maktabi M. H., Sobieski C., Zhou H., Konopko A. M., Martin B. R., Mennerick S. J., and Blumer K. J. (2014) A mechanism regulating G protein-coupled receptor signaling that requires cycles of protein palmitoylation and depalmitoylation. J. Biol. Chem. 289, 6249–6257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Panicker L. M., Zhang J.-H., Posokhova E., Gastinger M. J., Martemyanov K. A., and Simonds W. F. (2010) Nuclear localization of the G protein β5/R7-regulator of G protein signaling protein complex is dependent on R7 binding protein. J. Neurochem. 113, 1101–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Patikoglou G. A., and Koelle M. R. (2002) An N-terminal region of Caenorhabditis elegans RGS proteins EGL-10 and EAT-16 directs inhibition of Gαo versus Gαq signaling. J. Biol. Chem. 277, 47004–47013 [DOI] [PubMed] [Google Scholar]
- 26. Kozasa T., Hajicek N., Chow C. R., and Suzuki N. (2011) Signalling mechanisms of RhoGTPase regulation by the heterotrimeric G proteins G12 and G13. J. Biochem. 150, 357–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shu F. J., Ramineni S., and Hepler J. R. (2010) RGS14 is a multifunctional scaffold that integrates G protein and Ras/Raf MAP kinase signalling pathways. Cell. Signal. 22, 366–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang J., Stewart A., Maity B., Hagen J., Fagan R. L., Yang J., Quelle D. E., Brenner C., and Fisher R. A. (2014) RGS6 suppresses Ras-induced cellular transformation by facilitating Tip60-mediated Dnmt1 degradation and promoting apoptosis. Oncogene 33, 3604–3611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu Z., and Fisher R. A. (2004) RGS6 interacts with DMAP1 and DNMT1 and inhibits DMAP1 transcriptional repressor activity. J. Biol. Chem. 279, 14120–14128 [DOI] [PubMed] [Google Scholar]
- 30. Tayou J., Wang Q., Jang G.-F., Pronin A. N., Orlandi C., Martemyanov K. A., Crabb J. W., and Slepak V. Z. (2016) Regulator of G-protein signaling 7 (RGS7) can exist in a homo-oligomeric form that is regulated by Gαo and R7-binding protein. J. Biol. Chem. 291, 9133–9147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Orlandi C., Xie K., Masuho I., Fajardo-Serrano A., Lujan R., and Martemyanov K. A. (2015) Orphan receptor GPR158 is an allosteric modulator of regulator of G protein signaling 7 (RGS7) catalytic activity with essential role in dictating its expression and localization in the brain. J. Biol. Chem. 290, 13622–13639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gloeckner C. J., Boldt K., Schumacher A., and Ueffing M. (2009) Tandem affinity purification of protein complexes from mammalian cells by the Strep/FLAG (SF)-TAP tag. Methods Mol. Biol. 564, 359–372 [DOI] [PubMed] [Google Scholar]
- 33. Misra R. P., and Duncan S. A. (2002) Gene targeting in the mouse: advances in introduction of transgenes into the genome by homologous recombination. Endocrine 19, 229–238 [DOI] [PubMed] [Google Scholar]
- 34. Borchelt D. R., Davis J., Fischer M., Lee M. K., Slunt H. H., Ratovitsky T., Regard J., Copeland N. G., Jenkins N. A., Sisodia S. S., and Price D. L. (1996) A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet. Anal. 13, 159–163 [DOI] [PubMed] [Google Scholar]
- 35. Luker K. E., Smith M. C., Luker G. D., Gammon S. T., Piwnica-Worms H., and Piwnica-Worms D. (2004) Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc. Natl. Acad. Sci. U.S.A. 101, 12288–12293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Villalobos V., Naik S., Bruinsma M., Dothager R. S., Pan M. H., Samrakandi M., Moss B., Elhammali A., and Piwnica-Worms D. (2010) Dual-color click beetle luciferase heteroprotein fragment complementation assays. Chem. Biol. 17, 1018–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hughes T. E., Zhang H., Logothetis D. E., and Berlot C. H. (2001) Visualization of a functional Gαq-green fluorescent protein fusion in living cells. Association with the plasma membrane is disrupted by mutational activation and by elimination of palmitoylation sites, but not by activation mediated by receptors or AlF4−. J. Biol. Chem. 276, 4227–4235 [DOI] [PubMed] [Google Scholar]
- 38. Katoh H., Aoki J., Yamaguchi Y., Kitano Y., Ichikawa A., and Negishi M. (1998) Constitutively active Gα12, Gα13, and Gαq induce Rho-dependent neurite retraction through different signaling pathways. J. Biol. Chem. 273, 28700–28707 [DOI] [PubMed] [Google Scholar]
- 39. Kranenburg O., Poland M., van Horck F. P., Drechsel D., Hall A., and Moolenaar W. H. (1999) Activation of RhoA by lysophosphatidic acid and Gα12/13 subunits in neuronal cells: induction of neurite retraction. Mol. Biol. Cell. 10, 1851–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sayas C. L., Avila J., and Wandosell F. (2002) Glycogen synthase kinase-3 is activated in neuronal cells by Gα12 and Gα13 by Rho-independent and Rho-dependent mechanisms. J. Neurosci. 22, 6863–6875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moers A., Nürnberg A., Goebbels S., Wettschureck N., and Offermanns S. (2008) Gα12/Gα13 deficiency causes localized overmigration of neurons in the developing cerebral and cerebellar cortices. Mol. Cell. Biol. 28, 1480–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nürnberg A., Braüer A. U., Wettschureck N., and Offermanns S. (2008) Antagonistic regulation of neurite morphology through Gq/G11 and G12/G13. J. Biol. Chem. 283, 35526–35531 [DOI] [PubMed] [Google Scholar]
- 43. Yamazaki J., Katoh H., and Negishi M. (2008) Lysophosphatidic acid and thrombin receptors require both Gα12 and Gα13 to regulate axonal morphology in hippocampal neurons. Biol. Pharm. Bull. 31, 2216–2222 [DOI] [PubMed] [Google Scholar]
- 44. Sun Y., Kim N. H., Yang H., Kim S. H., and Huh S. O. (2011) Lysophosphatidic acid induces neurite retraction in differentiated neuroblastoma cells via GSK-3β activation. Mol. Cells 31, 483–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Choi J. W., and Chun J. (2013) Lysophospholipids and their receptors in the central nervous system. Biochim. Biophys. Acta. 1831, 20–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bhattacharyya R., Banerjee J., Khalili K., and Wedegaertner P. B. (2009) Differences in Gα12- and Gα13-mediated plasma membrane recruitment of p115-RhoGEF. Cell. Signal. 21, 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jordan J. D., He J. C., Eungdamrong N. J., Gomes I., Ali W., Nguyen T., Bivona T. G., Philips M. R., Devi L. A., and Iyengar R. (2005) Cannabinoid receptor-induced neurite outgrowth is mediated by Rap1 activation through Gαo/i-triggered proteasomal degradation of Rap1GAPII. J. Biol. Chem. 280, 11413–11421 [DOI] [PubMed] [Google Scholar]
- 48. He J. C., Gomes I., Nguyen T., Jayaram G., Ram P. T., Devi L. A., and Iyengar R. (2005) The Gαo/i-coupled cannabinoid receptor-mediated neurite outgrowth involves Rap regulation of Src and Stat3. J. Biol. Chem. 280, 33426–33434 [DOI] [PubMed] [Google Scholar]
- 49. Drenan R. M., Doupnik C. A., Jayaraman M., Buchwalter A. L., Kaltenbronn K. M., Huettner J. E., Linder M. E., and Blumer K. J. (2006) R7BP augments the function of RGS7-Gβ5 complexes by a plasma membrane-targeting mechanism. J. Biol. Chem. 281, 28222–28231 [DOI] [PubMed] [Google Scholar]
- 50. Zhou H., Chisari M., Raehal K. M., Kaltenbronn K. M., Bohn L. M., Mennerick S. J., and Blumer K. J. (2012) GIRK channel modulation by assembly with allosterically regulated RGS proteins. Proc. Natl. Acad. Sci. U.S.A. 109, 19977–19982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Masuho I., Xie K., and Martemyanov K. A. (2013) Macromolecular composition dictates receptor and G protein selectivity of regulator of G protein signaling (RGS) 7 and 9-2 protein complexes in living cells. J. Biol. Chem. 288, 25129–25142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Neubig R. R. (2015) RGS-insensitive G proteins as in vivo probes of RGS function. Prog. Mol. Biol. Transl. Sci. 133, 13–30 [DOI] [PubMed] [Google Scholar]
- 53. Anderson G. R., Semenov A., Song J. H., and Martemyanov K. A. (2007) The membrane anchor R7BP controls the proteolytic stability of the striatal specific RGS protein, RGS9-2. J. Biol. Chem. 282, 4772–4781 [DOI] [PubMed] [Google Scholar]
- 54. Deleted in proof.
- 55. Nini L., Waheed A. A., Panicker L. M., Czapiga M., Zhang J.-H., and Simonds W. F. (2007) R7-binding protein targets the G protein β5/R7-regulator of G protein signaling complex to lipid rafts in neuronal cells and brain. BMC Biochem. 8, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Blazer L. L., Roman D. L., Chung A., Larsen M. J., Greedy B. M., Husbands S. M., and Neubig R. R. (2010) Reversible, allosteric small-molecule inhibitors of regulator of G protein signaling proteins. Mol. Pharmacol. 78, 524–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Popov S. G., Krishna U. M., Falck J. R., and Wilkie T. M. (2000) Ca2+/calmodulin reverses phosphatidylinositol 3,4,5-trisphosphate-dependent inhibition of regulators of G protein-signaling GTPase-activating protein activity. J. Biol. Chem. 275, 18962–18968 [DOI] [PubMed] [Google Scholar]
- 58. Zhang J. H., and Simonds W. F. (2000) Copurification of brain G-protein β5 with RGS6 and RGS7. J. Neurosci. 20, RC59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Morgans C. W., Liu W., Wensel T. G., Brown R. L., Perez-Leon J. A., Bearnot B., and Duvoisin R. M. (2007) Gβ5-RGS complexes co-localize with mGluR6 in retinal ON-bipolar cells. Eur. J. Neurosci. 26, 2899–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gloeckner C. J., Boldt K., Schumacher A., Roepman R., and Ueffing M. (2007) A novel tandem affinity purification strategy for the efficient isolation and characterisation of native protein complexes. Proteomics 7, 4228–4234 [DOI] [PubMed] [Google Scholar]
- 61. Klarenbeek J., Goedhart J., van Batenburg A., Groenewald D., and Jalink K. (2015) Fourth-generation Epac-based FRET sensors for cAMP feature exceptional brightness, photostability and dynamic range: characterization of dedicated sensors for FLIM, for ratiometry and with high affinity. PLoS One 10, e0122513. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.