

Abstract
Small heat-shock proteins (sHSPs) are a conserved group of molecular chaperones with important roles in cellular proteostasis. Although sHSPs are characterized by their small monomeric weight, they typically assemble into large polydisperse oligomers that vary in both size and shape but are principally composed of dimeric building blocks. These assemblies can include different sHSP orthologues, creating additional complexity that may affect chaperone activity. However, the structural and functional properties of such hetero-oligomers are poorly understood. We became interested in hetero-oligomer formation between human heat-shock protein family B (small) member 1 (HSPB1) and HSPB6, which are both highly expressed in skeletal muscle. When mixed in vitro, these two sHSPs form a polydisperse oligomer array composed solely of heterodimers, suggesting preferential association that is determined at the monomer level. Previously, we have shown that the sHSP N-terminal domains (NTDs), which have a high degree of intrinsic disorder, are essential for the biased formation. Here we employed iterative deletion mapping to elucidate how the NTD of HSPB6 influences its preferential association with HSPB1 and show that this region has multiple roles in this process. First, the highly conserved motif RLFDQXFG is necessary for subunit exchange among oligomers. Second, a site ∼20 residues downstream of this motif determines the size of the resultant hetero-oligomers. Third, a region unique to HSPB6 dictates the preferential formation of heterodimers. In conclusion, the disordered NTD of HSPB6 helps regulate the size and stability of hetero-oligomeric complexes, indicating that terminal sHSP regions define the assembly properties of these proteins.
Keywords: chaperone, cysteine-mediated cross-linking, mass spectrometry (MS), site-directed mutagenesis, small heat-shock protein (sHsp), Hsp20, Hsp27
Introduction
Heat-shock proteins are an indispensable group of proteins in charge of maintaining cellular proteostasis. This protein superfamily ensures both the correct folding of newly synthesized proteins and prevents unfolding and aggregation under stress conditions (1). Small heat-shock proteins (sHSPs)4 are an important subfamily of this network and capture proteins in the early stages of unfolding, thereby preventing aberrant interactions leading to aggregation (2). sHSPs are capable of binding a wide variety of substrate proteins within the cell and are considered a first line of defense of the protein quality control network (3–6).
Small heat-shock proteins are notorious for assembling into large polydisperse complexes, composed of up to 40 subunits for some members (7–9). These assemblies are formed from dimeric building blocks, where the constituent monomeric subunits can freely exchange (10). Dimer association is mediated by the core structured region of sHSPs, the α-crystallin domain (ACD) (11–14). Crystal structures of the isolated ACD of a number of metazoan sHSPs show a 7-stranded β-sandwich that readily assembles into dimers via anti-parallel pairing of the β7-strand (15–17). Within this kingdom, higher-order assembly into the larger oligomeric species, as well as regulation of subunit exchange of the component protomers, is controlled via the N- and C-terminal arms that flank the ACD (18). Other than the presence of a tripeptide IX(I/V) motif, termed the C-terminal anchoring module (19), these domains are generally considered to be poorly conserved and are predicted to lack secondary structure making characterization of their role in assembly particularly challenging.
This multifaceted structural association and dynamics are further complicated by hetero-oligomerization, in which two or more orthologous sHSPs co-assemble into the typical high molecular weight complexes (20–24). This phenomenon has been described for bacterial, plant, and metazoan sHSPs (21, 25, 26). In humans, numerous members of the sHSP family have been shown to interact with each other (23), and the most well known complex is lens α-crystallin, which is composed of αA- and αB-crystallin in a 3:1 monomer ratio (27, 28). Although present in this ratio in most vertebrate lenses, when mixed in vitro they form a hetero-oligomer containing subunits consistent with the proportion used (29, 30). The α-crystallin heterocomplex therefore shows a ratio of the component sHSPs that likely reflects their relative expression levels. At the same time, there are a number of human orthologues that, when mixed together in various ratios, form hetero-oligomers with a specific subunit stoichiometry (20, 31).
In this study we focus on the biologically relevant complex formed between HSPB1 and HSPB6, two sHSPs that co-assemble in muscle tissue where they are both highly expressed (32, 33). Previous studies of the human HSPB1 and HSPB6 hetero-oligomers have shown that they form highly polydisperse complexes containing an equimolar amount of each sHSP (31, 34). Assuming both sHSPs can freely exchange subunits, random protomer turnover would be expected to result in oligomers containing both homo- and heterodimers. Recently, using native mass spectrometry, we have observed that although both sHSPs alone can exchange subunits in such a stochastic fashion, the hetero-oligomeric complex is composed solely of heterodimers (35). This result, in good agreement with earlier residue-specific cross-linking studies (21), supports a hetero-oligomer model where HSPB1 and HSPB6 preferentially associate at the core dimer level. Curiously, this preferred association, which is mediated by the ACD, requires the presence of the highly flexible NTDs (35). This latter region of human HSPB1 is 90 residues long and has low sequence homology with the 72-residue NTD of HSPB6, with the most prominent exception being a conserved RLFDQXFG motif close to the center of this domain in the two proteins. Because of these distinct differences, how this disordered region dictates the association properties of the structured ACD is not understood.
Here we have used a library of deletion constructs, previously employed to investigate the role of the NTD of HSPB6 in chaperoning (36), to identify the N-terminal residues in this sHSP that dictate hetero-oligomerization. The effect of these deletions, as well as additional point mutants, on the subunit exchange behavior with HSPB1 were analyzed by an array of techniques, including size-exclusion chromatography, mass spectrometry (MS), and disulfide-mediated cross-linking studies. The results point to a complex mechanism where different sequences within the NTD of HSPB6 influence the initial association of the two sHSPs, the size of the resultant hetero-oligomeric assemblies, and the preferential heterodimerization with HSPB1.
Results
Deletion mapping of the HSPB6 NTD defines regions involved in hetero-oligomerization
N-terminal deletion constructs of human HSPB6, used previously to identify substrate-binding sites of this sHSP (36), were employed to map the residues within the NTD that are required for the preferential association with HSPB1. Previous characterization of these HSPB6 constructs showed that they were all dimeric in solution, similar to the wild-type (WT) protein. This thus permits an investigation of the possible effect of truncation on the size distribution of formed hetero-oligomers without fear of interference of aberrant homo-oligomers of the deletion constructs.
The equimolar mixture of WT HSPB1 and HSPB6 prepared at 37 °C yielded a broad elution profile on analytical SEC, composed of two peaks with maxima corresponding to a molecular mass of 508 and 160 kDa, in line with previous studies (35). The corresponding eluted fractions contained equimolar amounts of both sHSPs when analyzed by SDS-PAGE (Fig. 1). All seven HSPB6 deletion constructs were capable of forming a hetero-oligomer with HSPB1, albeit with differing elution profiles for some (Fig. 1). Most notably, deletion of residues 51–60 led to the predominant formation of the smaller hetero-oligomeric species at the expense of the larger complex, despite these entities containing equal amounts of both sHSPs in SDS-PAGE analysis of the fractions. Another striking difference was observed with constructs that lack residues 21–30 and 31–40. In both cases, not all of the HSPB6 construct was incorporated into the complex, as evidenced by a dominance of HSPB1 in the SDS-PAGE analysis of fractions of the hetero-oligomer peak, as well as by a small remnant peak of the HSPB6 deletant alone in the size-exclusion chromatogram (Fig. 1).
Figure 1.
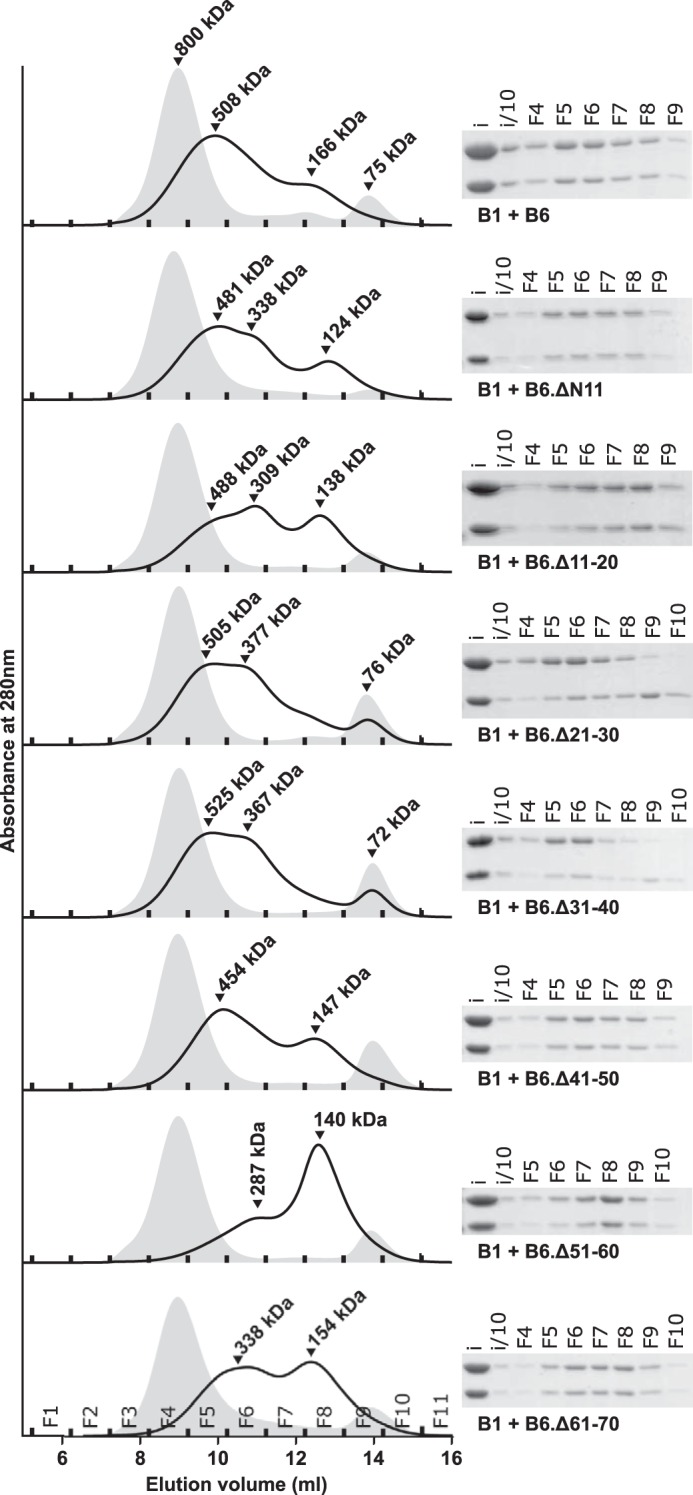
Role of the NTD of HSPB6 in hetero-oligomerization. Analytical SEC profiles of equimolar mixtures of HSPB1 and each HSPB6 deletion following overnight incubation at 37 °C. A 100-μl sample was loaded onto a Superdex 200 10/300 column equilibrated in 20 mm HEPES, 150 mm NaCl, and 2.5 mm DTT. The chromatogram of the control sample (the equimolar mixture stored at 4 °C) is shown as a gray-filled curve, and the heated mixture is shown as a black curve. Fractions (F) are denoted at the base of each curve, and their identities are shown on the bottom left panel. Right-hand side, corresponding SDS-PAGE analysis of the fractions from analytical SEC. An input sample (i) of the equimolar mix of both sHSPS taken prior to injection onto the column, and the same sample diluted 10-fold (i/10) were also loaded.
SEC-coupled SAXS experiments of the same mixtures showed a single elution peak, likely due to a lower resolving power of the Shodex column compared with the Superdex 200 column (supplemental Fig. 1). The calculated average mass of the complex between both WT proteins corresponds to 360 kDa and has an Rg value of 51 Å at the elution peak maxima, with a range of 39.5 to 52.4 Å (Table 1). This is smaller than the WT HSPB1 but larger than WT HSPB6, which have a Rg at the elution peak of 58 and 32 Å, respectively. Combined these values are in agreement with the hetero-oligomer being a polydisperse mixture containing species whose size ranges vary between those of the component homo-oligomers (Fig. 1 and supplemental Fig. 1). For the majority of complexes formed between the different HSPB6 deletions and HSPB1, the peak Rg value as well as the Rg range were similar to that of the WT complex. However, the deletion of residues 51–60 resulted in a clearly smaller species with a peak Rg of 41.6 Å. At the same time, the two truncations that did not form a complex with HSPB1 efficiently (Δ21–30 and Δ31–40) showed an increased peak Rg value of 57 Å as well as a wider range in Rg from 57 to 33 Å. This correlates well with the higher resolution SEC studies that showed the presence of both a larger species, containing predominantly HSPB1, as well the non-associated HSPB6 deletion dimeric species (Fig. 1).
Table 1.
SAXS analysis of the hetero-oligomeric complexes of HSPB1 and HSPB6 deletion constructs
The SAXS frames were processed using AUTORG and GNOM, both part of the ATSAS package (56). The 2nd to last column shows the Rg value at the elution peak. The last column shows the Rg range corresponding to the frames for which the quality measure in AUTORG was 50% or higher.
| Protein | Estimated average mass based on Porod volume | Calculated average number of subunits | Peak Rg | Rg range |
|---|---|---|---|---|
| (kDa) | Å | |||
| B1.WT | 647.6 | 28.4 | 58.2 | 63.0–56.0 |
| B6.WT | 45.9 | 2.7 | 32.4 | 32.9–27.5 |
| B1 + B6 | 342.4 | 17.2 | 51.1 | 52.4–39.5 |
| B1 + B6.ΔN11 | 311.7 | 16.1 | 49.6 | 50.9–39.7 |
| B1 + B6.Δ11–20 | 212.1 | 10.9 | 48.0 | 51.1–35.2 |
| B1 + B6.Δ21–30 | 490.8 | 25.2 | 56.9 | 57.1–31.5 |
| B1 + B6.Δ31–40 | 478.9 | 24.7 | 57.0 | 57.3–33.2 |
| B1 + B6.Δ41–50 | 356.4 | 18.3 | 52.2 | 52.9–44.9 |
| B1 + B6.Δ51–60 | 134.7 | 6.9 | 41.6 | 48.2–33.7 |
| B1 + B6.Δ61–70 | 337.6 | 17.3 | 51.2 | 51.8–42.5 |
To ascertain whether the hetero-oligomers formed between HSPB1 and the different HSPB6 truncations were composed primarily of heterodimers, disulfide cross-linking was used. To this end, an additional double mutation C46S/E116C (denoted by an asterisk in subsequent construct names) was introduced in every HSPB6 deletion construct. A single mutation E116C was introduced in Δ41–50 where the native cysteine is absent. Glu-116 and the corresponding residue Cys-137 in HSPB1 residue in the β7-strand of the ACD are located near the 2-fold axis of the APII dimer interface (Fig. 2A and supplemental Fig. 3A) (16, 18). This proximal position of the cysteines means that both the WT HSPB1 and the HSPB6* mutant can be readily cross-linked across the dimer interface by oxidation (16). Likewise, disulfide cross-linking of the heated HSPB1/HSPB6* mixture yields a species with a mass between that of the HSPB1 and HSPB6 cross-linked dimers when analyzed by non-reducing SDS-PAGE (Fig. 2, B and C). Importantly introduction of the C46S/E116C double mutation results in wild-type exchange behavior (21). In our hands, the additionally mutated HSPB6 truncations all behaved like their non-mutant equivalent during purification (data not shown).
Figure 2.
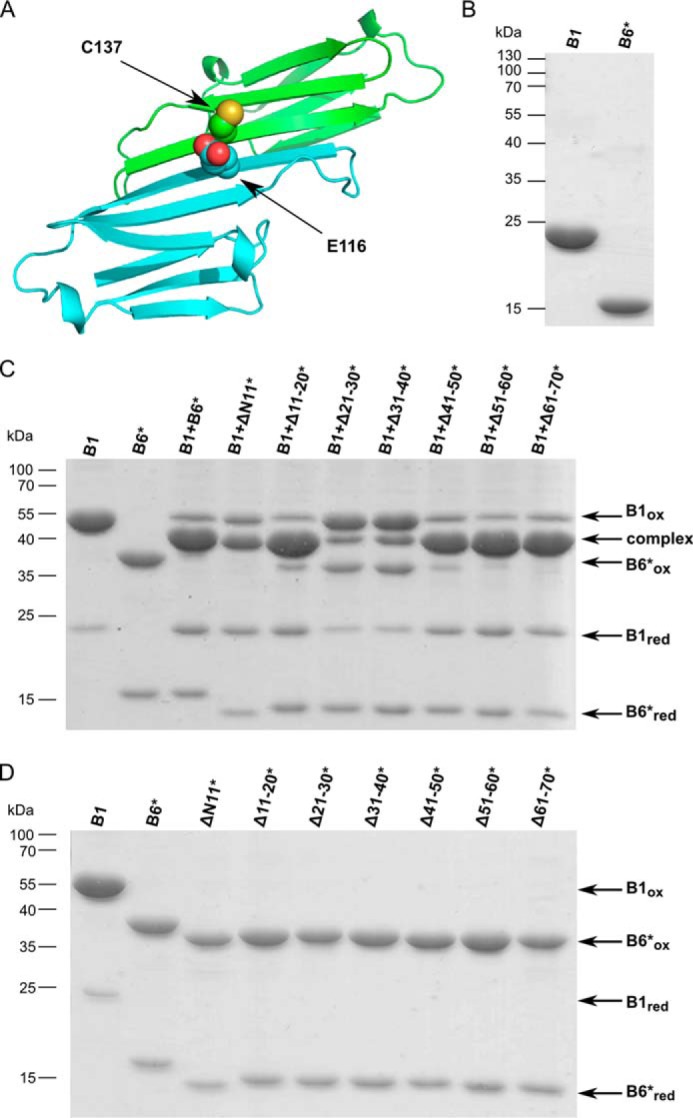
Disulfide cross-linking studies of HSBP1/HSPB6* heterocomplexes. A, cartoon representation of the putative ACD heterodimer interface. Residues Glu-116 of HSPB6 (green ribbons) and Cys-146 of HSPB1 (cyan ribbons) are shown in red as all-atom representations. B, reducing SDS-PAGE analysis of HSPB1 and HSPB6 C46S.E116C (HSPB6*) constructs. C, non-reducing SDS-PAGE analysis of the complexes between HSPB1 and the seven different HSPB6* deletion constructs. Samples were incubated at 37 °C overnight in 50 mm sodium phosphate, pH 7.5, 100 mm NaCl, and 5 mm DTT and then dialyzed against the same buffer without DTT to permit disulfide cross-linking. D, control experiment showing the non-reducing SDS-PAGE analysis of each HSPB6* deletion construct following extensive dialysis to remove DTT.
The results of the disulfide cross-linking clearly show that all HSPB6 truncations except for Δ21–30* and Δ31–40* are nearly fully engaged in a heterodimeric complex with HSPB1, just like the full-length proteins (Fig. 2B). In contrast, the Δ21–30* and Δ31–40* constructs, when incubated with HSPB1, reveal three bands, corresponding to cross-linked HSPB1 homodimer, the heterodimer, and the HSPB6* homodimers, respectively. As assessed by densitometry of the stained gels, the molar ratio of these species was close to 1:0.5:1 (corresponding to 20% heterodimer formation). This suggests that the deletion of these regions in HSPB6 hampers the heterodimer formation. To test whether the Δ21–30* and Δ31–40* constructs were capable of forming a cross-dimer disulfide when alone, a control oxidation experiment was performed that demonstrated that they oxidized as readily as all other constructs under the conditions employed (Fig. 2C). To rule out the further possibility that the Δ21–30 and Δ31–40 deletions limited HSPB6 subunit exchange, native MS analysis of 15N-labeled sample mixed in a 1:1 ratio with non-labeled sample was performed. The MS spectra showed that in both cases subunit turnover was not limited, as a stochastic 1:2:1 ratio of non-labeled, monolabeled, and dilabeled dimers was observed (supplemental Fig. 2).
In addition, native MS experiments with the preincubated 1:1 mixtures of HSPB1 and the various HSPB6 truncations were performed (Fig. 3). Just like the WT protein, all HSPB6 truncations except for Δ21–30 and Δ31–40 revealed almost exclusively the presence of heterodimers with HSPB1. The lack of detection of larger hetero-oligomeric species was previously observed and likely due to both the low protein concentrations used and less efficient detection of the resulting high m/z ions (35). Experiments involving the Δ11–20, Δ41–50, and Δ51–60 constructs additionally showed a small fraction of HSPB6 homodimers and dissociated monomers, which could be due to a slight molar excess of the HSPB6 construct due to inaccuracies in concentration determination. Crucially for the Δ21–30 and Δ31–40 HSPB6 constructs, no heterodimer was observed (HSPB1 is not visible in these spectra, as it occurs as larger oligomers, mainly tetramer, under these conditions). Thus both the cysteine cross-linking and the native MS experiments together suggest that the region found between residues 21 and 40 of HSPB6 is necessary for heterocomplex formation with HSPB1.
Figure 3.
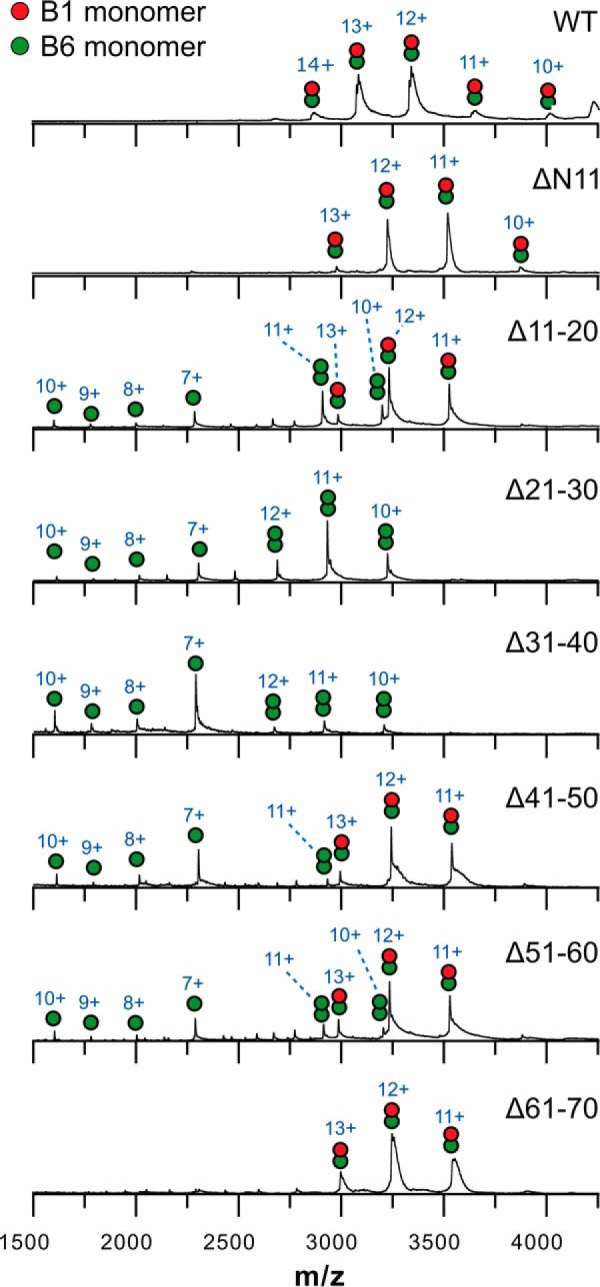
Native mass spectrometry analysis of mixtures of HSPB1 and the HSPB6 deletions. 20 μm (monomer concentration) of each preincubated mixture in 200 mm ammonium acetate, pH 6.9, containing 2.5 mm DTT was analyzed on a Synapt G2 HDMS (Waters). Charge states are indicated above each peak.
Specific contributions of several regions within the HSPB6 NTD in heterocomplex assembly
To investigate the regions that influence hetero-oligomerization in more detail, a series of HSPB6 constructs with shorter deletions were cloned and purified. First of all, we evaluated which amino acids in the region encompassing residues 51–60 are responsible for the formation of the larger hetero-oligomeric species (which are diminished upon deletion of this sequence, Fig. 1) by creating two 5-aa deletion constructs. Analytical SEC analysis of HSPB1 preincubated with the HSPB6 Δ56–60 construct showed a profile exactly like that of the mixture of the WT sHSPs, whereas deletion of residues 51–55 biased the hetero-oligomer profile to the smaller species (Fig. 4A). Residues within this latter region are found to be somewhat conserved in other HSPB6 homologues known to interact with this sHSP, the core of which is a tripeptide sequence where the terminal residues are typically proline and tyrosine (Fig. 4B) (26).
Figure 4.
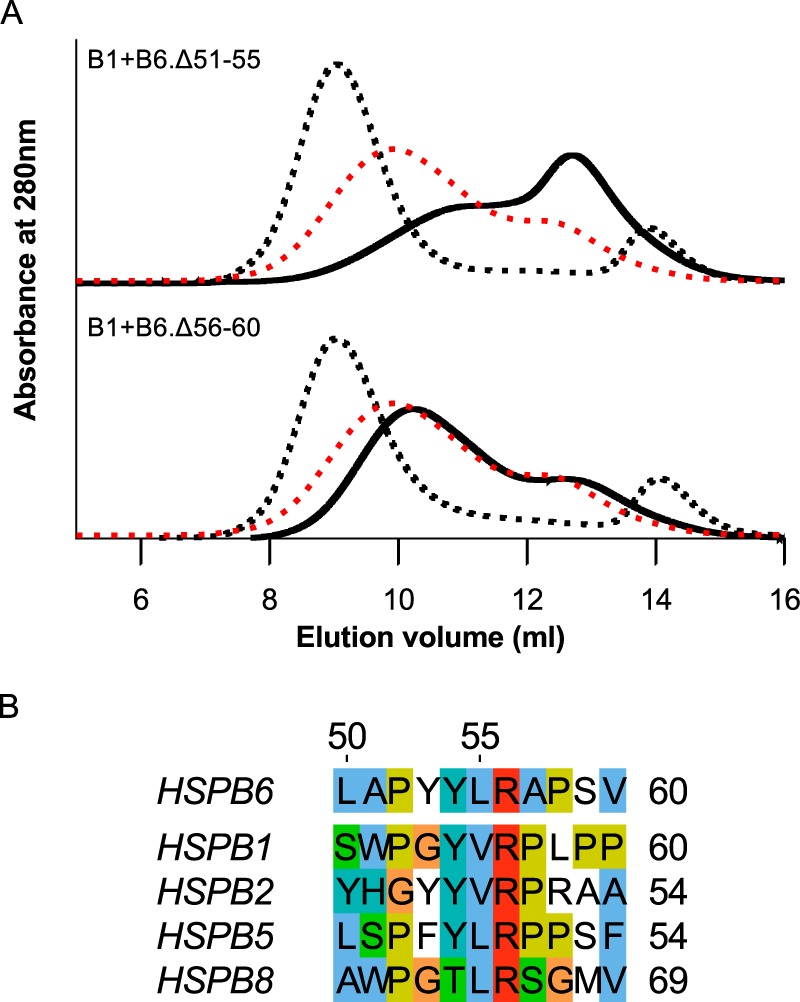
Iterative mapping of the region containing residues 51–60. A, analytical SEC profile of an equimolar mixture of HSPB1 and the HSPB6 Δ51–55 and Δ56–50 truncations following overnight incubation at 37 °C. The SEC profile of the control sample (mixture stored at 4 °C) is shown as a black dashed line, and the hetero-oligomer (incubated at 37 °C) is shown as a continuous black line. For each chromatogram, the profile of the hetero-oligomer for WT HSPB1 and HSPB6 is shown as a red dashed line. B, alignment of the 50–60 region of HSPB6 with equivalent sequences from human homologues reported to interact with this sHSP. Residues are highlighted using the Clustal color scheme. The numbering above the alignment corresponds to the residue position in human HSPB6. UniProt accession numbers are as follows: HSPB6, O14558; HSPB1, P04792; HSPB2, Q16082; HSPB5, P02511; HSPB8; Q9UJY1.
Next, we addressed the contribution of the HSPB6 region encompassing residues 21–40. The results from SEC and disulfide cross-linking on the four 5-aa HSPB6 deletions (Fig. 5) provided more detail on the involvement of this region in hetero-oligomer formation. The Δ26–30, Δ31–35, and Δ36–40 constructs demonstrated an SEC profile (Fig. 5A) similar to that obtained for the Δ21–30 and Δ31–40 deletions (Fig. 1). In particular, all featured the presence of a more-or-less resolved peak containing only HSPB6 homodimers. At the same time, the Δ21–25 construct behaved like the WT protein (Fig. 5A). As before, the C46S/E116C double mutation was introduced into each truncation to examine heterodimer formation by disulfide cross-linking (Fig. 5B). As expected, the Δ21–25* construct showed wild-type behavior, revealing almost exclusively cross-linked heterodimers with HSPB1 on a non-reducing SDS-PAGE. In contrast, the remaining three 5-aa deletion constructs revealed the presence of both homo- and heterodimers, with some specific differences between them. In the case of Δ26–30* and Δ31–35*, densitometry of the various disulfide-linked species showed ∼20% heterodimer formation. This suggests that these two 5-aa deletions are severely limited in their ability to form hetero-oligomers with HSPB1, similar to the corresponding 10-aa deletions (Figs. 2B and 5B). At the same time, these two smaller deletion constructs fully oxidize on their own (Fig. 5C). For Δ36–40*, the bands corresponding to HSPB1 homodimers, heterodimers, and HSPB6 homodimers had a molar ratio of 1:2:1, suggesting a stochastic exchange of monomers. For each of the 5-aa deletions, the ratios of the two homodimers and the heterodimer seen on a non-reducing SDS-PAGE could be further confirmed by analyzing the cross-linked samples using MS under denaturing conditions (Fig. 5E).
Figure 5.

Iterative mapping of residues 21–40 containing the conserved region. A, analytical SEC profiles of an equimolar mixture of HSPB1 with either HSPB6 Δ21–25, Δ26–30, Δ31–35, or Δ36–40 following overnight incubation at 37 °C. The control sample (kept at 4 °C) is shown with black dashed line, and the hetero-oligomer (incubated at 37 °C) is shown as a black line. For each chromatogram, the hetero-oligomer for WT HSPB1 and HSPB6 is shown as a red dashed line. B, non-reducing SDS-PAGE analysis of the disulfide cross-linked complexes between HSPB1 and the four HSPB6* truncations. C, control experiment showing the non-reducing SDS-PAGE analysis of all oxidized constructs on their own. D, sequence alignment and the corresponding consensus secondary structure (CSS) predictions of the 21–40 region of HSPB6 and known interacting homologues. Aligned residues are shaded using the Clustal color scheme. The numbering above the alignment corresponds to the residue position in human HSPB6. The database identity of each sequence is presented in the legend to Fig. 4. The consensus secondary structure for each sequence was determined using the GeneSilico meta-server (54). E, MaxEnt 1 deconvoluted mass spectra of oxidized samples.
As the disulfide cross-link is chemically labile and can exchange with non-oxidized species, we confirmed the observed differences between the four constructs using the chemical cross-linker bismaleimidoethane (BMOE) (supplemental Fig. 3). One of the preferred configurations of this compound results in the maleimide groups being at a suitable distance to cross-link the cysteine residues at the dimer interface. Incubation of the preheated mixtures with BMOE for only 15 min at 4 °C resulted in a pattern that was equivalent to that seen by disulfide-mediated cross-linking (Fig. 5B and supplemental Fig. 3C).
The differences between the Δ26–30/Δ31–35 constructs and the Δ36–40 construct point to a possible dual role played by the central part of the HSPB6 NTD. First, residues 26–35, which contain the highly conserved RLFDQXFG motif (Fig. 5D), are necessary for facilitating subunit exchange. Second, the residues 36–40 appear to define the preferential heterodimeric interaction between both sHSPs.
Asymmetric roles of equivalent NTD regions in HSPB1 and HSPB6
Because residues 27–34 within the NTD of HSPB6 are highly conserved in HSPB1 (Fig. 5D), we wondered whether homologous deletions in HSPB1 would have a similar effect on the hetero-oligomer formation. To this end, we created the HSPB1.Δ26–30 and HSPB1.Δ31–35 constructs, with each being an equivalent of the corresponding 5-aa deletant of HSPB6 (the residue numbering is consistent between the two proteins in this region). Both HSPB1 truncations readily formed hetero-oligomeric complexes with WT HSPB6 as observed by SEC (Fig. 6, A and B), and disulfide cross-linking experiments revealed that such complexes were predominantly built from heterodimers (Fig. 6C). These results are in strong contrast with the observations for the equivalent deletions in HSPB6, which had a negative effect on subunit exchange between the two orthologues (Fig. 5).
Figure 6.
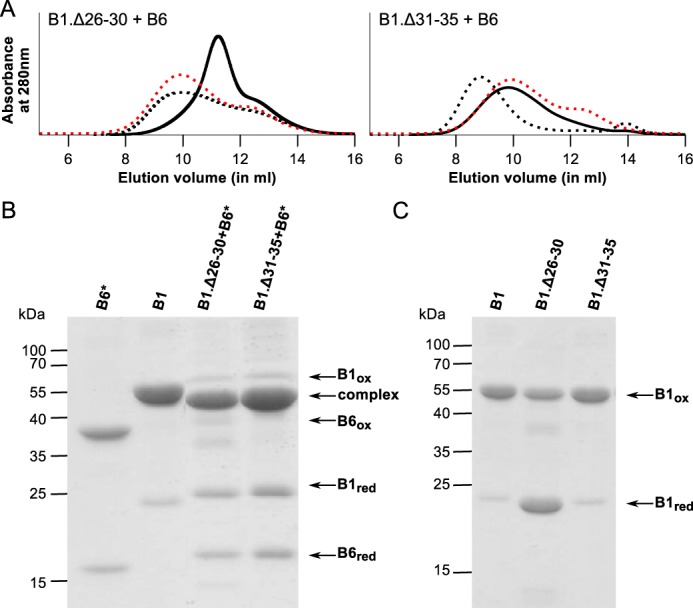
Deletion of the conserved region in HSPB1. A, analytical SEC profiles of an equimolar mixture of HSPB1 Δ26–30 or Δ31–35 deletion constructs and WT HSPB6 following overnight incubation at 37 °C. The control sample (sample kept at 4 °C) is shown with black dashed lines, and the hetero-oligomer (incubated at 37 °C) is shown as a black line. For each chromatogram the hetero-oligomer for WT HSPB1 and HSPB6 is shown as a red dashed line. B, non-reducing SDS-PAGE analysis of the complexes between the HSPB1 deletions and HSPB6*. C, control experiment showing the non-reducing SDS-PAGE analysis of HSPB1 Δ26–30 or Δ31–35 following extensive dialysis to remove DTT.
Curiously, the HSPB1.Δ26–30 construct readily formed hetero-oligomers with HSPB6 at 4 °C, yielding an SEC profile that was only seen for the WT sHSP mixture when heated to 37 °C (Fig. 6A). This observation can possibly be attributed to an increase in HSPB1 subunit turnover, thereby allowing interaction with HSPB6 even at low temperatures. It should be noted that HSPB1.Δ26–30 alone forms somewhat smaller oligomers than the WT HSPB1, evident from a shift of the SEC elution peak during purification (data not shown). In addition, HSPB1.Δ26–30 does not cross-link efficiently with itself (Fig. 6C), a result that suggests that residues 26–30 may be important for HSPB1 oligomerization or might somehow affect the register of the ACD dimer interface (16). When incubated at 37 °C, the hetero-oligomers formed by HSPB1.Δ26–30 and HSPB6 showed a major peak at 300 kDa, a value that is an intermediate of the two peaks usually observed for the WT HSPB1 and HSPB6 hetero-oligomers (Fig. 6A). In comparison, hetero-oligomerization of the HSPB1.Δ31–35 construct and HSPB6 occurred only upon incubation at 37 °C and led to the formation of hetero-oligomeric complexes, which revealed an asymmetric SEC profile with a single maximum at 500 kDa, corresponding to the larger hetero-oligomer species seen when mixing both WT proteins (Fig. 6A).
The results from deletion of the 26–35 region in HSPB1 suggest there is an asymmetry between HSPB1 and HSPB6 in the role of this highly conserved sequence for hetero-oligomer formation. To assess whether this depends on the small sequence differences in this region between the two sHSPs, or is driven in part by their context within the respective full-length proteins, two domain swaps of this region were created (Fig. 7A). These 10-residue swaps change the sequence GRLFDQRFGE of HSPB6 to SRLFDQAFGL of HSPB1 and vice versa, which corresponds to triple mutations indicated in bold. Swapping of these residues led to wild-type hetero-oligomerization behavior for HSPB6 containing the HSPB1 sequence (HSPB6.10swap) when mixed with WT HSPB1. The opposite swap in HSPB1 (HSPB1.10swap) also yielded hetero-oligomers, but with a preference for forming predominantly larger assemblies (Fig. 7B). In both cases, the complexes readily oxidized when mixing with the appropriate HSPB6* mutant yielding mainly heterodimers when analyzed by non-reducing SDS-PAGE showing that the mutations did not hamper heterodimerization (Fig. 7C).
Figure 7.

Domain swapping of conserved region in HSPB1 and HSPB6. A, schematic of the constructs used. The figures were created using DOG (55). B, analytical SEC profiles of an equimolar mixture of the various HSPB1 and HSPB6 swaps following overnight incubation at 37 °C. The control sample (stored at 4 °C) is shown as a black dashed line, and the hetero-oligomer (incubated at 37 °C) is shown as a continuous black line. For each chromatogram, the hetero-oligomer for WT HSPB1 and HSPB6 is shown as a red dashed line. C, non-reducing SDS-PAGE analysis of the complexes between the HSPB1 and HSPB6* swaps. D, control experiment showing the non-reducing SDS-PAGE analysis of different constructs following extensive dialysis to remove DTT.
An equimolar mixture of the two 10swap mutants resulted in an SEC profile similar to that seen with the HSPB1.10swap and WT HSPB6 (Fig. 7B). The asymmetric elution, with a single maximum at ∼500 kDa, was also observed with the HSPB1.Δ31–35 construct (Fig. 6B) and points to a role of either Ala-32 or Leu-35 of HSPB1 in the concentration-dependent formation of the second smaller hetero-oligomeric species typically observed with the mixtures of the WT proteins (Fig. 7B). The fact that the mixture of the two 10swap mutants did not result in a wild-type SEC profile, despite the HSPB6 chimera containing the HSPB1(26–35) region, suggests that the mode of action of Ala-32 or Leu-35 occurs only when they found within the full-length HSPB1 protein.
To investigate the role of residues 36–40 in driving preferential heterodimerization, we also swapped this region between the two sHSPs (Fig. 7A). These swaps correspond to four mutations in each construct, exchanging residues GLLEA of HSPB6 to PRLPE of HSPB1 (HSPB6.5swap) and vice versa (HSPB1.5swap). When mixed with HSPB1 the HSPB6.5swap chimera led to only stochastic subunit exchange as observed by SEC and cross-linking studies using the HSPB6.5swap* mutant (Fig. 7, B and C). This behavior was equivalent to that seen with the HSPB6.Δ36–40 deletion construct and points to the role of one or more of the mutated residues in heterodimer formation. The converse HSPB1.5swap with HSPB6 resulted in heterodimer formation, albeit yielding hetero-oligomers of a much higher molecular mass than seen with the WT proteins (Fig. 7, B and C). The fact that the HSPB1.5swap, which is effectively HSPB6-like in the 36–40 region, did not demonstrate stochastic exchange with HSPB6, as has been reported for the latter sHSP alone (35), suggests that the preferential heterodimerization between these two sHSPs is not driven by these positionally equivalent regions interacting with each other in the hetero-oligomer. Combined, these results support a model where residues 36–40 in HSPB6 likely interact with different residues in HSPB1 to drive the formation of the heterodimer. The asymmetric behavior of this region in both sHSPs was further evaluated by mixing the two 5swap constructs together. The presence of remnant HSPB6 in the SEC profile suggests that residues 36–40 only act within the context of the surrounding NTD residues in HSPB6 (Fig. 7B).
Mapping of specific residues involved in hetero-oligomerization
The slight bias in overall hetero-oligomer size for the HSPB1.10swap was further investigated by point mutations. Because most of the sequence is similar, we decided to mutate the differing residues in the conserved stretch. HSPB6 has an arginine at position 32 where HSPB1 has an alanine (Fig. 5D). A point mutation was created in both sHSPs to convert it to the sequence of the other and these confirmed the results seen using the larger swaps: HSPB1.A32R (B6-like) forms complexes with HSPB6 that are larger than with the WT HSPB1, whereas HSPB6.R32A (B1-like) demonstrates an SEC profile that is wild type (Fig. 8A). When mixing both mutations, the same profile was seen as for the HSPB1.A32R-HSPB6 complex, showing that this arginine is acting within the context of HSPB1 and is responsible for the larger size oligomers, whereas this residue in HSPB6 does not seem to be involved in interactions (results not shown).
Figure 8.

Point mutation analysis of the conserved region in HSPB1 and HSPB6. A, analytical SEC profile of an equimolar mixture of various HSPB1 and HSPB6 point mutations following overnight incubation at 37 °C. The control sample (stored at 4 °C) is shown as a black dashed line, and the hetero-oligomer (incubated at 37 °C) is shown as continuous black line. For each chromatogram, the hetero-oligomer for WT HSPB1 and HSPB6 is shown as a red dashed line. B, non-reducing SDS-PAGE analysis of the complex between HSPB1 and HSPB6* point mutants. C, control experiment showing the non-reducing SDS-PAGE analysis of different constructs following extensive dialysis to remove DTT.
As neither of the 10swap constructs mimicked the behavior seen for the Δ26–30 and Δ31–35 constructs of HSPB6, point mutations of the key conserved residues were also created. It has been shown previously that phenylalanines in the NTD play an important role in intersubunit contacts (37). This region in HSPB1 and HSPB6 contains two such residues. We therefore generated mutants of one (F33A) or both (F29A and F33A, termed FFAA) for each sHSP. Different permutations of these mutants, mixed with either the WT or mutant partner, were examined. Mixing HSPB6.F33A with WT HSPB1 led to a chromatogram similar to that obtained for HSPB6.Δ31–35 (Fig. 8A), suggesting that this phenylalanine residue in HSPB6 is important in hetero-oligomer formation, a result confirmed by disulfide cross-linking using the HSPB6.F33A* mutant (Fig. 8B). However, the opposite experiment, mixing WT HSPB6 with HSPB1.F33A, did not hamper the preferred heterodimerization of these two sHSPs as evaluated by the oxidation experiment (Fig. 8B). Unexpectedly, however, the HSPB1.F33A mutation led to exchange of subunits at 4 °C, as evidenced by a reduction of the HSPB6 peak, and the formation of smaller hetero-oligomeric species (∼300 kDa) following heating. This behavior was similar to that observed with the HSPB1.Δ26–30 deletion mutant (Figs. 8A and 6A). As seen before, this suggests an asymmetric role for this highly conserved residue in defining the interaction between these two sHSPs.
When mixing the HSPB1.F33A and HSPB6.F33A, together a unique chromatogram was observed (Fig. 8A). The broad profile at 4 °C showed two peaks overlapping, suggesting some mixing of the two mutants at low temperature. After incubation at 37 °C, a shift in the ratio of the peak maximum could be noticed, but the overall shape of the elution profile remained similar to the unheated sample. Disulfide cross-linking showed a minor fraction of heterodimers similar to that seen for the mixture of WT HSPB1 and HSPB6.F33A* (Fig. 8B). Combined, the data suggest that these mutants can associate at lower temperatures, but subunit exchange is severely hampered.
The results for the double mutations (FFAA) looked exactly like those for the single phenylalanine substitutions, alluding to a more important role for Phe-33 than Phe-29 in both proteins (Fig. 8A). It should be noted that the mutations in HSPB1 yielded smaller homo-oligomers than the WT protein as assessed by SEC during purification (results not shown), and that the FFAA construct did not cross-link to the same extent as the WT protein on its own (Fig. 8C). This suggests an important role for the phenylalanine residues in HSPB1 homo-oligomerization. These phenylalanines thus seem essential to allow the interaction between HSPB1 and HSPB6 and are involved in important intersubunit contacts in the oligomeric assemblies.
Discussion
Hetero-oligomerization of sHSPs has long been recognized in different organisms, although the exact function and benefit of such assemblies are still unknown. As many members of the family tend to form such mixed oligomers, information about their structure and the determinants of the assembly process should help to understand the chaperone activity of sHSPs and their interaction with different partner proteins. We have focused our attention on HSPB1 and HSPB6, two human sHSPs that are highly expressed in muscle tissue (33).
Previously, it has been shown that HSPB1 and HSPB6 preferentially form hetero-oligomers over interactions with other orthologues (21). This is most likely the result of their propensity to yield species composed solely of heterodimers when mixed together (35), suggesting a biased association at the level considered to be the basic building block of the larger oligomeric assemblies. Surprisingly, deletion constructs of the two proteins corresponding to the ACD, the only structured region that form the core dimer interface (15–17), were found to hetero-oligomerize in a stochastic fashion (35). It is the NTD, a region shown to be largely unstructured in HSPB6 and predicted to have similar properties in HSPB1 (18), that is essential for this preferred association. By employing a comprehensive set of deletion mutants of HSPB6, we have identified three regions of importance within its NTD that affect the hetero-oligomer assembly with HSPB1 (Table 2).
Table 2.
Summary of the hetero-oligomerization properties of HSPB6 deletions and point mutations with HSPB1
| 10 amino acid deletions |
5 amino acid deletions |
Point mutations |
||||||
|---|---|---|---|---|---|---|---|---|
| Construct | SECa | S–Sb | Construct | SECa | S–Sb | Constructc | SECa | S–Sb |
| ΔN11 | WT | 1 | Δ21–25 | WT | 1 | 10swap | WT | 1 |
| Δ11–20 | WT | 1 | Δ26–30 | Remnant HSPB6 | 1:0.5:1 | 5swap | Remnant HSPB6 | 1:0.5:1 |
| Δ21–30 | Remnant HSPB6 | 1:0.5:1 | Δ31–35 | Remnant HSPB6 | 1:0.5:1 | R32A | WT | 1 |
| Δ31–40 | Remnant HSPB6 | 1:0.5:1 | Δ36–40 | Remnant HSPB6 | 1:2:1 | F33A | Remnant HSPB6 | 1:0.5:1 |
| Δ41–50 | WT | 1 | Δ51–55 | Small | 1 | FFAA | Remnant HSPB6 | 1:0.5:1 |
| Δ51–60 | Small | 1 | Δ56–60 | WT | 1 | |||
| Δ61–70 | WT | 1 | ||||||
a SEC profile of each HSPB6 construct when mixed in equimolar amounts with HSPB1 and heated are shown. WT, closely resembling the mixture of WT HSPB6 and HSPB1. Remnant HSPB6, chromatograms contain an additional late eluting peak corresponding to the HSPB6 dimer position. Small, chromatograms show a profile biased to an approximately 140-kDa species.
b Ratio of disulfide cross-linked HSPB1 homodimer, HSPB1 and HSPB6 construct heterodimer, and HSPB6 construct homodimer as evaluated by non-reducing SDS-PAGE. A single value corresponds to heterodimers only.
c 10swap corresponds to the HSPB6 G26S/R32A/E35L mutant. The 5swap corresponds to the HSPB6 G36P/RL37R/E39P/A40E mutant. FFAA corresponds to the HSPB6 F29A/F33A mutant.
First, we have discovered that a conserved motif P(G/F/Y)Y, which is located at position 52–54 in the second half of the NTD of HSPB6 and found at an equivalent position in a number of other orthologues known to interact with this sHSP (Fig. 4B), seems to regulate the size of the resultant hetero-oligomer. Deletion of these residues in HSPB6 leads to the formation of smaller hetero-oligomeric species with HSPB1, albeit composed of the canonical heterodimers (Fig. 9). Predictions suggest that this region in both proteins, like the majority of the NTD, contain no secondary structural elements (supplemental Fig. 4). This is in good agreement with a recent X-ray crystallographic structure of the full-length HSPB6 bound to a 14-3-3 adapter protein that showed that this part of the NTD is highly disordered (38). However, studies of HSPB5 oligomers, using solid-state NMR, report that the homologous region in this sHSP shows chemical shifts resembling a β-strand (39). Therefore, it is possible that in the higher-order assemblies formed between HSPB1 and HSPB6 some structure is induced that is necessary for association. Indeed, the importance of this region in HSPB5 has been highlighted by pin array experiments, where peptides encompassing this sequence were identified to interact with HSPB4 (40), suggesting a possible conserved role in hetero-association.
Figure 9.

Schematic summarizing the regions of HSPB6 involved in hetero-oligomer formation. 1) CTD of HSPB1 recruits HSPB6 via patching of the hydrophobic groove formed between the β4- and β8-strands of the ACD (represented by semi-circle) of the latter sHSP. This interaction is blocked by mutation of Ser-134 in HSPB6 and by disulfide cross-linking (S–S) of both proteins (21, 43). 2) Residue Phe-33 of the NTD of HSPB6 destabilizes the NTD interactions in the HSPB1 homo-oligomer permitting subunit exchange and full incorporation of HSPB6. 3) Within the resultant heterocomplex, the individual subunits can freely exchange. 4) Preferential heterodimerization within the mixed oligomer is driven by residues 36–40 of HSPB6. 5) The hetero-oligomer is found in an equilibrium of two species, the populations of which are regulated by residues 51–55 in HSPB6.
Second, we have found that the highly conserved RLFDQXFG sequence (36) is a prerequisite of hetero-oligomerization. Intriguingly, this motif is always present in sHSPs reported to form hetero-oligomers with HSPB6 (Fig. 5D), but it is absent in a number of homologues, including HSPB3 (supplemental Fig. 4C), an sHSP that has been shown not to interact with HSPB6 (20). This predicted unstructured sequence (Fig. 5D) appears to be necessary for the exchange of HSPB6 subunits with HSPB1. This region was previously identified as a negative regulator of activity in HSPB6 and was also found to control both size and activity in αB-crystallin (36, 41). Specifically our results point to the highly conserved Phe-29 and Phe-33 as essential for the HSPB1/HSPB6 association. Despite their conservation between the two sHSPs, there appears to be an asymmetry in their importance. Just like the deletion of residues 31–35, mutation of Phe-33 in HSPB6 effectively blocked hetero-association, whereas mutation of the equivalent residue in HSPB1 led to the formation of smaller oligomers but still composed of heterodimers. The involvement of phenylalanines of the NTD in controlling sHSP association and size has also been observed in the fission yeast SpHSP16.0 where the mutation of Phe-6 or Phe-7 led to an impairment of oligomer formation (37). The residue bias of phenylalanines in the NTD in genome-wide studies suggests this is likely a common phenomenon (42).
Third, we have identified a region in the NTD of HSPB6, with no functional equivalent in HSPB1, which is responsible for the preferential heterodimerization of these two sHSPs. Specifically, deletion or mutation of residues 36–40 of HSPB6 resulted in a protein that could form hetero-oligomers with HSPB1, but contained HSPB1 and HSPB6 homodimers alongside the heterodimers. Importantly, the observed molar ratio of the three species suggest a stochastic exchange of subunits at the monomer level. The same behavior was seen for the hetero-oligomerization of HSPB1 and HSPB6 truncations each missing the whole NTD (35). Because swapping of this sequence into HSPB1 had no observable effect on subunit exchange, which is that the HSPB1.5swap chimera did not show a preferred association with itself over HSPB6, we conclude that these residues function within the context of the heterodimer. Thus, the 36–40 region is somehow responsible for stabilizing the interface of the ACDs occurring within a heterodimer. The aforementioned structure of HSPB6 in complex with a 14-3-3 adapter protein has shown that regions of the NTD can thread through the shared groove formed between the β3-strands of the ACD dimer (38). Patching of this same groove in the heterodimer, by one of more residues in the 36–40 region of HSPB6, could possibly stabilize this new interface and explain the asymmetric behavior of this interaction.
A recent study of hetero-oligomer formation between HSPB5 and HSPB6 has proposed a model in which the C-terminal IX(I/V) motif of HSPB5 is required for the initial capture of the HSPB6 dimer, whereupon a new heterodimer interface is formed and stabilized by the NTD of HSPB6 (43). Assuming a similar model of association between HSPB1 and HSPB6, the results presented here can extend our understanding of this association further (Fig. 9). Initially, HSPB6 is recruited to the HSPB1 homo-oligomer via the interaction of its ACD with the C-terminal IX(I/V) motif of HSPB1. Although an equivalent C-terminal localized motif is missing in HSPB6, its ACD has been shown to readily bind sequences containing this tripeptide sequence (18, 38, 43, 44). Following this capture step, incorporation of HSPB6 into the HSPB1 oligomer is dependent, first, on the formation of HSPB1/HSPB6 heterodimers. Indeed, Mymrikov et al. (21) have shown that hetero-oligomerization is blocked by disulfide cross-linking of both sHSPs prior to mixing. Therefore, stable incorporation of HSPB6 is dependent on localized dissociation of both the HSPB1 and HSPB6 homodimers and subunit exchange, in agreement with the earlier model (43). Second our data show the conserved NTD motif, and in particular residue Phe-33, plays an important role in this initial subunit exchange. In the case of HSPB1, mutation of this residue to alanine enhances the recruitment of HSPB6, whereas the equivalent mutation in HSPB6 severely hampers this process. This suggests that the conserved motif of HSPB6 must displace an interaction made by the same sequence in the HSPB1 homo-oligomers to permit homodimer dissociation and subunit exchange (Fig. 9). Finally, our experiments show that, uniquely for the HSPB1 and HSPB6 mixed complex, residues 36–40 of the latter sHSP enhance preferential hetero-association at the ACD interface in the hetero-oligomers. Whether this HSPB6-mediated biased association already occurs during initial subunit incorporation, or appears as a result of the free exchange of subunits in the hetero-oligomeric species as presented in Fig. 9, cannot be ascertained from the equilibrium experiments performed here.
The involvement of the NTD in defining the strength and specificity of the newly formed ACD dimer interface is quite remarkable. Indeed, we have found this intrinsically disordered region to be highly implicated in dictating the properties of the sole structured region in these proteins. Similar involvement of the NTD and CTD have been published for HSPB5, where binding of the CTD destabilizes the dimer interface (45), and phosphorylation of N-terminal serine residues leads to loss of dimeric substructure in the homo-oligomers (46). In summary, modifications such as phosphorylation within the NTD may have a profound effect on the structure of sHSPs and can even lead to a destabilization of the ACD dimer interface, suggesting that indeed this region must interact with the ACD core and function either as a stabilizer or destabilizer, dependent on the modifications present.
Many reviews have acknowledged the potential of sHSPs both as drug targets and for biotechnological applications (47–51). To create inhibitors or enhancers of sHSP activity, a detailed understanding of their function and the sequences involved is necessary. Similarly, to use sHSPs as nanomaterials for activities such as targeted drug delivery or imaging purposes, it is crucial that we understand the sequence properties that define the size and shape of these oligomers. Studies focusing on sHSP hetero-oligomerization could bring us one step closer to eventually designing sHSPs with tailored properties.
This work is the first to fully explore the sequence properties in defining hetero-oligomerization of two human sHSPs. The results described here show a complicated yet important interplay between the NTD, which features a high degree of intrinsic disorder, and the structured ACD, in regulating size and stability of the complexes, proving that the terminal regions in sHSPs play an important role in defining their assembly properties. As these same regions also have a role in chaperone activity (35, 36), a correlation between the ability to hetero-oligomerize and to protect substrates from aggregating should be logically expected. A systematic investigation of this interplay, which is clearly relevant in vivo, is necessary.
Experimental procedures
Mutagenesis and cloning
The previously described small ubiquitin modifier (SUMO) fusions of HSPB1 and HSPB6 (16) were used as a template for the generation of additional deletion constructs and point mutations. Point mutations were created using site-directed mutagenesis. DpnI-treated PCR products were transformed into Escherichia coli NEB5α (New England Biolabs), and positive clones were verified by sequencing. A double mutant C46S/E116C of HSPB6, in which the native cysteine 46 is mutated to serine and glutamate 116 at the dyad axis of the ACD dimer is mutated to cysteine, was created using two rounds of mutagenic PCR. This mutant was also used as a template to create additional N-terminal deletions using a PCR-based overlap mutagenesis method described previously (36). A similar approach was employed to generate the HSPB1 and HSPB6 sequence swaps. All constructs were designed such that, upon cleavage of the linearly fused SUMO chimera with recombinant SUMO-hydrolase, no additional non-native residues were present on the target protein (52).
Expression and purification
All constructs were transformed into the E. coli Rosetta 2 (DE3) pLysS strain (Novagen), and clones were cultured in ZYP-5052 auto-inducing medium (53) using described conditions (16). Cell were harvested by centrifugation at 8000 × g, resuspended in buffer (50 mm sodium phosphate, 250 mm sodium chloride, and 12.5 mm imidazole, pH 7.5), and stored at −80 °C. For the expression of 15N-labeled proteins, transformed clones were cultured in 2 ml of LB medium for 7 h. This culture was then transferred to 50 ml of P0.5G-medium (53) and grown overnight at 25 °C. 10 ml of this culture was spun down at 3000 relative centrifugal force, and the pellet was transferred to 200 ml of auto-inducing minimal medium containing [15N]ammonium chloride (53). Cells were grown and harvested using the same conditions as above.
Cells were thawed and diluted in the same buffer complemented with 1 unit/ml of Cryonase Cold Active Nuclease (Clontech) and 10 mm MgCl2. Cells were lysed by sonication in three cycles with 20 min between each cycle. Further purification by immobilized metal affinity chromatography, ion exchange, and size exclusion was performed as described previously (36, 52). 15N-Labeled proteins were purified using the same protocol except the final SEC was performed using a Superdex 200 10/300 GL column pre-equilibrated in 200 mm ammonium acetate, pH 6.9, containing 2.5 mm DTT.
Analytical size-exclusion chromatography
Prior to analysis, 220 μm (corresponding to 5 mg/ml for HSPB1) of each protein was mixed in 20 mm HEPES, pH 7.4, 150 mm NaCl, and 10 mm DTT to produce hetero-oligomeric complexes. The mixture was incubated overnight at 37 °C to allow complete subunit exchange. 100 μl of each protein or complex was loaded onto a Superdex 200 10/300 GL column (GE Healthcare), pre-equilibrated at 4 °C in 20 mm HEPES, pH 7.2, 150 mm NaCl, and 2.5 mm DTT using a flow-rate of 0.5 ml/min. The column was calibrated using standards from the Molecular Weight Calibration kit (GE Healthcare) including blue dextran, ferritin, aldolase, conalbumin, ovalbumin, carbonic anhydrase, ribonuclease A, and aprotinin. The standards were diluted in the same buffer and run under the same conditions.
Disulfide cross-linking
For oxidation cross-linking experiments, 50 μm of each protein was mixed in 50 mm sodium phosphate, pH 7.5, 100 mm NaCl, and 5 mm DTT, incubated overnight at 37 °C, and then subjected to dialysis for 48 h at 20 °C with the same buffer without DTT. 10 μl from this mixture was analyzed on a non-reducing SDS-PAGE containing 15% acrylamide. Reduced controls were obtained by diluting the samples with buffer with DTT before loading. Densitometry using ImageJ of the Coomassie-stained gels was used to quantify the molar amounts of protein present in each band, whereby the reference bands containing the known molar amounts of fully oxidized HSPB6*, HSPB1, or the HSPB6*/HSPB1 heterodimers were used for calibration.
Chemical cross-linking
The purified proteins were dialyzed against 50 mm sodium phosphate, 100 mm NaCl, 1 mm EDTA, and 2 mm tris(2-carboxyethyl)phosphine (TCEP), pH 7.5 at 4 °C. The buffer was exchanged three times to ensure the complete removal of DTT. A solution containing 50 μm of each construct (i.e. 100 μm total sHSP for the 1:1 mixtures of HSPB1 and each HSPB6* mutant) was prepared and heated at 42 °C for 1.5 h. TCEP was then removed from each reaction using a Zeba spin desalting column with a 40-kDa molecular mass cutoff (Thermo Fisher Scientific) pre-equilibrated with 50 mm sodium phosphate, 100 mm NaCl, 1 mm EDTA, pH 7.5. The samples, containing no reducing agent, were split in half and placed at 4 °C. To one tube a 1.1 m excess of BMOE, solubilized in DMSO, was added. Following a 15-min incubation period, 10 μl of each reaction was taken, and cross-linking was stopped by the addition of SDS-PAGE loading buffer containing 100 mm β-mercaptoethanol. The second tube, containing no cross-linker, was incubated for 1 h at 4 °C. At this point a 10-μl sample was taken and diluted in SDS-PAGE sample buffer containing no reducing agent. All samples were analyzed by SDS-PAGE using a 12–15% acrylamide gradient.
Mass spectrometry
All MS measurements were performed on a quadrupole/ion mobility/time-of-flight instrument with ion mobility capabilities (Synapt G2 HDMS, Waters, Wilmslow, UK), operated in positive ion mode. Data acquisition and processing were performed using MassLynx (version 4.1) and external calibration up to 5000 m/z was performed with CsI solution.
For native MS analyses of the complexes, 200 μm (monomer concentration) of each protein was mixed together in 200 mm ammonium acetate, pH 6.9, 2.5 mm DTT and incubated overnight at 37 °C. The samples were further dialyzed against the same buffer in three exchanges, over a 24-h period, to ensure complete removal of non-volatile salt. Approximately 5 μl of solution containing 20 μm protein (monomer concentration) diluted with the 200 mm ammonium acetate, pH 6.9, 2.5 mm DTT buffer was transferred to gold-coated glass capillaries prepared in-house and infused into the mass spectrometer using the nanoflow version of the Z-spray ion source. A capillary voltage of 1.0–1.3 kV and minimal (<0.2 bar) nanoflow gas pressure were used, and the instrument was operated in Mobility/Sensitivity mode. Instrument parameters were as follows, unless stated otherwise: sample cone 80 V, extraction cone 1 V, backing pressure 3.2–4.5 mbar, source pressure 4.6e-3–5.8e-3 mbar, trap collision energy 10 V, trap DC bias 50 V, transfer collision energy 5 V. The IM cell was filled with 3.5 mbar of N2 (He cell gas flow 180 ml/min, IMS gas flow 60 ml/min), and IM wave height and velocity were 40 V and 1000 m/s, respectively.
MS analyses of cross-linked species were performed in the denatured state on the same instrument, but in this case the protein concentration was reduced to 2 μm (monomer concentration) in 50% acetonitrile and 1% formic acid. The instrument was operated in TOF/sensitivity mode for these experiments, with the following settings: capillary voltage 1.0 kV, sampling cone 40 V, extraction cone 2 V, backing pressure 2.5 mbar, source pressure 4.1e-3 mbar, trap collision energy 4 V, trap DC bias 5 V, transfer collision energy 0 V. Spectra were subsequently deconvoluted using the MaxEnt 1 module of the MassLynx software to determine relative abundances.
Small-angle X-ray scattering
The measurements were performed at Synchrotron Soleil (Saint-Aubin, France) as described previously (36). An 80-μl sample containing 5 mg/ml HSPB1 (equivalent to ∼220 μm) and the molar equivalent of HSPB6, incubated at 45 °C for 1 h to form complexes, which has been shown to be sufficient for complex formation (31), was loaded onto a Shodex KW-404F column at 0.2 ml/min pre-equilibrated with 50 mm sodium phosphate, pH 7.5, 100 mm NaCl, and 2.5 mm DTT. During each run, 100 frames were recorded prior to the start of the protein elution toward evaluating the buffer scattering. Thereafter, 250 frames were recorded while the complex eluted from the column. The exposure time per frame was 750 ms, with a dead time of 1500 ms between frames. Data processing was done as described previously (35).
Author contributions
M. H. and S. D. W. conceived and designed the experiments. M. H., F. L., E. M. M., S. B., and S. D. W. contributed various reagents, performed experiments, and analyzed the data. M. H., F. L., F. S., S. V. S., and S. D. W. were involved in data interpretation and preparation of the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank The Hercules Foundation Flanders for funding the Synapt instrument. We acknowledge support from the European Community's Seventh Framework Programme under the BioStruct-X initiative (Project Number 6131) toward the Synchrotron SAXS measurements. We greatly appreciate the excellent support from the beamline scientists on the SWING beamline (Soleil Synchrotron, France).
This work was supported by Research Foundation Flanders (FWO) Grants G093615N and WO03315N, KU Leuven Grant OT13/097 (all to S. V. S.), and FWO Grant 11L4115N for Ph.D. (to F. L.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4 and Refs. 1–4.
- sHSP
- small heat-shock protein
- ACD
- α-crystallin domain
- CTD
- C-terminal domain
- NTD
- N-terminal domain
- SAXS
- small-angle X-ray scattering
- SEC
- size-exclusion chromatography
- TCEP
- tris(2-carboxyethyl)phosphine
- aa
- amino acid
- BMOE
- bismaleimidoethane
- SUMO
- small ubiquitin modifier.
References
- 1. Kim Y. E., Hipp M. S., Bracher A., Hayer-Hartl M., and Hartl F. U. (2013) Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 82, 323–355 [DOI] [PubMed] [Google Scholar]
- 2. Haslbeck M., and Vierling E. (2015) A first line of stress defense: small heat-shock proteins and their function in protein homeostasis. J. Mol. Biol. 427, 1537–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Haslbeck M. (2002) sHsps and their role in the chaperone network. Cell. Mol. Life Sci. 59, 1649–1657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Basha E., Lee G. J., Breci L. A., Hausrath A. C., Buan N. R., Giese K. C., and Vierling E. (2004) The identity of proteins associated with a small heat-shock protein during heat stress in vivo indicates that these chaperones protect a wide range of cellular functions. J. Biol. Chem. 279, 7566–7575 [DOI] [PubMed] [Google Scholar]
- 5. Andley U. P., Malone J. P., and Townsend R. R. (2014) In vivo substrates of the lens molecular chaperones αA-crystallin and αB-crystallin. PLoS ONE 9, e95507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mymrikov E. V., Daake M., Richter B., Haslbeck M., and Buchner J. (2017) The chaperone activity and substrate spectrum of human small heat-shock proteins. J. Biol. Chem. 292, 672–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clauwaert J., Ellerton H. D., Koretz J. F., Thomson K., and Augusteyn R. C. (1989) The effect of temperature on the renaturation of α-crystallin. Curr. Eye Res. 8, 397–403 [DOI] [PubMed] [Google Scholar]
- 8. Baldwin A. J., Lioe H., Hilton G. R., Baker L. A., Rubinstein J. L., Kay L. E., and Benesch J. L. (2011) The polydispersity of αB-crystallin is rationalized by an interconverting polyhedral architecture. Structure 19, 1855–1863 [DOI] [PubMed] [Google Scholar]
- 9. Lermyte F., Williams J. P., Brown J. M., Martin E. M., and Sobott F. (2015) Extensive charge reduction and dissociation of intact protein complexes following electron transfer on a quadrupole-ion mobility-time-of-flight MS. J. Am. Soc. Mass Spectrom. 26, 1068–1076 [DOI] [PubMed] [Google Scholar]
- 10. Haslbeck M., Franzmann T., Weinfurtner D., and Buchner J. (2005) Some like it hot: the structure and function of small heat-shock proteins. Nat. Struct. Mol. Biol. 12, 842–846 [DOI] [PubMed] [Google Scholar]
- 11. Haley D. A., Horwitz J., and Stewart P. L. (1998) The small heat-shock protein, αB-crystallin, has a variable quaternary structure. J. Mol. Biol. 277, 27–35 [DOI] [PubMed] [Google Scholar]
- 12. Aquilina J. A., Benesch J. L., Bateman O. A., Slingsby C., and Robinson C. V. (2003) Polydispersity of a mammalian chaperone: mass spectrometry reveals the population of oligomers in αB-crystallin. Proc. Natl. Acad. Sci. U.S.A. 100, 10611–10616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mymrikov E. V., Seit-Nebi A. S., and Gusev N. B. (2011) Large potentials of small heat-shock proteins. Physiol. Rev. 91, 1123–1159 [DOI] [PubMed] [Google Scholar]
- 14. Basha E., O'Neill H., and Vierling E. (2012) Small heat-shock proteins and α-crystallins: dynamic proteins with flexible functions. Trends Biochem. Sci. 37, 106–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bagnéris C., Bateman O. A., Naylor C. E., Cronin N., Boelens W. C., Keep N. H., and Slingsby C. (2009) Crystal structures of α-crystallin domain dimers of αB-crystallin and Hsp20. J. Mol. Biol. 392, 1242–1252 [DOI] [PubMed] [Google Scholar]
- 16. Baranova E. V., Weeks S. D., Beelen S., Bukach O. V., Gusev N. B., and Strelkov S. V. (2011) Three-dimensional structure of α-crystallin domain dimers of two human small heat-shock proteins, HSPB1 and HSPB6. J. Mol. Biol. 411, 110–122 [DOI] [PubMed] [Google Scholar]
- 17. Hochberg G. K., Ecroyd H., Liu C., Cox D., Cascio D., Sawaya M. R., Collier M. P., Stroud J., Carver J. A., Baldwin A. J., Robinson C. V., Eisenberg D. S., Benesch J. L., and Laganowsky A. (2014) The structured core domain of αB-crystallin can prevent amyloid fibrillation and associated toxicity. Proc. Natl. Acad. Sci. U.S.A. 111, E1562–E1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weeks S. D., Baranova E. V., Heirbaut M., Beelen S., Shkumatov A. V., Gusev N. B., and Strelkov S. V. (2014) Molecular structure and dynamics of the dimeric human small heat-shock protein HSPB6. J. Struct. Biol. 185, 342–354 [DOI] [PubMed] [Google Scholar]
- 19. Poulain P., Gelly J.-C., and Flatters D. (2010) Detection and architecture of small heat-shock protein monomers. PLoS ONE 5, e9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. den Engelsman J., Boros S., Dankers P. Y., Kamps B., Vree Egberts W. T., Böde C. S., Lane L. A., Aquilina J. A., Benesch J. L., Robinson C. V., de Jong W. W., and Boelens W. C. (2009) The small heat-shock proteins HSPB2 and HSPB3 form well-defined hetero-oligomers in a unique 3 to 1 subunit ratio. J. Mol. Biol. 393, 1022–1032 [DOI] [PubMed] [Google Scholar]
- 21. Mymrikov E. V., Seit-Nebi A. S., and Gusev N. B. (2012) Hetero-oligomeric complexes of human small heat-shock proteins. Cell Stress Chaperones 17, 157–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aquilina J. A., Shrestha S., Morris A. M., and Ecroyd H. (2013) Structural and functional aspects of hetero-oligomers formed by the small heat-shock proteins αB-crystallin and HSP27. J. Biol. Chem. 288, 13602–13609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Arrigo A.-P. (2013) Human small heat-shock proteins: protein interactomes of homo- and hetero-oligomeric complexes: An update. FEBS Lett. 587, 1959–1969 [DOI] [PubMed] [Google Scholar]
- 24. Sobott F., Benesch J. L., Vierling E., and Robinson C. V. (2002) Subunit exchange of multimeric protein complexes. Real-time monitoring of subunit exchange between small heat-shock proteins by using electrospray mass spectrometry. J. Biol. Chem. 277, 38921–38929 [DOI] [PubMed] [Google Scholar]
- 25. Studer S., and Narberhaus F. (2000) Chaperone activity and homo- and hetero-oligomer formation of bacterial small heat-shock proteins. J. Biol. Chem. 275, 37212–37218 [DOI] [PubMed] [Google Scholar]
- 26. Basha E., Jones C., Wysocki V., and Vierling E. (2010) Mechanistic differences between two conserved classes of small heat-shock proteins found in the plant cytosol. J. Biol. Chem. 285, 11489–11497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Spector A., Li L. K., Augusteyn R. C., Schneider A., and Freund T. (1971) α-Crystallin. The isolation and characterization of distinct macromolecular fractions. Biochem. J. 124, 337–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Augusteyn R. C. (2004) α-Crystallin: a review of its structure and function. Clin. Exp. Optom. 87, 356–366 [DOI] [PubMed] [Google Scholar]
- 29. Srinivas P. N., Reddy P. Y., and Reddy G. B. (2008) Significance of α-crystallin heteropolymer with a 3:1 αA/αB ratio: chaperone-like activity, structure and hydrophobicity. Biochem. J. 414, 453–460 [DOI] [PubMed] [Google Scholar]
- 30. Sun T. X., and Liang J. J. (1998) Intermolecular exchange and stabilization of recombinant human αA- and αB-crystallin. J. Biol. Chem. 273, 286–290 [DOI] [PubMed] [Google Scholar]
- 31. Bukach O. V., Glukhova A. E., Seit-Nebi A. S., and Gusev N. B. (2009) Hetero-oligomeric complexes formed by human small heat-shock proteins HspB1 (Hsp27) and HspB6 (Hsp20). Biochim. Biophys. Acta 1794, 486–495 [DOI] [PubMed] [Google Scholar]
- 32. Kato K., Goto S., Inaguma Y., Hasegawa K., Morishita R., and Asano T. (1994) Purification and characterization of a 20-kDa protein that is highly homologous to αB crystallin. J. Biol. Chem. 269, 15302–15309 [PubMed] [Google Scholar]
- 33. Sugiyama Y., Suzuki A., Kishikawa M., Akutsu R., Hirose T., Waye M. M., Tsui S. K., Yoshida S., and Ohno S. (2000) Muscle develops a specific form of small heat-shock protein complex composed of MKBP/HSPB2 and HSPB3 during myogenic differentiation. J. Biol. Chem. 275, 1095–1104 [DOI] [PubMed] [Google Scholar]
- 34. Bukach O. V., Seit-Nebi A. S., Marston S. B., and Gusev N. B. (2004) Some properties of human small heat-shock protein Hsp20 (HspB6). Eur. J. Biochem. 271, 291–302 [DOI] [PubMed] [Google Scholar]
- 35. Heirbaut M., Lermyte F., Martin E. M., Beelen S., Verschueren T., Sobott F., Strelkov S. V., and Weeks S. D. (2016) The preferential heterodimerization of human small heat-shock proteins HSPB1 and HSPB6 is dictated by the N-terminal domain. Arch. Biochem. Biophys. 610, 41–50 [DOI] [PubMed] [Google Scholar]
- 36. Heirbaut M., Beelen S., Strelkov S. V., and Weeks S. D. (2014) Dissecting the functional role of the N-terminal domain of the human small heat-shock protein HSPB6. PLoS ONE 9, e105892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hanazono Y., Takeda K., Oka T., Abe T., Tomonari T., Akiyama N., Aikawa Y., Yohda M., and Miki K. (2013) Nonequivalence observed for the 16-meric structure of a small heat-shock protein, SpHsp16.0, from Schizosaccharomyces pombe. Structure 21, 220–228 [DOI] [PubMed] [Google Scholar]
- 38. Sluchanko N. N., Beelen S., Kulikova A. A., Weeks S. D., Antson A. A., Gusev N. B., and Strelkov S. V. (2017) Structural basis for the interaction of a human small heat-shock protein with the 14-3-3 universal signaling regulator. Structure 25, 305–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jehle S., Vollmar B. S., Bardiaux B., Dove K. K., Rajagopal P., Gonen T., Oschkinat H., and Klevit R. E. (2011) N-terminal domain of αB-crystallin provides a conformational switch for multimerization and structural heterogeneity. Proc. Natl. Acad. Sci. U.S.A. 108, 6409–6414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ghosh J. G., and Clark J. I. (2005) Insights into the domains required for dimerization and assembly of human αB crystallin. Protein Sci. 14, 684–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pasta S. Y., Raman B., Ramakrishna T., and Rao ChM. (2003) Role of the conserved SRLFDQFFG region of α-crystallin, a small heat-shock protein. Effect on oligomeric size, subunit exchange, and chaperone-like activity. J. Biol. Chem. 278, 51159–51166 [DOI] [PubMed] [Google Scholar]
- 42. Heirbaut M., Strelkov S. V., and Weeks S. D. (2015) in The Big Book on Small Heat-shock proteins (Tanguay R. M., and Hightower L. E., eds) pp. 197–227, Springer International Publishing, New York [Google Scholar]
- 43. Delbecq S. P., Rosenbaum J. C., and Klevit R. E. (2015) A mechanism of subunit recruitment in human small heat-shock protein oligomers. Biochemistry 54, 4276–4284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Delbecq S. P., Jehle S., and Klevit R. (2012) Binding determinants of the small heat-shock protein, αB-crystallin: recognition of the “IxI” motif. EMBO J. 31, 4587–4594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hilton G. R., Hochberg G. K., Laganowsky A., McGinnigle S. I., Baldwin A. J., and Benesch J. L. (2013) C-terminal interactions mediate the quaternary dynamics of αB-crystallin. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368, 20110405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aquilina J. A., Benesch J. L., Ding L. L., Yaron O., Horwitz J., and Robinson C. V. (2004) Phosphorylation of αB-crystallin alters chaperone function through loss of dimeric substructure. J. Biol. Chem. 279, 28675–28680 [DOI] [PubMed] [Google Scholar]
- 47. James M., Crabbe C., and Hepburne-Scott H. W. (2001) Small heat-shock proteins (sHSPs) as potential drug targets. Curr. Pharm. Biotechnol. 2, 77–111 [DOI] [PubMed] [Google Scholar]
- 48. Sun Y., and MacRae T. H. (2005) The small heat-shock proteins and their role in human disease. FEBS J. 272, 2613–2627 [DOI] [PubMed] [Google Scholar]
- 49. Flenniken M. L., Uchida M., Liepold L. O., Kang S., Young M. J., and Douglas T. (2009) A library of protein cage architectures as nanomaterials. Curr. Top. Microbiol. Immunol. 327, 71–93 [DOI] [PubMed] [Google Scholar]
- 50. Maham A., Tang Z., Wu H., Wang J., and Lin Y. (2009) Protein-based nanomedicine platforms for drug delivery. Small 5, 1706–1721 [DOI] [PubMed] [Google Scholar]
- 51. Furnish E. J., Brophy C. M., Harris V. A., Macomson S., Winger J., Head G. A., and Shaver E. G. (2010) Treatment with transducible phosphopeptide analogues of the small heat shock-related protein, HSP20, after experimental subarachnoid hemorrhage: prevention and reversal of delayed decreases in cerebral perfusion. J. Neurosurg. 112, 631–639 [DOI] [PubMed] [Google Scholar]
- 52. Weeks S. D., Drinker M., and Loll P. J. (2007) Ligation independent cloning vectors for expression of SUMO fusions. Protein Expr. Purif. 53, 40–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 54. Kurowski M. A., and Bujnicki J. M. (2003) GeneSilico protein structure prediction meta-server. Nucleic Acids Res. 31, 3305–3307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ren J., Wen L., Gao X., Jin C., Xue Y., and Yao X. (2009) DOG 1.0: illustrator of protein domain structures. Cell Res. 19, 271–273 [DOI] [PubMed] [Google Scholar]
- 56. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D., Konarev P. V., and Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.