

Abstract
Endoplasmic reticulum (ER) stress and unfolded protein response (UPR) are involved in anti‐human immunodeficiency virus (HIV) drugs and alcohol‐induced liver disease in a significant number of patients infected with HIV. However, the precise mechanism by which the drugs and alcohol cause ER stress remains elusive. We found that ritonavir‐boosted lopinavir (RL) activated two canonical UPR branches without activation of the third canonical activating transcription factor 6 (ATF6) branch in either HepG2 cells or primary mouse hepatocytes. In the RL‐treated cells, ATF6 localization in the Golgi apparatus required for its activation was reduced; this was followed by Golgi fragmentation and dislocation/redistribution of Golgi‐resident enzymes. Severities of Golgi fragmentation induced by other anti‐HIV drugs varied and were correlated with the ER stress response. In the liver of mice fed RL, alcohol feeding deteriorated the Golgi fragmentation, which was correlated with ER stress, elevated alanine aminotransferase, and liver steatosis. The Golgi stress response (GSR) markers GCP60 and HSP47 were increased in RL‐treated liver cells, and knockdown of transcription factor for immunoglobulin heavy‐chain enhancer 3 of the GSR by small interfering RNA worsened RL‐induced cell death. Cotreatment of pharmacological agent H89 with RL inhibited the RL‐induced Golgi enzyme dislocation and ER stress. Moreover, the coat protein complex II (COPII) complexes that mediate ER‐to‐Golgi trafficking accumulated in the RL‐treated liver cells; this was not due to interference of RL with the initial assembly of the COPII complexes. RL also inhibited Golgi fragmentation and reassembly induced by short treatment and removal of brefeldin A. Conclusion: Our study indicates that ER‐to‐Golgi trafficking is disrupted by anti‐HIV drugs and/or alcohol, and this contributes to subsequent ER stress and hepatic injury. (Hepatology Communications 2017;1:122‐139)
Abbreviations
- AIDS
acquired immune deficiency syndrome
- ALT
alanine aminotransferase
- ANOVA
analysis of variance
- APV
amprenavir
- ATF6
activated transcription factor 6
- BFA
brefeldin A
- CHOP
CCAAT/enhancer binding protein‐homologous protein
- COPI
coat protein complex I
- COPII
coat protein complex II
- CREB3
cyclic adenosine monophosphate responsive element binding protein 3
- DAV
darunavir
- eIF2α
eukaryotic translation initiation factor 2A
- ER
endoplasmic reticulum
- FASN
sterol regulatory element binding protein 1c (SREBP1c)‐mediated fatty acid synthase
- GADD34
growth arrest and DNA‐damage‐inducible 34
- GCP60
Golgi resident protein 60
- GM130
Golgi matrix protein 130
- GRP78
glucose regulated protein 78
- GSR
Golgi stress response
- H&E
hematoxylin and eosin staining
- HIV
human immunodeficiency virus
- HSP47
heat shock protein 47
- IRE1
the serine/threonine‐protein kinase/endoribonuclease inositol‐requiring enzyme 1
- LPV
lopinavir
- MAN2A1
Golgi‐resident enzyme α‐mannosidase II
- mRNA
messenger RNA
- NFV
nelfinavir
- OCT
optimum cutting temperature
- p58IPK
protein kinase inhibitor p58
- PBS
phosphate‐buffered saline
- PBST
Triton‐X 100 in PBS
- PDIA4
protein disulfide isomerase family A member 4
- PERK
protein kinase RNA‐like ER kinase
- PMH
primary mouse hepatocytes
- qPCR
quantitative polymerase chain reaction
- RL
ritonavir‐boosted lopinavir
- RLA
RL plus alcohol
- RT‐PCR‐
reverse‐transcription polymerase chain reaction
- RTV
ritonavir
- S1P, S2P
site‐1‐protease, site‐2‐protease
- SAR1
secretion‐associated and Ras‐related factor 1
- Sec31
a COPII coat complex component
- siRNA
small interfering RNA
- sXBP1
spliced X‐box binding protein 1
- TFE3
transcription factor for immunoglobulin heavy‐chain enhancer 3
- TG
thapsigargin
- TM
tunicamycin
- UPR
unfolded protein response
Introduction
There are 35 million human immunodeficiency virus (HIV)‐infected individuals worldwide, and 2 million persons acquire HIV infection every year.1, 2 The situation demands development of antiretroviral drugs. While anti‐HIV vaccines have yet to be developed, anti‐HIV drugs are effective.3, 4 For instance, HIV protease inhibitors that inhibit HIV proteinase or protease are used in highly active antiretroviral therapies. Patients with HIV/acquired immune deficiency syndrome (AIDS) under lifetime therapies live longer; however, there are reports that certain antivirals singly or in combination increase the risk of comorbidities, which undermine the quality of life.5, 6 Although most side effects of the antivirals are manageable, some can be very serious and fatal. For instance, liver disease has emerged as the most common non‐AIDS‐related cause of death among patients with HIV, accounting for 15% of all deaths.7, 8 Nearly 50% of patients infected with HIV abuse or consume alcohol, which not only impairs patients' adherence to highly active antiretroviral therapies but also deteriorates anti‐HIV drug‐induced hepatotoxicity, leading to greater morbidity and mortality.9, 10 In addition, there are 10 million patients with HIV/AIDS worldwide co‐infected with other viruses (e.g., hepatitis C virus, hepatitis B virus).7, 11, 12 Antivirals against the co‐infections could further increase the severity of liver toxicities and injuries. Currently, there is no consensus on how to manage patients with AIDS suffering from liver disease because the mechanisms underlying the hepatotoxicity of alcohol abuse and drugs are still under investigation.
Major mechanisms underlying the hepatotoxicity are idiosyncratic hypersensitive reactions mediated by the adaptive immune system and cellular stress responses that either induce cell death directly or generate danger signals costimulating the immune system.7, 11, 13 The cellular stress responses are most relevant because both alcohol and anti‐HIV drugs are metabolized in the liver, and this is bound to stress the liver. We and others reported that alcohol and/or anti‐HIV drugs induced endoplasmic reticulum (ER) stress, lipid accumulation, and hepatic cell death.13, 14, 15, 16 The ER stress initially triggers a protective unfolded protein response (UPR), which involves three canonical ER stress sensors, serine/threonine‐protein kinase/endoribonuclease inositol‐requiring enzyme 1 (IRE1), protein kinase RNA‐like ER kinase (PERK), and activated transcription factor 6 (ATF6), to restore ER homeostasis. However, prolonged UPR resulting from chronic alcohol and/or long‐term anti‐HIV therapies induces hepatosteatosis, inflammation, cell death, and development of fibrosis and cirrhosis.13, 17, 18, 19 The ER stress/UPR has thus been an important therapeutic target in liver disease. Xenobiotics, including molecular chaperones (e.g., 4‐phenylbutyrate and tauroursodeoxycholic acid) and UPR enhancers (e.g., salubrinal and valproate) to ensure proper ER homeostasis have been developed and tested in a variety of in vitro and in vivo models.20 However, these compounds for restoration of ER homeostasis only yield limited protective effects. The incomplete protections through targeting the ER suggest that although ER stress is involved in alcohol and drug‐induced hepatotoxicity, there are other drug and alcohol‐targeted cellular components that either enhance or are upstream of the ER stress response.21
The organelle that is closely associated with the ER is the Golgi apparatus. There is bidirectional ER–Golgi trafficking transporting proteins and lipids between the two organelles.22 The anterograde route of ER‐to‐Golgi is mediated by coat protein complex II (COPII) complexes, and the retrograde route of Golgi‐to‐ER is mediated by COPI complexes. Like the ER, the structure and capacity of the Golgi fluctuate according to physiological demands or pathological stress conditions. Abnormal Golgi morphologies occur when the protein load and modifications exceed Golgi capacity.21, 22, 23 For instance, enlargement or hypertrophy of the Golgi has been observed in human liver due to amyloidosis, toxic hepatitis, and other conditions, including drug‐induced cholestasis and malnutrition.24 Atrophy or degeneration occurs during starvation, viral hepatitis, and choline or protein deficiency.25 Under viral infection, the Golgi is partially fragmented.26 In man and animal, chronic alcohol feeding with nutritionally adequate diets induced ultrastructural abnormalities of hepatocytes.27 There is also an adaptive Golgi stress response (GSR) that involves Golgi resident protein 60 (GCP60), heat shock protein 47 (HSP47), and transcription factor for immunoglobulin heavy‐chain enhancer 3 (TFE3) to cope with disrupted homeostasis of the Golgi.21, 22 However, persistent stress on the Golgi disturbs metabolism in the liver, and dysfunction of the Golgi apparatus is associated with various stress‐induced liver injuries.21, 22, 23, 24, 25, 26, 27, 28 The role that the Golgi plays in drug and alcohol‐induced ER stress and what underlying molecular mechanism links Golgi dysfunction with the ER stress response and injury are not known. In this study, we observed that antivirals induced Golgi morphological changes in cultured liver cells and mouse livers. These changes linked to disrupted ER‐to‐Golgi trafficking of ATF6 and other factors that potentiated the ER stress response and liver injury.
Materials and Methods
CELL CULTURES
HepG2 cells were purchased from ATCC and maintained in Dulbecco's modified Eagle's medium with 4.5 g/mL glucose, 10% fetal bovine serum, nonessential amino acids, and 100 U/mL of penicillin‐streptomycin. The cells were treated with ritonavir (RTV)‐boosted lopinavir (LPV) or other anti‐HIV drugs at 0‐20 μg/mL. Dimethyl sulfoxide (0.05%, weight [wt]/volume [vol]) plus ethanol (0.05%, vol/vol) in Dulbecco's modified Eagle's medium was used as the vehicle control. Primary mouse hepatocytes (PMH) were isolated by the University of Southern California Liver Cell Culture Core.13 The PMH were either treated with or without 85 mM alcohol for 16 hours. The next day, the cells were treated with the antivirals. After the treatments, the cells were washed with ice‐cold phosphate‐buffered saline (PBS) and subjected to fixing or protein and RNA extractions. In vitro assays were repeated at least 3 times with three to four duplicates for each treatment.
PLASMID AND SMALL INTERFERING RNA TRANSFECTION
p3xFLAG‐ATF6 was a gift from Ron Prywes, Columbia University Biological Science, New York, NY (Addgene plasmid #11975). The liver cells were transfected with 1 μg of p3xFLAG‐ATF6 with Lipofectamine 3000 following the manufacturer's instructions and were treated with the drugs or pharmaceutical inhibitors/activators 24 hours after the transfection. TFE3 small interfering RNA (siRNA) and scrambled siRNA control were purchased from ThermoFisher Scientific. Briefly, siRNA was diluted at 30 pmol/200 μL OptiMEM and mixed with 3 μL of Lipofectamine 3000 at room temperature for 15 minutes; to this solution, 0.1 million HepG2 cells in 1 mL of antibiotic‐free complete medium was added. The transfected cells were incubated in 12‐well plates coated with collagen 1 for 40 hours. The gene knockdown was confirmed by real time polymerase chain reaction (RT‐PCR).
EXPERIMENTAL ANIMALS
Male C57BL/6 mice were purchased from The Jackson Laboratory. Mice were given a 5% alcohol liquid diet (Dyets, Inc., Bethlehem, PA) or an isocaloric control diet for 10 days. Mice were intraperitoneally injected with RTV, LPV, or other anti‐HIV drugs at concentrations of 0‐50 μg/mL from day 5 of feeding. On day 10, mice were gavaged with 30% alcohol in PBS (5 mg/g body weight) or the same volume of isocaloric maltose solution. The mice were killed for serum and liver sampling 8 hours after the alcohol gavage. All animals were treated in accordance with the Guide for Care and Use of Laboratory Animals, and the study was approved by the local animal care committee.
LIVER PATHOLOGICAL PARAMETERS
Analyses of serum alanine aminotransferase (ALT) and liver steatosis have been described.17, 18 For hematoxylin and eosin (H&E) staining, liver tissues were fixed in 10% formalin overnight at 4°C and washed with and stored in 80% ethanol. The fixed tissues were embedded in paraffin, sectioned at 5 μm, and then stained with H&E. For Oil Red O staining, liver tissues were embedded in optimum cutting temperature (OCT) compound, snap frozen, sectioned at 5 μm, and mounted on glass slides, which were fixed in 10% formalin and stained with an Oil Red O isopropanol solution (Electron Microscopy Sciences, Hatfield, PA).
IMMUNOBLOTTING, RT‐PCR, AND QUANTITATIVE PCR
Methods for cytosolic/nuclear protein and RNA extractions, immunoblotting, and RT‐PCR have been described.13, 17, 18 For quantitative PCR (qPCR analysis), approximately 20 mg tissue was ground in QIAzol and extracted with the DirectZol RNA extraction kit (Zymo). All primers for Golgi and ER stress markers are listed in Supporting Table 1.
IMMUNOFLUORESCENT STAINING, CONFOCAL MICROSCOPY, AND QUANTIFICATION
For in vitro experiments, cells were attached to 18‐mm‐round glass‐cover slides coated with collagen 1 and cultured in 12‐well plates. After the drug and/or alcohol treatments, cells were washed with ice‐cold PBS, fixed in 4% paraformaldehyde solution for 15 minutes, permeabilized in 2% Triton‐X 100 in PBS (PBST) for 15 minutes, and then blocked with 10% goat serum in PBS with 0.05% Tween 20. The fixed cells were probed with rabbit polyclonal anti‐Golgi matrix protein 130 (GM130) antibodies (1:250; Genetex) and/or mouse monoclonal anti‐Golgi‐resident enzyme α‐mannosidase II (MAN2A1) antibodies (1:50; Santa Cruz), which were diluted in the blocking buffer overnight. The cells on the slides were then labeled with secondary antibodies conjugated with fluorescence and/or nuclear counterstained with 10 ng/mL Hoechst blue in PBS. After being sequentially washed with PBST, PBS, and double‐distilled water, the cover slides were mounted on glass slides with prolonged gold mounting media. For in vivo experiments, liver tissues were embedded in OCT compound, snap frozen, sectioned at 5 μm, and mounted on glass slides, which were fixed and permeabilized in ice‐cold acetone at –20oC for 10 minutes, air dried at room temperature for 30 minutes, and then processed with the same staining method as in vitro experiments. The cis‐Golgi structure of liver cells was indicated by the immunostaining of GM130, whereas Golgi regions (mainly medial) were indicated by the immunostaining of MAN2A1.29, 30 Color images were taken with a Zeiss 510 LS confocal microscope and processed with Zen 2.1 using the same parameters. Changes in Golgi morphology were based on positive immunostaining and quantified with the Particle Analysis function of ImageJ according to published methods.31 For counting cells under Golgi fragmentation, 10 random microscopic fields, including approximately 50 primary mouse hepatocytes or 100 HepG2 cells, were taken. For colocalization analysis on proteins and Golgi structure components, channels of red, green, blue colors were separated with ImageJ and analyzed with Just Another Colocalization Plugin (JACoP).32
STATISTICS
Data are presented as means ± SEM unless otherwise indicated. Statistical analyses were performed with GraphPad Prism 6 using one‐way analysis of variance (ANOVA) for comparing multiple groups and two‐way ANOVA for comparing trends between different treatments. P values of 0.05 or less are considered significant.
Results
COPRESENCE OF ER STRESS AND ABNORMAL GOLGI LOCALIZATION OF ATF6 INDUCED BY RTV AND LPV
Both the anti‐HIV drugs (RTV plus LPV [RL]) and the ER stress‐inducing agent tunicamycin (TM) induced an ER stress response in HepG2 cells indicated by the expression of ER stress markers of CCAAT/enhancer binding protein‐homologous protein (CHOP), spliced X‐box binding protein 1 (sXBP1), and phosphorylation of eukaryotic translation initiation factor 2A (eIF2α) and IRE1α, representing each branch of the UPR (Fig. 1A‐C; Supporting Fig. S1). However, compared to 10 μg/mL, RL at 20 μg/mL resulted in significantly faster increases in CHOP expression while the increases by TM were relatively equal at both concentrations (Fig. 1A). In the dose‐response tests with concentrations of 5‐20 μg/mL, RL increased CHOP messenger RNA (mRNA) 11‐fold to 123‐fold compared to the control. CHOP expression was dramatically increased when the concentrations of RL were higher than 12.5 μg/mL (Fig. 1B). In contrast, TM induced smoother CHOP increases of 22‐fold to 31‐fold at concentrations of 5‐20 μg/mL. The patterns and trends of increased sXBP1 in response to RL were similar to those of CHOP (Supporting Fig. S2A,B). However, up‐regulation of glucose‐regulated protein 78 (GRP78) was undetectable under RL in either time‐course or dose‐response experiments (Supporting Fig. S2C,D). In immunoblotting assays, TM but not RL increased CHOP and GRP78 in a dose‐dependent manner (Fig. 1C). In mammalian cells, up‐regulation of GRP78 relies mainly on activation of the ATF6 branch.33, 34 To know whether RL specifically suppresses GRP78 via the ATF6 branch, effects of RL on expression of protein disulfide isomerase family A member 4 (PDIA4) and protein kinase inhibitor p58 (p58IPK) downstream of ATF614, 33, 34, 35 were investigated. PDIA4, p58IPK (Protein Kinase Inhibitor P58), and Grp78 were suppressed 2‐fold, 5‐fold, and 14‐fold, respectively, in RL+TM compared to TM, whereas suppression was not observed in growth arrest and DNA‐damage‐inducible (Gadd)34, Gadd45, or TRIB3 (Tribbles homolog 3), which are not regulated by ATF6 (Supporting Fig. S3), indicating that RL inactivates specifically the ATF6 branch. Activation of ATF6 requires transorganelle proteolytic processing and translocation from the ER to the Golgi and from the Golgi to the nucleus.33, 34 To confirm the interrupted ATF6 activation, HepG2 cells were transfected with FLAG‐ATF6 and treated with RL and/or thapsigargin (TG). RL did not affect levels of cytosolic or nuclear ATF6. TG reduced cytosolic ATF6 and increased nuclear ATF6, which were blocked by RL (Fig. 1D). Nuclear localizations of ATF6 were observed in more than 90% of the cells treated with TG but in less than 20% of the cells treated with RL (Fig. 1E). Moreover, cytosolic ATF6 was less colocalized with the Golgi structural protein GM130 in RL‐treated cells than in TG‐treated cells. RL reduced the TG‐induced Golgi localization of ATF6 and GM130 by 40% (Fig. 1F; Supporting Fig. S4).
Figure 1.
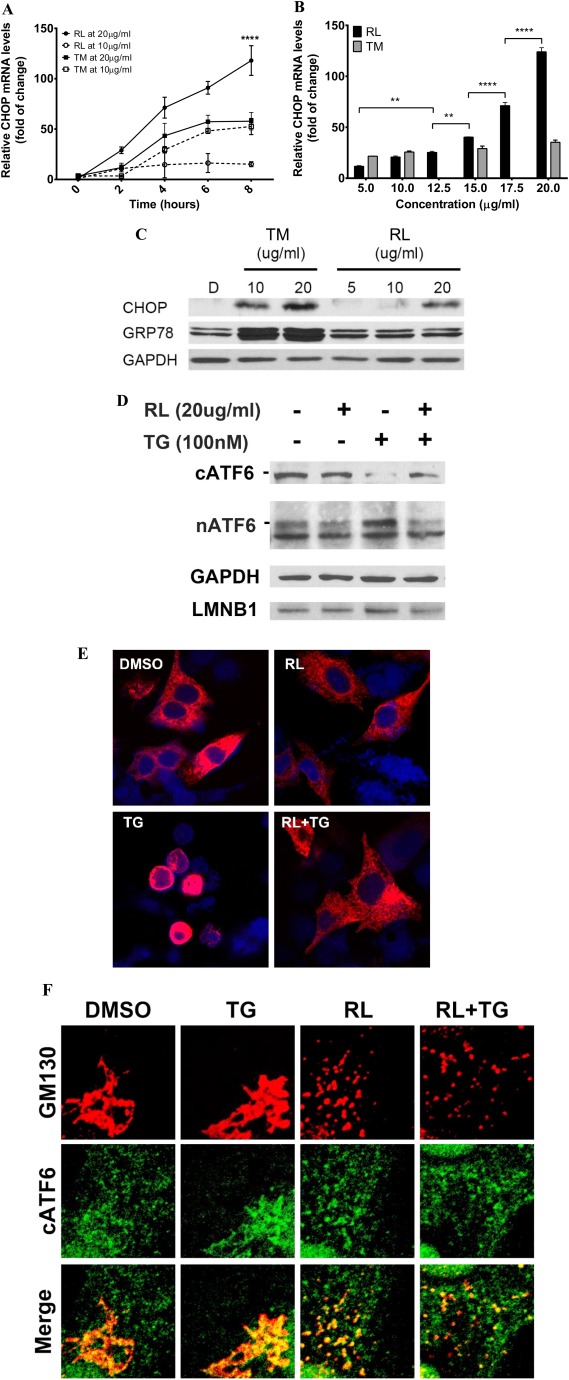
Effects of anti‐HIV drugs on ER stress response and cellular localization of ATF6. (A) Time course of mRNA of ER stress marker CHOP. HepG2 cells were treated with RL or TM. (B) Dose response of CHOP mRNA in the RL‐ or TM‐treated cells. (C) GRP78 and CHOP protein expression in TM‐ and RL‐treated cells. HepG2 cells were treated with TM or RL for 8 hours at different concentrations; D, DMSO as vehicle control. (D) Effect of RL on the processing of ATF6 protein in HepG2 cells. Cells were transfected with FLAG‐ATF6 for 24 hours and treated with either DMSO or RL at 20 μg/mL for 2 hours followed by treatment with 100 nM TG for 2 hours. GAPDH and LMNB1 were used as loading controls for cytoplasmic and nuclear proteins, respectively. (E) Blockage of TG‐induced nuclear ATF6 by RL. Confocal images (×630) showing nuclear localization of ATF6 in the cells expressing ATF6. (F) RL‐induced dispersal of ATF6 and Golgi components. Confocal images (×1000) showing changes of colocalization and dispersal of ATF6 (green) and GM130 (red) in the cells exposed to the drugs. **P < 0.01; ****P < 0.0001 compared to control or indicated otherwise; n = 5‐7. Error bars indicate standard error mean (SEM). Abbreviations: cATF6, cytoplasmic ATF6 precursor; DMSO, dimethyl sulfoxide; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; LMNB1, lamina B1; nATF6, activated nuclear ATF6.
INDUCTION OF GOLGI FRAGMENTATION, ENZYME DISLOCATION, GSR, AND CELL DEATH
Translocation of ATF6 to the Golgi requires ER‐to‐Golgi trafficking and functional Golgi.33, 34, 35 Interruptions of Golgi function are commonly associated with changes in the Golgi morphology.22 In HepG2 cells, immunofluorescent staining with GM130 and MAN2A1 demonstrated dramatic effects of the anti‐HIV drugs on the Golgi. RL at 20 μg/mL caused complete dispersal of the Golgi matrix GM130 (Fig. 2A) and dislocation/redistribution of MAN2A1 (Fig. 2B) to ER or other cytosolic locations, some of which were costained with the ER marker protein disulfide isomerase (PDI) (Supporting Fig. S5A); however, the Golgi structures remained as perinuclear‐located and tightly organized ribbons in the control cells. The Golgi fragmentation and enzyme dislocation were comparable to those caused by brefeldin A (BFA), which is known to interfere with ER–Golgi trafficking (Fig. 2A,B).26, 35 In dose‐dependent experiments, the minimal dose to induce observable Golgi fragmentation was 15 μg/mL (Supporting Fig. S5B); at this dose 81% of cells were with Golgi fragmentation and the size of the fragmented Golgi that reflected severity of the Golgi fragmentation was 50% of that of the control (Supporting Fig. S5C). In addition, individual treatment with either RTV or LPV induced Golgi fragmentation (Supporting Fig. S5D). The average size of the fragmented Golgi (from GM130 staining) was decreased by 60% in the cells treated with LPV and by 80% with RL at 20 μg/mL for 4 hours (Supporting Fig. S5E). To demonstrate whether Golgi function in addition to the morphological changes was affected by RL, three GSR molecular markers, GCP60, HSP47, and TFE3 were examined. In qPCR analysis of gene expression at the transcriptional level, treatment with RL at 20 μg/mL up‐regulated GCP60 and HSP47 3‐fold and 10‐fold, respectively (Fig. 2C,D). These levels were comparable with those induced by modeled Golgi stress inducers sodium monensin and benzyl‐GalNAc. Effects of TFE3 knockdown (TFE3KD) by siRNA on ER stress and cell death injury were further investigated. In the TFE3KD cells with TFE3 protein being abolished (Supporting Fig. S6), RL‐induced cell death was increased by 30% (Fig. 2E) whereas expression of CHOP remained unchanged (Fig. 2F), indicating that the drug‐induced GSR caused additional injury.
Figure 2.
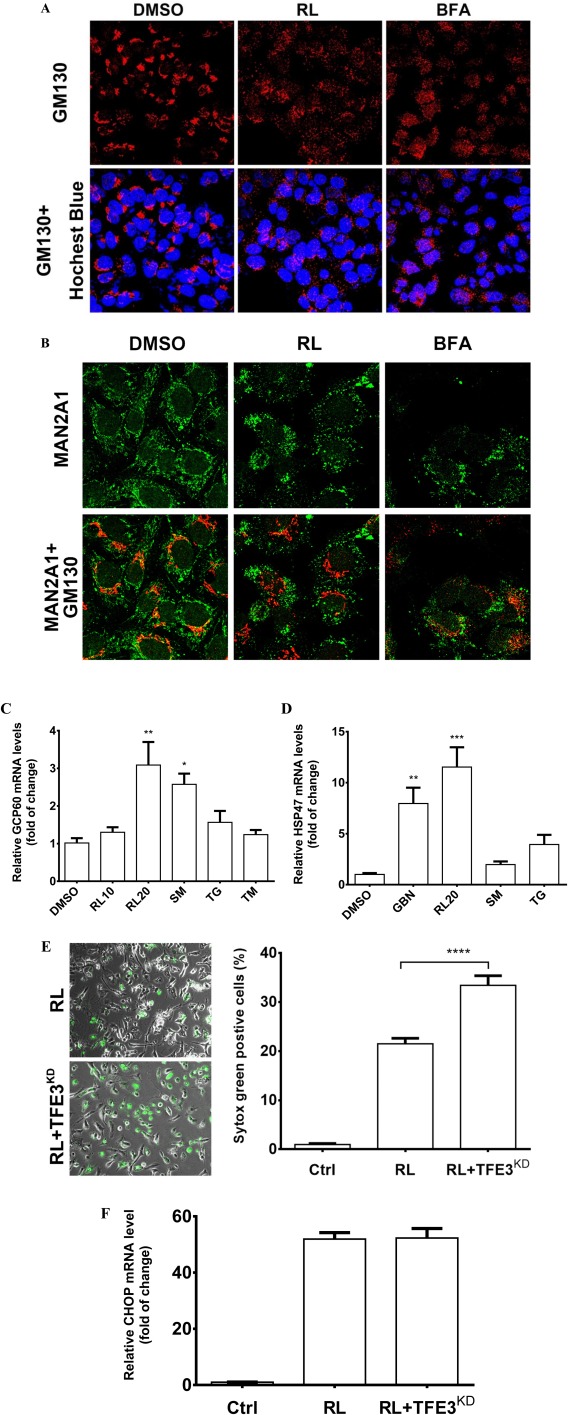
Golgi fragmentation, stress response, and death of liver cells treated with the anti‐HIV drugs. (A) Golgi fragmentation in HepG2 cells. The cells were treated with RL (20 μg/mL), DMSO (0.1%) as vehicle control, or BFA (20 nM) as positive control for 4 hours. The Golgi matrix was labeled with anti‐GM130 antibodies (red), and nuclei were stained with Hoechst blue (blue, ×400). (B) Cellular distribution of MAN2A1 and GM130. The cells were treated with RL, DMSO, and BFA for 2 hours. MAN2A1 and GM130 were labeled by green and red fluorescence, respectively (×630). (C) Expression of Golgi stress marker GCP60 and (D) HSP47 at 8 hours. RL10, RL at 10 μg/mL; RL20, RL at 20 μg/mL. (E) Cell death in the cells deficient in GSR. Cell death was revealed by Syntox green staining (left panel, ×400) and quantified by ImageJ (right panel) 16 hours after the treatments. RL, RL treatment in cells transfected with control siRNA; RL+TFE3KD, RL treatment in cells transfected with TFE3 siRNA. (F) mRNA levels of CHOP at 4 hours. Ctrl, control cells without any transfection or drug treatment; *P < 0.05; **P < 0.01; ***P < 0.005; ****P < 0.0001 compared to control or indicated otherwise; n = 5‐6. Error bars indicate standard error mean (SEM). Abbreviations: GBN, benzyl‐GalNAc; SM, sodium monensin.
GOLGI FRAGMENTATION, ER STRESS, AND HEPATIC INJURIES IN PRIMARY HEPATOCYTES AND MOUSE LIVER
The presence of ER stress and Golgi fragmentation was also reproduced in PMH. Expression of sXBP1 and CHOP was increased in PMH in response to RL at concentrations from 12.5 μg/mL to 20 μg/mL (Fig. 3A; Supporting Fig. S7A,B). Expression of GRP78 at either mRNA or protein levels was not changed in RL‐treated PMH (Supporting Fig. S7C,D), indicating that the ATF6 branch was inhibited. Alcohol did not increase sXBP1 expression within the short period of treatment. Golgi fragmentation was observed in RL‐treated PMH (Fig. 3B). The degree of Golgi fragmentation increased as the RL concentration and exposure time increased. Compared to the HepG2 cells, PMH appeared more sensitive to RL in terms of Golgi fragmentation. Under treatment at 10 μg/mL for 4 hours, half of the PMH were under Golgi fragmentation (Fig. 3C) whereas less than 5% of the HepG2 cells were under the fragmentation (Supporting Fig. S5B). At 20 μg/mL for 2 hours, Golgi fragmentation occurred in 50% of the HepG2 cells and in greater than 80% of PMH (Fig. 3C; Supporting Fig. S7E). Most importantly, Golgi fragmentation was also observed in liver sections from mice treated with RL or RL plus alcohol (RLA) (Fig. 4A). The size of Golgi fragments was reduced by 20% in the liver of mice treated with RL and by 45% in the liver of mice treated with RLA (Fig. 4B). Along with Golgi fragmentation, ALT levels were elevated 4‐fold in RL‐treated mice and 6‐fold in RLA‐treated mice (Fig. 4C). CHOP expression was increased 10‐fold in mice with RLA (Fig. 4D). In H&E and Oil Red O staining, accumulation of fatty droplets in the mouse livers was observed under RL treatment, but this was more severe under RLA treatment (Fig. 4E,F; Supporting Fig. S8). The fatty droplets surrounded the central vein, indicating the presence of macrovascular steatosis (Fig. 4F).
Figure 3.
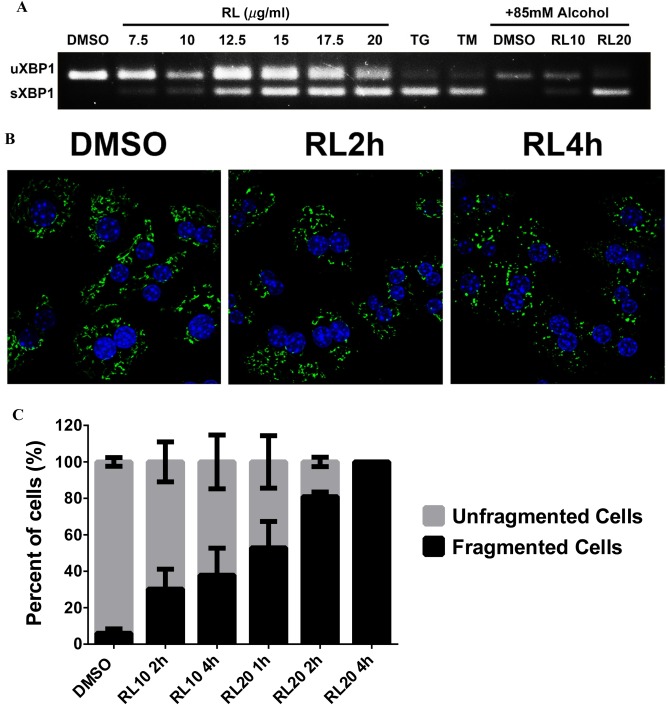
Association of ER stress with Golgi dysfunction in primary mouse hepatocytes. (A) Induction of sXBP1 in PMH treated with anti‐HIV drugs and alcohol. RL10, RL at 10 μg/mL; RL20, RL at 20 μg/mL. (B) Golgi fragmentation in PMH treated with RL. Left panel, confocal images (×630) showing Golgi fragmentation (indicated by GM130 labeled with green fluorescence) in PMH treated with the drug for 2 hours and 4 hours. (C) Quantitation of drug‐induced Golgi fragmentation at different time points and concentrations. Error bars indicate standard error mean (SEM). Abbreviation: DMSO, dimethyl sulfoxide; uXBP1, un‐spliced XBP1.
Figure 4.
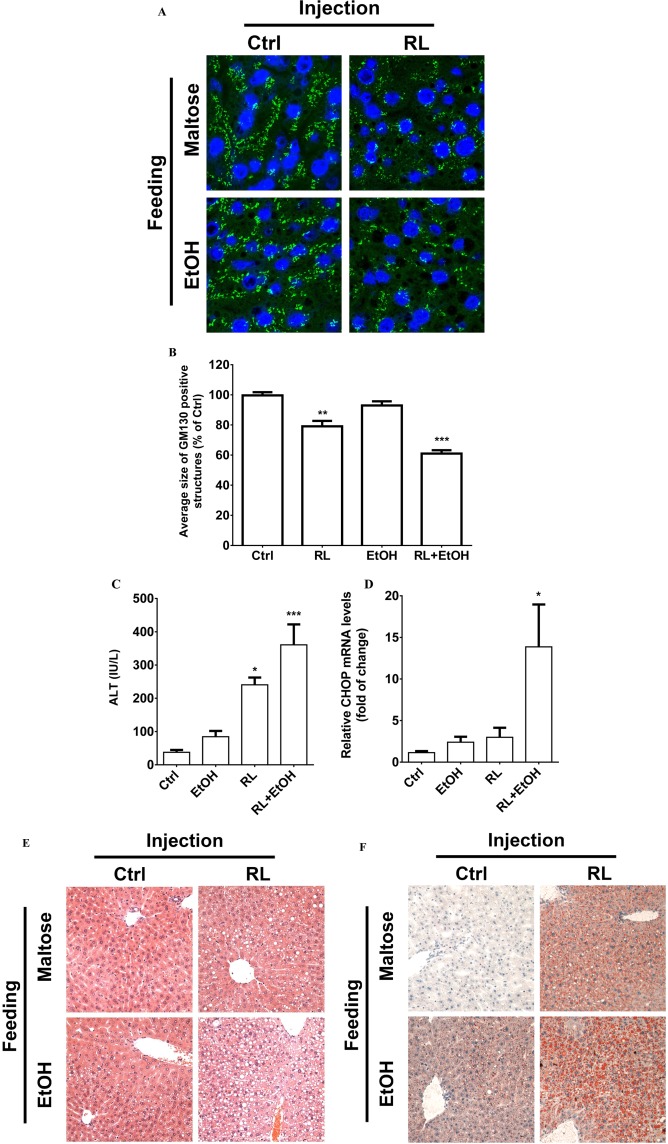
Association of Golgi fragmentation with liver injuries in mice injected with anti‐HIV drugs and/or fed with alcohol. (A) Confocal images (×1000) showing Golgi fragmentation of hepatocytes of liver sections. The Golgi morphology was probed with immunofluorescence (green) using anti‐GM130 antibodies. The nuclei of hepatocytes were revealed with Hoechst blue staining. Ctrl, sample from mice injected with vehicle control and fed with isocaloric control diet; RL, mice injected with RTV+LPV and fed with control diet; EtOH, sample from mice injected with vehicle control and fed with alcohol diet. The image at the lower right corner represents samples from mice injected with RL and fed with an alcohol diet (i.e., RL+EtOH). (B) Quantitation of the Golgi fragmentation. (C) Levels of serum ALT in mice treated with the drugs and/or alcohol. (D) CHOP expression in liver samples from RL‐injected mice with or without alcohol feeding. (E) H&E staining of liver tissues showing fatty liver injury (×200). (F) Oil Red O staining of liver tissues showing fat accumulation in mice treated with the drugs and/or alcohol (×200). *P < 0.05; **P < 0.01; ***P < 0.005 compared to control; n = 4‐6. Error bars indicate standard error mean (SEM). Abbreviation: EtOH, ethanol.
CONTRIBUTION OF GOLGI ABNORMALITY TO THE DOWNSTREAM ER STRESS RESPONSE
The RL‐induced Golgi enzyme dislocation occurred earlier than the ER stress response regulated by the ATF6 branch. The Golgi‐resident enzyme (e.g., MAN2A1) dislocation known to be a consequence of the loss of Golgi integrity22, 26, 36 was observed in HepG2 cells at 30 minutes after RL treatment (Fig. 5A). CHOP was not significantly increased until 1 hour after the drug treatment, and no change of GRP78 was detected in the first hour after RL (Fig. 5B). Consistent with H89 alleviating BFA‐induced Golgi abnormality and ER stress,37, 38, 39 cotreatments with H89 also reduced RL‐induced MAN2A1 dislocation/redistribution and the ER stress response (Fig. 5C,D). H89 reduced expression of CHOP and sXBP1 20‐fold and 2‐fold, respectively in RL‐treated cells but not in TM‐treated cells (Fig. 5D). Furthermore, there was a contribution of the drug‐induced Golgi fragmentation to downstream ER stress. Different degrees of Golgi fragmentation could be induced by different anti‐HIV drugs, including amprenavir (APV), darunavir (DAV), LPV, nelfinavir (NFV), RTV, and RL. At 20 μg/mL, RL treatment for 4 hours induced fragmented Golgi in 100% of the cells and the strongest ER stress response whereas DAV, which did not cause Golgi fragmentation, induced a minimal ER stress response (Supporting Fig. S9). The Golgi fragmentation and ER stress response by APV, RTV, LPV, and NFV fell in between those by DAV and RL. Golgi fragmentation by APV, RTV, LPV, NFV, and RL were well associated with downstream increases of CHOP expression (r 2 = 0.972, P < 0.0001).
Figure 5.
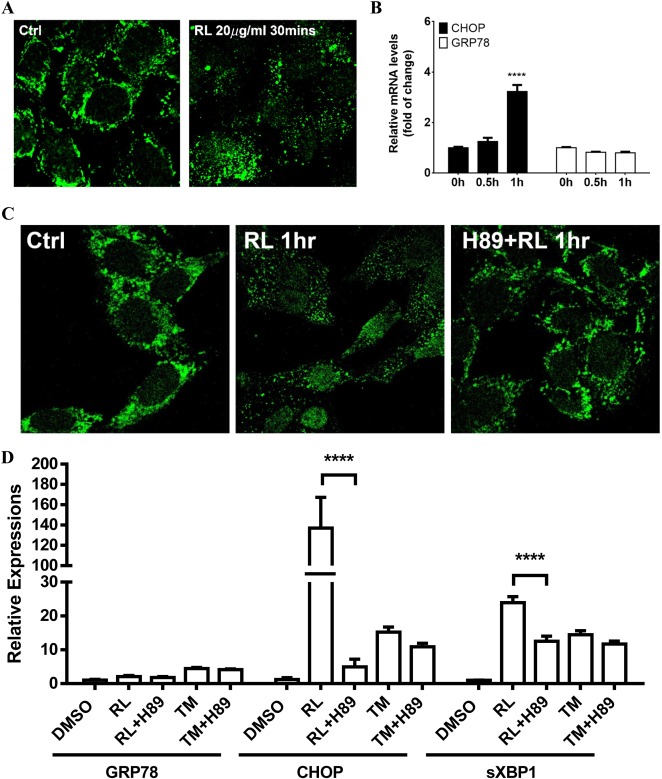
Association of drug‐induced Golgi fragmentation with the onset of the ER stress response. (A) Confocal images showing the time point of Golgi enzyme dislocation/redistribution revealed by immunofluorescent staining with anti‐MAN2A1 antibodies. (B) Onset of the ER stress response indicated by expression changes of CHOP and GRP78 measured with qPCR. (C) Inhibition of RL‐induced MAN2A1 dislocation by a PKA inhibitor, H89 (×630). (D) Inhibition of the drug‐induced ER stress by H89. **** P < 0.0001 compared to control or indicated otherwise; n = 4‐5. Error bars indicate standard error mean (SEM). Abbreviation: PKA, protein kinase A.
CONTRIBUTION OF IMPAIRED ER‐TO‐GOLGI TRAFFICKING TO RL‐INDUCED GOLGI FRAGMENTATION
We next investigated which of the two routes of ER–Golgi trafficking was affected by RL. Results presented in Fig. 2 indicated that both BFA and RL induced Golgi fragmentation but RL tended to cause more MAN2A1 dislocation than BFA. Since BFA has been known to target the COPI route39 and ATF6 trafficking is COPII dependent,33, 34, 35 RL was hypothesized to target the COPII route. To support this, the Sec31 protein of the COPII vesicles22 was examined with immunofluorescence. Under a confocal microscope, cytosolic retention of the COPII complexes indicated by Sec31 was stronger in RL‐treated HepG2 cells than in the vehicle control or BFA‐treated cells (Fig. 6A). Sec31‐positive foci were highly overlapped with intermediate structures between the ER and the Golgi, which was indicated by immunofluorescence of the ER–Golgi intermediate compartment marker ERGIC53. The average size of Sec31‐positive foci was increased 3‐fold in RL‐treated cells compared to the control cells, indicating an interruption of COPII complexes or their trafficking (Supporting Fig. S10). The protein levels of Sec31 were not significantly changed among the treatments (Fig. 6B). In addition, RL did not affect the assembly/initiation of COPII complexes as H89 treatment uniformly eliminated the Sec31‐containing COPII vesicles in the cells treated with vehicle control, RL, or BFA (Fig. 6A). RL then mainly affected the COPII‐mediated ER‐to‐Golgi trafficking. An assay of Golgi disintegration and reassembly was further conducted to assess this possibility based on the fact that the Golgi reassembly requires returning Golgi materials through COPII‐dependent ER‐to‐Golgi trafficking.22, 26, 36 Treatment of BFA at a high concentration resulted in rapid dispersion of the Golgi matrix, and removal of BFA resulted in reassembly of the Golgi (Fig. 6C). RL clearly blocked the reassembly (Fig. 6C,D), indicating that ER‐to‐Golgi trafficking was affected. In addition, similar phenomena were observed in RL‐treated PMH (Fig. 6E).
Figure 6.
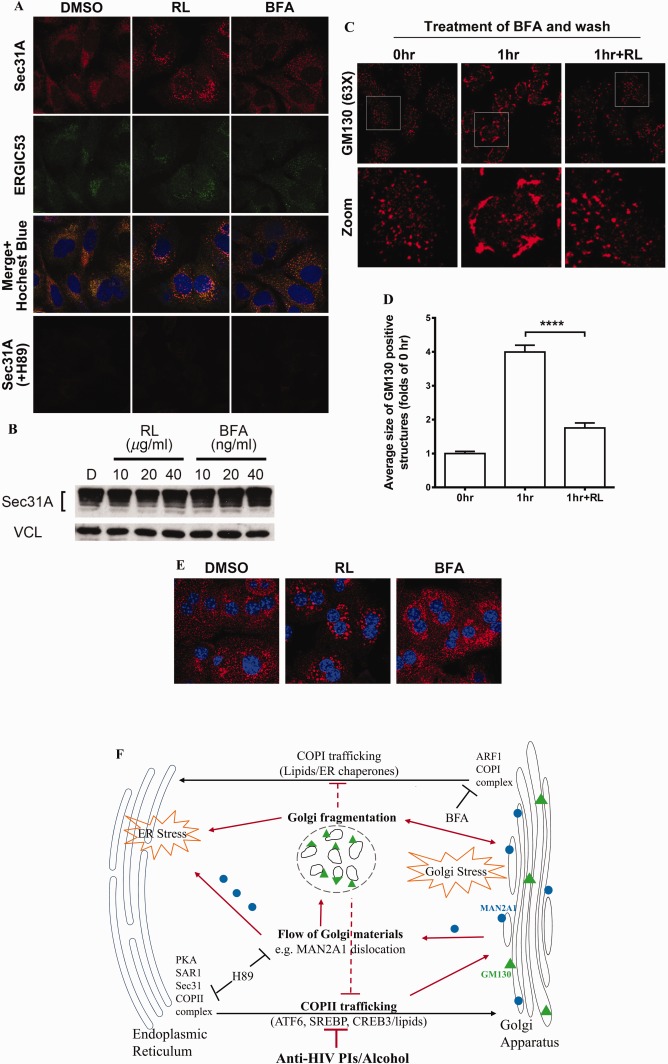
Effects of anti‐HIV drugs on ER–Golgi trafficking. (A) Accumulation of COPII in the drug‐treated HepG2 cells and inhibition of COPII assembly by H89. The COPII complex was revealed by immunofluorescent staining with anti‐Sec31A antibody as shown in the confocal images (red). Anti‐ERGIC53 antibody was used to label the ERGIC structure by immunofluorescent staining (green, ×630). (B) Expression of Sec31A protein in RL‐ and BFA‐treated HepG2 cells. Cells were treated with RL or BFA at different concentrations for 8 hours. (C) Recovery of Golgi morphology after removal of BFA, which was inhibited by RL (×630). The cells were treated first with BFA (100 nM) for 1 hour and then treated with RL for 1 hour after BFA had been removed from the media. (D) Quantitation of Golgi fragmentation. (E) Accumulation of COPII complex in PMH treated with DMSO, RL, and BFA. COPII was probed with immunofluorescence (red) using anti‐Sec31 antibodies (×630). (F) Diagram depicting sites of the ER‐to‐Golgi trafficking targeted by the drugs. The red lines depict that the drugs disrupt COPII trafficking, causing Golgi stress, Golgi fragmentation, enzyme dislocation/redistribution, and ER stress. Error bars indicate standard error mean (SEM). Abbreviations: ARF1, ADP‐ribosylation factor 1; DMSO, dimethyl sulfoxide; ERGIC, ER–Golgi intermediate compartment.
Discussion
Despite ER stress and UPR being well established to play an important role in various forms of liver injury, how liver stress conditions (e.g., drugs, alcohol, high‐fat diet, and HCV/HBV infections) cause ER stress is largely unknown. In this study, we followed the ER stress model of anti‐HIV drugs and alcohol‐induced liver injury established by us and others and investigated early and/or upstream molecular events of ER stress and UPR. Time‐course and dose‐dependent tests revealed for the first time that the three canonical UPR branches IRE‐XBP1, PERK‐eIF2α‐CHOP, and ATF6 were differentially expressed in HepG2 cells or primary hepatocytes in response to RL. The ATF6 branch was inactivated while the other two branches of UPR were highly up‐regulated. This was confirmed as RL specifically inhibited genes/factors (e.g., GRP78, p58IPK, and PDIA4) downstream of the ATF6 pathway. Since the activation of ATF6 requires interorganelle proteolytic processing that involves the Golgi apparatus, abnormal changes of the Golgi were noticed under the drug treatment. Colocalization of ATF6 and the Golgi in the liver cells under RL treatment was significantly lower than the colocalization in the cells under the specific ER stress‐inducing agents. In parallel to the reduction of ATF6 in the Golgi, marked Golgi fragmentation was observed in the liver cells; this was not seen in the TM‐treated and TG‐treated cells. Presence or absence of ATF6 inhibition by the RL treatment was highly correlated with Golgi fragmentation, both of which were concentration and time dependent. It is unlikely that the Golgi fragmentation was due to mitosis as RL‐induced fragmentation was observed in both HepG2 cells and nondividing or rarely dividing primary hepatocytes and liver tissues. The specific Golgi fragmentation by RL started to occur within 30 minutes, which was earlier than the ER stress response indicated by CHOP expression and alternative splicing of XBP1. Apoptosis did not cause the fragmentation as increased caspase activities were not detected until 12 hours after the RL treatments and the pan‐caspase inhibitor QVD‐OPh did not show any rescue effects on the Golgi fragmentation (Supporting Fig. S11). Further, variations of severity of the fragmentation were observed in response to other anti‐HIV drugs, including APV, DAV, and NFV, that are also in use or listed in the Department Health and Human Services guidelines and which were associated with downstream ER stress and cell death injury. In in vivo mouse models, alcohol feeding deteriorated the RL‐induced Golgi fragmentation, which was associated with ER stress, elevated ALT, and liver steatosis. All the above pieces of evidence from this study suggest for the first time that the Golgi dysfunction is a robust mechanism underlying or at least contributing to the anti‐HIV drugs and/or alcohol‐induced ER stress and liver injury.
The Golgi apparatus is part of the cellular endomembrane system closely associated with the ER.22, 40 The structure and function of the Golgi are intimately linked. There is bidirectional ER–Golgi trafficking responsible for biogenesis and intracellular distribution of biomolecules.22, 37 ER‐to‐Golgi trafficking or anterograde transportation moves newly synthesized proteins and lipids to the Golgi for processing, sorting, and redistribution. Meanwhile, Golgi‐to‐ER trafficking or retrograde transportation ensures recycling of lipids, fluids, and ER‐escaped proteins.41 Impairment of either anterograde or retrograde eventually collapses the whole ER–Golgi trafficking and causes cellular stresses and injury.21, 22, 23 Thus, integrity of ER–Golgi trafficking is essential for maintaining rigid Golgi morphology.22, 36, 41 The Golgi fragmentation observed by us indicates that the drugs stress the Golgi and disrupt ER–Golgi trafficking (Fig. 6F). Indeed, GSR markers of GCP60 and HSP47 were increased in the drug‐treated liver cells, and knockdown of TFE3 by siRNA worsened the drug‐induced cell death. We also believe that it was the anterograde ER‐to‐Golgi trafficking that was mostly affected by the drugs for two reasons. First, BFA is known to specifically inhibit the assembly of COPI and ADP‐ribosylation factor 1 (ARF1) complexes;39 this disrupts the retrograde route. The effects of RL on the Golgi appeared different from those of BFA. It took 2 hours for RL to induce fragmentation in 50% of the cells (Supporting Fig. S7E), whereas within 2 hours after BFA almost all the cells were with fragmented Golgi (Fig. 6C). Second, the BFA‐induced fragmentation was primarily associated with abnormal distributions of the Golgi matrix protein GM130, whereas the RL‐induced fragmentation was primarily associated with dislocation/redistribution of the Golgi‐resident MAN2A1.29, 30, 40 The observed enzyme dislocation might be due to blocked ER‐to‐Golgi transportation by RL, which is similar to the reported results that the overexpressing, dominant, negative form of secretion‐associated and Ras‐related factor 1 (SAR1DN), a key component of the COPII complexes, affected Golgi morphology significantly in rat kidney cells.42, 43 While other COPI‐ or COPII‐independent routes may also be involved, our observations support the fact that the effects of drugs on COPII trafficking contribute to Golgi fragmentation, which has also been linked to downstream ER stress and apoptosis in pancreatic beta cells and patients with amyotrophic lateral sclerosis.44, 45 The observed lipid accumulation in the RL‐treated hepatocytes may be a direct consequence of the blocking. In addition, we further addressed what stage of the ER‐to‐Golgi trafficking was affected by RL. Initiation and assembly of the anterograde route requires SAR1, protein kinase A, COPII, Sec proteins (e.g., Sec31), and guanosine triphosphatases,22 which can be blocked by H89. Since H89 did not exert the same effects as RL on the retention of COPII complexes, the possibility of preventing the assembly of COPII complexes by RL was ruled out. Thus, we have narrowed the drug‐targeted molecular sites to COPII‐mediated vesicle secretion, which involves different Rab proteins, Rab effectors controlling vesicle docking, and paired sets of soluble NSF (N‐ethylmateimide‐sensitive factor) attachment receptor proteins mediating fusion of vesicles with target membranes.22 Molecular details on COPII vesicle secretion in relation to RL‐induced Golgi stress, UPR, and liver injury will be our future research goals.
A few additional aspects are worthy of discussion. First, in linking the impaired ATF6 processing to Golgi fragmentation, we considered the possibility that RL might suppress the proteolytic cleavage of ATF6 by inhibiting site‐1‐protease (S1P) and site‐2‐protease (S2P).33, 34, 35 However, neither potent S1P inhibitor PF‐429242 nor potent S2P inhibitor 1,10‐phenanthroline reproduced comparable ER stress, cell death, and Golgi fragmentation in HepG2 cells. In addition, other factors, such as sterol regulatory element binding protein (SREBP) and cyclic adenosine monophosphate response element‐binding protein 3 (CREB), that are also known to be processed through trafficking to the Golgi33, 38 were also suppressed by the RL treatments (Supporting Fig. S12), suggesting that the RL effects on Golgi fragmentation and processing of the transcription factors in the Golgi are quite specific. Second, in the RTV‐boosted LPV regimen, plasma concentrations of LPV ranged widely from 7.2 μg/mL to 13.9 μg/mL.46, 47 Liver injuries occur in 3%‐10% of patients infected with HIV under LPV‐based treatments. However, due to drug–drug interactions, the drug concentrations in the liver can increase dramatically, resulting in multiplied hepatotoxicity as degrees of Golgi fragmentation and ER stress are very sensitive to the treatment doses based on this study (Supporting Fig. S5). We observed that alcohol feeding increased the blood concentration of LPV in mice more than 5‐fold (Supporting Fig. S13)13 and reduction of blood alcohol levels by applying nano‐enzyme particles of alcohol metabolism alleviated alcohol‐induced organelle stress and injury (to be published separately).48 Third, chronic alcohol alone has been linked to Golgi dysfunction and injury. Alcohol alters glycosyl transferase activities and interferes with protein modifications in the Golgi of hepatocytes.28, 49 Accumulations of lipid and carbohydrates were observed in the Golgi of alcohol‐treated primary hepatocytes,50 which was associated with production of anticytoplasmic autoantibodies. Significantly, high titers of anti‐Golgi antibodies were detected in humans with alcoholism and hepatocellular carcinoma.51 Thus, our discovery of anti‐HIV drug and alcohol‐induced Golgi stress and pathological consequences are clinically relevant.
In summary, we have discovered that certain currently used anti‐HIV drugs and/or alcohol disrupt ER‐to‐Golgi trafficking, which contributes to ER stress and subsequent hepatic injury (Fig. 6F). This study may provide precise therapeutic targets for HIV‐infected patients who are under anti‐HIV therapies and suffer from liver injury.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1030/suppinfo.
Supporting Information
Acknowledgment
We thank Dr. James Ou and Dr. Laurie DeLeve for useful comments and the University of Southern California Liver Research Center for technical assistance.
Potential conflict of interest: Nothing to report.
Supported in part by National Institutes of Health grants AA023952 (to C. JI) and DK048522 (USC Research Center for Liver Disease).
REFERENCES
- 1. Faria NR, Rambaut A, Suchard MA, Baele G, Bedford T, Ward MJ, et al. HIV epidemiology. The early spread and epidemic ignition of HIV‐1 in human populations. Science 2014;346:56‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Piot P, Quinn TC. Response to the AIDS pandemic‐‐a global health model. N Engl J Med 2013;368:2210‐2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Granich R, Crowley S, Vitoria M, Smyth C, Kahn JG, Bennett R, et al. Highly active antiretroviral treatment as prevention of HIV transmission: review of scientific evidence and update. Curr Opin HIV AIDS 2010;5:298‐304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Günthard HF, Saag MS, Benson CA, del Rio CD, Eron JJ, Gallant JE, et al. Antiretroviral drugs for treatment and prevention of HIV infection in adults. Jama 2016;316:191‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith CJ, Ryom L, Weber R, Morlat P, Pradier C, Reiss P, et al. Trends in underlying causes of death in people with HIV from 1999 to 2011 (D:A:D): a multicohort collaboration. Lancet 2014;384:241‐248. [DOI] [PubMed] [Google Scholar]
- 6. Wang Y, Lv Z, Chu Y. HIV protease inhibitors: a review of molecular selectivity and toxicity. HIV AIDS (Auckl) 2015;7:95‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sherman KE, Rockstroh J, Thomas D. Human immunodeficiency virus and liver disease: an update. Hepatology 2015;62:1871‐1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Neuman MG, Schneider M, Nanau RM, Parry C. HIV‐antiretroviral therapy induced liver, gastrointestinal, and pancreatic injury. Int. J Hepatology 2012;2012:1‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bilal U, Lau B, Lazo M, Mccaul ME, Hutton HE, Sulkowski MS, et al. Interaction Between alcohol consumption patterns, antiretroviral therapy type, and liver fibrosis in persons living with HIV. AIDS Patient Care and STDs 2016;30:200‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kahler CW, Liu T, Cioe PA, Bryant V, Pinkston MM, Kojic EM, et al. Direct and indirect effects of heavy alcohol use on clinical outcomes in a longitudinal study of HIV patients on ART. AIDS Behav 2016; [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Esposito I, Labarga P, Barreiro P, Fernandez‐Montero JV, de Mendoza C, Benítez‐Gutiérrez L, et al. Dual antiviral therapy for HIV and hepatitis C‐drug interactions and side effects. Expert Opin Drug Saf 2015;14:1421‐1434. [DOI] [PubMed] [Google Scholar]
- 12. Muga R, Sanvisens A, Fuster D, Tor J, Martínez E, Pérez‐Hoyos S, et al. Unhealthy alcohol use, HIV infection and risk of liver fibrosis in drug users with hepatitis C. PLoS One 2012;7:e46810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kao E, Shinohara M, Feng M, Lau MY, Ji C. Human immunodeficiency virus protease inhibitors modulate Ca2+ homeostasis and potentiate alcoholic stress and injury in mice and primary mouse and human hepatocytes. Hepatology 2012;56:594‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Y, Zhang L, Wu X, Gurley EC, Kennedy E, Hylemon PB, et al. The role of CCAAT enhancer‐binding protein homologous protein in human immunodeficiency virus protease‐inhibitor‐induced hepatic lipotoxicity in mice. Hepatology 2013;57:1005‐1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu J, Han H, Lau MY, Lee H, Macveigh‐Aloni M, Ji C. Effects of combined alcohol and anti‐HIV drugs on cellular stress responses in primary hepatocytes and hepatic stellate and kupffer cells. Alcohol Clin Exp Res 2015;39:11‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. De Gassart A, Bujisic B, Zaffalon L, Decosterd LA, Di Micco A, Frera G, et al. An inhibitor of HIV‐1 protease modulates constitutive eIF2α dephosphorylation to trigger a specific integrated stress response. Proc Natl Acad Sci USA 2016;113: E117‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol‐fed mice. Gastroenterology 2003;124:1488‐1499. [DOI] [PubMed] [Google Scholar]
- 18. Ji C, Kaplowitz N, Lau MY, Kao E, Petrovic LM, Lee AS. Liver‐specific loss of glucose‐regulated protein 78 perturbs the unfolded protein response andexacerbates a spectrum of liver diseases in mice. Hepatology 2011;54: 229‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016;529:326‐335. [DOI] [PubMed] [Google Scholar]
- 20. Lafleur MA, Stevens JL, Lawrence JW. Xenobiotic perturbation of ER stress and the unfolded protein response. Toxicol Pathol 2013;41:235‐262. [DOI] [PubMed] [Google Scholar]
- 21. Ji C. Advances and new concepts in alcohol‐induced organelle stress, unfolded protein responses and organ damage. Biomolecules 2015;5:1099‐1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brandizzi F, Barlowe C. Organization of the ER‐Golgi interface for membrane traffic control. Nat Rev Mol Cell Biol 2013;14:382‐392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sasaki K, Yoshida H. Organelle autoregulation‐stress responses in the ER, Golgi, mitochondria and lysosome. J Biochem 2015;157:185‐195. [DOI] [PubMed] [Google Scholar]
- 24. Rubin E, Rybak BJ, Lindenbaum J, Gerson CD, Walker G, Lieber CS. Ultrastructural changes in the small intestine induced by ethanol. Gastroenterology 1972;63:801‐814. [PubMed] [Google Scholar]
- 25. Chang PL, Sharma RN, Sturgess JM, Moscarello MA. Characterization of intracisternal and membrane subfractions from sonically disrupted rough microsomal and Golgi‐rich fractions of the rat liver. Exp Cell Res 1978;112:187‐197. [DOI] [PubMed] [Google Scholar]
- 26. Avitabile E, Di Gaeta S, Torrisi MR, Ward PL, Roizman B, Campadelli‐Fiume G. Redistribution of microtubules and Golgi apparatus in herpes simplex virus‐infected cells and their role in viral exocytosis. J Virol 1995;69:7472‐7482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Petrosyan A, Cheng PW, Clemens DL, Casey CA. Downregulation of the small GTPase SAR1A: a key event underlying alcohol‐induced Golgi fragmentation in hepatocytes. Sci Rep 2015;5:17127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gang H, Lieber CS, Rubin E. Ethanol increases glycosyl transferase activity in the hepatic Golgi apparatus. Nat New Biol 1973;243:123‐125. [PubMed] [Google Scholar]
- 29. Velasco A. Cell type‐dependent variations in the subcellular distribution of alpha‐mannosidase I and II. J Cell Biol 1993;122:39‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Corda D, Barretta ML, Cervigni RI, Colanzi A. Golgi complex fragmentation in G2/M transition: an organelle‐based cell‐cycle checkpoint. IUBMB Life 2012;64:661‐670. [DOI] [PubMed] [Google Scholar]
- 31. Jensen EC. Quantitative analysis of histological staining and fluorescence using ImageJ. Anat. Rec 2013;296:378‐381. [DOI] [PubMed] [Google Scholar]
- 32. Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol 2011;300:C723‐742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011;334:1081‐1086. [DOI] [PubMed] [Google Scholar]
- 34. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 2012;13:89‐102. [DOI] [PubMed] [Google Scholar]
- 35. Oslowski CM, Urano F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol 2011;:71‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sengupta P, Satpute‐Krishnan P, Seo AY, Burnette DT, Patterson GH, Lippincott‐Schwartz J. ER trapping reveals Golgi enzymes continually revisit the ER through a recycling pathway that controls Golgi organization. Proc Natl Acad Sci U S A 2015;112: E6752‐6761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee TH, Linstedt AD. Potential role for protein kinases in regulation of bidirectional endoplasmic reticulum‐to‐Golgi transport revealed by protein kinase inhibitor H89. Mol Biol Cell 2000;11:2577‐2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reiling JH, Olive AJ, Sanyal S, Carette JE, Brummelkamp TR, Ploegh HL, et al. A CREB3‐ARF4 signalling pathway mediates the response to Golgi stress and susceptibility to pathogens. Nat Cell Biol 2013;15:1473‐1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Helms JB, Rothman JE. Inhibition by brefeldin A of a Golgi membrane enzyme that catalyses exchange of guanine nucleotide bound to ARF. Nature 1992;360:352‐354. [DOI] [PubMed] [Google Scholar]
- 40. Dunphy WG, Fries E, Urbani LJ, Rothman JE. Early and late functions associated with the Golgi apparatus reside in distinct compartments. Proc Natl Acad Sci U S A 1981;78:7453‐7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Borgese N. Getting membrane proteins on and off the shuttle bus between the endoplasmic reticulum and the Golgi complex. J Cell Sci 2016;129:1537‐1545. [DOI] [PubMed] [Google Scholar]
- 42. Seemann J, Jokitalo E, Pypaert M, Warren G. Matrix proteins can generate the higher order architecture of the Golgi apparatus. Nature 2000;407:1022‐1026. [DOI] [PubMed] [Google Scholar]
- 43. Ward TH, Polishchuk RS, Caplan S, Hirschberg K, Lippincott‐Schwartz J. Maintenance of Golgi structure and function depends on the integrity of ER export. J Cell Biol 2001;155:557‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fang J, Liu M, Zhang X, Sakamoto T, Taatjes DJ, Jena BP, et al. COPII‐dependent ER export: a critical component of insulin biogenesis and β‐cell ER homeostasis. Mol Endocrinol 2015;29:1156‐1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Soo KY, Halloran M, Sundaramoorthy V, Parakh S, Toth RP, Southam KA, et al. Rab1‐dependent ER‐Golgi transport dysfunction is a common pathogenic mechanism in SOD1, TDP‐43 and FUS‐associated ALS. Acta Neuropathol 2015;130:679‐697. [DOI] [PubMed] [Google Scholar]
- 46. Jackson A, Hill A, Puls R, Else L, Amin J, Back D, et al. Pharmacokinetics of plasma lopinavir/ritonavir following the administration of 400/100 mg, 200/150 mg and 200/50 mg twice daily in HIV‐negative volunteers. J Antimicrob Chemother 2011;66:635‐640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Canta F. Pharmacokinetics and hepatotoxicity of lopinavir/ritonavir in non‐cirrhotic HIV and hepatitis C virus (HCV) co‐infected patients. J Antimicrob Chemother 2005;55:280‐281. [DOI] [PubMed] [Google Scholar]
- 48. Liu Y, Du J, Yan M, Lau MY, Hu J, Han H, et al. Biomimetic enzyme nanocomplexes and their use as antidotes and preventive measures for alcohol intoxication. Nat Nanotechnol 2013;8:187‐1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cottalasso D, Gazzo P, Dapino D, Domenicotti C, Pronzato MA, Traverso N, et al. Effect of chronic ethanol consumption on glycosylation processes in rat liver microsomes and Golgi apparatus. Alcohol Alcohol 1996;31:51‐59. [DOI] [PubMed] [Google Scholar]
- 50. Slomiany A, Piotrowski E, Grabska M, Piotrowski J, Slomiany BL. Chronic ethanol‐initiated apoptosis in hepatocytes is induced by changes in membrane biogenesis and intracellular transport. Alcohol Clin Exp Res 1999;23:334‐343. [PubMed] [Google Scholar]
- 51. Mozo L, Simó A, Suárez A, Rodrigo L, Gutiérrez C. Autoantibodies to Golgi proteins in hepatocellular carcinoma: case report and literature review. Eur J Gastroenterol Hepatol 2002;14:771‐774. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1030/suppinfo.
Supporting Information