

Abstract
We employed a recently introduced class of sterol-modified lipids (SML) to produce m-PEG-DSPE containing liposome compositions with a range of cis-platinum content release rates. SML have a cholesterol succinate attached to the phosphatidylglycerol head group and a fatty acid at the 2 position. These compositions were compared to the well-studied liposome phospholipid compositions: mPEG-DSPE/Hydrogenated Soy PC/cholesterol or mPEG-DSPE/POPC/cholesterol to determine the effect of the cis-platinum release extent on C26 tumor proliferation in the BALB/c colon carcinoma mouse model. The release rates of cis-platinum from liposomes composed of SML are a function of the acyl chain length. SML-liposomes with shorter acyl chain lengths C-8 provided more rapid cisplatin release, lower in vitro IC50, and were easier to formulate compared to liposomes using traditional phospholipid compositions. Similar to other liposome cis-platinum formulations, the half-life of m-PEG-DSPE SML liposome cisplatin is substantially longer than the free drug. This resulted in a higher tumor cisplatin concentration at 48 h post-dosing compared to the free drug and higher Pt-DNA adducts in the tumor. Moreover, the maximum tolerated dose of the liposome formulations where up to four fold greater than the free drug. Using X-ray fluorescence spectroscopy on tumor sections, we compared the location of platinum, to the location of a fluorescence lipid incorporated in the liposomes. The liposome platinum co-localized with the fluorescent lipid and both were non-uniformly distributed in the tumor. Non-encapsulated Cis-platinum, albeit at a low concentration, was more uniformly distributed thorough the tumor. Three liposome formulations, including the well-studied hydrogenated HSPC composition, had better antitumor activity in the murine colon 26 carcinoma model as compared to the free drug at the same dose but the SML liposome platinum formulations did not perform better than the HSPC formulation.
Keywords: Cisplatin, C26 colon carcinoma, Liposome, Sterol-modified phospholipid, X-ray fluorescence microscopy
1. Introduction
Cisplatin is a potent chemotherapeutic used to treat a variety of cancers including ovarian, lung, and colon; but treatment dosage and thus efficacy has been hampered by severe toxicities (Oberoi et al., 2013; Kieler-Ferguson et al., 2013). Limited success has been achieved in designing new platinum derivatives or carrier mediated delivery. Polymer platinum delivery has focused on the design of chelators that release platinum slowly enough to reduce toxicity, but quickly enough to maintain activity (Kieler-Ferguson et al., 2013; Hang et al., 2016). In contrast, liposomal delivery has focused on formulation conditions, which includes methods of encapsulation as well as lipid formulations that would provide appropriate platinum release in vivo. The instability of platinates makes drug delivery more challenging. Cisplatin activity is maintained or increased by limiting light exposure, high NaCl concentrations to prevent the loss of chloride and limit isomerization, or exposure to deactivating sulfur nucleophiles (Kieler-Ferguson et al., 2013; Hang et al., 2016). To this end, a variety of different lipid formulations have been explored for cisplatin (Newman et al., 1999; Woo et al., 2008; Schroeder et al., 2009; Hirai et al., 2010; Stathopoulos, 2010; Zisman et al., 2011), oxaliplatin (Dragovich et al., 2006; Suzuki et al., 2008; Tippayamontri et al., 2011) and other platinates (Mori et al., 1996; Wheate et al., 2010). Currently there are no FDA-approved platinum liposomes and four formulations are in clinical trials: Lipoplatin (cisplatin) and Lipoxal (oxaliplatin), developed by Regulon (Fantini et al., 2011) and two others (Hang et al., 2016).
SPI-077 (SEQUUS) is a well-known cisplatin formulation removed from clinical trials due to lack of activity. SPI-077 is composed of hydrogenated soy phosphatidylcholine (HSPC), cholesterol, and m-PEG-DSPE at a 51:44:5 M ratio; this formulation exhibited superior results in animal models compared to both cisplatin and other cisplatin liposome formulations (Newman et al., 1999). Despite SPI-077 resulting in appreciable Pt uptake in tumor tissue and improved survival outcomes in mice, there was minimal activity in Phase I/II human clinical trials (Harrington et al., 2001; Rosenthal et al., 2002; Meerum Terwogt et al., 2002; White et al., 2006). Zamboni et al. used microdialysis in mice to track the bioavailability of cisplatin and found that only a small percentage of the drug was freely diffusible, despite the improved tumor accumulation (Zamboni et al., 2004). These results were confirmed in a similar efficacy study by Bandak and coworkers, where <10% of cisplatin was released from liposomes and no improvement in efficacy was observed in lung, lymphoma and melanoma models (Bandak et al., 1999).
Additional clinical trials with liposome based platinum compounds are recently reviewed although here again some of these formulations are well tolerated but have not yet exhibited high antitumor activity compared to the non-encapsulated platinum in either animal models or in human trial (Oberoi et al., 2013; Hang et al., 2016).
These data suggested to us that a new liposome formulation which increases the bioavailability of cisplatin in the tumor, is still needed. Thus, we explored the potential of sterol modified lipids or SMLs (Huang and Szoka, 2008; Huang et al., 2009; Foglia et al., 2011; Kohli et al., 2014a; Kohli et al., 2014b) to deliver cisplatin, as previous studies have shown variable content release based upon chain length and lipid composition. The SMLs were designed to improve bilayer properties by covalently linking cholesterol the PC lipid head group, replacing one of the acyl chains, and eliminating cholesterol transfer between the liposome and biomembranes which may contribute to catastrophic instability of traditional fluid phospholipid/cholesterol compositions such as POPC/cholesterol mixtures. This study illustrates how modifications to lipid composition can influence drug release, animal toxicity, tumor uptake, and anti-tumor efficacy. The intratumoral distribution of the liposome and the platinum reinforces the previous findings that the liposome carrier and the encapsulated platinum are co-localized in the tumor periphery.
2. Experimental methods
2.1. Materials
Materials were used as obtained from commercial sources unless otherwise noted. Cisplatin was purchased from Sigma-Aldrich. Lipids (Hydrogenated Soy Phosphatidylcholine (HSPC)), PalmitoylOleoyl Phosphatidyl choline (POPC), dipalmitoylphosphatidylcholine (DPPC) and methoxypolyethyleneglycol-2000-distearoylphosphatidylethanolamine (m-PEG-DSPE) were purchased from Avanti Polar Lipids, kindly synthesized by Dr. Zhaohua Huang, or synthesized as previously described (Huang and Szoka, 2008; Huang et al., 2009). Cholesterol (Chol) was re-crystallized from methanol prior to use. SPEX CertiPrep Ultralene Film (4 μm) was purchased from Fisher Scientific. The platinum ICP standard was purchased from VHG Labs. Chloroform was removed under reduced pressure using a rotary evaporator.
2.2. General liposome preparation
Prior to liposome formation, lipids were dissolved in chloroform, evaporated to form a thin film, and dried overnight at room temperature under a high vacuum. Lipid mixtures are abbreviated as listed in Table 1. Various formulation conditions, as described below and in the supplemental materials, were explored to make liposomes. Following formation, liposomes were sequentially extruded through a 200 nm and 100 nm polycarbonate membrane 11 times each at 60 °C, held at 60 °C for 15 min and then cooled to room temperature. Any non-encapsulated cisplatin that precipitated and was removed by filtration through a 0.4 Spectrum 25 mm PTFE sterile syringe filter, and the liposomes were then dialyzed for 24 h against 100 volumes of HEPES buffer (10 mM HEPES, 150 mM NaCl. pH 7.4). Average liposome diameter and zeta potential were determined by dynamic light scattering measurements (Malvern Instruments Zetasizer Nano ZS) (Table 1).
Table 1.
Lipid formulations and physical characterization.
| Name | Lipid Composition | Molar Ratio | Diameter (nm) | PDI | Zeta potential (mV) |
|---|---|---|---|---|---|
| HSPC | HSPC:Chol:m-PEG-DSPE | 51:44:5 | 125 | 0.04 | −2.2 |
| POPC 51 | POPC:Chol:m-PEG-DSPE | 51:44:5 | 132 | 0.06 | −2.2 |
| POPC 80 | POPC:Chol:m-PEG-DSPE | 80:15:5 | 110 | 0.06 | −2.3 |
| C8Chems | C8Chems:m-PEG-DSPE | 95:5 | 100 | 0.08 | −2.4 |
| C18.1Chems | C18.1Chems:m-PEG-DSPE | 95.5 | 115 | 0.04 | −1.1 |
| DChems | DiChems:m-PEG-DSPE | 95:5 | 130 | 0.09 | −2.4 |
| DChems/DPPC | DiChems:DPPC:m-PEG-DSPE | 32:63:5 | 119 | 0.1 | −2.6 |
2.3. Quantification of platinum in liposomes
Cisplatin loading in the liposomes was measured by inductively coupled plasma-atomic emission spectroscopy (ICP-AES; Perkin Elmer Optima 7000 DV Optical Emission Spectrometer). Liposomes (50 μL) were diluted to 6 mL with a 5% HNO3 solution and allowed to stand at room temperature for 1 h. Samples were measured in triplicate for their Pt emission at 214.423 nm.
2.3.1. Liposome formulations
2.3.1.1. Ethanol injection method (Peleg-Shulman et al., 2001)
This method was used to prepare liposomes for all in vitro and in vivo studies. Cisplatin (8.5 mg/mL) was dissolved in 0.9% NaCl at 60 °C, while the lipids were dissolved in ethanol (100 mg/mL) at 60–70 °C. With a pre-warmed syringe, the ethanolic mixture (100 μL) was rapidly injected into the cisplatin solution (900 μL) and allowed to stir covered for 1 h at 60 °C. In certain instances, the volume of the preparation was increased to 5 mL total. Formulation continued as described above. Size: 100–132 nm, Platinum loading: 1–1.7 mg cisplatin/10 mg lipids. Other methods including: DMSO, Chaotropic solvents and remote loading were employed to improve the solubility or encapsulation of Pt in liposomes but the ethanol procedure (Peleg-Shulman et al., 2001) was the most reliable and efficient of the variations we examined (Supplemental Information).
2.3.1.2. Platinum release experiments
Cisplatin liposomes diluted into fetal bovine serum (FBS) final concentration 30 vol% were aliquoted into Eppendorf tubes and placed in a 37 °C shaker. At various times over 48 h, a sample was passed through a Sepharose GL-6B column and the liposome fraction collected. We used this separation method because platinum can interact with serum proteins, it was important to separate both free cisplatin and the cisplatin that had interacted with serum proteins from the liposomes. The liposome fraction was diluted with 5% HNO3 and the platinum content measured by ICP as described above.
2.3.1.3. Toxicity of cisplatin liposomes in C26 cells
Cells were seeded onto a 96-well plate at a density of 5.0 × 103 cells per well in 100 μL of medium and incubated overnight (37 °C, 5% CO2, and 80% humidity). An additional 100 μL of new medium (RPMI medium 1640/10% FBS/1% penicillin-streptomycin) containing either cisplatin or cisplatin liposomes with concentrations ranging from 50 nM to 500 μM Pt equivalents was added to the cells. The tests were conducted in replicates of three for each concentration. After incubation for 72 h, 40 μL of media containing thiazolyl blue tetrazolium bromide solution (2.5 mg/mL) was added. The cells were incubated for 2 h, after which time the medium was carefully removed. To the resulting purple crystals was added 200 μL of DMSO, followed by 25 μL of pH 10.5 glycine buffer (0.1 M glycine/0.1 M NaCl). The optical densities at 570 nm and 690 nm (background) were measured on a SpectraMAX M3 microplate reader (Molecular Devices, Sunnyvale, CA). Wells containing cells that received neither liposome nor drug were considered to represent 100% viability. IC50 values were obtained from sigmoidal fits of semilogarithmic plots of the percentage of viability versus platinum concentration by using Origin 7 SR4 8.0552 software (OriginLab, Northhampton, MA). And had standard deviations between ±10% of the stated IC50 value.
2.3.1.4. Cisplatin degradation
Cisplatin was dissolved in 7 M Urea/150 mM NaCl, 6 M Urea/1 M NaCl, or 6 M GuCl/150 mM NaCl at 15 mg/mL and heated in the dark at 65 °C. At various time points, samples were removed, diluted (1:50), and stored until use. A standard MTT assay in C26 cells was used to assess loss of activity. Cisplatin in PBS represented 100% viability and the chaotropic solutions were the negative control (see supplemental information).
2.3.1.5. Animal and tumor models
All animal experiments were performed in compliance with National Institutes of Health guidelines for animal research under a protocol approved by the Committee on Animal Research at the University of California (San Francisco, CA) (UCSF). C26 colon carcinoma cells obtained from the UCSF cell culture facility were cultured in RPMI medium 1640 containing 10% FBS. Female BALB/c mice were obtained from Simonsen Laboratories, Inc. (Gilroy, CA).
2.3.1.6. Toxicity in healthy mice
Female BALB/C mice were injected via tail vein injection with cisplatin or cisplatin liposomes. Mice weights and general health were monitored over 9 days. When gross toxicity was observed, such as loss of N15% of initial body weight, lethargy or ruffled fur, the animal would be removed from the study.
2.3.1.7. Pharmacokinetic analysis of formulation elimination in healthy mice
Six- to eight week-old female BALB/C mice, three per group were dosed via tail vein injection with liposomes (3 mg Pt/mL). Blood was collected in heparinized tubes (10 μL heparin (50 mg/mL)) by submandibular bleeds 5, 20, 60, 180, 240, and 480 min post injection and by heart puncture 24, 48, and 72 h post injection. The blood was centrifuged at 5000 rpm for 3 min, the serum separated, and stored at 4 °C until use. Samples were diluted to 2 mL with 5% HNO3, incubated for 1 h at 40 °C, and then centrifuged to remove insoluble particles. Platinum content was analyzed by ICP. The PK data for each group as a whole was fit to either a one or two-compartment model using GraphPad Prism. The standard deviation for circulation half-lives was calculated by fitting the PK data of each individual animal to the two-compartment model and determining the standard deviation within each group. Once circulation half-life were computed, p values were determined using the Student’s t-test. A p value ≤ 0.05 was considered significant.
2.3.1.8. Tissue sections and fluorescence imaging
While under anesthesia, female BALB/C mice were shaved, and C26 cells (3 × 105 cells in 50 μL) were injected subcutaneously in the right hind flank. At ten days post-tumor implantation, mice were injected with cisplatin (10 mg/kg) or cisplatin liposomes (10 mg/kg) labeled with 0.2% membrane dye DiD. After two days, mice were sacrificed and the tumors collected. The tumor volume was approximately 200 mm3 when the animals were sacrificed and the tumors removed. Tissue was mounted in O.C.T. embedding compound (Sakura Tissue-TEK) and flash frozen with dry ice cooled isopentane. Using a cryostat, 10 μm tissue samples were cut and transferred to Ultralene film which had been gently secured to a glass slide. Sections were air dried before being transported to Argonne National Lab for X-ray fluorescent microscopy. Sections for H&E were transferred directly to a glass slide, air dried, and then preserved with 100% EtOH for 1 min. Slides were then stained with hematoxylin for 30 s, rinsed, stained with Eosin for 15 s, rinsed thoroughly, and mounted with Permount. Adjacent sections to the ones used for the X-ray fluorescent microscopy were examined using a confocal microscope.
2.3.1.9. Tumor imaging and analysis
Tumor sections were imaged on a Nikon Ti-E microscope and processed using NIS Elements 4.20 (Nikon) and the Fiji program. Three-channel images were captured on the microscope: nuclear stain Hoechst 3342, CD31 stain FITC; and lipid membrane dye DiD. Imaging and exposure settings remained consistent through all experiments. Tumors were imaged at 20× and large composite images were stitched together using the NIS Elements software. When collecting large images, the microscope was refocused on every second image and exposure times and microscope settings remained constant across all samples. Each channel was saved in a separate TIFF file and loaded in Fiji. Background was subtracted from images using a Rolling Ball background subtraction with a radius of 50. Then an absolute background subtraction of a pixel value of 1500 was applied. Only the DiD signal for an adjacent section to the X-ray Fluorescent image is shown in the photomicrograph (Fig. 4).
Fig. 4.
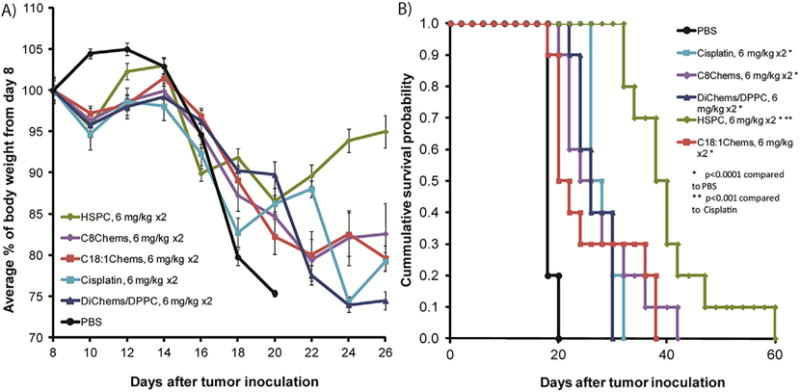
A) Average change in body weight from day of injection. B) Survival probability for mice treated with cisplatin and liposomal cisplatin. Mice (n = 10 per group) dosed on day 8 and day 15 at 6 mg/kg cis-platinum.
2.3.1.10. X-ray fluorescence microscopy
Sections were imaged with the scanning X-ray microprobe at beamline 8-BM-B at the Advanced Photon Source (Argonne, IL). Undulator-generated x-rays of 15-keV incident energy were monochromatized with a double monochromator and focused to a measured spot size of ~100 μm pinhole. Tissues were rasterscanned in steps of 0.1 mm and fluorescence spectra were collected for 4–8 s per pixel with a four-element silicon drift detector (Vortex-EX, SII Nanotechnology, CA). Quantitation and image-processing of the x-ray fluorescence (XRF) datasets were performed with MAPS software (Paunesku et al., 2006; Vogt, 2003).
2.3.1.11. Isolation of genomic DNA and measurement of Pt-DNA adducts
The procedure of Yamada et al., (Yamada et al., 2005) was used to quantify the Pt-DNA adducts. Genomic DNA was extracted from cells digested with proteinase K using a phenol–chloroform method and dissolved in a 10 mM Tris-EDTA buffer consisting of 10 mM Tris–HCl (pH 7.4) and The purity at this step was checked by measuring the absorbance at 260, 280, and 320 nm. The 260/280 ratio was N1.8. Samples were treated with ribonuclease A (Sigma–Aldrich) for 2 h extracted again. The Pt concentration was quantified using ICP as described above.
2.3.1.12. Chemotherapy experiment in tumored mice
While under anesthesia, female BALB/C mice were shaved, and C26 cells (3 × 105 cells in 50 μL) were injected subcutaneously in the right hind flank. At eight days post-tumor implantation, mice were randomly distributed into treatment groups of 10 animals. Mice were injected by means of the tail vein with cisplatin (6 mg/kg once a week for 2 weeks) or cisplatin liposomes (6 mg/kg once a week for 2 weeks) in approximately 200 μL of solution. Five days post-treatment, blood was collected by submandibular bleeds and analyzed for 19 toxicity markers (UC Davis Comparative Pathology Dept). Blood urea nitrogen levels were quantified with Quantichrome Urea Assay Kit (BioAssay Systems, Hayward, CA). Mice were weighed and tumors measured every other day. The tumor volume was estimated by measuring the tumor volume in three dimension with calipers and calculated using the formula tumor volume = length × width × height. Mice were removed from the study when (i) a mouse lost 15% of its initial weight, (ii) any tumor dimension was >20 mm, or (iii) the mouse was found dead. The mice were followed until day 60 post-tumor inoculation. Statistical analysis was performed using MedCalc 8.2.1.0 for Windows (MedCalc Software, Mariakerke, Belgium). The tumor growth delay was calculated based upon a designated tumor volume of 400 mm3.
3. Results and discussion
3.1. Formulation strategies
A variety of formulation methods (see supplemental material for alternative methods used to increase the solubility of cis-platinum to improve Pt encapsulation in liposomes) were explored to encapsulate cisplatin. The original EtOH injection method (Newman et al., 1999; Peleg-Shulman et al., 2001) was the most reproducible in our laboratory and provided stable liposome formulations. All of the liposome compositions incorporated PEG-DSPE at 5 mol% and were prepared from a number of different lipids including: C8Chems, C10Chems, C16Chems, C18:1Chems, DChems, DChems/DPPC, HSPC, and POPC (molar ratios given in Table 2). The advantage for preparation of liposomes for the formulations that contain the SML lipids is that only one other component, the mPEG-PEG2000-DSPE needs to be added. The liposome diameters and Zeta potentials used in the reported experiments were measured using a Malvern Zetasizer and had diameters between ~ 100–132 nm and Zeta potentials of about −2.0 mV ± 1.0 mV. (Table 1). Cisplatin loading in the liposome preparations was quantified by ICP analysis and ranged from 1 to 1.7 mg cisplatin/mL.
Table 2.
Cisplatin release in the presence of serum and cytotoxicity against the C26 cells in culture.
| Name | Formulation | % Release after 72 h | IC50 (uM) |
|---|---|---|---|
| C8Chems | Chems:PEG (95:5) | 26% | 5.0 |
| DChems/DPPC | DChems:DPPC:PEG (32:63:5) | 20% | 5.8 |
| C16Chems | Chems:PEG (95:5) | <5% | 79 |
| C18:1Chems | Chems:PEG (95:5) | <5% | >115 |
| DChems | DChems:PEG (95:5) | <5% | 53 |
| HSPC | HSPC:Chol:PEG (51:44:5) | <5% | 260 |
| POPC 51 | POPC:Chol:PEG (51:44:5) | 14% | 130 |
| POPC 80 | POPC:Chol:PEG (80:15:5) | 37% | 5.9 |
3.2. Liposomal cisplatin leakage and toxicity
After exploring a variety of loading conditions, leakage rate of cisplatin from the various formulations was determined. To replicate some aspects of the in vivo conditions, liposomes were heated at 37 °C in 30% FBS. At various times, liposomes were removed from the incubation container and liposome encapsulated Pt was separated from Pt that had leaked from the liposome and the cisplatin concentration analyzed by ICP. Cisplatin can interact with serum proteins; therefore we separated cisplatin liposomes from Pt-proteins and free cisplatin using a SEC Sepharose GL-6B column rather than a dialysis method. By quantifying liposomal cisplatin, the early time release Pt amounts were never below the Pt detection limit. As expected, the shorter chain SML had a faster Pt release than the longer chain SML lipids. Likewise, the more fluid oleoyl (POPC) traditional lipid formulations had faster release profiles than did liposomes composed of saturated lipids.
The formulations prepared had a range of Pt release rates so the cytotoxicity of the liposome preparations was screened against the C26 cell line. The IC50 values are shown in Table 2. When the liposomes were sonicated for 5 min prior to adding to them to the cells, thus releasing cisplatin, toxicity was comparable to the free drug control. The leakier liposome formulations were the most cytotoxic, while the less leaky formulations were relatively non-toxic.
3.3. In vivo toxicity and platinum distribution
To identify candidate formulations for in vivo evaluation, we used the cisplatin release rate and cytotoxicity values. Four formulations were chosen for further study to identify formulations that might be better than HSPC. We included the. C18:1Chems since it behaved in a similar manner to the HSPC formulations in the Pt release and cytotoxicity assays. DChems/DPPC was selected as an intermediate release rate liposome formulation. Finally, the leakiest liposome, C8Chems, was chosen as a fast release formulation. We first measured the single dose, maximum tolerated dose of the selected formulations in healthy animals.
Due to the loading limitations, a maximum tolerated dose was never established for the HSPC formulation (Newman et al., 1999). While we didn’t expect to observe any gross toxicity, the HSPC formulation was included in the toxicity screen as a comparison to the non-leaky C18:1Chems and to the leaky C8Chems. All three formulations were dosed at the highest allowable volume < 500 μL. this set the dose administered to between 30 and 45 mg/kg for the three formulations and no significant toxicity was observed (Fig. 1). In comparison, 15 mg cisplatin/kg was lethal to healthy, tumor free BALB/C mice; as in previous studies in BALB/c mice (Newman et al., 1999) the MTD was established at 10.
Fig. 1.
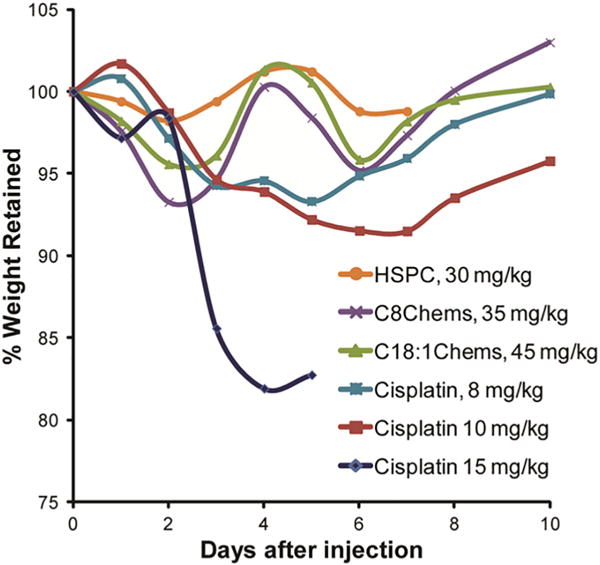
Retention of body weight after single injection of cisplatin and cisplatin liposomes in healthy, tumor free mice. The lines are not a fit and are only provided to enable the identification of the weight change with the formulation.
We compared the pharmacokinetics/distribution of the C8Chems SML formulation to the HSPC formulation and free drug in animals. Mice were dosed with either HSPC, C8Chems, or free cisplatin at 3 mg cisplatin/kg. Blood was collected over 72 h and analyzed for platinum by ICP. Cisplatin is rapidly excreted from the body (Newman et al., 1999) and the plasma concentrations at the 3 mg/Kg dose were below the Pt detection limit for the non-encapsulated cisplatin. Given the rapid clearance of non-encapsulated cisplatin, the platinum detected in plasma is encapsulated in liposomes. Although the C8Chems liposome was more leaky than the HSPC liposome the plasma platinum concentration is only modestly lower than that found in the HSPC liposomes (Fig. 2).
Fig. 2.
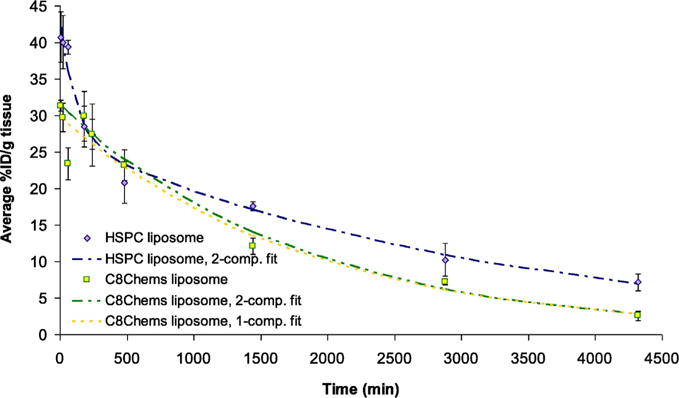
Blood circulation of C8Chems and HSPC liposomes in healthy, tumor free mice. Each time included three samples from individual mice (n = 3).
3.3.1. The T1/2, V1 and AUC were significantly different p < 0.05
In general, the liposome circulation time of both the HSPC and C8Chems were prolonged in part because of the stability of the two formulations, and protection from phagocytosis of the liposomes afforded to the liposomes by the PEG corona [38,]. As shown in Table 4, both formulations had elimination phase half-lives > 20 h, favorable area-under-the-curve for the Pt (AUC0 → ∞), which resulted in almost 10% of the injected platinum dose in circulation at 48 h, which is advantageous for increased tumor uptake.
Table 4.
Platinum-DNA adducts isolated from five individual C26 tumors for each liposome Pt composition quantified by ICP-MS. Values represent the mean ± the standard deviation, n = 5.
| ng Pt/μg DNA | % Injected dose (×10−3) | % ID/μg DNA (×10−5) | |
|---|---|---|---|
| Cisplatin | 0.022 ± 0.013 | 0.5 ± 0.04 | 1.2 ± 0.7 |
| C8Chems | 0.078 ± 0.076 | 3.7 ± 1.0 | 3.9 ± 3.8 |
| POPC 80 | 0.195 ± 0.220 | 2.6 ± 2.1 | 10.8 ± 12.2 |
| POPC 51 | 0.093 ± 0.052 | 2.1 ± 0.7 | 4.8 ± 2.3 |
| HSPC | 0.221 ± 0.227 | 5.1 ± 0.1 | 11.8 ± 12.1 |
3.4. Location of platinum and lipid label in C26 tumors
To investigate the intratumoral disposition of platinum from various formulations, a microfluorescence (X-ray fluorescence microscopy, or XFM) experiment was conducted at Argonne National Lab. This technique maps the spatial distribution of multiple elements within a sample and is amenable to both single cells and tissue slices (Vogt, 2003). To aid in the design of new platinate drugs, XFM has been used to better understand the fate of platinates in tumor cells (Paunesku et al., 2006; Hall et al., 2003; Hall et al., 2006). C26 tumors were excised (after 54 h) from mice treated with cisplatin, C8Chems, POPC 80 (the more leaky POPC formulation), and HSPC at 10 mg/kg. Ten μm tissue sections were mounted on a thin film for analysis. As expected for the non-encapsulated cisplatin, the Pt concentration was low and was distributed throughout the tumor section (Fig. 4). In contrast, the liposomal formulations tended to accumulate along the tumor periphery, often in clusters. Generally these overlay well with the fluorescent lipid signal, which is indicative of the liposome distribution. As expected, liposome distribution within the tumor, as observed using DiD fluorescence intensity, was highest along the tissue periphery. There are subtle differences between the formulations. C8Chems appeared to have higher localized distribution at the tumor periphery, which is correlated with the higher local Pt concentration as detected using X-ray fluorescence (Fig. 3). The HSPC had lower levels of Pt in any one location throughout the tumor, and the Pt signal was more uniformly distributed as indicated by the DiD fluorescent label intensity. The POPC 80 formulation, which is a formulation prepared from traditional lipids that released Pt and had a IC50 value similar to the C8Chems composition, had a distribution pattern (both DiD and Pt) that was similar to the C8Chems liposome delivered Pt. It is a more punctate distribution, suggesting that in this tumor model, the leaky liposomes do not become as uniformly distributed throughout the tumor as does the very stable HSPC composition and the distribution of the platinum mirrors the distribution of the liposome. This suggests that the leakiness of the liposome composition may be associated with how uniformly the liposome can penetrate into the tumor.
Fig. 3.
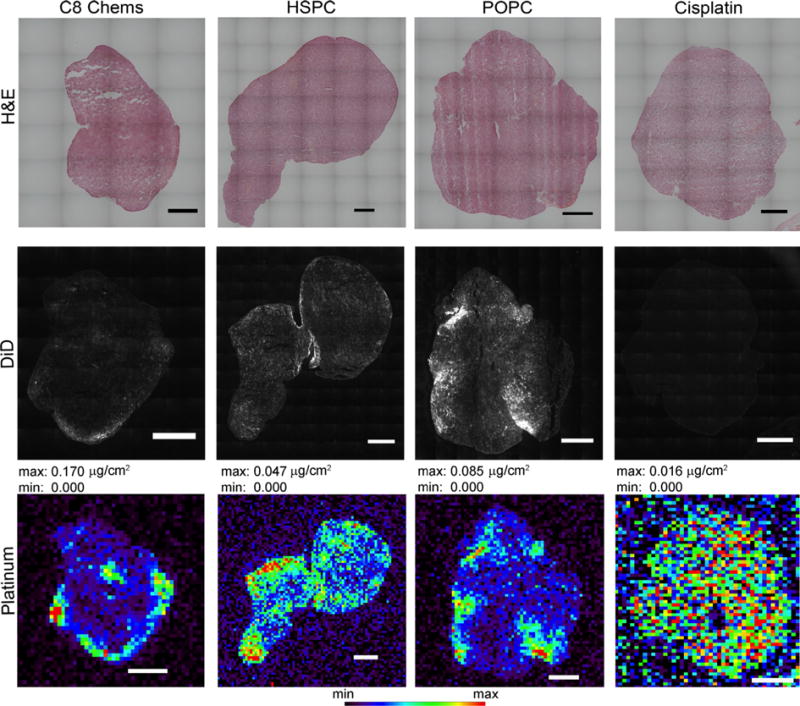
Localization of liposome-associated fluorescent dye, encapsulated platinum or non-encapsulated platinum in tumor sections at 54 h post dosing. Microfluorescence of platinum in C26 tumor the scale inserted into the images represents 1 mm. Maximum and minimum threshold values are given which correspond with the rainbow color scale at the bottom of the image.
3.5. Platinum-DNA adducts in C26 tumors treated with platinum of liposome-encapsulated platinum
To learn if the tumor cis-platinum content, as measured by tumor associated Pt or from the X-ray fluorescent images, had in fact leaked from the liposome and reached tumor cells, we isolated tumor DNA from five tumors in each group and quantified DNA-Pt adducts by ICP-MS (Table 4). As expected, cisplatin had the lowest concentration of DNA bound platinum, while HSPC had the highest. The POPC 80 formulation, which released Pt faster than the POPC 51 composition, had higher DNA-Pt levels than the POPC 51 formulation. A broad range of Pt concentrations were measured in the five samples. The imaging results support the hypothesis that more stable liposomal cisplatin was more uniformly distributed across the tumor, but both non-leaky and leaky liposome platinum compositions deliver Pt into tumor cells in vivo, than does the non-encsulated drug. We can conjecture that the platinum leakage from the more leakier formulations prior to the cis-platinum reaching the tumor cell may explain the lower platinum adducts on the DNA because of the inactivation of cis-platinum by nucleophiles prior to reaching the nucleus (Florea and Büsselberg, 2011).
3.6. Efficacy of liposomal cisplatin in C26 tumor model
To learn if the formulations, which had a low IC50 in cell culture and equivalent tumor uptake to the HSPC liposome, have better antitumor activity compared to cisplatin, the antitumor efficacy of four cisplatin liposomal formulations was compared to the free drug. BALB/C mice were tumored subcutaneously with C26 colon carcinoma cells and treated with two doses, the first dose 8 days after tumor inoculation and the second dose 15 days after tumor inoculation. The treatment groups included PBS, cisplatin (6 mg/kg, once a week for 2 weeks), and liposomes C8Chems, DChems/DPPC, C18:1Chems, and HSPC at 6 mg cisplatin/kg once a week for 2 weeks. Although weight loss was observed and increased after the second dose on day 15 (Fig. 4A), a blood panel was normal and the mice showed no other signs of toxicity such as lethargy or ruffled fur. All of the treatment groups had statistically significant improved survival and tumor regression as compared to mice treated with PBS (Fig. 4B). The C8 and C18:1 SML formulations were modestly better than non-encapsulated drug. The HSPC formulation, despite the low cisplatin release in vitro, performed better than cisplatin and the other liposome formulations (p < 0.001) (Fig. 4B). This result is consistent with the higher level of DNA-Pt adducts measured in the tumors (Table 4).
The results of the chemotherapy experiment suggest that the sterol-modified lipids may not improve delivery of cisplatin, as neither the leaky C8Chems nor non-leaky C18:1 Chems liposomes substantially improved antitumor activity.
We were interested in learning if the presence of cholesterol that could be exchanged from the liposome at the tumor site, might explain this result because the cholesterol can transfer from the traditional formulations but not from the SML formulations. This might cause traditional formulations to release Pt faster in the tumor than in the in vitro release assay. Cholesterol is added to liposomes to stabilize the bilayer during circulation and control content release, but can exchange with other biomembranes causing increased leakage (Kohli et al., 2014b; Kan et al., 1992; Drummond et al., 1999; Hamilton, 2003; Dos Santos et al., 2002). Likewise, unsaturated lipids such as POPC can provide more fluidity to a membrane and affect drug release. When these two parameters were combined (Table 3), the unsaturated POPC 80 formulation (15% cholesterol) had excellent Pt release and cytotoxicity, while POPC 51 (44% cholesterol) was more stable and had lesser cytotoxicity. Thus in the tumor microenvironment and more rapid removal of cholesterol might facilitate Pt release from the HSPC and POPC formulations in the tumor which would not have been evident in the in vitro cisplatin release experiment.
Table 3.
Pharmacokinetic parameters of mPEG-DSPE HSPC and mPEG-DSPE C8Chems cis-platinum containing liposomes based upon the circulating platinum levels in healthy mice.
| PK parameters | HSPC | C8Chems |
|---|---|---|
| t1/2, α (hr) | 1.3 | – |
| t1/2, β (hr) | 36.8 | 20.7 |
| V1 (g blood) | 0.77 | 1.2 |
| AUC0 → ∞ (%ID ∗ h/g blood) | 1456 | 902 |
| kel (1/min) | 0.0005 | 0.0006 |
mg/kg in healthy mice.
We did not have a direct way to test this conjecture so we decided to compare the antitumor activity of platinum delivered in two leaky formulations, C8Chems and the POPC 80 that had similar in vitro IC50 values circa 5 uM (Table 2) to the less leaky HSPC and POPC 51 formulations, IC50 values 260 uM and 130 uM respectively at the same 6 mg cisplatin/kg × 2 dose schedule (Fig. 5). On day 20, a blood sample was collected and blood urea nitrogen concentration determined as an indicator of kidney damage and decreased kidney function.
Fig. 5.
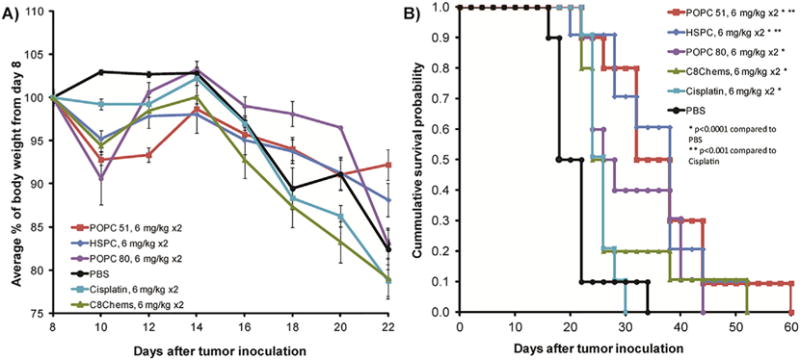
A) Average weight change following treatment at days 8 and 15. B) Survival plot of mice treated with four leaky formulations and the less leaky HSPC formulation. Mice (n=10/group) were dosed on day 8 and 15 at 6 mg/kg.
As with the previous chemotherapy studies, the mice exhibited weight loss (Fig. 5A) without other signs of toxicity and did not develop elevated serum urea levels. All of the formulations had statistically significant tumor regression and increased survival compared to PBS p < 0.0001 (Fig. 5B). Interestingly, both leaky formulation minimally improved survival as compared to cisplatin despite the favorable Pt release and tumor accumulation, while both non-leaky formulations did significantly better than cisplatin (Fig. 4B, p < 0.001). These results suggest that while the leaky formulations devised in this study increase the availability of cisplatin in vitro, this does not translate to increased anti-tumor activity in the C26 tumor model compared to the non-leaky formulations.
The results reported here are consistent with an extensive literature describing carefully performed formulation and characterization experiments, that investigated if modest adjustments of the liposome compositions could provide better anti-tumor activity compared to the HSPC formulation, SPI-077 (Newman et al., 1999). For instance, Alavizadeh and coworkers (Alavizadeh et al., 2014) determined that DPPC/mPEG2000–DSPE/Chol gave a slight increase in median survival time growth delay compared to the SPI-077-like formulation. A similar improvement was observed by Marzban and colleagues (Marzban et al., 2015) in which a SPI-077 like formulation that contained 10% DPPG yielded a modest improvement in tumor progression time over the standard HSPC formulation.
Approaches that attempt to increase platinum delivery by exploiting environmental or external triggering such as pH-sensitive liposome formulations or heat-sensitive liposomes that release more drug under the triggered conditions, in most cases, also lead to modest improvements. For instance Zhou et al. (Zhou et al., 2015) prepared a fatty acid linked cis-platinum and found it provided a slight MST improvement over the non-encapsulated cisplatin. At this juncture, therapeutic approaches that apply light triggered or heat triggered relatively rapid increases in permeability such as described by Dou et al. (Dou et al., 2016) who used a formulation that contained a lysolipid, monostearylphosphatidyl choline, provided a two fold increase in cisplatin uptake in the tumor when the tumor was heated with a laser based heating system. This resulted in a substantial decrease in tumor proliferation rate. Photoactivated systems such as devised in the group of Urtti using indo-cyanine green-doped liposomes, (Lajunen et al., 2016) can also mediate rapid contents release when illuminated. Such systems have the potential to provide a route to liposome compositions that can circulate a sufficient long time, deliver a substantial fraction of the dose into the tumor as the encapsulated Pt and be induced to rapidly deliver the contents.
4. Conclusions
We initiated this project with the hypothesis that the slower release of Pt from the HSPC formulation would make it easier for nucleophiles, such as tissue resident sulfhydryl compounds, to inactive the Pt (Florea and Büsselberg, 2011) to a greater extent, than would happen if the Pt was released more rapidly from the liposome. A number of formulation strategies were explored to improve cisplatin loading and release in liposomes composed of a variety of compositions. The SML, C8Chems gave the fastest in vitro release and the best cytotoxicity profile and was compared to the standard HSPC formulation in the C26 colon carcinoma model. All of the formulations had favorable circulation times and resulted in the delivery of a substantial amount of cis-platinum into the tumor. In the C26 tumor model the SML liposomes were efficacious but did not outperform the HSPC formulation. Our results are consistent with many prior studies with other liposome compositions. The cis-platinum is much better tolerated and the antitumor activity in a murine tumor is better than the free compound. However, it seems that more creative approaches are required before we will achieve substantially superior cis-platinum antitumor activity compared to the free compound.
Supplementary Material
Acknowledgments
Funding sources. We would like to thank Prof. Walter Finkbeiner for assistance with tissue preparation, sectioning and histochemical staining of the tumor sections. This work was funded by NIH R01GM061851 and 3R01GM061851-09S1 (FCS).
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.ejps.2017.03.003.
References
- Alavizadeh SH, Badiee A, Golmohammadzadeh S, Jaafari MR. The influence of phospholipid on the physicochemical properties and anti-tumor efficacy of liposomes encapsulating cisplatin in mice bearing C26 colon carcinoma. Int J Pharm. 2014;473:326–333. doi: 10.1016/j.ijpharm.2014.07.020. [DOI] [PubMed] [Google Scholar]
- Bandak S, Goren D, Horowitz A, Tzemach D, Gabizon A. Pharmacological studies of cisplatin encapsulated in long-circulating liposomes in mouse tumor models. Anti-Cancer Drugs. 1999;10:911–920. doi: 10.1097/00001813-199911000-00007. [DOI] [PubMed] [Google Scholar]
- Dos Santos N, Mayer LD, Abraham SA, Gallagher RC, Cox KAK, Tardi PG, Bally MB. Improved retention of idarubicin after intravenous injection obtained for cholesterol-free liposomes. Biochim Biophys Acta Biomembr. 2002;1561:188–201. doi: 10.1016/s0005-2736(02)00345-0. [DOI] [PubMed] [Google Scholar]
- Dou YN, Dunne M, Huang H, Mckee T, Chang MC, Jaffray DS, Allen C. Thermosensitive liposomal cisplatin in combination with local hyperthermia results in tumor growth delay and changes in tumor microenvironment in xenograft models of lung carcinoma. J Drug Target. 2016;24:865–877. doi: 10.1080/1061186X.2016.1191079. [DOI] [PubMed] [Google Scholar]
- Dragovich T, Mendelson D, Kurtin S, Richardson K, Von Hoff D, Hoos A. A Phase 2 trial of the liposomal DACH platinum L-NDDP in patients with therapy-refractory advanced colorectal cancer. Cancer Chemother Pharmacol. 2006;58:759–764. doi: 10.1007/s00280-006-0235-4. [DOI] [PubMed] [Google Scholar]
- Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol Rev. 1999;51:691–744. [PubMed] [Google Scholar]
- Fantini M, Gianni L, Santelmo C, Drudi F, Castellani C, Affatato A, Nicolini M, Ravaioli A. Lipoplatin treatment in lung and breast cancer. Chemoth Res Pract. 2011;2011:1–7. doi: 10.1155/2011/125192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florea AM, Büsselberg D. Cisplatin as an anti-tumor drug: cellular mechanisms of activity, drug resistance and induced side effects. Cancer. 2011;3:1351–1371. doi: 10.3390/cancers3011351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foglia F, Barlow DJ, Szoka FC, Huang Z, Rogers SE, Lawrences MJ. Structural studies of the monolayers and bilayers formed by a novel cholesterol-phospholipid chimera. Langmuir. 2011;27:8275–8281. doi: 10.1021/la200739y. [DOI] [PubMed] [Google Scholar]
- Hall MD, Dillon CT, Zhang M, Beale P, Cai Z, Lai B, Stampfl APJ, Hambley TW. The cellular distribution and oxidation state of platinum(II) and platinum(IV) antitumour complexes in cancer cells. J Biol Inorg Chem. 2003;8:726–732. doi: 10.1007/s00775-003-0471-6. [DOI] [PubMed] [Google Scholar]
- Hall MD, Alderden RA, Zhang M, Beale PJ, Cai Z, Lai B, Stampfl APJ, Hambley TW. The fate of platinum(II) and platinum(IV) anti-cancer agents in cancer cells and tumours. J Struct Biol. 2006;155:38–44. doi: 10.1016/j.jsb.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Hamilton JA. Fast flip-flop of cholesterol and fatty acids in membranes: implications for membrane transport proteins. Curr Opin Lipidol. 2003;14:263–271. doi: 10.1097/00041433-200306000-00006. [DOI] [PubMed] [Google Scholar]
- Hang Z, Cooper MA, Ziora ZM. Platinum-based anticancer drugs encapsulated liposome and polymeric micelle formulation in clinical trials. Comput Biol Chem. 2016;4:1–10. [Google Scholar]
- Harrington KJ, Lewanski CR, Northcote AD, Whittaker J, Wellbank H, Vile RG, Peters AM, Stewart JS. Phase I–II study of pegylated liposomal cisplatin (SPI-077) in patients with inoperable head and neck cancer. Ann Oncol. 2001;12:493–496. doi: 10.1023/a:1011199028318. [DOI] [PubMed] [Google Scholar]
- Hirai M, Minematsu H, Hiramatsu Y, Kitagawa H, Otani T, Iwashita S, Kudoh T, Chen L, Li Y, Okada M, Salomon DS, Igarashi K, Chikuma M, Seno M. Novel and simple loading procedure of cisplatin into liposomes and targeting tumor endothelial cells. Int J Pharm. 2010;391:274–283. doi: 10.1016/j.ijpharm.2010.02.030. [DOI] [PubMed] [Google Scholar]
- Huang Z, Szoka FC. Sterol-modified phospholipids: cholesterol and phospholipid chimeras with improved biomembrane properties. J Am Chem Soc. 2008;130:15702–15712. doi: 10.1021/ja8065557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Jaafari MR, Szoka FC. Disterolphospholipids: nonexchangeable lipids and their application to liposomal drug delivery. Angew Chem Int Ed. 2009;121:4210–4213. doi: 10.1002/anie.200900111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan CC, Yan J, Bittman R. Rates of spontaneous exchange of synthetic radiolabeled sterols between lipid vesicles. Biochemistry. 1992;31:1866–1874. doi: 10.1021/bi00121a040. [DOI] [PubMed] [Google Scholar]
- Kieler-Ferguson HM, Fréchet JM, Szoka FC., Jr Clinical developments of chemotherapeutic nanomedicines: polymers and liposomes for delivery of camptothecins and platinum (II) drugs. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2013;5:130–138. doi: 10.1002/wnan.1209. [DOI] [PubMed] [Google Scholar]
- Kohli AG, Kieler-Ferguson HM, Chan D, Szoka FC. A robust and quantitative method for tracking liposome contents after intravenous administration. J Control Release. 2014a;176:86–93. doi: 10.1016/j.jconrel.2013.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli AG, Kierstead PH, Venditto VJ, Walsh CL, Szoka FC. Designer lipids for drug delivery: from heads to tails. J Control Release. 2014b;90:274–287. doi: 10.1016/j.jconrel.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajunen T, Kontturi LS, Viitala L, Manna M, Cramariuc O, Róg T, Bunker A, Laaksonen T, Viitala T, Murtomäki L, Urtti A. Indocyanine green-loaded liposomes for light-triggered drug release. Mol Pharm. 2016;13:2095–2107. doi: 10.1021/acs.molpharmaceut.6b00207. [DOI] [PubMed] [Google Scholar]
- Marzban E, Alavizadeh SH, Ghiadi M, Khoshangosht M, Khashayarmanesh Z, Abbasi A, Jaafari MR. Optimizing the therapeutic efficacy of cisplatin PEGylated liposomes via incorporation of different DPPG ratios: In vitro and in vivo studies. Colloids Surf B: Biointerfaces. 2015;136:885–891. doi: 10.1016/j.colsurfb.2015.10.046. [DOI] [PubMed] [Google Scholar]
- Meerum Terwogt JM, Groenewegen G, Pluim D, Maliepaard M, Tibben MM, Huisman A, ten Bokkel Huinink WW, Schot M, Welbank H, Voest EE, Beijnen JH, Schellens JM. Phase I and pharmacokinetic study of SPI-77, a liposomal encapsulated dosage form of cisplatin. Cancer Chemother Pharmacol. 2002;49:201–210. doi: 10.1007/s002800100371. [DOI] [PubMed] [Google Scholar]
- Mori A, Wu SP, Han I, Khokhar AR, Perez-Soler R, Huang L. In vivo antitumor activity of cis-bis-neodecanoato-trans-R, Rl, 2-diaminocyclohexane platinum (II) formulated in long-circulating liposomes. Cancer Chemother Pharmacol. 1996;37:435–444. doi: 10.1007/s002800050409. [DOI] [PubMed] [Google Scholar]
- Newman MS, Colbern GT, Working PK, Engbers C, Amantea MA. Comparative pharmacokinetics, tissue distribution, and therapeutic effectiveness of cisplatin encapsulated in long-circulating, pegylated liposomes (SPI-077) in tumor-bearing mice. Cancer Chemother Pharmacol. 1999;43:1–7. doi: 10.1007/s002800050855. [DOI] [PubMed] [Google Scholar]
- Oberoi HS, Nukolova NV, Kabanov AV, Bronich TK. Nanocarriers for delivery of platinum anticancer drugs. Adv Drug Deliv Rev. 2013;65:1667–1685. doi: 10.1016/j.addr.2013.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paunesku T, Vogt S, Maser J, Lai B, Woloschak G. X-ray fluorescence microprobe imaging in biology and medicine. J Cell Biochem. 2006;99:1489–1502. doi: 10.1002/jcb.21047. [DOI] [PubMed] [Google Scholar]
- Peleg-Shulman T, Gibson D, Cohen R, Abra R, Barenholz Y. Characterization of sterically stabilized cisplatin liposomes by nuclear magnetic resonance. Biochim Biophys Acta Biomembr. 2001;1510:278–291. doi: 10.1016/s0005-2736(00)00359-x. [DOI] [PubMed] [Google Scholar]
- Rosenthal DI, Yom SS, Liu L, Machtay M, Algazy K, Weber RS, Weinstein GS, Chalian AA, Miller LK, Rockwell K, Tonda M, Schnipper E, Hershock D. A Phase I study of SPI-077 (Stealth® liposomal cisplatin) concurrent with radiation therapy for locally advanced head and neck cancer. Investig New Drugs. 2002;20:343–349. doi: 10.1023/a:1016201732368. [DOI] [PubMed] [Google Scholar]
- Schroeder A, Honen R, Turjeman K, Gabizon A, Kost J, Barenholz Y. Ultrasound triggered release of cisplatin from liposomes in murine tumors. J Control Release. 2009;137:63–68. doi: 10.1016/j.jconrel.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Stathopoulos GP. Liposomal cisplatin: a new cisplatin formulation. Anti-Cancer Drugs. 2010;21:732–736. doi: 10.1097/CAD.0b013e32833d9adf. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Takizawa T, Kuwata Y, Mutoh M, Ishiguro N, Utoguchi N, Shinohara A, Eriguchi M, Yanagie H, Maruyama K. Effective anti-tumor activity of oxaliplatin encapsulated in transferrin-PEG-liposome. Int J Pharm. 2008;346:143–150. doi: 10.1016/j.ijpharm.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Tippayamontri T, Kotb R, Paquette B, Sanche L. Cellular uptake and cytoplasm/DNA distribution of cisplatin and oxaliplatin and their liposomal formulation in human colorectal cancer cell HCT116. Investig New Drugs. 2011;29:1321–1327. doi: 10.1007/s10637-010-9494-3. [DOI] [PubMed] [Google Scholar]
- Vogt S. MAPS: a set of software tools for analysis and visualization of 3D X-ray fluorescence data sets. J Phys IV. 2003;104:635–638. [Google Scholar]
- Wheate NJ, Walker S, Craig GE, Oun R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010;39:8113–8127. doi: 10.1039/c0dt00292e. [DOI] [PubMed] [Google Scholar]
- White SC, Lorigan P, Margison GP, Margison JM, Martin F, Thatcher N, Anderson H, Ranson M. Phase II study of SPI-77 (sterically stabilised liposomal cisplatin) in advanced non-small-cell lung cancer. Br J Cancer. 2006;95:822–828. doi: 10.1038/sj.bjc.6603345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo J, Chiu GNC, Karlsson G, Wasan E, Ickenstein L, Edwards K, Bally MB. Use of a passive equilibration methodology to encapsulate cisplatin into preformed thermosensitive liposomes. Int J Pharm. 2008;349:38–46. doi: 10.1016/j.ijpharm.2007.07.020. [DOI] [PubMed] [Google Scholar]
- Yamada K, Kato N, Takagi A, Koi M, Hemmi H. One-milliliter wet-digestion for inductively coupled plasma mass spectrometry (ICP-MS): determination of platinum-DNA adducts in cells treated with platinum(II) complexes. Anal Bioanal Chem. 2005;382:1702–1707. doi: 10.1007/s00216-005-3339-5. [DOI] [PubMed] [Google Scholar]
- Zamboni WC, Gervais AC, Egorin MJ, Schellens JHM, Zuhowski EG, Pluim D, Joseph E, Hamburger DR, Working PK, Colbern G, Tonda ME, Potter DM, Eiseman JL. Systemic and tumor disposition of platinum after administration of cisplatin or STEALTH liposomal-cisplatin formulations (SPI-077 and SPI-077 B103) in a preclinical tumor model of melanoma. Cancer Chemother Pharmacol. 2004;53:329–336. doi: 10.1007/s00280-003-0719-4. [DOI] [PubMed] [Google Scholar]
- Zhou X, Wang J, Wu X, Yang X, Yung BC, Lee LJ, Lee RJ. Preparation and evaluation of a novel liposomal formulation of cisplatin. Eur J Pharm Sci. 2015;66:90–95. doi: 10.1016/j.ejps.2014.10.004. [DOI] [PubMed] [Google Scholar]
- Zisman N, Dos Santos N, Johnstone S, Tsang A, Bermudes D, Mayer L, Tardi P. Optimizing liposomal cisplatin efficacy through membrane composition manipulations. Chemoth Res Pract. 2011;2011:1–7. doi: 10.1155/2011/213848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.