

Abstract
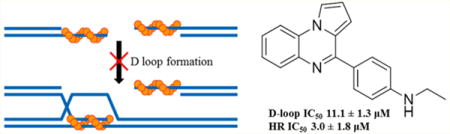
RAD51 is the central protein in homologous recombination (HR) DNA repair and represents a therapeutic target in oncology. Herein we report a novel class of RAD51 inhibitors that were identified by high throughput screening. In contrast to many previously reported RAD51 inhibitors, our lead compound 1 is capable of blocking RAD51-mediated D-loop formation (IC50 21.3 ± 7.8 μM) at concentrations that do not influence RAD51 binding to ssDNA. In human cells, 1 inhibits HR (IC50 13.1 ± 1.6 μM) without blocking RAD51’s ability to assemble into subnuclear foci at sites of DNA damage. We determined that the active constituent of 1 is actually an oxidized derivative (termed RI(dl)-1 or 8) of the original screening compound. Our SAR campaign also yielded RI(dl)-2 (hereafter termed 9h), which effectively blocks RAD51’s D-loop activity in biochemical systems (IC50 11.1 ± 1.3 μM) and inhibits HR activity in human cells (IC50 3.0 ± 1.8 μM).
Graphical abstract
INTRODUCTION
Homologous recombination (HR) is an evolutionarily conserved DNA repair process, which repairs DNA double-strand breaks (DSBs) and promotes tolerance of interstrand DNA cross-links (ICLs). Unlike the error-prone nonhomologous end-joining (NHEJ) pathway of DSB rejoining, HR faithfully repairs DNA damage by utilizing an undamaged homologous DNA template to guide the repair process.1,2 An initial step of HR involves nuclease processing that generates a 3′ single-stranded DNA (ssDNA) tail at the site of DNA damage (Figure 1). The ssDNA is then coated with RAD51 protein to form a helical nucleoprotein filament. This resulting nucleoprotein filament subsequently searches for a homologous DNA sequence and invades it to form a joint molecule intermediate, termed a D-loop. Finally, with the assistance of other related HR proteins, accurate DNA synthesis is performed using the undamaged homologous sequence as a template.
Figure 1.
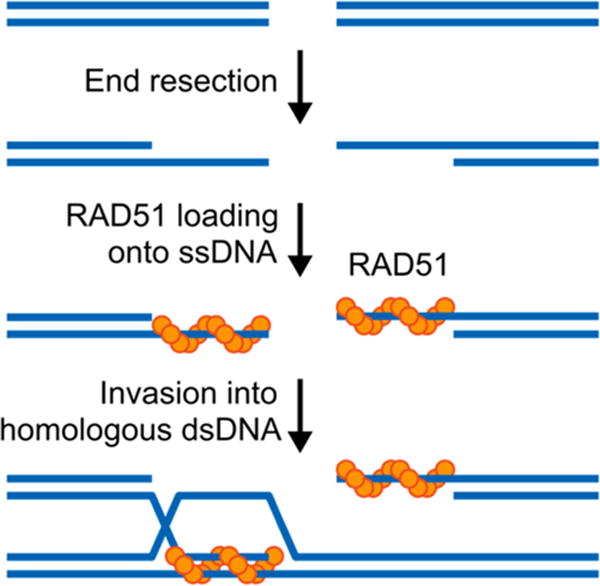
Schematic representation of the mechanism of DNA double-strand breaks repair by homologous recombination. Following a DSB, the DNA ends are resected to generate 3′ ssDNA overhangs onto which RAD51 loads. The resulting RAD51-ssDNA nucleoprotein filament is capable of invading homologous dsDNA and base-pairing with complementary DNA, thereby forming a heteroduplex and displacing a loop (D-loop) of ssDNA.
Because HR facilitates cellular recovery from harmful DNA lesions, cells with deficient HR functionality are especially vulnerable to radiotherapy and chemotherapeutic agents that generate DSBs or DNA replication-blocking lesions.2–4 RAD51, the central protein of HR, is overexpressed in various types of human cancer cells.5,6 These high expression levels of RAD51 can elevate HR efficiency in cancer cells, thereby inducing cellular resistance to DNA-damaging chemotherapy and radiotherapy.7–10 Inhibition of RAD51 by antisense RNAs or RAD51 inhibitors are reported to enhance the sensitivity of cancer cells to chemotherapy and radiotherapy.11–13 These observations support the concept of RAD51 as a therapeutic target and the development of small molecule therapeutics to sensitize tumors.14–16 Although several small molecule RAD51 inhibitors have been developed, most of these inhibitors act by preventing the formation of RAD51-ssDNA nucleoprotein filaments.13,14,16–19 One possible exception is halenaquinone, a chemical purported to specifically inhibit D-loop formation by RAD51.20 However, halenaquinone also reduces the subnuclear appearance of RAD51 foci at DSB sites in cells, which raises questions as to its mechanistic interaction with RAD51 in cells.
In addition to its function in HR, RAD51 filaments also serve an important function at stalled DNA replication forks.21,22 These RAD51 nucleoprotein filaments protect the ssDNA from extensive nuclease processing, thereby aiding in replication and preserving genomic integrity. Because most RAD51 inhibitors act by preventing the formation of RAD51 nucleoprotein filaments on ssDNA, they are expected to inhibit both HR and replication fork stabilization activities of RAD51. As such, their use is predicted to generate unintended toxicity to normal cells by interrupting this centrally important function in normal replication.
We reasoned that specifically inhibiting RAD51-mediated D-loop formation while preserving RAD51’s ability to form nucleoprotein filaments may target DNA repair while minimizing replication-associated toxicity in normal tissue. Therefore, we carried out a two-part high-throughput (HT) screen of the ASDI diversity library and LOPAC1280 compound library for drug candidates that selectively target RAD51’s D-loop function. The preferred lead compound resulting from this screen, compound 1, inhibits RAD51’s D-loop activity while having minimal effects on RAD51’s ssDNA binding activity. Compound 1 is a commercially obtained sample labeled as 4-(7-methoxy-4,5-dihydropyrrolo[1,2-a]-quinoxalin-4-yl)-N,N-dimethylaniline, but its structure was found to be inconsistent with the named compound. This report details our HT screen and subsequent structure–activity relationship (SAR) optimizations performed to improve the potency and selectivity of 1.
RESULTS AND DISCUSSION
Screen for RAD51 D-Loop Inhibitors
We carried out two parallel HT screens to identify small molecules that inhibit RAD51 D-loop activity without affecting its ssDNA binding activity. A previously described fluorescence polarization-based microplate assay was used to assess the compounds’ effects on RAD51-ssDNA binding23 (Figure 2a). To enable a screen for D-loop specific RAD51 inhibitors, we previously developed a microplate-based assay that can accurately quantify D-loop formation in a high-throughput fashion24 (Figure 2b). This method makes use of two substrates: a closed linear double-hairpin dsDNA and an ssDNA fused with a black hole quencher. The sequence homology of the two substrates enables RAD51 to catalyze the formation of D-loops, which is quantified as a function of quenched fluorescence. Among the screening compounds tested, we focused on those that prevented RAD51 D-loop formation while exerting little effect on RAD51-ssDNA binding (Figure 2c). Potential hits were confirmed using secondary low-throughput retesting, which involved a gel-based D-loop assay to exclude the possibility that the observed inhibition of D-loop activity was due to artifactual interference by compound autofluorescence or fluorescence quenching.
Figure 2.
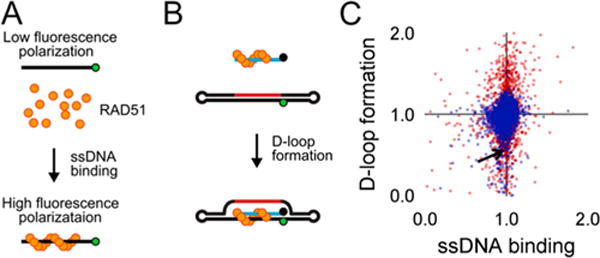
High-throughput (HT) screen for compounds that inhibit RAD51’s D-loop activity. (A) One biochemical assay monitors RAD51 binding to fluorescently labeled ssDNA, which is detected as an increase in fluorescence polarization. (B) A second parallel biochemical assay monitors fluorescence intensity, which decreases upon pairing of Black Hole Quencher 1-labeled ssDNA with a fluorescein-labeled complementary double-hairpin duplex. (C) Compounds were tested for the ability to influence the efficiency of these two RAD51-mediated processes, and the results of this HT are displayed. Blue symbols = ASDI library compounds, red symbols = LOPAC library compounds. Arrow indicates the position of compound 1.
By using this screening platform, the ASDI diversity library (6800 compounds of diverse chemical structures) and the LOPAC library of known drugs (library of 1280 pharmacologically active compounds) were screened, leading to the identification of 10 hit compounds that were capable of inhibiting D-loop activities with minimal effects on ssDNA binding. The best of these 10 compounds is shown in Figure 3 and hereon referred to as 1. Compound 1 inhibits the D-loop activity of RAD51 by up to 74% while exerting minimal dose-dependent inhibition of RAD51 filament assembly in the same concentration range (Figure 3). As expected, parallel experiments demonstrate that the more generalized inhibitors of RAD51 (like RI-1) do generate dose-dependent inhibition of RAD51 filament assembly using identical assay conditions (see Figure 7b for reference).
Figure 3.
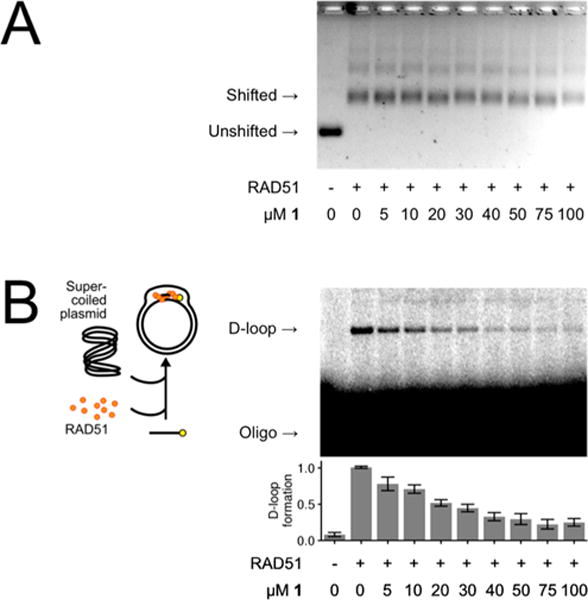
Compound 1 inhibits RAD51’s D-loop activity with minimal interference of RAD51-ssDNA binding. (A) Electromobility shift assay showing that 1 does not inhibit interaction with RAD51 and ssDNA. (B) Gel-based D-loop formation assay showing that 1 inhibits RAD51-mediated assimilation of a radiolabeled ssDNA oligonucleotide into the homologous region of a supercoiled dsDNA plasmid by up to 74%. Error bars indicate the standard error for four replicates with a representative gel image shown.
Figure 7.
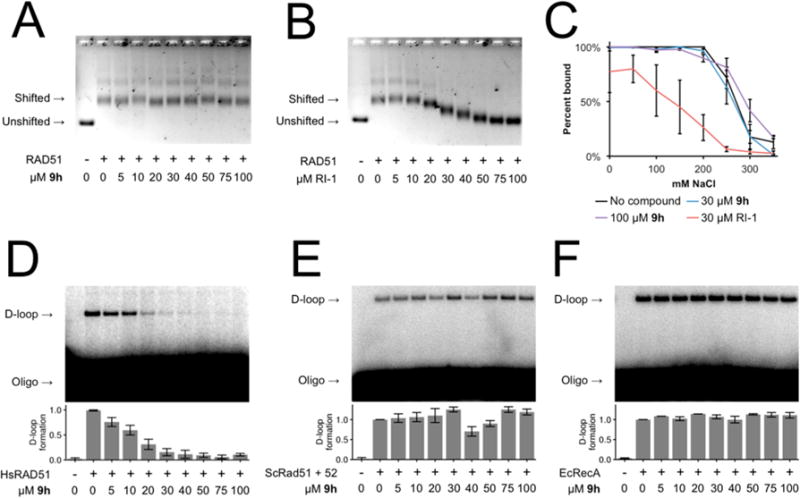
(A,B) Electromobility shift assay showing RAD51-ssDNA binding in the presence of 9h or RI-1, respectively. (C) 9h does not destabilize RAD51-ssDNA nucleoprotein filaments as shown by salt titration midpoint. RI-1 serves as a positive control for disruption of RAD51-ssDNA nucleoprotein filament stability. Error bars denote the standard error for three replicates. (D,E,F) Representative D-loop assay gel images showing recombinase-mediated assimilation of a radiolabeled ssDNA oligonucleotide into the homologous region of a supercoiled dsDNA plasmid in the presence of 9h by human RAD51 protein, S. cerevisiae Rad51 and Rad52 proteins, or E. coli RecA protein, respectively. Error bars denote the standard error for three replicates.
Synthesis and Activities of the Reduced and Oxidized Forms of 1
As compound 1 was identified from a commercial compound library, we decided to first confirm its chemical structure by carrying out our own independent synthesis as detailed in Scheme 1. The starting material 2a was first condensed with dimethoxytetrahydrofuran (3) in refluxing acetic acid to afford 4a.29 The nitro group of 4a was then reduced, and the aniline 5a29 was reacted with benzaldehyde 6a in the presence of 1,2,3-benzotriazole and AlCl3 to provide the desired product 7 in moderate yield.
Scheme 1. Synthesis of 7a.
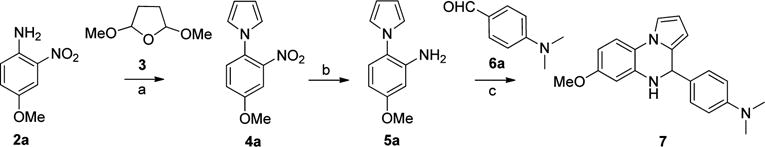
aReagents and conditions: (a) AcOH, reflux, 3 h, 90%; (b) H2, Pd/C, MeOH, 5 h, 97%; (c) 1,2,3-benzotriazole, AlCl3, THF, 4 h, 32%.
The newly synthesized compound 7 was then subjected to biological evaluation as shown in Table 1. Unexpectedly, compound 7 displayed a much weaker D-loop inhibitory activity in comparison to the compound 1 that we obtained from the commercial source (maximum inhibition of 17% for 7 vs 74% for 1). Moreover, compound 7 also showed significant stimulatory activity in RAD51-ssDNA binding when compared to 1. These discrepancies suggested that compound 7 and 1 might be different substances. Interestingly, we observed that compound 7 was not stable when exposed to air at room temperature; instead, it slowly oxidized over time to compound 8 (Scheme 2). Additionally, we found during the preparation of compound 7, some of the oxidized product 8 was always produced. These observations raised the possibility that the commercially obtained material 1 might have undergone oxidation during its storage and that the oxidized form 8 might be the active constituent responsible for the D-loop inhibitory activity of 1.
Table 1.
RAD51 D-Loop Inhibitory and RAD51-ssDNA Binding Stimulatory/Inhibitory Activities of 1, 7, and 8a
| compd | D-loops
|
ssDNA binding
|
||
|---|---|---|---|---|
| max inhibition effect (%) | IC50 (μM) | max effect (%) | IC50 (μM) | |
| 1 | 74 ± 2 | 21.3 ± 7.8 | 33 ± 10↓ | >100 |
| 7 | 17 ± 3 | >100 | 47 ± 33↑ | Stim |
| 8 | 67 ± 11 | 13.0 ± 4.7 | 40 ± 28↑ | Stim |
↑:Indicates that the compound stimulates RAD51-ssDNA binding. ↓:Compound inhibits RAD51-ssDNA binding. Stim: Compound is a stimulator.
Scheme 2. Synthesis of 8a.
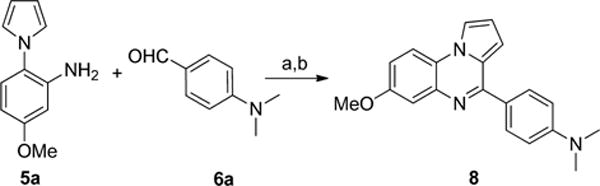
aReagents and conditions: (a) 1,2,3-benzotriazole, AlCl3, THF, overnight; (b) MnO2, acetone, 55 °C, 24 h, 59% in two steps.
To confirm this hypothesis, the structural identity of compound 1 was determined. According to the 1H NMR spectrum, the commercially obtained material 1 was impure, with the oxidized compound 8 as the major component (Supporting Information, Figure S1). The presence of compound 7 was not observed. The mass spectrum also confirmed the presence of compound 8 rather than compound 7. To further confirm the D-loop inhibitory activity of this oxidized compound, compound 8 was synthesized by following the route as shown in Scheme 2. The aniline 5a and benzaldehyde 6a were reacted in the presence of 1,2,3-benzotriazole and AlCl3 at room temperature until the conversion was complete, which resulted in a mixture of the reduced (7) and oxidized (8) forms. This crude mixture was then heated in acetone with MnO2 to induce further oxidization, thereby yielding the oxidized form 8 as the exclusive product. Subsequent testing in our D-loop assay (Table 1) showed that compound 8 exhibits similar activity to 1 in terms of maximal inhibition (67% vs 74%, respectively) and IC50 values (13.0 vs 21.3 μM, respectively). These results thus confirm that the oxidized form 8 is the actual substance that generates the D-loop inhibitory activity of the commercially obtained compound 1. Therefore, we have named compound 8 as “RI(dl)-1”, for D-loop activity of RAD51 Inhibitor no. 1.
Structure Optimization of 8
To improve upon the D-loop inhibitory activity, medicinal chemical studies were undertaken to optimize 8. An overview of the optimization strategy is summarized in Figure 4. First, different functional groups were appended to rings A and B as substituents, including alkyl groups of different sizes, electron withdrawing/donating groups, and hydrogen bond donor/acceptor groups. Next, replacement of the phenyl ring B with different heterocycles or other saturated/unsaturated rings was investigated in order to probe its role. For each of the prepared compounds, we used the gel-based D-loop assay and the microplate-based DNA binding assay to characterize the biochemical activities.
Figure 4.
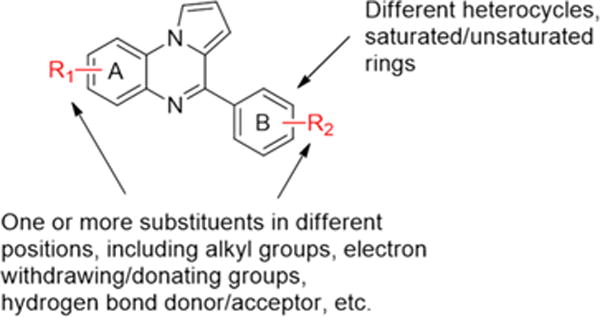
An overview is shown for the optimization strategy of 8.
Optimization of Ring B
To synthesize analogues with different substituents in the B-ring, the aniline 5e was treated with different benzaldehydes 6a–h in the presence of 1,2,3-benzotriazole and AlCl3 (in Scheme 3). Then the crude products were oxidized with MnO2 to provide 9a–j in good yield. Interestingly, when benzaldehyde 6f was reacted with the aniline 5e under these conditions, partial de-ethylation was observed and the monoethylated product 9h was isolated as the major product. The analogues 9c and 9g were obtained by reducing the nitro group of 9b and 9f31 via hydrogenation. To synthesize analogues in which the B-ring was replaced with other aromatic or saturated rings, the aniline 5e was reacted with different aldehydes 10a–d under similar conditions (in Scheme 4) to afford the products 11a–d31–33 in good yield.
Scheme 3. Synthesis of Analogues 9a–ja.
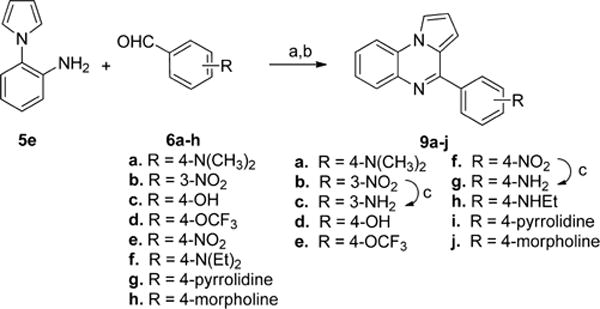
aReagents and conditions: (a) 1,2,3-benzotriazole, AlCl3, THF, overnight; (b) MnO2, acetone, 55 °C, 24 h, 19–70% in two steps; (c) H2, Pd/C, methanol, 5 h, 57–87%.
Scheme 4. Synthesis of Analogues 11a–da.
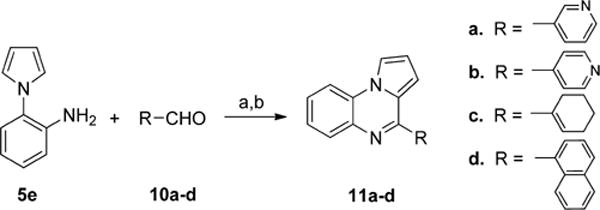
aReagents and conditions: (a) 1,2,3-benzotriazole, AlCl3, THF, overnight; (b) MnO2, acetone, 55 °C, 24 h, 35–71% in two steps.
The D-loop inhibitory activities measured for compounds 9a–j and 11a–d are shown in Table 2 (see D-loop gels in Supporting Information, Figure S2). Results indicate that a dimethylamino group (9a), amino group (9g), ethylamino group (9h). or morpholine ring (9j) in the para position of the B-ring allow for D-loop inhibition and that 9g and 9h display potencies that are similar to that of compound 8. By contrast, introducing a hydroxyl group (9d), trifluoromethoxyl group (9e), nitro group (9f), or pyrrolidine group (9i) in the para-position significantly decreased the D-loop inhibitory activities. Moving the amino group from the para- (9g) to the meta-position (9c) also decreased the D-loop inhibitory activity. Replacing the B-ring with other aromatic or saturated rings only resulted in moderate (11a, 11b) or weak (11c, 11d) D-loop inhibitors. The ability of these analogues to modulate RAD51’s ssDNA binding was also evaluated as shown in Table 2. Many of these compounds had only a weak influence on RAD51-ssDNA binding. On the other hand, compounds 9a, 9b, 9i, 11c, and 11d had a modest influence on RAD51-ssDNA binding.
Table 2.
RAD51 D-Loop Inhibitory and RAD51-ssDNA Binding Stimulatory/Inhibitory Activities of 9a–j and 11a–da
| |||||
|---|---|---|---|---|---|
| Cpd | D-Loops
|
ssDNA binding
|
|||
| R | Max inhibition effect | IC50 (μM) | Max effect | IC50 (μM) | |
| 9a | 4-N(CH3)2 | 61 ±4% | 27.6±6.0 | 88±22%↑ | Stim |
| 9b | 3-NO2 | 20±8% | >100 | 52±14%↓ | >100 |
| 9c | 3-NH2 | 79±8% | 91.1±10.3 | 11±9%↓ | >100 |
| 9d | 4-OH | 38±3% | >100 | 14±16%↓ | >100 |
| 9e | 4-OCF3 | 22±8% | >100 | 30±21%↑ | Stim |
| 9f | 4-NO2 | 2±21% | >100 | 16±13%↓ | >100 |
| 9g | 4-NH2 | 100±2% | 22.1±2.4 | 18±11%↓ | >100 |
| 9h | 4-NHEt | 91±1% | 11.1±1.3 | 2±24%↑ | >100 |
| 9i |
|
8±7% | >100 | 69±35%↑ | Stim |
| 9j |
|
68±7% | 27.2±3.0 | 11±19%↓ | >100 |
| 11a |
|
70±13% | 71.7±18.1 | 6±10%↓ | >100 |
| 11b |
|
63±4% | 79.4±6.0 | 7±38%↓ | >100 |
| 11c |
|
5±11% | >100 | 42±17%↑ | Stim |
| 11d |
|
42±11% | >100 | 56±31%↑ | Stim |
↑: Compound stimulates RAD51-ssDNA binding. ↓: Compound inhibits RAD51-ssDNA binding. Stim: Compound is a stimulator.
Optimization of Ring A
To synthesize analogues with different substituents in the A-ring, the substituted anilines 5b–d were first prepared according to the route shown in Scheme 5. The starting materials 2b–d were condensed with dimethoxytetrahydrofuran (3) in refluxing acetic acid to afford 4b–d.30 Then, the nitro group was reduced to provide the products 5b–d. Subsequently, the anilines 5a–d were reacted with benzaldehydes (6a, 6c, and 6e) to provide the analogues 12a–m in good yield (in Scheme 6). For analogues with an amino group (12c, 12f, 12i, and 12l), compounds were prepared from the corresponding nitro analogues (12b, 12e, 12h, and 12k) via hydrogenation. Two analogues (13a–b) with a 3-pyridine ring were also prepared from anilines 5a–b and aldehyde 10a as shown in Scheme 7.
Scheme 5. Synthesis of 5b–da.
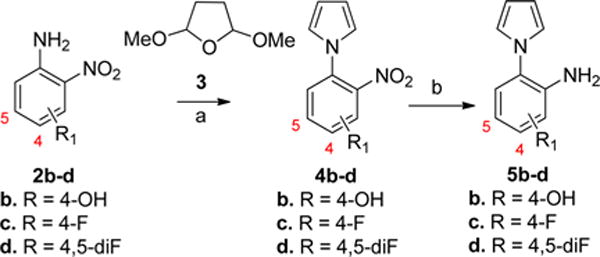
aReagents and conditions: (a) AcOH, reflux, 3 h, 45–63%; (b) H2, Pd/C, MeOH, 5 h, 37–97%.
Scheme 6. Synthesis of Analogues 12a–ma.
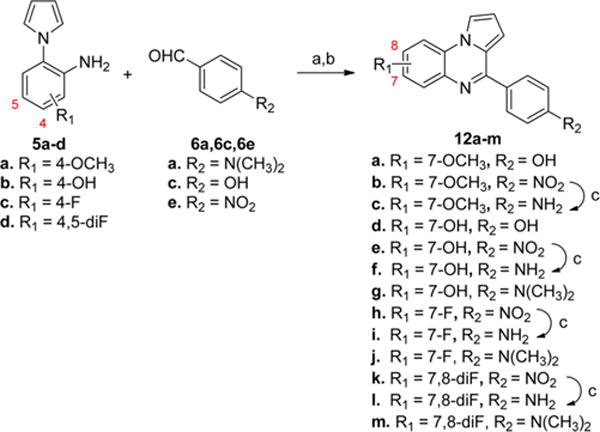
aReagents and conditions: (a) 1,2,3-benzotriazole, AlCl3, THF, overnight; (b) MnO2, acetone, 55 °C, 24 h, 7–77% in two steps; (c) H2, Pd/C, MeOH, 5 h, 50–93%.
Scheme 7. Synthesis of Analogues 13a–ba.
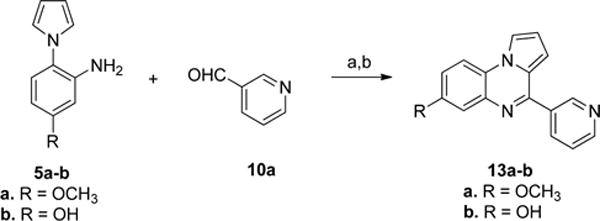
aReagents and conditions: (a) 1,2,3-benzotriazole, AlCl3, THF, overnight; (b) MnO2, acetone, 55 °C, 24 h, 52–78% in two steps.
To explore the effects of replacing the hydroxyl group of compound 12f with different alkyl groups, compound 12e was first treated with allyl bromide to introduce the allyl group (14a), and then the nitro group was reduced by hydrogenation (in Scheme 8). The side chain double bond was also reduced under these conditions to afford the n-propoxy product 15a. The fluoroethyl compound 15b was prepared in a similar fashion but employed the Mitsunobu reaction to introduce the side chain (in Scheme 8).25,26 An analogue 16 with a 2-methylpropionamide side chain was also prepared as shown in Scheme 9.
Scheme 8. Synthesis of Analogues 15a–ba.
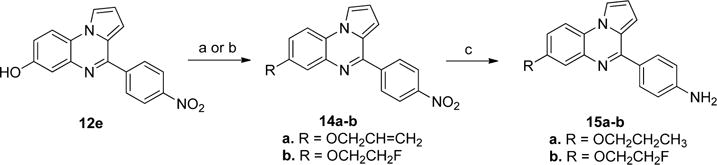
aReagents and conditions: (a) allyl bromide, Cs2CO3, DMF, microwave, 80 °C, 30 min, 58%; (b) FCH2CH2OH, DEAD, Ph3P, THF, microwave, 60 °C, 40 min; (c) H2, Pd/C, MeOH, overnight, 36–45%.
Scheme 9. Synthesis of Analogues 16a.
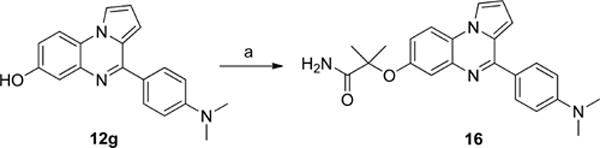
aReagents and conditions: (a) Br(CH3)2CHCONH2, NaOH, DMA, 50 °C, 3 h, 41%.
The D-loop inhibitory activities for compounds 12a–m, 13a–b, 15a–b, and 16 are shown in Table 3. These results indicate that a methoxyl group in the 7 position of A-ring improves its D-loop inhibitory activity, which can be seen by comparing compound 12c (IC50 13.2 μM) against 9g (IC50 22.1 μM). A similar improvement was observed when a hydroxyl group was introduced at the same position (compare 12f, IC50 8.4 μM, and 12g, IC50 9.8 μM, against 9g, IC50 22.1 μM, and 9a, IC50 27.6 μM). In contrast, the introduction of a fluorine atom in the 7 position or two fluorine atoms in the 7 and 8 positions of the A-ring yielded compounds whose D-loop inhibitory potential was moderately diminished (compounds 12i, IC50 25.0 μM, and 12l, IC50 34.0 μM) or nearly absent (compound 12j and 12m). For compounds with 3-pyridyl substituents (compounds 13a and 13b), the incorporation of a methoxy or hydroxyl group in the A-ring yielded a slight improvement in D-loop inhibition compared with 11a. The appendage of larger alkoxy groups at the 7 position of the A-ring (compounds 15a–b and 16) did not provide more potent D-loop inhibitors (except for compound 15b) when compared to 9g.
Table 3.
RAD51 D-Loop Inhibitory and RAD51-ssDNA Binding Stimulatory/Inhibitory Activities of 12a–m, 13a–b, 15a–b, and 16a
| ||||||
|---|---|---|---|---|---|---|
| Cpd | R1 | R2 | D-Loops
|
ssDNA binding
|
||
| Max inhibition effect | IC50 (μM) | Max effect | IC50 (μM) | |||
| 12a | 7-OCH3 | -OH | 13±12% | >100 | 100±18%↓ | 35.0±14.0 |
| 12b | 7-OCH3 | -NO2 | 17±7% | >100 | 105±47%↑ | Stim |
| 12c | 7-OCH3 | -NH2 | 100±4% | 13.2±2.8 | 29±8%↓ | >100 |
| 12d | 7-OH | -OH | 97±8% | 27.6±4.9 | 72±11%↓ | >100 |
| 12e | 7-OH | -NO2 | 13±10% | >100 | 92±21%↓ | 57.8±3.0 |
| 12f | 7-OH | -NH2 | 100±3% | 8.4±3.4 | 82±23%↓ | 96.9±44.9 |
| 12g | 7-OH | -N(CH3)2 | 91±8% | 9.8±3.5 | 34±32%↑ | Stim |
| 12i | 7-F | -NH2 | 95±6% | 25.0±5.1 | 60±18%↓ | 81.5±16.6 |
| 12j | 7-F | -N(CH3)2 | 31±4% | >100 | 57±28%↑ | Stim |
| 12l | 7,8-diF | -NH2 | 59±4% | 34.0±2.9 | 42±8%↓ | >100 |
| 12m | 7,8-diF | -N(CH3)2 | 19±15% | >100 | 65±22%↓ | Stim |
| 13a | 7-OCH3 | 74±5% | 38.6±6.2 | 16±28%↓ | >100 | |
| 13b | 7-OH | 53±30% | 64.9±31.6 | 87±10%↓ | >100 | |
| 15a |
|
-NH2 | 75±7% | 46.1±2.8 | 117±18%↑ | Stim |
| 15b |
|
-NH2 | 98±5% | 20.0±4.1 | 79±23%↑ | Stim |
| 16 |
|
-NI(CH3)2 | 38±4% | >100 | 131±28%↑ | Stim |
“↑” Compound stimulates RAD51-ssDNA binding. “↓”Compound inhibits RAD51-ssDNA binding. “Stim” Compound is a stimulator.
The influence of these alterations on RAD51-ssDNA binding activity was also evaluated for these analogues (Table 3). Most of the compounds influenced RAD51-ssDNA binding weakly or modestly, except for compounds 12b, 15a, 15b, and 16, which were stimulators of RAD51-ssDNA binding, and 12a, which showed relatively strong ssDNA binding inhibition. Among the most potent D-loop inhibitors in this series, 12c and 12g only weakly influenced ssDNA-RAD51 binding; 12f and 15b exerted modest effects on ssDNA-RAD51 binding.
Cellular Effects of 1 and 9h
We used the DR-GFP assay to test the ability of select compounds to inhibit HR in human cells.27 Briefly, cells containing the chromosomal DR-GFP reporter were transfected with a plasmid that expresses I-SceI, a rare-cutting endonuclease that makes a DSB within the DR-GFP cassette. Compounds were added at the time of transfection, and the cells were outgrown for 24 h. We noted that for longer outgrowth periods such as 48 h, cells regained some HR activity (data not shown). This suggests the possibility of compensatory responses that oppose RAD51 inhibition by compounds; these effects will be detailed in a separate report. To overcome this technical limitation, we selected the relatively early time point of 24 h to describe the in vivo effects of these compounds.
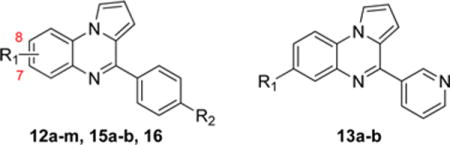
We demonstrated that 1 is able to inhibit HR activity in human cells (Figure 5a). Importantly, 1 yields 50% inhibition of cellular HR activity at 13.1 μM, which is comparable to its observed activity in biochemical assays. The HR inhibitory activity of 1 reaches 83% at concentrations of 40 μM, which is a dose where the RAD51-ssDNA binding activity remains unaffected in biochemical assays. Next, we compared 1 against its pure oxidized and pure reduced forms. In contrast to our results using biochemical assays, the inhibitory activity of 7 in the cell-based HR assay was comparable to that of 1 and 8. As noted earlier, 7 spontaneously oxidizes to 8. As such, this observed inhibitory activity of 7 is likely caused by its oxidation into the active constituent (8) during the HR assay; such would be expected given the relatively long (24 h) incubation of 7 in an aerated tissue culture medium.
Figure 5.
(A) Inhibition of cellular HR activity by 1 and optimized analogues of 1 using the DR-GFP assay. Data were collected at 24 h following transfection with the I-SceI expressing plasmid pCBASce. Error bars denote the standard error for three replicates. (B) Inhibition of cellular HR and SSA activity by 9h at 24 h post-transfection. Error bars denote the standard error for three replicates.
Next we used this cell-based HR quantification method to evaluate analogues that exhibited IC50 values less than 20 μM in biochemical D-loop assays. While compounds 12f and 12g had the lowest IC50 values in the D-loop assay, they both yielded comparable or slightly less inhibitory activity in the DR-GFP assay relative to 1 and 8. The most potent inhibitor of cellular HR was 9h, having an IC50 of 3.0 ± 1.8 μM, which is over 4-fold lower than the IC50 concentrations of the starting lead compound 1.
Compound 9h was tested further using cells containing the chromosomal DR-SSA reporter, which quantifies the ability of the cells to perform single-strand annealing (SSA) as a means to repair I-SceI induced DSBs. SSA is an alternative method of religating DNA when HR cannot be completed. SSA efficiency is known to be elevated in situations where RAD51 function has been generally disrupted.27,28 Similarly, we previously demonstrated that both RI-1 and RI-2 induce elevated levels of SSA activity.19 By contrast, we found that 9h does not stimulate SSA, using a relevant concentration range that inhibits HR (Figure 5b). This suggests that 9h stabilizes nucleoprotein filaments in a nonfunctional state, which are incapable of D-loop activity and simultaneously shielded from related (e.g., RAD52-mediated) pathways that promote SSA.
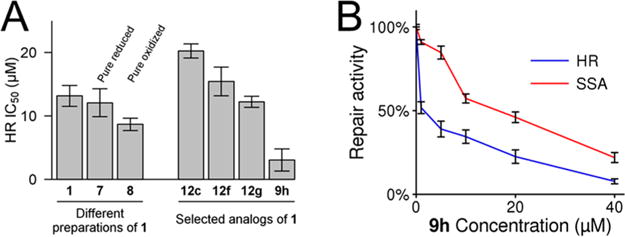
We tested whether 9h permits the timely assembly of RAD51 into subnuclear foci in response to DNA damage. As expected, 9h does not inhibit the appearance of RAD51 into subnuclear foci in response to radiation (Figure 6a), even at concentrations of 9h that had inhibited cellular HR by 75% in prior DR-GFP assays (Figure 5b). Importantly, 9h does not affect the colocalization of RAD51 foci with RPA, an HR associated protein that binds to ssDNA prior to the recruitment of RAD51 (Figure 6b). We observed 57% colocalization in 9h treated cells and 60% colocalization in vehicle-treated cells. This important control confirms that our imaged subnuclear RAD51 puncta represent true DSB-associated foci rather than off-target accumulations of RAD51 protein. Additionally, 9h does not affect RAD51 protein levels (cytosolic, nuclear, or chromatin-bound fractions) at comparable time points (Supporting Information, Figure S3). Our interpretation is further substantiated by quantitative gel-shift assays (Figure 7a–c, Supporting Information, Figure S4). We also tested the selectivity of 9h as a specific inhibitor of human RAD51 by comparing its ability to inhibit the D-loop activity of human RAD51 versus the distantly related Saccharomyces cerevisiae Rad51 and Escherichia coli RecA recombinase proteins and found no significant inhibition of these proteins by 9h (Figure 7d–f). Unlike the generalized RAD51 inhibitor RI-1, 9h does not modulate the affinity of RAD51 to ssDNA or the stability of preformed RAD51-ssDNA complexes when challenged with high concentrations of salt.
Figure 6.
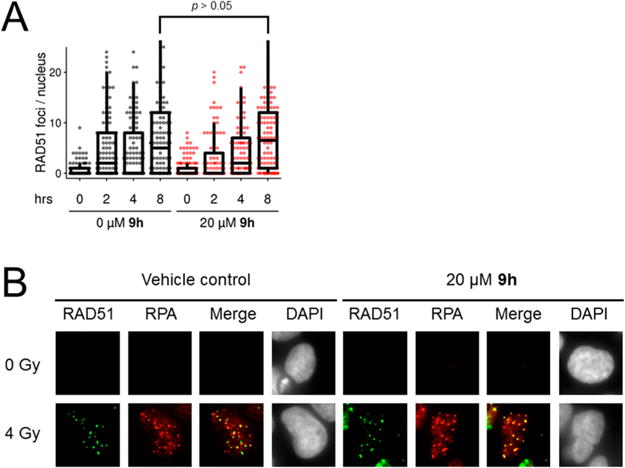
(A) Compound 9h does not inhibit the appearance of IR-induced RAD51 foci in 293-DR-GFP cells over an 8 h time course. At least 100 nuclei per condition are represented. (B) Representative micrographs showing colocalization of RAD51 foci with replication protein A (RPA) foci in DAPI-counterstained 293-DR-GFP nuclei at 8 h postirradiation in 293-DR-GFP cells treated with 9h or the DMSO vehicle control.
Because 9h inhibits HR-mediated repair of DSBs, we next investigated whether 9h could sensitize cancer cells to radiation damage. Indeed, we observed that 9h significantly sensitizes three different cancer cell lines when 9h was administered immediately after following irradiation and allowed to incubate with cells for the duration of clonogenic outgrowth thereafter (Figure 8). Given the substantial improvements of 9h over the starting compound identified in our high throughput screen, we renamed compound 9h as RI(dl)-2, for D-loop activity of RAD51 Inhibitor no. 2.
Figure 8.
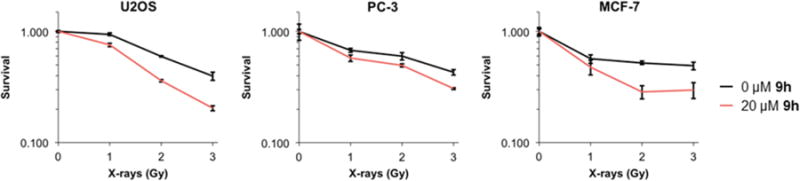
Clonogenic survival of (A) U2OS, (B) PC-3, and (C) MCF-7 tumor cell lines treated with ionizing radiation followed by outgrowth in the presence of 9h or the vehicle-only control. Error bars denote the standard error for three replicates.
CONCLUSION
A common hallmark of tumor cells is the overexpression of the RAD51 recombinase protein. Numerous lines of evidence indicate that tumor cells are especially dependent upon the HR function of RAD51.11,12,29 This well-established feature of cancer cells has made RAD51 an attractive drug target, and several small molecules that inhibit cellular RAD51 function have been described as reviewed by Huang and Mazin.16 In addition to its classical role in catalyzing homologous strand exchange, RAD51 additionally plays a central role in protecting stalled replication forks from nucleolytic degradation.21,22 This protective function in replication requires that RAD51 is able to bind ssDNA, however, it may not require the strand invasion activity of RAD51. Therefore, we carried out a drug development campaign to inhibit RAD51’s D-loop activity while sparing its ssDNA binding activity. Compound 1 was identified as a promising lead compound, and subsequent SAR efforts yielded compound 9h with improved biochemical and cell-based activities.
We believe that this series of compounds represents a major step forward in the development of RAD51-targeting small molecules. To our current knowledge, these are the only known compounds that specifically inhibit RAD51’s D-loop activity without interfering with its ability to bind ssDNA both in vitro and in cells. Halenaquinone was previously reported to strongly inhibit RAD51’s D-loop activity while sparing its ssDNA binding activity in biochemical assays.20 However, halenaquinone was shown to inhibit the formation of RAD51 foci in human cells following irradiation, suggesting that in a cellular context this compound may prevent sufficient loading of RAD51 onto damaged DNA at the site of radiation-induced DSBs to form visible foci. Other characterized RAD51 inhibitors including our previously described inhibitors RI-1 and RI-2 act by preventing RAD51 from loading onto damaged DNA.13,19
Compound 1 and its analogues represent potential cancer therapeutics aimed at sensitizing tumors to DNA-damaging therapies. Considering its high potency for inhibiting both D-loop formation and HR, compound 9h provides an important candidate for further investigation. Meanwhile, because compound 9h has almost no impact on RAD51-ssDNA binding, it also presents a novel tool for basic scientists who wish to study the different functions of RAD51.
EXPERIMENTAL SECTION
General
The nuclear magnetic resonance (1H and 13C NMR) spectra were obtained using a Bruker spectrometer with TMS as an internal standard. Automated column chromatography was performed using the CombiFlash Rf apparatus loaded with Merck silica gel (40–60 mesh). Preparative HPLC was carried out using a Shimadzu preparative liquid chromatograph with a column from ACE 5 AQ (150 mm × 21.2 mm) with 5 μm particle size. All solvents used in preparative HPLC were spiked with 0.05% TFA. The purities of biologically important compounds are ≥95% as determined by analytical HPLC (ACE 3AQ C18 column (150 mm × 4.6 mm, particle size 3 μm), 0.05% TFA in H2O/0.05% TFA in MeOH gradient eluting system).
General Procedure for Preparation of 4a–d
A solution of 2a–d (20 mmol) and 3 (20 mmol) in acetic acid (30 mL) was heated to reflux for 3 h. After cooling down, the mixture was poured into water (150 mL) and extracted with diethyl ether (150 mL × 3). The organic layers were combined and dried over Na2SO4. Remove solvent provided the crude products of 4a–d.
1-(4-Methoxy-2-nitrophenyl)-1H-pyrrole (4a).30
The crude product of 4a was further purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 1:4) to provide the product as orange solid (90%). 1H NMR (400 MHz, CDCl3) δ 7.41–7.38 (m, 2 H), 7.19–7.16 (m, 1 H), 6.76 (s, 2 H), 6.35 (s, 2 H), 3.92 (s, 3 H). 13C NMR (100 MHz, CDCl3) δ 158.3, 145.6, 129.0, 126.9, 121.4, 118.8, 110.0, 109.1, 55.8.
3-Nitro-4-(1H-pyrrol-1-yl)phenol (4b).31
The crude product of 4b was further purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 1:2) to provide the product as orange solid (59%). 1H NMR (400 MHz, methanol-d4 and CDCl3) δ 7.25–7.22 (m, 2 H), 7.05–7.02 (m, 1 H), 6.68 (s, 2 H), 6.25 (s, 2 H). 13C NMR (100 MHz, methanol-d4 and CDCl3) δ 156.5, 145.6, 129.0, 125.6, 121.5, 119.8, 110.7, 109.5.
1-(4-Fluoro-2-nitrophenyl)-1H-pyrrole (4c)
The crude product of 4c was further purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 1:7) to provide the product as yellow oil (63%). 1H NMR (400 MHz, CDCl3) δ 7.64–7.61 (m, 1 H), 7.52–7.48 (m, 1 H), 7.43–7.39 (m, 1 H), 6.80–6.78 (m, 2 H), 6.40–6.38 (m, 2 H).
1-(4,5-Difluoro-2-nitrophenyl)-1H-pyrrole (4d)
The crude product of 4d was further purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 1:9) to provide the product as yellow oil (45%). 1H NMR (400 MHz, CDCl3) δ 7.85–7.80 (m, 1 H), 7.36–7.32 (m, 1 H), 6.78–6.76 (m, 2 H), 6.40–6.38 (m, 2 H).
General Procedure for Preparation of 5a–d
A suspension of 4a–d (3 mmol) and palladium on charcoal (10%, 60 mg) in methanol (10 mL) was stirred under a hydrogen balloon for 5 h. The black solid was removed by filtration, and the filtrate was concentrated to provide the crude product of 5a–d.
5-Methoxy-2-(1H-pyrrol-1-yl)aniline (5a).30
The crude product of 5a was further purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 1:4) to provide the product as pale-yellow oil (97%). 1H NMR (400 MHz, CDCl3) δ 7.13–7.09 (m, 1 H), 6.83–6.82 (m, 2 H), 6.40–6.37 (m, 4 H), 3.84 (s, 3 H). 13C NMR (100 MHz, CDCl3) δ 159.9, 143.4, 128.2, 122.1, 121.2, 109.2, 103.6, 101.1, 55.4.
3-Amino-4-(1H-pyrrol-1-yl)phenol (5b)
The crude product of 5b was further purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 1:2) to provide the product as yellow solid (37%). 1H NMR (400 MHz, methanol-d4 and CDCl3) δ 6.93–6.90 (m, 1 H), 6.71–6.69 (m, 2 H), 6.26–6.24 (m, 3 H), 6.22–6.19 (m, 1 H). 13C NMR (100 MHz, methanol-d4 and CDCl3) δ 156.7, 142.9, 127.8, 121.8, 120.1, 108.4, 105.1, 102.0.
5-Fluoro-2-(1H-pyrrol-1-yl)aniline (5c)
The crude product of 5c was further purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 1:7) to provide the product as white solid (97%). 1H NMR (400 MHz, CDCl3) δ 7.16–7.12 (m, 1 H), 6.85–6.83 (m, 2 H), 6.55–6.50 (m, 2 H), 6.42–6.41 (m, 2 H).
4,5-Difluoro-2-(1H-pyrrol-1-yl)aniline (5d)
The crude product 5d was further purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 1:7) to provide the product as white solid (87%). 1H NMR (400 MHz, CDCl3) δ 7.06–7.00 (m, 1 H), 6.82–6.80 (m, 2 H), 6.62–6.56 (m, 1 H), 6.39–6.38 (m, 2 H).
4-(7-Methoxy-4,5-dihydropyrrolo[1,2-a][13]quinoxalin-4-yl)-N,N-dimethylaniline (7)
A mixture of the aniline 5a (94 mg, 0.50 mmol), the benzaldehydes 6a (75 mg, 0.50 mmol), 1,2,3-benzotriazole (59 mg, 0.50 mmol), and AlCl3 (18 mg, 0.13 mmol) in THF (4 mL) was stirred at room temperature for 4 h. The mixture was diluted with water (15 mL) and extracted with ethyl acetate (15 mL × 3). The organic layers were combined, washed with saturated NaHSO3 solutions (15 mL) three times, and then washed with 2 N NaOH aqueous solution (15 mL) and brine (15 mL) sequentially and dried over Na2SO4. The solvent was evaporated, and the residue was further purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 1:3). The purified product was recrystallized in methanol–acetonitrile (1:1, 3 mL) solutions to provide the pure product of 7 as white solid (52 mg, 32%). 1H NMR (400 MHz, CDCl3) δ 7.35–7.33 (m, 2 H), 7.27–7.24 (m, 1 H), 7.15–7.13 (m, 1 H), 6.76–6.73 (m, 2 H), 6.42–6.38 (m, 1 H), 6.31–6.30 (m, 1 H), 6.25–6.23 (m, 1 H), 5.62–5.60 (m, 1 H), 5.44 (s, 1 H), 4.11 (s, 1 H), 3.79 (s, 3 H), 2.98 (s, 6 H). 13C NMR (100 MHz, CDCl3) δ 156.6, 150.2, 137.2, 129.8, 128.6, 128.4, 119.5, 115.0, 113.4, 112.0, 109.1, 104.8, 103.7, 100.8, 55.4, 55.0, 40.2. HRESIMS m/z calcd for C20H22N3O (MH+) 320.1763, found 320.1768. HPLC purity 98.3%, 98.9% (C-18 reverse phase, MeOH–H2O).
General Procedure for Preparation of 8, 9a–j, 11a–d, 12a–m, and 13a–b
A mixture of the aniline (5a–d, 1 mmol), the aldehyde (6a–h or 10a–d, 1 mmol), 1,2,3-benzotriazole (1 mmol), and AlCl3 (0.2 mmol) in THF (4 mL) was stirred at room temperature overnight. The mixture was diluted with water (15 mL) and extracted with ethyl acetate (15 mL × 3). The organic layers were combined, washed with 2 N NaOH solution (15 mL) and brine (15 mL) sequentially, and dried over Na2SO4 (for preparation of compounds with phenolic hydroxyl groups, including 9d, 12a, 12d–g, and 13b, the organic layers were not washed with NaOH solution, only washed with brine). The solvent was evaporated, and the residue was combined with MnO2 (10 mmol) and dissolved with acetone (5 mL). The suspension was heated to 55 °C for 24 h. MnO2 was removed by filtration, and the solvent was evaporated to provide the crude products of 8, 9a–j, 11a–d, 12a–m, and 13a–b.
4-(7-Methoxypyrrolo[1,2-a]quinoxalin-4-yl)-N,N-dimethylaniline (8)
The aniline 5a and benzaldehyde 6a were reacted according to the general procedure, and the crude product was first purified by flash silica gel column chromatography (ethyl acetate–dichloromethane, gradient up to 3:17), and then further purified by preparative HPLC (with methanol–water, gradient eluting system) to afford the products 8 as TFA salt, orange solid (59%). 1H NMR (400 MHz, methanol-d4 and CDCl3) δ 8.50–8.49 (m, 1 H), 8.06–8.03 (m, 1 H), 7.86–7.83 (m, 2 H), 7.57–7.55 (m, 1 H), 7.43–7.42 (m, 1 H), 7.21–7.16 (m, 2 H), 6.83–6.80 (m, 2 H), 3.88 (s, 3 H), 3.12 (s, 6 H). 13C NMR (100 MHz, methanol-d4 and CDCl3) δ 158.3, 153.7, 150.0, 131.0, 126.4, 122.2, 121.9, 119.2, 119.0, 117.9, 117.2, 116.0, 114.3, 111.4, 102.3, 55.2, 39.1. HRESIMS m/z calcd for C20H20N3O (MH+) 318.1606, found 318.1598. HPLC purity 98.8%, 99.0% (C-18 reverse phase, MeOH–H2O).
N,N-Dimethyl-4-(pyrrolo[1,2-a]quinoxalin-4-yl)aniline (9a)
The aniline 5e and benzaldehyde 6a were reacted according to the general procedure, and the crude product was purified by flash silica gel column chromatography (ethyl acetate–dichloromethane, gradient up to 1:9) to provide the product 9a as yellow solid (28%). 1H NMR (400 MHz, methanol-d4) δ 8.06–8.04 (m, 1 H), 7.89–7.87 (m, 1 H), 7.82–7.80 (m, 1 H), 7.76–7.73 (m, 2 H), 7.40–7.33 (m, 2 H), 6.94–6.92 (m, 1 H), 6.79–6.76 (m, 3 H), 2.96 (s, 6 H). 13C NMR (100 MHz, methanol-d4) δ 154.4, 151.9, 135.4, 129.5, 127.9, 126.7, 126.6, 125.1, 124.8, 115.3, 113.7, 111.3, 109.4, 39.0. HRESIMS m/z calcd for C19H18N3 (MH+) 288.1501, found 288.1506. HPLC purity 98.1%, 98.4% (C-18 reverse phase, MeOH–H2O).
4-(3-Nitrophenyl)pyrrolo[1,2-a]quinoxaline (9b)
The aniline 5e and benzaldehyde 6b were reacted according to the general procedure, and the crude product was purified by flash silica gel column chromatography (ethyl acetate–dichloromethane, gradient up to 1:19) to provide the product 9b as light-yellow solid (65%). 1H NMR (400 MHz, CDCl3) δ 8.93–8.91 (m, 1 H), 8.41–8.38 (m, 2 H), 8.07–8.04 (m, 2 H), 7.94–7.91 (m, 1 H), 7.76–7.72 (m, 1 H), 7.61–7.57 (m, 1 H), 7.53–7.49 (m, 1 H), 7.02–7.00 (m, 1 H), 6.98–6.95 (m, 1 H). 13C NMR (100 MHz, CDCl3) δ 151.1, 148.1, 139.7, 135.5, 134.1, 130.0, 129.3, 127.9, 126.8, 125.2, 124.3, 124.1, 123.3, 114.8, 114.1, 113.4, 107.8. HRESIMS m/z calcd for C17H12N3O2 (MH+) 290.0930, found 290.0918. HPLC purity 99.5%, 99.4% (C-18 reverse phase, MeOH–H2O).
4-(Pyrrolo[1,2-a]quinoxalin-4-yl)phenol (9d)
The aniline 5e and benzaldehyde 6c were reacted according to the general procedure, and the crude product was first purified by flash silica gel column chromatography (ethyl acetate–dichloromethane, gradient up to 2:3) and then further purified by preparative HPLC (with methanol–water, gradient eluting system) to afford the products 9d as TFA salt, yellow solid (57%). 1H NMR (400 MHz, methanol-d4) δ 8.87 (s, 1 H), 8.40–8.37 (m, 1 H), 8.03–8.01 (m, 1 H), 7.96–7.94 (m, 2 H), 7.82–7.78 (m, 1 H), 7.73–7.67 (m, 2 H), 7.35–7.33 (m, 1 H), 7.15–7.12 (m, 2 H). 13C NMR (100 MHz, methanol-d4) δ 163.0, 151.5, 131.6, 129.6, 127.3, 126.0, 125.8, 124.1, 123.1, 121.0, 120.3, 120.0, 118.6, 116.2, 115.5. HRESIMS m/z calcd for C17H13N2O (MH+) 261.1028, found 261.1025. HPLC purity 99.5%, 99.5% (C-18 reverse phase, MeOH–H2O).
4-(4-(Trifluoromethoxy)phenyl)pyrrolo[1,2-a]quinoxaline (9e)
The aniline 5e and benzaldehyde 6d were reacted according to the general procedure, and the crude product was purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 1:9) to provide the product 9e as yellow solid (70%). 1H NMR (400 MHz, CDCl3) δ 8.08–8.03 (m, 3 H), 7.97 (s, 1 H), 7.85–7.82 (m, 1 H), 7.53–7.40 (m, 4 H), 6.97–6.96 (m, 1 H), 6.90–6.89 (m, 1 H). 13C NMR (100 MHz, CDCl3) δ 152.7, 150.3, 137.1, 136.1, 130.2, 130.1, 129.9, 127.7, 127.1, 125.3, 125.0, 124.4, 121.0, 120.5 (q, CF3), 116.7, 114.8, 114.1, 113.6, 108.4. HRESIMS m/z calcd for C18H12N2OF3 (MH+) 329.0902, found 329.0908. HPLC purity 97.3%, 97.1% (C-18 reverse phase, MeOH–H2O).
4-(4-Nitrophenyl)pyrrolo[1,2-a]quinoxaline (9f).32
The aniline 5e and benzaldehyde 6e were reacted according to the general procedure, and the crude product was purified by flash silica gel column chromatography (ethyl acetate–dichloromethane, gradient up to 1:9) to provide the product 9f as yellow solid (51%). 1H NMR (400 MHz, CDCl3) δ 8.43–8.40 (m, 2 H), 8.22–8.19 (m, 2 H), 8.07–8.05 (m, 2 H), 7.94–7.92 (m, 1 H), 7.62–7.57 (m, 1 H), 7.53–7.49 (m, 1 H), 6.97 (s, 2 H). 13C NMR (100 MHz, CDCl3) δ 151.4, 148.2, 144.0, 135.5, 130.1, 129.3, 128.0, 126.8, 125.3, 124.4, 123.4, 114.8, 114.1, 113.4, 107.9. HRESIMS m/z calcd for C17H12N3O2 (MH+) 290.0930, found 290.0922. HPLC purity 99.0%, 98.1% (C-18 reverse phase, MeOH–H2O).
N-Ethyl-4-(pyrrolo[1,2-a]quinoxalin-4-yl)aniline (9h)
The aniline 5e and benzaldehyde 6f were reacted according to the general procedure, and the crude product was first purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 1:3), and then further purified by preparative HPLC (with methanol–water, gradient eluting system) to afford the products 9h as TFA salt, red oil (23%). 1H NMR (400 MHz, methanol-d4) δ 8.64 (s, 1 H), 8.19–8.16 (m, 1 H), 7.89–7.87 (m, 1 H), 7.80–7.78 (m, 2 H), 7.65–7.61 (m, 2 H), 7.56–7.52 (m, 1 H), 7.21–7.18 (m, 1 H), 6.80–6.77 (m, 2 H), 3.25 (q, J = 7.2 Hz, 2 H), 1.31 (t, J = 7.2 Hz, 3 H). 13C NMR (100 MHz, methanol-d4) δ 154.1, 150.6, 131.6, 128.7, 127.0, 125.5, 125.3, 123.3, 122.4, 120.2, 119.9, 118.0, 115.2, 114.7, 112.0, 37.1, 13.0. HRESIMS m/z calcd for C19H18N3 (MH+) 288.1501, found 288.1504. HPLC purity 98.5%, 98.4% (C-18 reverse phase, MeOH–H2O).
4-(4-(Pyrrolidin-1-yl)phenyl)pyrrolo[1,2-a]quinoxaline (9i)
The aniline 5e and benzaldehyde 6g were reacted according to the general procedure, and the crude product was first purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 1:3) and then further purified by preparative HPLC (with methanol–water, gradient eluting system) to afford the products 9i as TFA salt, orange solid (61%). 1H NMR (400 MHz, methanol-d4) δ 8.63 (s, 1 H), 8.17–8.14 (m, 1 H), 7.88–7.85 (m, 1 H), 7.77–7.74 (m, 2 H), 7.63–7.58 (m, 2 H), 7.54–7.50 (m, 1 H), 7.20–7.17 (m, 1 H), 6.60–6.58 (m, 2 H), 3.31 (s, 4 H), 2.07 (s, 4 H). 13C NMR (100 MHz, methanol-d4) δ 151.7, 150.3, 131.4, 128.6, 127.0, 125.4, 125.2, 123.1, 122.3, 120.2, 119.9, 118.0, 115.1 113.9, 111.7, 47.3, 24.9. HRESIMS m/z calcd for C21H20N3 (MH+) 314.1657, found 314.1651. HPLC purity 95.6%, 97.7% (C-18 reverse phase, MeOH–H2O).
4-(4-(Pyrrolo[1,2-a]quinoxalin-4-yl)phenyl)morpholine (9j)
The aniline 5e and benzaldehyde 6h were reacted according to the general procedure. The crude product was combined with NaBH4 (68 mg, 1.8 mmol), dissolved with methanol (3 mL)–dichloromethane (0.5 mL) mixture solvent, and stirred at room temperature for 3 h. The mixture was diluted with water (10 mL) and extracted with ethyl acetate (10 mL × 3). The organic layers were combined and dried over Na2SO4. Solvent was evaporated and the residues were further purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 1:2) and the product was recrystallized in acetonitrile to provide the product 9j as yellow solid (19%). 1H NMR (400 MHz, CDCl3) δ 8.04–7.97 (m, 4 H), 7.87–7.84 (m, 1 H), 7.47–7.44 (m, 2 H), 7.06–7.04 (m, 3 H), 6.90–6.88 (m, 1 H), 3.92–3.87 (m, 4 H), 3.32–3.27 (m, 4 H). 13C NMR (100 MHz, CDCl3) δ 153.5, 152.0, 136.1, 129.6, 129.4, 129.3, 126.6, 126.5, 125.0, 124.8, 114.5, 114.0, 113.4, 113.2, 108.1, 66.4, 48.3. HRESIMS m/z calcd for C21H20N3O (MH+) 330.1606, found 330.1577. HPLC purity 98.4%, 98.9% (C-18 reverse phase, MeOH–H2O).
4-(Pyridin-3-yl)pyrrolo[1,2-a]quinoxaline (11a)
The aniline 5e and aldehyde 10a were reacted according to the general procedure, and the crude product was purified by flash silica gel column chromatography (hexane–ethyl acetate, gradient up to 2:3) to provide the product 11a as white solid (64%). 1H NMR (400 MHz, CDCl3) δ 9.26–9.25 (m, 1 H), 8.77–8.74 (m, 1 H), 8.31–8.29 (m, 1 H), 8.02–7.96 (m, 2 H), 7.84–7.81 (m, 1 H), 7.52–7.41 (m, 3 H), 6.96–6.94 (m, 1 H), 6.89–6.87 (m, 1 H). 13C NMR (100 MHz, CDCl3) δ 150.9, 150.2, 149.0, 135.8, 135.6, 133.8, 129.8, 127.6, 126.7, 125.1, 124.5, 123.2, 114.7, 113.9, 113.3, 107.9. HRESIMS m/z calcd for C16H12N3 (MH+) 246.1031, found 246.1038. HPLC purity 99.1%, 98.9% (C-18 reverse phase, MeOH–H2O).
4-(Pyridin-4-yl)pyrrolo[1,2-a]quinoxaline (11b).32
The aniline 5e and aldehyde 10b were reacted according to the general procedure, and the crude product was further purified by flash silica gel column chromatography (ethyl acetate–dichloromethane, gradient up to 7:3) to provide the product 11b as yellow solid (64%). 1H NMR (400 MHz, CDCl3) δ 8.83–8.81 (m, 2 H), 8.05–8.02 (m, 2 H), 7.91–7.87 (m, 3 H), 7.58–7.54 (m, 1 H), 7.50–7.46 (m, 1 H), 7.00–6.98 (m, 1 H), 6.94–6.92 (m, 1 H). 13C NMR (100 MHz, CDCl3) δ 151.3, 149.9, 145.3, 135.5, 130.1, 127.9, 126.9, 125.1, 124.3, 122.5, 114.7, 114.0, 113.3, 107.8. HRESIMS m/z calcd for C16H12N3 (MH+) 246.1031, found 246.1025. HPLC purity 98.0%, 98.9% (C-18 reverse phase, MeOH–H2O).
4-(Cyclohex-1-en-1-yl)pyrrolo[1,2-a]quinoxaline (11c).33
The aniline 5e and aldehyde 10c were reacted according to the general procedure, and the crude product was further purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 1:9) to provide the product 11c as pale-yellow oil (35%). 1H NMR (400 MHz, CDCl3) δ 7.97–7.94 (m, 1 H), 7.92–7.91 (m, 1 H), 7.82–7.79 (m, 1 H), 7.48–7.40 (m, 2 H), 6.98–6.96 (m, 1 H), 6.85–6.83 (m, 1 H), 6.59–6.57 (m, 1 H), 2.69–2.67 (m, 2 H), 2.35–2.32 (m, 2 H), 1.90–1.86 (m, 2 H), 1.82–1.77 (m, 2 H). 13C NMR (100 MHz, CDCl3) δ 156.2, 136.3, 135.6, 130.8, 129.5, 126.7, 126.6, 124.8, 124.6, 113.8, 113.1, 113.0, 107.8, 26.6, 25.3, 22.4, 21.7. HRESIMS m/z calcd for C17H17N2 (MH+) 249.1392, found 249.1392. HPLC purity 98.8%, 98.9% (C-18 reverse phase, MeOH–H2O).
4-(Naphthalen-1-yl)pyrrolo[1,2-a]quinoxaline (11d).34
The aniline 5e and aldehyde 10d were reacted according to the general procedure and the crude product was first purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 3:17) and then further purified by preparative HPLC (with methanol–water, gradient eluting system) to afford the products 11d as TFA salt, pale-yellow oil (71%). 1H NMR (400 MHz, methanol-d4 and CDCl3) δ 8.50–8.49 (m, 1 H), 8.19–8.16 (m, 1 H), 8.13–8.11 (m, 2 H), 7.99–7.96 (m, 1 H), 7.84–7.82 (m, 1 H), 7.79–7.72 (m, 2 H), 7.67–7.61 (m, 2 H), 7.58–7.54 (m, 1 H), 7.48–7.44 (m, 1 H), 7.13–7.10 (m, 1 H), 7.03–7.02 (m, 1 H). 13C NMR (100 MHz, methanol-d4 and CDCl3) δ 151.6, 133.3, 131.8, 130.4, 129.6, 128.3, 128.2, 128.1, 128.0, 127.2, 127.0, 126.5, 126.2, 124.8, 124.6, 124.1, 121.0, 117.5, 117.2, 114.6. HRESIMS m/z calcd for C21H15N2 (MH+) 295.1235, found 295.1228. HPLC purity 99.4%, 99.7% (C-18 reverse phase, MeOH–H2O).
4-(7-Methoxypyrrolo[1,2-a]quinoxalin-4-yl)phenol (12a)
The aniline 5a and benzaldehyde 6c were reacted according to the general procedure and the crude product was first purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 2:3) and then further purified by preparative HPLC (with methanol–water, gradient eluting system) to afford the product 12a as TFA salt, yellow solid (10 mg, 7%). 1H NMR (400 MHz, methanol-d4 and CDCl3) δ 8.74–8.73 (m, 1 H), 8.49–8.48 (m, 1 H), 8.23–8.19 (m, 1 H), 8.15–8.12 (m, 1 H), 7.48–7.46 (m, 1 H), 7.44–7.41 (m, 1 H), 7.34–7.31 (m, 2 H), 7.17–7.15 (m, 1 H), 3.96 (s, 3 H). HRESIMS m/z calcd for C18H15N2O2 (MH+) 291.1134, found 291.1144. HPLC purity 98.3%, 98.1% (C-18 reverse phase, MeOH–H2O).
7-Methoxy-4-(4-nitrophenyl)pyrrolo[1,2-a]quinoxaline (12b)
The aniline 5a and benzaldehyde 6e were reacted according to the general procedure and the crude product was further purified by flash silica gel column chromatography eluted with pure dichloromethane to provide the product 12b as yellow solid (63%). 1H NMR (400 MHz, CDCl3) δ 8.41–8.38 (m, 2 H), 8.20–8.17 (m, 2 H), 7.99–7.98 (m, 1 H), 7.83–7.81 (m, 1 H), 7.51–7.50 (m, 1 H), 7.21–7.18 (m, 1 H), 6.94–6.91 (m, 2 H), 3.94 (s, 3 H). 13C NMR (100 MHz, CDCl3) δ 157.0, 151.7, 148.2, 144.1, 136.6, 129.2, 124.2, 123.4, 121.1, 117.3, 114.4, 114.3, 113.8, 111.1, 107.5, 55.4. HRESIMS m/z calcd for C18H14N3O3 (MH+) 320.1035, found 320.1020. HPLC purity 96.4%, 97.1% (C-18 reverse phase, MeOH–H2O).
4-(4-Hydroxyphenyl)pyrrolo[1,2-a]quinoxalin-7-ol (12d)
The aniline 5b and benzaldehyde 6c were reacted according to the general procedure, and the crude product was further purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 3:2) to provide the product 12d as yellow solid (40%). 1H NMR (400 MHz, methanol-d4) δ 8.14 (s, 1 H), 7.93–7.91 (m, 1 H), 7.80–7.77 (m, 2 H), 7.31–7.30 (m, 1 H), 7.06–7.03 (m, 1 H), 6.99–6.94 (m, 3 H), 6.87–6.85 (m, 1 H). 13C NMR (100 MHz, methanol-d4) δ 158.9, 154.7, 154.3, 136.1, 129.5, 128.7, 124.2, 120.1, 115.6, 114.8, 114.5, 114.3, 113.0, 111.9, 108.6. HRESIMS m/z calcd for C17H13N2O2 (MH+) 277.0977, found 277.0983. HPLC purity 96.4%, 96.3% (C-18 reverse phase, MeOH–H2O).
4-(4-Nitrophenyl)pyrrolo[1,2-a]quinoxalin-7-ol (12e)
The aniline 5b and benzaldehyde 6e were reacted according to the general procedure and the crude product was first purified by flash silica gel column chromatography (ethyl acetate-dichloromethane, gradient up to 1:3) and then further purified by preparative HPLC (with methanol–water, gradient eluting system) to afford the products 12e as TFA salt, yellow solid (49%). 1H NMR (400 MHz, methanol-d4 and CDCl3) δ 8.50–8.47 (m, 2 H), 8.43 (s, 1 H), 8.17–8.14 (m, 2 H), 8.06–8.03 (m, 1 H), 7.42–7.41 (m, 1 H), 7.26–7.21 (m, 2 H), 7.14–7.13 (m, 1 H). HRESIMS m/z calcd for C17H12N3O3 (MH+) 306.0879, found 306.0876. HPLC purity 99.4%, 99.5% (C-18 reverse phase, MeOH–H2O).
4-(4-(Dimethylamino)phenyl)pyrrolo[1,2-a]quinoxalin-7-ol (12g)
The aniline 5b and benzaldehyde 6a were reacted according to the general procedure and the crude product was first purified by flash silica gel column chromatography (ethyl acetate-dichloromethane, gradient up to 2:3) and then further purified by preparative HPLC (with methanol–water, gradient eluting system) to afford the products 12g as TFA salt, orange solid (25%). 1H NMR (400 MHz, methanol-d4 and CDCl3) δ 8.46 (s, 1 H), 8.01–7.99 (m, 1 H), 7.88–7.86 (m, 2 H), 7.59–7.57 (m, 1 H), 7.39–7.37 (m, 1 H), 7.19–7.17 (m, 2 H), 6.91–6.88 (m, 2 H), 3.16 (s, 6 H). 13C NMR (100 MHz, methanol-d4 and CDCl3) δ 156.6, 153.7, 150.2, 130.9, 126.5, 122.0, 121.9, 118.9, 118.5, 117.8, 116.0, 114.6, 111.5, 104.7, 39.2. HRESIMS m/z calcd for C19H18N3O (MH+) 304.1450, found 304.1465. HPLC purity 99.0%, 99.5% (C-18 reverse phase, MeOH–H2O).
7-Fluoro-4-(4-nitrophenyl)pyrrolo[1,2-a]quinoxaline (12h)
The aniline 5c and benzaldehyde 6e were reacted according to the general procedure, and the crude product was further purified by flash silica gel column chromatography (ethyl acetate–dichloromethane, gradient up to 1:19) to provide the product 12h as yellow solid (77%). 1H NMR (400 MHz, CDCl3) δ 8.44–8.42 (m, 2 H), 8.22–8.20 (m, 2 H), 8.05 (s, 1 H), 7.92–7.90 (m, 1 H), 7.76–7.73 (m, 1 H), 7.36–7.32 (m, 1 H), 7.00–6.98 (m, 2 H). HRESIMS m/z calcd for C17H11N3O2F (MH+) 308.0835, found 308.0837.
4-(7-Fluoropyrrolo[1,2-a]quinoxalin-4-yl)-N,N-dimethylaniline (12j)
The aniline 5c and benzaldehyde 6a were reacted according to the general procedure, and the crude product was purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 1:2) to provide the product 12j as yellow oil (77%). 1H NMR (400 MHz, CDCl3) δ 8.01–7.98 (m, 2 H), 7.91–7.89 (m, 1 H), 7.79–7.75 (m, 1 H), 7.71–7.67 (m, 1 H), 7.20–7.15 (m, 1 H), 7.10–7.08 (m, 1 H), 6.88–6.84 (m, 3 H), 3.07 (s, 6 H). HRESIMS m/z calcd for C19H17N3F (MH+) 306.1406, found 306.1399. HPLC purity 96.4%, 97.4% (C-18 reverse phase, MeOH–H2O).
7,8-Difluoro-4-(4-nitrophenyl)pyrrolo[1,2-a]quinoxaline (12k)
The aniline 5d and benzaldehyde 6e were reacted according to the general procedure, and the crude product was further purified by flash silica gel column chromatography (dichloromethane, 100%) to provide the product 12k as yellow solid (41%). 1H NMR (400 MHz, CDCl3) δ 8.44–8.41 (m, 2 H), 8.20–8.18 (m, 2 H), 7.93–7.92 (m, 1 H), 7.89–7.84 (m, 1 H), 7.74–7.69 (m, 1 H), 7.01–6.99 (m, 2 H). HRESIMS m/z calcd for C17H10N3O2F2 (MH+) 326.0741, found 326.0738.
4-(7,8-Difluoropyrrolo[1,2-a]quinoxalin-4-yl)-N,N-dimethylaniline (12m)
The aniline 5d and benzaldehyde 6a were reacted according to the general procedure and the crude product was purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 1:2) to provide the product 12m as yellow solid (50%). 1H NMR (400 MHz, CDCl3) δ 7.98–7.95 (m, 2 H), 7.81–7.76 (m, 2 H), 7.61–7.56 (m, 1 H), 7.09–7.07 (m, 1 H), 6.89–6.87 (m, 1 H), 6.85–6.83 (m, 2 H), 3.07 (s, 6 H). HRESIMS m/z calcd for C19H16N3F2 (MH+) 324.1312, found 324.1294. HPLC purity 97.3%, 97.2% (C-18 reverse phase, MeOH–H2O).
7-Methoxy-4-(pyridin-3-yl)pyrrolo[1,2-a]quinoxaline (13a)
The aniline 5a and aldehyde 10a were reacted according to the general procedure, and the crude product was purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 95:5) to afford the products 13a as yellow solid (78%). 1H NMR (400 MHz, methanol-d4 and CDCl3) δ 9.09–9.08 (m, 1 H), 8.69–8.67 (m, 1 H), 8.30–8.28 (m, 1 H), 7.97–7.96 (m, 1 H), 7.77–7.74 (m, 1 H), 7.56–7.52 (m, 1 H), 7.37–7.35 (m, 1 H), 7.09–7.05 (m, 1 H), 6.87–6.83 (m, 2 H), 3.86 (s, 3 H). 13C NMR (100 MHz, methanol-d4 and CDCl3) δ 156.9, 150.8, 149.6, 148.3, 136.4, 136.1, 133.9, 124.0, 123.5, 121.0, 116.7, 115.0, 114.4, 113.8, 110.3, 107.9, 54.9. HRESIMS m/z calcd for C17H14N3O (MH+) 276.1137, found 276.1121. HPLC purity 99.4%, 98.7% (C-18 reverse phase, MeOH–H2O).
4-(Pyridin-3-yl)pyrrolo[1,2-a]quinoxalin-7-ol (13b)
The aniline 5b and aldehyde 10a were reacted according to the general procedure, and the crude product was further purified by flash silica gel column chromatography (first eluted with ethyl acetate–hexanes 1:1 and then switched to methanol–dichloromethane 5:95) to afford the products 13b as yellow solid (52%). 1H NMR (400 MHz, methanol-d4 and CDCl3) δ 9.09–9.08 (s, 1 H), 8.70–8.68 (m, 1 H), 8.32–8.29 (m, 1 H), 8.04–8.03 (m, 1 H), 7.84–7.81 (m, 1 H), 7.59–7.55 (m, 1 H), 7.36–7.34 (m, 1 H), 7.13–7.09 (m, 1 H), 6.90–6.88 (m, 2 H). 13C NMR (100 MHz, methanol-d4 and CDCl3) δ 154.7, 150.9, 149.6, 148.3, 136.4, 136.2, 134.0, 124.1, 123.5, 120.5, 117.0, 115.0, 114.5, 113.7, 112.8, 108.1. HRESIMS m/z calcd for C16H12N3O (MH+) 262.0980, found 262.0963. HPLC purity 97.0%, 97.0% (C-18 reverse phase, MeOH–H2O).
General Procedure for Preparation of 9c, 9g, 12c, 12f, 12i, and 12l
A suspension of the nitro compound 9b, 9f, 12b, 12e, 12h, or 12k (0.3 mmol) and palladium on charcoal (10%, 20 mg) in methanol (10 mL) was stirred under a hydrogen balloon for 5 h. The solid was removed and the filtrate was concentrated to provide the crude product of 9c, 9g, 12c, 12f, 12i, and 12l.
3-(Pyrrolo[1,2-a]quinoxalin-4-yl)aniline (9c)
The compound 9b was reduced by hydrogenation according the general procedure, and the crude product was further purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 3:2) to provide the product 9c as pale-yellow oil (57%). 1H NMR (400 MHz, methanol-d4) δ 8.14 (s, 1 H), 7.97–7.94 (m, 1 H), 7.86–7.84 (m, 1 H), 7.47–7.43 (m, 1 H), 7.40–7.35 (m, 1 H), 7.29–7.23 (m, 2 H), 7.18–7.15 (m, 1 H), 6.95–6.90 (m, 2 H), 6.85–6.83 (m, 1 H). 13C NMR (100 MHz, methanol-d4) δ 147.9, 138.5, 135.1, 128.9, 128.3, 127.4, 126.9, 124.9, 117.8, 116.6, 115.6, 114.9, 113.9, 113.8, 109.6. HRESIMS m/z calcd for C17H14N3 (MH+) 260.1188, found 260.1187. HPLC purity 99.6%, 98.8% (C-18 reverse phase, MeOH–H2O).
4-(Pyrrolo[1,2-a]quinoxalin-4-yl)aniline (9g)
The compound 9f was reduced by hydrogenation according the general procedure, and the crude product was purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 3:2) to provide the product 9g as yellow solid (87%). 1H NMR (400 MHz, methanol-d4 and CDCl3) δ 8.03–8.02 (m, 1 H), 7.89–7.85 (m, 2 H), 7.74–7.69 (m, 2 H), 7.49–7.36 (m, 2 H), 7.02–7.00 (m, 1 H), 6.87–6.81 (m, 3 H). 13C NMR (100 MHz, methanol-d4 and CDCl3) δ 154.1, 149.2, 134.5, 129.6, 127.5, 126.8, 126.4, 126.1, 125.0, 124.6, 115.3, 114.2, 113.9, 113.4, 109.9. HRESIMS m/z calcd for C17H14N3 (MH+) 260.1188, found 260.1170. HPLC purity 99.0%, 98.3% (C-18 reverse phase, MeOH–H2O).
4-(7-Methoxypyrrolo[1,2-a]quinoxalin-4-yl)aniline (12c)
The compound 12b was reduced by hydrogenation according the general procedure, and the crude product was further purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 3:2) to provide the product 12c as yellow oil (79%). 1H NMR (400 MHz, methanol-d4) δ 7.87–7.86 (m, 1 H), 7.66–7.63 (m, 3 H), 7.22–7.21 (m, 1 H), 6.90–6.87 (m, 1 H), 6.85–6.80 (m, 2 H), 6.70–6.68 (m, 1 H), 3.78 (s, 3 H). 13C NMR (100 MHz, methanol-d4) δ 156.8, 154.3, 149.8, 135.8, 129.3, 126.0, 124.1, 120.5, 114.7, 114.6, 114.1, 113.7, 112.9, 109.1, 108.7, 54.2. HRESIMS m/z calcd for C18H16N3O (MH+) 290.1293, found 290.1286. HPLC purity 98.7%, 99.5% (C-18 reverse phase, MeOH–H2O).
4-(4-Aminophenyl)pyrrolo[1,2-a]quinoxalin-7-ol (12f)
The compound 12e was reduced by hydrogenation according the general procedure, and the crude product was purified by preparative HPLC (with methanol–water, gradient eluting system) to afford the products 12f as TFA salt, brown oil (50%). 1H NMR (400 MHz, methanol-d4) δ 8.54–8.53 (m, 1 H), 8.05–8.02 (m, 1 H), 7.78–7.75 (m, 2 H), 7.57–7.56 (m, 1 H), 7.29–7.28 (m, 1 H), 7.17–7.15 (m, 1 H), 7.12–7.09 (m, 1 H), 6.93–6.90 (m, 2 H). 13C NMR (100 MHz, methanol-d4) δ 156.6, 153.5, 150.2, 131.1, 126.3, 122.4, 121.8, 118.7, 118.3, 117.4, 117.3, 116.0, 115.8, 113.9, 104.2. HRESIMS m/z calcd for C17H14N3O (MH+) 276.1137, found 276.1146. HPLC purity 98.4%, 95.6% (C-18 reverse phase, MeOH–H2O).
4-(7-Fluoropyrrolo[1,2-a]quinoxalin-4-yl)aniline (12i)
The compound 12h was reduced by hydrogenation according the general procedure and the crude product was first purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 1:2) and then further purified by preparative HPLC (with methanol–water, gradient eluting system) to afford the product 12i as TFA salt, yellow solid (74%). 1H NMR (400 MHz, methanol-d4) δ 8.75–8.74 (m, 1 H), 8.38–8.34 (m, 1 H), 7.87–7.84 (m, 2 H), 7.73–7.68 (m, 2 H), 7.54–7.48 (m, 1 H), 7.28–7.26 (m, 1 H), 6.93–6.90 (m, 2 H). HRESIMS m/z calcd for C17H13N3F (MH+) 278.1094, found 278.1099. HPLC purity 97.0%, 96.2% (C-18 reverse phase, MeOH-H2O).
4-(7,8-Difluoropyrrolo[1,2-a]quinoxalin-4-yl)aniline (12l)
The compound 12k was reduced by hydrogenation according the general procedure, and the crude product was further purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 2:3) to provide the product 12l as yellow solid (93%). 1H NMR (400 MHz, methanol-d4 and CDCl3) δ 7.88–7.85 (m, 2 H), 7.82–7.77 (m, 2 H), 7.65–7.60 (m, 1 H), 7.06–7.05 (m, 1 H), 6.92–6.89 (m, 1 H), 6.84–6.81 (m, 2 H). HRESIMS m/z calcd for C17H12N3F2 (MH+) 296.0999, found 296.1003. HPLC purity 96.5%, 96.2% (C-18 reverse phase, MeOH–H2O).
7-(Allyloxy)-4-(4-nitrophenyl)pyrrolo[1,2-a]quinoxaline (14a)
A suspension of 12e (86 mg, 0.282 mmol), allyl bromide (78 mg, 0.64 mmol), and cesium carbonate (136 mg, 0.42 mmol) in dry DMF (2 mL) was heated to 80 °C for 30 min in a microwave reactor under argon. After cooling down, the mixture was diluted with water (20 mL) and extracted with ethyl acetate (20 mL × 3). The organic layers were combined and dried over Na2SO4. The solvent was evaporated, and the residues were further purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 1:3) to afford the product 14a as yellow solid (56 mg, 58%). 1H NMR (400 MHz, CDCl3) δ 8.41–8.38 (m, 2 H), 8.20–8.17 (m, 2 H), 7.98–7.97 (m, 1 H), 7.83–7.81 (m, 1 H), 7.51–7.50 (m, 1 H), 7.24–7.21 (m, 1 H), 6.94–6.91 (m, 2 H), 6.18–6.08 (m, 1 H), 5.52–5.47 (m, 1 H), 5.37–5.33 (m, 1 H), 4.68–4.66 (m, 2 H). 13C NMR (100 MHz, CDCl3) δ 156.0, 151.7, 148.2, 144.1, 136.6, 132.4, 129.2, 124.2, 123.4, 121.2, 117.8, 117.7, 114.4, 114.3, 113.8, 112.2, 107.5, 68.9. HRESIMS m/z calcd for C20H16N3O3 (MH+) 346.1192, found 346.1162.
4-(7-Propoxypyrrolo[1,2-a]quinoxalin-4-yl)aniline (15a)
A suspension of 14a (56 mg, 0.162 mmol) and palladium on charcoal (10%, 18 mg) in methanol (5 mL) was stirred under a hydrogen balloon overnight. The solid was removed, and the filtrate was concentrated. The residue was further purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 3:2) to provide the product 15a as yellow oil (23 mg, 45%). 1H NMR (400 MHz, methanol-d4) δ 8.07–8.06 (m, 1 H), 7.86–7.84 (m, 1 H), 7.71–7.68 (m, 2 H), 7.30–7.29 (m, 1 H), 7.05–7.02 (m, 1 H), 6.96–6.95 (m, 1 H), 6.87–6.82 (m, 3 H), 4.00–3.96 (m, 2 H), 1.85–1.80 (m, 2 H), 1.09–1.05 (m, 3 H). 13C NMR (100 MHz, methanol-d4) δ 156.4, 154.4, 149.9, 135.6, 129.3, 125.8, 124.1, 120.5, 115.4, 115.0, 114.3, 113.8, 113.2, 109.8, 109.1, 69.3, 21.8, 9.1. HRESIMS m/z calcd for C20H20N3O (MH+) 318.1606, found 318.1588. HPLC purity 97.7%, 98.2% (C-18 reverse phase, MeOH–H2O).
4-(7-(2-Fluoroethoxy)pyrrolo[1,2-a]quinoxalin-4-yl)aniline (15b)
A solution of 12e (69 mg, 0.226 mmol), 2-fluoroethanol (89 mg, 1.4 mmol), and triphenylphosphine (183 mg, 0.70 mmol) in dry THF (4 mL) was stirred under argon and cooled to 0 °C. DEAD (145 mg, 0.83 mmol) was added dropwise. The mixture was warmed to room temperature and heated to 60 °C for 40 min in a microwave reactor. The solvent was evaporated, and the residues were further purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 3:7) to provide the crude product 14b. The crude product 14b was dissolved in methanol (5 mL), and palladium on charcoal (10%, 29 mg) was added. The suspension was stirred under a hydrogen balloon overnight. The solid was removed, and the filtrate was concentrated. The residue was further purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 2:1) to provide the product 15b as yellow oil (26 mg, 36%). 1H NMR (400 MHz, methanol-d4) δ 8.02–8.00 (m, 1 H), 7.82–7.79 (m, 1 H), 7.69–7.67 (m, 2 H), 7.29–7.28 (m, 1 H), 7.05–7.01 (m, 1 H), 6.94–6.92 (m, 1 H), 6.85–6.82 (m, 2 H), 6.81–6.78 (m, 1 H), 4.82–4.80 (m, 1 H), 4.70–4.68 (m, 1 H), 4.29–4.27 (m, 1 H), 4.22–4.20 (m, 1 H). 13C NMR (100 MHz, methanol-d4) δ 155.7, 154.4, 150.0, 135.6, 129.4, 125.7, 124.1, 120.9, 115.2, 115.0, 114.4, 113.7, 113.2, 109.9, 109.1, 82.2, 80.5, 67.3, 67.1. HRESIMS m/z calcd for C19H17N3OF (MH+) 322.1356, found 322.1330. HPLC purity 97.6%, 97.7% (C-18 reverse phase, MeOH–H2O).
2-((4-(4-(Dimethylamino)phenyl)pyrrolo[1,2-a]quinoxalin-7-yl)-oxy)-2-methylpropanamide (16)
A solution of 12g (84 mg, 0.28 mmol) and NaOH (180 mg, 4.5 mmol) in DMA (3 mL) was stirred at room temperature for 1 h, 2-bromo-2-methylpropanamide (131 mg, 0.789 mmol) was added, and the mixture was heated to 50 °C for 3 h. After cooling down, the mixture was diluted with water (12 mL) and extracted with ethyl acetate (20 mL × 3). The organic layers were combined, washed with brine (20 mL), and dried over Na2SO4. The solvent was evaporated and the residues were further purified by flash silica gel column chromatography (ethyl acetate–hexanes, gradient up to 19:1) and the crude product was further purified by preparative HPLC (with methanol–water, gradient eluting system) to afford the product 16 as TFA salt, yellow solid (55 mg, 41%). 1H NMR (400 MHz, methanol-d4) δ 8.63–8.62 (m, 1 H), 8.20–8.17 (m, 1 H), 7.91–7.88 (m, 2 H), 7.67–7.65 (m, 1 H), 7.54–7.53 (m, 1 H), 7.36–7.32 (m, 1 H), 7.24–7.22 (m, 1 H), 6.94–6.91 (m, 2 H), 3.14 (s, 6 H), 1.65 (s, 6 H). 13C NMR (100 MHz, methanol-d4) δ 177.3, 153.9, 153.5, 150.3, 131.0, 125.9, 122.8, 122.0, 121.5, 120.5, 119.3, 117.8, 115.9, 114.3, 111.3, 109.6, 81.2, 38.4, 23.8. HRESIMS m/z calcd for C23H25N4O2 (MH+) 389.1978, found 389.1956. HPLC purity 98.6%, 97.1% (C-18 reverse phase, MeOH–H2O).
Cells, Media, Plasmids, and Proteins
The 293-DR-GFP and 293-SA-GFP reporter cell lines along with the pBASce I-SceI expressing and pCAGGS empty vector control plasmids were kindly provided by Jeremy Stark and described previously.27 All cell lines except for PC-3 were maintained in complete DMEM (DMEM + 4.5 g/L D-glucose + L-glutamine + 10% fetal calf serum + penicillin/streptomycin); PC-3 cells were maintained in DMEM:F12 (1:1) with 10% fetal calf serum and penicillin/streptomycin. Human RAD51 protein was purified as described previously,23 and the purity was confirmed by SDS-PAGE (Supporting Information, Figure S5). Rad51 and Rad52 proteins from Saccharomyces cerevisiae were kindly provided by Yuen-Ling Chan and purified as described previously.35 RecA protein from Escherichia coli was purchased from New England Biolabs.
Screening Libraries
We conducted high-throughput screening of the ASDI Diversity library of 6800 compounds and the Library of Pharmacologically Active Compounds (LOPAC1280). From each of the library master plates, 2.5 nmol of compound was acoustically transferred to individual assay plates at Nextval, Inc. This gave a final concentration of 50 μM for each library compound in 50 μL per well of final reaction volume for both the RAD51-DNA binding and RAD51-mediated strand pairing assays.
Fluorescence Polarization-Based RAD51-DNA Binding Assay
The fluorescence polarization-based DNA binding assay was performed as previously described with a few modifications.23 Reactions were carried out in black 384-well polystyrene plates. In each well, RAD51 protein was combined with reaction buffer and varying concentrations of compound in DMSO in an initial volume of 40 μL and incubated at 37 °C for 40 min. Then 10 μL of reaction buffer containing fluorescently labeled ssDNA substrate (5′-Alexa488-oligo-dT45) was added and the reaction incubated at 37 °C for another 40 min, after which the plate was read on a Tecan Infinite 200 Pro plate reader equipped with two sets of 485 ± 25/535 ± 35 nm with parallel or perpendicularly oriented polarization filters. In both steps of the reaction setup, buffer components were maintained at 20 mM HEPES-NaOH (pH 7.5), 2 mM ATP, 10 mM MgCl2, 0.1 mM TCEP-HCl (tris(2-carboxyethyl)phosphine), and 0.25 μM BSA. The final reaction contained 4% DMSO, 0.2 μM RAD51, 30 mM NaCl, 2% glycerol, and 2.22 nM ssDNA substrate. Compound IC50 values were obtained by least-squares regression fitting of appropriate linear, log, or sigmoid dose–response models to the data, with the standard error indicated.
Solution Assay for RAD51-Mediated Strand Pairing
The FRET-based solution assay for RAD51-mediated strand pairing was performed as described previously using the 51-nt Black Hole Quencher (BHQ1)-tagged ssDNA substrate DHD-HQ and the 162-bp Alexa488-tagged dsDNA substrate DHD162.24 Reactions were performed in black 384-well polystyrene plates. RAD51 in reaction buffer and compound in DMSO were combined in 20 μL and incubated at room temperature for 40 min. Then 10 μL of solution containing the ssDNA substrate and CaCl2 was added to each well, and the reaction was incubated at 37 °C for 5 min to allow RAD51-ssDNA filaments to form. Finally, 20 μL of solution was added containing the dsDNA double hairpin substrate with homology to the ssDNA substrate and the plate was incubated at 37 °C for 50 min to allow RAD51-mediated strand invasion, which is measured as a decrease in fluorescence intensity as the BHQ1-tagged ssDNA is paired with the Alexa488-tagged dsDNA. The final reaction contained 25 mM HEPES-NaOH (pH 7.5), 3 mM ATP, 1 mM TCEP-HCl, 1.5 μM BSA, 4% DMSO, 0.1 μM RAD51, 2 mM NaCl, 0.1% glycerol, 10 nM ssDNA, and 5 nM dsDNA. The concentration of CaCl2 was maintained at 5 mM throughout all three steps of the reaction setup. Fluorescence measurements were taken on a Tecan Infinite 200 Pro plate reader equipped with 485 ± 20/535 ± 35 nm filters.
Gel-Based RAD51-DNA Binding Assays
Human RAD51 protein in reaction buffer was combined with compound in DMSO in a total volume of 9 μL, incubated at 37 °C for 40 min, and then 1 μL of 4,373-nt closed circular pRS306 virion ssDNA was added and the reaction was incubated at 37 °C for an additional 5 min. At this point, the reaction consisted of 25 mM HEPES-NaOH (pH 7.0), 3 mM ATP, 5 mM MgCl2, 1 mM TCEP, 100 μg/mL BSA, 0.5 μM RAD51, and 1.5 μM nucleotide concentration ssDNA. For experiments to determine the stability of RAD51-ssDNA complexes in salt, 1 μL of NaCl was added to achieve the indicated concentration and the reaction was incubated for an additional 5 min at 37 °C. RAD51-ssDNA complexes were fixed by addition of glutaraldehyde in 2 μL to 0.25%, incubated for 5 min at 37 °C, and run on a 1% agarose/1× TAE gel, which was stained in SYBR Gold (Molecular Probes) and photographed under UV light.
Gel-Based D-Loop Assay
Recombinase protein in reaction buffer was combined with compound in DMSO in a total volume of 8 μL, incubated at 37 °C for 10 min, and then 1 μL of 5′-32P-labeled 90-mer ssDNA (5′-TACGAATGCACACGGTGTGGTGGGCCCAGGTATTGTTAGCGGTTTGAAGCAGGCGGCAGAAGAAGTAACAAAGGAACCTAGAGGCCTTTT) with homology to the plasmid pRS30636 was added and the reaction was incubated at 37 °C for an additional 5 min. Then 1 μL of supercoiled pRS306 dsDNA plasmid was added to the reaction and incubated at 37 °C for 20 min. At this point, reactions containing human RAD51 protein consisted of 25 mM HEPES-NaOH (pH 7.0), 1 mM ATP, 1 mM MgCl2, 1 mM DTT, 1.5 μM BSA, 4.5% DMSO, 0.5 μM RAD51, 11 mM NaCl, 1% glycerol, 20 nM ssDNA, and 5 nM pRS306 dsDNA. For reactions containing Saccharomyces cerevisiae Rad51 + Rad52 proteins or Escherichia coli RecA protein, the reactions were carried out in a manner similar to the one described for the human proteins, except MgCl2 was present at 1 mM during the incubation with ssDNA and then increased to 10 mM during the incubation with dsDNA; the reaction buffer during incubation with dsDNA consisted of 20 mM HEPES–NaOH (pH 7.5), 2 mM ATP, 10 mM MgCl2, 1 mM DTT, 100 μg/mL BSA 10 nM ssDNA, and 4.5 nM dsDNA. HsRAD51 was present at 0.5 to 1.5 μM depending on the specific D-loop activity of the protein prep, ScRad51 and ScRad52 were both present at 1 and 2 μM respectively, and EcRecA was present at 0.4 μM. The reactions were deproteinized with 0.8% SDS and 0.8 mg/mL proteinase K, mixed with gel loading buffer, and run on a 0.9% agarose/1× TAE gel. The gel was dried under vacuum on a positively charged nylon membrane for 2 h at 80 °C, exposed to a phosphor screen overnight, and imaged on a Storm 860 PhosphorImager. Quantitation of free oligo and D-loop bands was performed using ImageJ software (NIH, Bethesda, MD), and compound IC50 values were obtained by least-squares regression fitting of appropriate linear, log, or sigmoid dose–response models to the data, with the standard error indicated.
Detection of RAD51 Protein in Subcellular Fractions
Subconfluent dishes of 293-DR-GFP cells were grown in the presence of compound for the indicated times, washed once with ice-cold PBS, and processed for subcellular fractionation at 4 °C. Soluble nuclear fractions were obtained by incubating cells in 5 pellet volumes of hypotonic buffer (10 mM HEPES–NaOH (pH 7.9), 10 mM KCl, 1.5 mM MgCl2, 1 mM DTT, 10 mM NaF, 2 mM sodium orthovanadate, 2 mM sodium pyrophosphate, Complete Mini protease inhibitors (Roche)) on ice for 15 min, pelleting the cells, and saving the supernatant as the cytosolic fraction. The pellet was then resuspended in 1.5 pellet volumes of hypotonic buffer +0.5 M NaCl, incubated on ice for 15 min, and the supernatant containing the soluble nuclear fraction was saved. Chromatin fractions were obtained from cells by extracting three times for 5 min each at 4 °C in 5 pellet volumes of RIPA buffer (50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 10 mM NaF, 1 mM sodium orthovanadate, 2 mM sodium pyrophosphate, Complete Mini protease inhibitors (Roche)) to remove total soluble proteins and saving the pellet containing the insoluble chromatin fraction. Samples of subcellular fractions were boiled for 5 min in SDS-PAGE loading buffer (25 mM Tris-HCl (pH 6.8), 1% SDS, 2.5% β-mercaptoethanol, 5 mM DTT, 0.0015% bromophenol blue, 5% glycerol), separated on a 10% SDS-PAGE, and transferred to a PVDF membrane by overnight electrophoretic transfer. The membrane was probed with primary antibody (rabbit antihuman RAD51 polyclonal antibody from Pacific Immunology at a 1:2000 dilution, or rabbit antihuman PCNA polyclonal antibody from Millipore at a 1:1000 dilution), followed by secondary antibody (HRP-conjugated donkey antirabbit IgG from GE Healthcare at a 1:2000 dilution) and developed with Western Lightning Plus-ECL reagent (PerkinElmer).
Quantitation of DNA Repair Efficiency in Cells
293-DR-GFP or 293-SA-GFP cells were electroporated in Opti-MEM with 37.5 μg/mL of the I-Sce-I endonuclease bearing pCBASce plasmid or the pCAGGS empty vector control in 0.4 cm cuvettes at 325 V, 975 μF, and seeded into 6-well plates with 2.5 mL of complete DMEM + 0.5% DMSO with compound and allowed to outgrow for 24 h. Following outgrowth, cells were harvested and suspended in PBS with 1 μg/mL 7-aminoactinomycin D (7-AAD) and analyzed on a BD FACSCalibur flow cytometer. Dead and apoptotic cells were gated out based on size, shape, and 7-AAD staining. The fraction of GFP-positive cells was determined within the population of live cells.
Clonogenic Survival Assay
Cells were grown in flasks to subconfuence, irradiated with X-rays at 1.5 Gy/min using a Philips RT250 Maxitron, trypsinized, and seeded at 1000–1500 cells per well in 6-well cluster plates (Corning 3516) in complete media with 0.5% DMSO and the indicated dose of compound. The plates were incubated for 7–11 days until colonies of at least 50 cells formed, washed once with PBS, fixed, and stained for at least 30 min at room temperature in 6% glutaraldehyde + 0.5% crystal violet, washed twice in cold tap water, dried, photographed, and scored using a custom macro set in ImageJ for automatic colony counting.
RAD51 Cytological Focus Formation
Cells were grown on glass coverslips for 2 days prior to irradiation with X-rays at 1.5 Gy/min using a Philips RT250 Maxitron. Following the indicated outgrowth time of 0, 2, 4, or 8 h, coverslips to be costained with RAD51 and RPA were washed once in PBS, fixed in ice-cold fixative solution (3% paraformaldehyde, 3.4% sucrose in 1× PBS) for 10 min at room temperature, washed three times in PBS, incubated in permeabilization buffer (20 mM HEPES-NaOH (pH 7.4), 50 mM NaCl, 3 mM MgCl2, 0.5% Triton X-100, 300 mM sucrose) for 10 min at room temperature, washed three times in PBS, and incubated for at least 15 min in blocking buffer (1% BSA in 1× PBS) at 4 °C. For coverslips to be costained with RAD51 and γH2AX, the same procedure was carried out with the fixation and permeabilization steps reversed. The coverslips were incubated with primary antibody (rabbit antihuman RAD51 polyclonal antibody from Pacific Immunology, mouse anti-γH2AX monoclonal antibody clone JBW301 from Millipore or mouse anti-RPA monoclonal antibody clone RPA34-19 from Calbiochem) at a 1:1000 dilution in blocking buffer for 18 h at 4 °C, washed 3 times in PBS, incubated with secondary antibody (Alexa488-conjugated goat antiraddit IgG polyclonal antibody from Invitrogen, or Alexa594-conjugated goat antimouse IgG polyclonal antibody from Invitrogen) at a 1:1000 dilution in blocking buffer for 1 h at room temperature, washed 3 times in PBS, air-dried, and mounted onto glass slides in Vectashield with DAPI H-1200 (Vector laboratories). Slides were viewed on a Zeiss Axio Imager.M1 epifluorescence microscope equipped with a CCD camera for acquiring images of at least 100 randomly selected nuclei per test condition at a representative focal plane within the nuclear volume. Subnuclear RAD51, RPA, and γH2AX foci were quantified using ImageJ using a custom macro set that automatically counts foci. Focus colocalization was scored for overlapping RAD51 and RPA foci in nuclei that contained at least five RPA foci and for RAD51 and γH2AX foci in nuclei that contained at least five γH2AX foci and did not exhibit pan-nuclear γH2AX staining. Plots of RAD51 foci were generated using R software with the optional Bee Swarm package.37,38
Acknowledgments
We thank Yuen-Ling Chan for providing recombinase proteins and DNA substrates used in the D-loop gel assay and gel shift assays. This work was funded by NIH grants 2R01CA142642 to P.P.C and A.P.K, as well as funding from the Wendy Will Cancer Fund.
ABBREVIATIONS
- HR
homologous recombination
- DSBs
DNA double strand breaks
- ICLs
interstrand cross-links
- ssDNA
single-stranded DNA
- NHEJ
nonhomologous end-joining
- SAR
structure activity relationship
- HT
high-throughput
Footnotes
Author Contributions
W.L. and B.B. contributed equally and are considered as cofirst authors. P.P.C. and A.P.K. contributed equally and are considered as colast authors.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.5b01762.
NMR spectrum of compounds 1, 7, and 8, mass spectrum of compound 1, representative D-loop gel images used to generate D-loop IC50 values in Tables 1–3, RAD51 protein levels in cytosolic, soluble nuclear, and chromatin subcellular fractions of cells treated with or without 9h, representative gel images of salt-titration midpoint experiments from Figure 7e, coomassie-stained SDS-PAGE of the human RAD51 preparation used for D-loop and DNA binding assays (PDF)
SMILES molecular formula strings (CSV)
Notes
The authors declare no competing financial interest.
References
- 1.Thompson LH, Schild D. Homologous Recombinational Repair of DNA Ensures Mammalian Chromosome Stability. Mutat Res, Fundam Mol Mech Mutagen. 2001;477:131–153. doi: 10.1016/s0027-5107(01)00115-4. [DOI] [PubMed] [Google Scholar]
- 2.Tebbs RS, Zhao Y, Tucker JD, Scheerer JB, Siciliano MJ, Hwang M, Liu N, Legerski RJ, Thompson LH. Correction of Chromosomal Instability and Sensitivity to Diverse Mutagens by a Cloned Cdna of the Xrcc3 DNA Repair Gene. Proc Natl Acad Sci U S A. 1995;92:6354–6358. doi: 10.1073/pnas.92.14.6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu N, Lamerdin JE, Tebbs RS, Schild D, Tucker JD, Shen MR, Brookman KW, Siciliano MJ, Walter CA, Fan W, Narayana LS, Zhou ZQ, Adamson AW, Sorensen KJ, Chen DJ, Jones NJ, Thompson LH. Xrcc2 and Xrcc3, New Human Rad51-Family Members, Promote Chromosome Stability and Protect against DNA Cross-Links and Other Damages. Mol Cell. 1998;1:783–793. doi: 10.1016/s1097-2765(00)80078-7. [DOI] [PubMed] [Google Scholar]
- 4.Takata M, Sasaki MS, Tachiiri S, Fukushima T, Sonoda E, Schild D, Thompson LH, Takeda S. Chromosome Instability and Defective Recombinational Repair in Knockout Mutants of the Five Rad51 Paralogs. Mol Cell Biol. 2001;21:2858–2866. doi: 10.1128/MCB.21.8.2858-2866.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klein HL. The Consequences of Rad51 Overexpression for Normal and Tumor Cells. DNA Repair. 2008;7:686–693. doi: 10.1016/j.dnarep.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hine CM, Seluanov A, Gorbunova V. Use of the Rad51 Promoter for Targeted Anti-Cancer Therapy. Proc Natl Acad Sci U S A. 2008;105:20810–20815. doi: 10.1073/pnas.0807990106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vispe S, Cazaux C, Lesca C, Defais M. Overexpression of Rad51 Protein Stimulates Homologous Recombination and Increases Resistance of Mammalian Cells to Ionizing Radiation. Nucleic Acids Res. 1998;26:2859–2864. doi: 10.1093/nar/26.12.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slupianek A, Schmutte C, Tombline G, Nieborowska-Skorska M, Hoser G, Nowicki MO, Pierce AJ, Fishel R, Skorski T. Bcr/Abl Regulates Mammalian Reca Homologs, Resulting in Drug Resistance. Mol Cell. 2001;8:795–806. doi: 10.1016/s1097-2765(01)00357-4. [DOI] [PubMed] [Google Scholar]
- 9.Bello VE, Aloyz RS, Christodoulopoulos G, Panasci LC. Homologous Recombinational Repair Vis-a-Vis Chlorambucil Resistance in Chronic Lymphocytic Leukemia. Biochem Pharmacol. 2002;63:1585–1588. doi: 10.1016/s0006-2952(02)00954-1. [DOI] [PubMed] [Google Scholar]
- 10.Hansen LT, Lundin C, Spang-Thomsen M, Petersen LN, Helleday T. The Role of Rad51 in Etoposide (Vp16) Resistance in Small Cell Lung Cancer. Int J Cancer. 2003;105:472–479. doi: 10.1002/ijc.11106. [DOI] [PubMed] [Google Scholar]
- 11.Russell JS, Brady K, Burgan WE, Cerra MA, Oswald KA, Camphausen K, Tofilon PJ. Gleevec-Mediated Inhibition of Rad51 Expression and Enhancement of Tumor Cell Radiosensitivity. Cancer Res. 2003;63:7377–7383. [PubMed] [Google Scholar]
- 12.Ito M, Yamamoto S, Nimura K, Hiraoka K, Tamai K, Kaneda Y. Rad51 Sirna Delivered by Hvj Envelope Vector Enhances the Anti-Cancer Effect of Cisplatin. J Gene Med. 2005;7:1044–1052. doi: 10.1002/jgm.753. [DOI] [PubMed] [Google Scholar]
- 13.Budke B, Logan HL, Kalin JH, Zelivianskaia AS, Cameron McGuire W, Miller LL, Stark JM, Kozikowski AP, Bishop DK, Connell PP. Ri-1: A Chemical Inhibitor of Rad51 That Disrupts Homologous Recombination in Human Cells. Nucleic Acids Res. 2012;40:7347–7357. doi: 10.1093/nar/gks353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Renodon-Cornière A, Weigel P, Le Breton M, Fleury F. New Potential Therapeutic Approaches by Targeting Rad51-Dependent Homologous Recombination. In: Chen C, editor. New Research Directions in DNA Repair. InTech; Rijeka, Croatia: 2013. [Google Scholar]
- 15.Carvalho JF, Kanaar R. Targeting Homologous Recombination-Mediated DNA Repair in Cancer. Expert Opin Ther Targets. 2014;18:427–458. doi: 10.1517/14728222.2014.882900. [DOI] [PubMed] [Google Scholar]
- 16.Huang F, Mazin AV. Targeting the Homologous Recombination Pathway by Small Molecule Modulators. Bioorg Med Chem Lett. 2014;24:3006–3013. doi: 10.1016/j.bmcl.2014.04.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang F, Mazina OM, Zentner IJ, Cocklin S, Mazin AV. Inhibition of Homologous Recombination in Human Cells by Targeting Rad51 Recombinase. J Med Chem. 2012;55:3011–3020. doi: 10.1021/jm201173g. [DOI] [PubMed] [Google Scholar]
- 18.Ishida T, Takizawa Y, Kainuma T, Inoue J, Mikawa T, Shibata T, Suzuki H, Tashiro S, Kurumizaka H. Dids, a Chemical Compound That Inhibits Rad51-Mediated Homologous Pairing and Strand Exchange. Nucleic Acids Res. 2009;37:3367–3376. doi: 10.1093/nar/gkp200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Budke B, Kalin JH, Pawlowski M, Zelivianskaia AS, Wu M, Kozikowski AP, Connell PP. An Optimized Rad51 Inhibitor That Disrupts Homologous Recombination without Requiring Michael Acceptor Reactivity. J Med Chem. 2013;56:254–263. doi: 10.1021/jm301565b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takaku M, Kainuma T, Ishida-Takaku T, Ishigami S, Suzuki H, Tashiro S, van Soest RW, Nakao Y, Kurumizaka H. Halenaquinone, a Chemical Compound That Specifically Inhibits the Secondary DNA Binding of Rad51. Genes Cells. 2011;16:427–436. doi: 10.1111/j.1365-2443.2011.01494.x. [DOI] [PubMed] [Google Scholar]
- 21.Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-Strand Break Repair-Independent Role for Brca2 in Blocking Stalled Replication Fork Degradation by Mre11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ying S, Hamdy FC, Helleday T. Mre11-Dependent Degradation of Stalled DNA Replication Forks Is Prevented by Brca2 and Parp1. Cancer Res. 2012;72:2814–2821. doi: 10.1158/0008-5472.CAN-11-3417. [DOI] [PubMed] [Google Scholar]
- 23.Jayathilaka K, Sheridan SD, Bold TD, Bochenska K, Logan HL, Weichselbaum RR, Bishop DK, Connell PP. A Chemical Compound That Stimulates the Human Homologous Recombination Protein Rad51. Proc Natl Acad Sci U S A. 2008;105:15848–15853. doi: 10.1073/pnas.0808046105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Budke B, Chan YL, Bishop DK, Connell PP. Real-Time Solution Measurement of Rad51- and Reca-Mediated Strand Assimilation without Background Annealing. Nucleic Acids Res. 2013;41:e130. doi: 10.1093/nar/gkt362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng J, Giguere PM, Onajole OK, Lv W, Gaisin A, Gunosewoyo H, Schmerberg CM, Pogorelov VM, Rodriguiz RM, Vistoli G, Wetsel WC, Roth BL, Kozikowski AP. Optimization of 2-Phenylcyclopropylmethylamines as Selective Serotonin 2c Receptor Agonists and Their Evaluation as Potential Antipsychotic Agents. J Med Chem. 2015;58:1992–2002. doi: 10.1021/jm5019274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng J, Giguere PM, Lv W, Roth BL, Kozikowski AP. Design and Synthesis of (2-(5-Chloro-2,2-Dimethyl-2,3-Dihydrobenzofuran-7-Yl)Cyclopropyl)Methanamine as a Selective Serotonin 2c Agonist. Tetrahedron Lett. 2015;56:3420–3422. doi: 10.1016/j.tetlet.2015.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bennardo N, Cheng A, Huang N, Stark JM. Alternative-Nhej Is a Mechanistically Distinct Pathway of Mammalian Chromosome Break Repair. PLoS Genet. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. Genetic Steps of Mammalian Homologous Repair with Distinct Mutagenic Consequences. Mol Cell Biol. 2004;24:9305–9316. doi: 10.1128/MCB.24.21.9305-9316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raderschall E, Stout K, Freier S, Suckow V, Schweiger S, Haaf T. Elevated Levels of Rad51 Recombination Protein in Tumor Cells. Cancer Res. 2002;62:219–225. [PubMed] [Google Scholar]
- 30.Guillon J, Grellier P, Labaied M, Sonnet P, Leger JM, Deprez-Poulain R, Forfar-Bares I, Dallemagne P, Lemaitre N, Pehourcq F, Rochette J, Sergheraert C, Jarry C. Synthesis, Antimalarial Activity, and Molecular Modeling of New Pyrrolo[1,2-a]Quinoxalines, Bispyrrolo[1,2-a]Quinoxalines, Bispyrido[3,2-E]-Pyrrolo[1,2-a]Pyrazines, and Bispyrrolo[1,2-a]Thieno[3,2-E]-Pyrazines. J Med Chem. 2004;47:1997–2009. doi: 10.1021/jm0310840. [DOI] [PubMed] [Google Scholar]
- 31.Campiani G, Morelli E, Gemma S, Nacci V, Butini S, Hamon M, Novellino E, Greco G, Cagnotto A, Goegan M, Cervo L, Dalla Valle F, Fracasso C, Caccia S, Mennini T. Pyrroloquinoxaline Derivatives as High-Affinity and Selective 5-Ht(3) Receptor Agonists: Synthesis, Further Structure-Activity Relationships, and Biological Studies. J Med Chem. 1999;42:4362–4379. doi: 10.1021/jm990151g. [DOI] [PubMed] [Google Scholar]
- 32.Preetam A, Nath M. An Eco-Friendly Pictet-Spengler Approach to Pyrrolo- and Indolo[1,2-a]Quinoxalines Using P-Dodecylbenzenesulfonic Acid as an Efficient Bronsted Acid Catalyst. RSC Adv. 2015;5:21843–21853. [Google Scholar]
- 33.Guillon J, Forfar I, Mamani-Matsuda M, Desplat V, Saliege M, Thiolat D, Massip S, Tabourier A, Leger JM, Dufaure B, Haumont G, Jarry C, Mossalayi D. Synthesis, Analytical Behaviour and Biological Evaluation of New 4-Substituted Pyrrolo[1,2-a]-Quinoxalines as Antileishmanial Agents. Bioorg Med Chem. 2007;15:194–210. doi: 10.1016/j.bmc.2006.09.068. [DOI] [PubMed] [Google Scholar]
- 34.Wang C, Li Y, Guo R, Tian J, Tao C, Cheng B, Wang H, Zhang J, Zhai H. Iodine-Catalyzed Facile Synthesis of Pyrrolo- and Indolo[1,2-a]Quinoxalines. Asian J Org Chem. 2015;4:866–869. [Google Scholar]
- 35.Cloud V, Chan YL, Grubb J, Budke B, Bishop DK. Rad51 Is an Accessory Factor for Dmc1-Mediated Joint Molecule Formation During Meiosis. Science. 2012;337:1222–1225. doi: 10.1126/science.1219379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sikorski RS, Hieter P. A System of Shuttle Vectors and Yeast Host Strains Designed for Efficient Manipulation of DNA in Saccharomyces Cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2015. [Google Scholar]
- 38.Eklund A. The Bee Swarm Plot, an Alternative to Stripchart. 2015 [Google Scholar]