

Abstract
Pioglitazone is an FDA-approved PPAR-γ agonist drug used to for treat diabetes, and it has demonstrated neuroprotective effects in multiple models of central nervous system (CNS) injury. Acute treatment after spinal cord injury (SCI) in rats is reported to suppress neuroinflammation, rescue injured tissues, and improve locomotor recovery. In the current study, we additionally assessed the protective efficacy of pioglitazone treatment on acute mitochondrial respiration, as well as functional and anatomical recovery after contusion SCI in adult male C57BL/6 mice. Mice received either vehicle or pioglitazone (10 mg/kg) at either 15 min or 3 hr after injury (75 kDyn at T9) followed by a booster at 24 hr post-injury. At 25 hr, mitochondria were isolated from spinal cord segments centered on the injury epicenters and assessed for their respiratory capacity. Results showed significantly compromised mitochondrial respiration 25 hr following SCI, but pioglitazone treatment that was initiated either at 15 min or 3 hr post-injury significantly maintained mitochondrial respiration rates near sham levels. A second cohort of injured mice received pioglitazone at 15 min post injury, then once a day for 5 days post-injury to assess locomotor recovery and tissue sparing over 4 weeks. Compared to vehicle, pioglitazone treatment resulted in significantly greater recovery of hind-limb function over time, as determined by serial locomotor BMS assessments and both terminal BMS subscores and gridwalk performance. Such improvements correlated with significantly increased grey and white matter tissue sparing, although pioglitazone treatment did not abrogate long-term injury-induced inflammatory microglia/macrophage responses. In sum, pioglitazone significantly increased functional neuroprotection that was associated with remarkable maintenance of acute mitochondrial bioenergetics after traumatic SCI. This sets the stage for dose-response and delayed administration studies to maximize pioglitazone’s efficacy for SCI while elucidating the precise role that mitochondria play in governing its neuroprotection; the ultimate goal to develop novel therapeutics that specifically target mitochondrial dysfunction.
Keywords: PPAR, mitoNEET, neuroprotection, bioenergetics, locomotor recovery
INTRODUCTION
There are approximately 17,000 new traumatic spinal cord injury (SCI) cases in the United States each year yet, despite numerous clinical trials, there are currently no comprehensively accepted therapies to treat the pathophysiology of acute traumatic SCI (Center, 2016; Rabchevsky et al., 2011). The exception is methylprednisolone sodium succinate (MPSS), the only clinically approved compound reported to show modest efficacy on certain functional outcome measures when administered within the first 8 h after SCI (Bracken, 1990, 2001; Bracken et al., 1984; Bracken et al., 1992; Bracken et al., 1990; Bracken et al., 1985; Bracken et al., 1997). Traumatic SCI is characterized by an initial mechanical insult followed by a series of destructive pathophysiological cascades that disrupt a constellation of biochemical and cellular signaling pathways (Hall and Springer, 2004; McEwen et al., 2011). Mitochondrial dysfunction is thought to be one of the primary cause/effect components of these secondary events since they are the principal energy producers for the cell, as well as the main site for reactive oxygen/nitrogen species (ROS/RNS) formation which initiate many cell death mechanisms (Finkel, 2001; Patel et al., 2012; Patel et al., 2010; Sullivan et al., 2007; Sullivan et al., 2005; Yonutas et al., 2015). Accordingly, there is emerging evidence that preventing mitochondrial dysfunction acutely with pharmacotherapeutics following experimental neurotrauma results in significant neuroprotection and functional recovery at later stages (Pandya et al., 2014; Patel et al., 2012; Patel et al., 2010; Patel et al., 2014; Sauerbeck et al., 2011).
Pioglitazone is an FDA approved treatment for type-2 diabetes and belongs to a class of drugs called thiazolidinediones (TZDs), which are peroxisome proliferating activating receptor (PPAR) agonists (Michalik et al., 2004; Michalik and Wahli, 1999; Sood et al., 2000). The three isoforms of PPAR (PPRA-α, PPAR-β/δ, and PPAR-γ) are part of the nuclear receptor superfamily and perform a wide array of tasks, such that their function has not been systematically defined (Berger and Moller, 2002; Michalik and Wahli, 1999; Yonutas and Sullivan, 2013). Nevertheless, it is widely accepted that PPAR activity plays a critical role in lipid metabolism, modulates expression patterns of pro-inflamatory cytokines, and increases expression of antioxidant proteins (Chen et al., 2007; Frazier-Wood et al., 2013; Kapadia et al., 2008; Martin et al., 2012; Sood et al., 2003; Yi et al., 2008). The TZDs which bind PPAR-γ are particularly germane to SCI therapeutics, notably pioglitazone. Pioglitazone can rapidly cross the blood-brain barrier (Berger and Moller, 2002) and has demonstrated more robust neuroprotection than other TZDs such as rosiglitazone which has higher binding affinity for PPAR-γ (Kapadia et al., 2008; Thal et al., 2011). This may be due to emerging evidence that pioglitazone’s therapeutic effects may also depend on PPAR-independent mechanisms that ameliorate mitochondrial dysfunction and which stem from its interactions with the mitoNEET, an iron-sulfur domain-containing protein (Colca et al., 2004).
mitoNEET is a protein localized in the brain, liver and skeletal muscles of rodents (Colca et al., 2004). This finding came long after the discovery that pioglitazone had a binding affinity for the mitochondrial membrane. After further investigation, it was determined that this binding was mediated through a new m-17 kDa protein which was later termed mitoNEET (Colca et al., 2004). At the time of its discovery, mitoNEET was proposed to be a pivotal protein for mitochondrial metabolism which had the potential of being modulated by pioglitazone. Since its initial discovery, the exact role of mitoNEET in the cell remains uncertain. However, a handful of groups have studied mitoNEET’s protein dynamics and suggested one possible role is to be a shuttle protein for the mitochondria (Conlan et al., 2009; Hou et al., 2007; Lin et al., 2007; Paddock et al., 2007). Pioglitazone binding can then inhibit the transfer of [2Fe-2S] clusters making mitoNEET unable to dimerize and perform its necessary task. Additionally, through the use of mitoNEET knockout mice, it was shown that mitochondria without mitoNEET have a decreased oxidative capacity which suggests that this iron-containing protein may be pivotal in controlling the rate of mitochondrial respiration (Geldenhuys et al., 2011; Kusminski et al., 2012; Pedada et al., 2014; Wiley et al., 2007). Finally, a recent study has highlighted that mitoNEET is a redox-sensitive protein and can be reduced by biological thiols such as gluthathione (GSH), reversing the effect of mitoNEET oxidation (Landry and Ding, 2014) in accordance that GSH precursors can reverse mitochondrial dysfunction and afford neuroprotection following CNS injuries (Pandya et al., 2014; Patel et al., 2014).
Pioglitazone has been shown to reduce neuroinflammation, restore mitochondrial homeostasis, spare neurons, and promote functional recovery following traumatic brain injury (TBI) (Liu et al., 2016; Sauerbeck et al., 2011; Thal et al., 2011). However, much of the protection appears to be independent of PPAR, given the concentrations used and the incomplete reversal of protection in the presence of PPAR antagonists, again indicating a potential role for mitoNEET in mediating pioglitazone neuroprotection. Despite its use in TBI, pioglitazone has a limited but promising history of experimental therapeutic application for SCI, studied exclusively in rats (McTigue et al., 2007; Park et al., 2007). Furthermore, its effects on mitochondrial bioenergetics after SCI remain uncharacterized. While pioglitazone has been shown to preserve mitochondrial function following TBI in mice (Sauerbeck et al., 2011), it is not known if pioglitazone elicits similar outcomes following SCI. Accordingly, in the current study we assessed the protective efficacy of pioglitazone treatment on mitochondrial bioenergetics acutely and long-term functional recovery and tissue sparing following contusion SCI in mice.
MATERIALS AND METHODS
Animals
Adult male C57BL/6 mice (n = 64) (Jackson Laboratories, Bar Harbor, Maine) were housed in a vivarium at 25°C under a 12 hour light/dark cycle and were allowed access to food and water ad libidum. All experimental procedures occurred during the light cycle. All procedures were approved by and in accordance with the University of Kentucky Institutional Animal Care and Use Committee.
Spinal Cord Injury Surgery and Treatments
Before surgery, animals were deeply anesthetized via ketamine (150 mg/kg) and xylazine (10 mg/kg) injection (i.p.). The surgical site was shaved and disinfected with isopropyl alcohol and betadine. A small incision was made on the dorsal surface, and the muscle and subcutaneous tissue overlaying the thoracic spine were retracted. A T9 laminectomy was performed to expose the mid-thoracic spinal cord. Severe contusions were produced using the Infinite Horizons impactor device (75 kdyn; Precision Systems and Instrumentation, LLC, Lexington KY, USA) as we have described previously (Zhang et al., 2015b). The hit parameters (mean ± SD) for mice included in final analyses were equivalent between vehicle (n=19) and pioglitazone (n=29) treated injury cohorts for impact force [vehicle group (78.26 kdyn ± 2.56) vs pioglitazone group (78.48 kdyn ± 3.0)], cord displacement [vehicle group (586.2 μm ± 91.74) vs pioglitazone group (559.3 μm ± 80.53)], and velocity at impact [vehicle group (120.9 m/s ± 2.77) vs pioglitazone group (122.0 m/s ± 2.26)]. Any injured animals with abnormalities in the force vs time curve, which are indicative of bone hits or instability of the spinal cord, were excluded from analysis. For biochemical assessment of mitochondrial integrity, sham mice (n = 7) received laminectomy only and displayed normal hind-limb function post-surgery. The wound was closed with monofilament suturing of the muscle and skin incisions. Afterwards, animals were returned to their home cage placed a heating pad and administered injections (s.c.) of Buprenorphine SR (0.1 mg/kg, Reckitt Benckisser Pharmaceuticals Inc., Richmond, VA) and Baytril (2.5 ml/kg, mixed with 2 ml Sterile Saline), then allowed to regain consciousness.
Injured mice being assessed for acute mitochondrial respiration (n=27 total) received an injection (i.p) of either pioglitazone (10 mg/kg, dissolved in DMSO at 3mg/ml) at 15 min (n = 9) or 3 hr (n = 10), versus vehicle (DMSO) 15 min after injury (n = 7), and they also received a booster of pioglitazone or DMSO at 24 hr post-injury, one hour prior to tissue harvesting. Due to postsurgical complications, one mouse died prior to acute biochemical assessments. Long-term behavioral experiments were carried out using two independent cohorts (n=30 total). Injured mice received an injection (i.p) of either 10mg/kg pioglitazone (n = 10) or 100% DMSO (n = 12) 15 min after injury, and then daily injections (i.p.) of either pioglitazone or DMSO for 5 days post-injury, followed by injection (s.c.) of Baytril (2.5 ml/kg, mixed with 1 ml sterile saline). Manual bladder expression was performed twice daily on injured mice. Data for 8 injured mice are not presented because they were either excluded due to post-injury complications or died. Specifically, three mice were euthanized due to abnormal contusions, one due to severe bladder infection, two due to autophagia, and one mouse died during long-term behavioral experiments for unknown reasons.
Mitochondrial Isolation
Mitochondria from sham and injured spinal cords were isolated by modifying our previously reported method (Patel et al., 2014; Patel et al., 2009). At 24 hr post-injury, animals were decapitated and a 0.5 cm thoraco-lumbar spinal cord segment centered on injury site was rapidly dissected from all injured as well as naive mice and homogenized in an ice-cold dissecting plate containing isolation buffer with 1 mM EGTA (215 mM mannitol, 75 mM sucrose, 0.1% BSA, 20 mM HEPES, 1 mM EGTA, and pH adjusted to 7.2 with KOH). The homogenate was centrifuged at 1400 ×g for 3 min at 4 °C. Supernatant was transferred to a new 2 ml tube (S1, containing mitochondria and cytosol). The pellet was resuspended in 2 ml isolation buffer and centrifuged at 1400 ×g for 3 min and this supernatant was transferred to a new tube (S1′, containing mitochondria and cytosol) and the pellet was discarded. Both supernatant fractions (S1 & S1′) were then topped off with isolation buffer and centrifuged at 13,000 ×g for 10 min. The resultant two pellets were pooled and resuspended in 500 μl isolation buffer (with EGTA) and burst in a nitrogen cell disruption bomb at 4 °C for 10 min at 1200 psi to release synaptic mitochondria from synaptosomes. Resultant differential mitochondrial suspension was then centrifuged at 10,000 ×g for 10 min. Pellets were re-suspended in ~20ul of isolation buffer (without EGTA). All samples were kept on ice throughout the isolation process. The protein concentrations were determined using BCA protein assay kit (Thermo Scientific, Rockford, IL) by measuring absorbance at 560 nm with a Biotek Synergy HT plate reader (Winooski, Vermont). Note: isolation and measurements of OCRs were completed as quickly as possible after dissecting the tissues (within 5–6 hrs).
Assessment of Mitochondrial Respiration
Respiration of mitochondrial samples was measured in terms of mitochondrial oxygen consumption rate (OCR) using a Seahorse Bioscience XF24 Extracellular Flux Analyzer as described previously with slight modifications (Sauerbeck et al., 2011). Briefly, 24 hr prior to each experiment, a 24 well dual-analyzer sensor cartridge (Seahorse Bioscience) was incubated with 1 ml/well of XF Calibrant solution (Seahorse Bioscience) at 37 °C in a CO2-free incubator (Seahorse Bioscience). On the days of experimentation, either A) pyruvate plus malate plus ADP, B) oligomycin, C) FCCP, or D) rotenone plus succinate were added in ports A–D, respectively, in the Seahorse Flux Pak cartridges to yield final concentrations of 5 mM (pyruvate), 2.5 mM (malate), 1 mM (ADP), 1 μg/ml (oligomycin), 3 μM (FCCP), 100 nM (rotenone), and 10 mM (succinate) and placed into the Seahorse XF24 Flux Analyzer for automated calibration. During the sensor calibration, 50 μl volume of respiration buffer containing 5 μg of mitochondrial protein was added to each well of XF24 V7 cell culture microplates and centrifuged at 2000 rpm for 4 min at 4 °C. This was followed by addition of 450 μl of respiration buffer (125 mM KCl, 2 mM MgCl2, 2.5 mM KH2PO4, 20 mM HEPES and 0.1% BSA, pH 7.2) at 37 °C (gently added to each well) to make the final assay volume 500 μl per well. Plates were immediately placed into the calibrated Seahorse XF24 Flux Analyzer for assessment of mitochondrial OCR in the presence of substrates and inhibitors in respective ports. The OCR data were analyzed using Excel software package (Microsoft), point by point rates were generated using the AKOS algorithm written by AKOS Gerencser for Seahorse Bioscience. For State III and State V—Complex I, the “middle point” (average) of OCR was generated from 4 time-dependent point-by-point rates for each well sample. Then, three independent middle points were averaged for each sample to generate n = 1 per sample.
Behavioral Analyses
Recovery of hind limb motor function was assessed as previously described using the Basso Mouse Scale (BMS) (Basso et al., 2006; Zhang et al., 2015b). Mice were assessed in an open field apparatus pre-surgically and at 1, 3, 7, 14, 21, and 28 days post-injury (DPI) by two trained observers. Each hind limb was rated independently for movement coordination, and trunk stability, and then averaged to generate a single score on a scale of 0–9. Subscores were also assigned based on frequency of plantar stepping, paw placement, coordination, trunk stability, and tail position. Further assessments of hindlimb function were conducted using the gridwalk test (Cummings et al., 2007; Zhang et al., 2015a). Briefly, the gridwalk apparatus consists of an elevated horizontal ladder (92 cm × 16.5 cm), with rungs (4mm in diameter) spaced 1cm apart, and with a covered black goal box at one end. A high speed camera (Basler scA640) was placed 32cm under the middle of the ladder. Animals were trained on the apparatus prior to surgery. At 28 DPI, mice were acclimated to the dark box for three minutes, then removed and placed at the opposite, uncovered end of the apparatus and recorded as they crossed the length of the ladder back into the dark box. At least three passes were recorded per animal. Each animal’s best pass was chosen for analysis, as determined by overall coordination and uninterrupted movement. The total number of missed steps across 30 rungs was then calculated for each animal’s best pass. A missed step was counted when an animals’ hindlimb slipped below the horizontal plane of the rungs.
Spinal Cord Tissue Processing
Tissues were processed according to our previously established methods (Zhang et al., 2015b). Briefly, mice were anesthetized via ketamine (150 mg/kg) and xylazine (10 mg/kg) injection (i.p.). The chest cavity was then opened and animals were perfused transcardially with 0.1M phosphate-buffered saline (PBS, 7.4 pH) followed by 4% paraformaldehyde (PFA) in 0.1M PBS. A 1cm segment of spinal cord centered on the injury site was dissected and post-fixed in PFA for 2 hours before being washed overnight in 0.2M phosphate buffer (7.4 pH). Tissues were cryoprotected in 20% sucrose in 0.1M phosphate buffer saline at 4°C until the cords sank (3–5 days). Spinal cords were then trimmed to a 6mm segment centered on the injury epicenter and evenly distributed by experimental group into blocks containing optimal cutting temperature (OCT) compound. The blocks were rapidly frozen then stored at −80°C. A total of 6 mm spinal cord segment was serially cryosectioned at 10 μm thick coronal sections and every section was collected for a total of 10 slide series, with 6 slides in each series. Therefore, adjacent sections on a slide were separated by 100 μm.
Histology
A modified Eriochrome Cyanine (EC) staining protocol for myelin was used to differentiate between damaged tissue and spared white and grey matter (Rabchevsky et al., 2002; Zhang et al., 2015a). Briefly, slides were hydrated in dH20, submerged in acetone for 2 minutes, rehydrated in dH20, and then exposed to serial dilutions of decreasing concentration of EtOH before being incubated in EC for 30 minutes at room temperature. Excess stain was removed by dH20 washes and slides were differentiated in 0.3% Ammonium Hydroxide. All histological analyses were conducted blindly with respect to treatment condition using a Nikon microscope (Nikon Corporation, Tokyo, Japan) and Scion imaging analysis software (Scion Corporation, Frederick, MD, USA). Identification of intact white and grey matter was based on positive myelin staining as well as comparisons to normal cytoarchitecture. The cross-sectional area of lesion and spared tissues were quantified at 200μm intervals adjacent to the injury epicenter using the Cavalieri method (Rabchevsky et al., 2002), for a total of 17 sections per animal.
Immunohistochemistry
Prior to staining, frozen slide-mounted spinal cord sections were thawed for 1 hr at 37°C and rinsed with 0.1M PBS 3 × 5 min. Slides were then incubated at room temperature for 1hr in blocking buffer (0.1M PBS containing 1% bovine serum albumin, Fisher Scientific, Cat# BP1605), 0.1% Triton X-100 (Sigma-Aldrich, Cat# X-100), 0.1% fish gelatin (Sigma-Aldrich, Cat# G7765), and 5% normal goat (SigmaeAldrich, Cat# G9203). Double immunolabeling for ionized calcium-binding adapter molecule 1 (IBA-1), to evaluate microglia/brain macrophages, and glial fibrillary acidic protein (GFAP), to visualize the astrocytic scar, was performed by incubating slides in chicken anti-GFAP (10ug/ml; Aves, Cat# GFAP) and rabbit anti-IBA-1 (1ug/ml; Wako, Cat# 019-19741) primary antibodies overnight at 4°C in blocking buffer. On the second day, slides were rinsed in 0.1M PBS followed by a 1 hr incubation at room temperature of goat anti-chicken Alexa Flour (AF) 488 (2ug/ml; Life Technologies A-11039) and goat anti-rabbit AF546 (2ug/ml; Life Technologies A-11010) secondary antibodies in blocking buffer. Slides were then rinsed in 0.1M PBS and then were incubated in DAPI (1:95 in 0.1M PBS; Invitrogen D1306) for 30 min, then rinsed and coverslipped with Immu-Mount (Thermo-Scientific, Waltham, MA).
Microscopy and Densitometry
Fluorescent images were captured using a C2+ laser scanning confocal microscope (Nikon Instruments Inc., Melville, NY). For each animal, images were taken at the SCI epicenter (determined by lesion volume estimates and visually confirmed by GFAP staining) as well as 0.6mm and 1.2mm, rostral and caudal to the epicenter. A montaged maximum-intensity projection image, consisting of 9 adjacent z-stacks, was generated for each tissue section. When necessary, laser powers were adjusted to capture the full fluorescent intensity range of lightly stained sections. To quantify the proportional area of activated microglia/macrophages, threshold-based analysis of IBA-1 immunoreactivity was performed as described previously with modifications (Donnelly et al., 2009). Captured RGB images were opened in the MetaMorph analysis program (Molecular Devices, Sunnyvale, CA) and the perimeter of each section was digitally outlined referencing the cytoarchitecture revealed by GFAP and IBA-1 staining. Next, the AF546 channel was isolated and a threshold selected which identified positive IBA-1 staining above background. For each tissue section, the density of IBA-1 staining was calculated as the ratio of immunoreactive area/region of interest area (entire cross section).
Statistical Analyses
All statistical analyses were performed using GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA). For mitochondrial bioenergetics, differences between groups were investigated via analysis of variance (ANOVA). Two-way ANOVA were run to analyze differences in tissue morphology across spinal cord levels and to determine changes in BMS data over time. The 28-day BMS subscores and histological assessments were analyzed by one-tailed unpaired Student’s t-tests. Correlational analyses were run between histological measures and terminal BMS. When warranted, post-hoc analyses (Bonferroni) were performed. Significance was identified at p<0.05.
RESULTS
Effects of pioglitazone on mitochondrial bioenergetics
Quantification of mitochondrial respiration in terms of OCR demonstrated that SCI result in significantly decreased State III OCR (F(3,28)=14.16, p<0.0001) and State V-I OCR (F(3,28)=16.10, p<0.0001) in all injured groups compared to Sham (Figure 1). Post hoc analysis showed a significant (p<0.05) decrease in all State III and State V-I in vehicle treated animals compared to Sham. Conversely, treatment with pioglitazone at 15 min and 3 hr post-injury significantly maintained State III and State V-I OCRs. However, State III and State V-I OCRs remained significantly lower than Sham (Figure 1).
Figure 1.
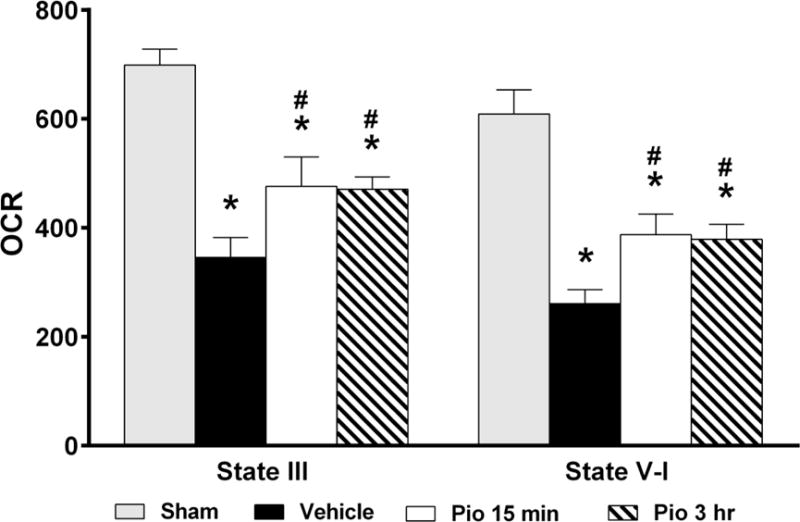
Effect of pioglitazone (Pio) treatment on mitochondrial respiration in terms of oxygen consumption rate (OCR) 25 hrs following SCI. Compared to the sham group, SCI significantly reduced mitochondrial OCR [State III, State V-I and State V-II)] for vehicle treated animals. Conversely, Pio treatment significantly (p<0.05) preserved mitochondrial OCR compared to vehicle. However, no differences were observed between groups when comparing respiration measurements for State V (complex II) enzyme activity. Bars represent means ± SEM, n =7–10 per group. *p<0.05 compared to Sham, #p<0.05 compared to Vehicle.
Effects of pioglitazone on functional recovery
We next assessed the effects of pioglitazone on long-term functional recovery. The data revealed that while both vehicle-and pioglitazone-treated animals demonstrated severe hindlimb paralysis following injury, pioglitazone-treated mice showed improved hindlimb function as early as 7 DPI, (Figure 2A). A two-way repeated measures ANOVA revealed main effects of time (F(6,120)=296.04, p<0.0001) and treatment condition (F(1,20)=7.90, p<0.05). Throughout post-injury assessments pioglitazone-treated mice outperformed vehicle-treated animals, precluding an interaction of the variables (F(5,100)=1.030, p=0.4). Furthermore, pioglitazone-treated animals outperformed vehicle-treated mice on multiple terminal assessments of fine locomotor skills. Specifically, Student’s t-tests revealed that pioglitazone treatment was associated with substantially higher terminal BMS subscores (p<0.01, Figure 2B). Specifically, differences in terminal BMS subscores are associated with higher number of animals with plantar stepping and coordinated walking pattern, better paw position and tail position above the ground compared to vehicle-treatment, which was revealed in significantly improved performance on the gridwalk assessment (p<0.05, Figure 2C).
Figure 2.
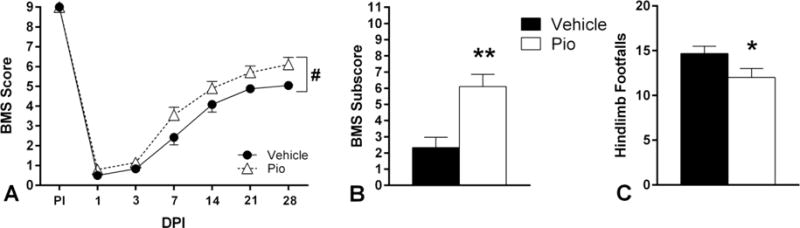
Effect of daily pioglitazone (Pio) treatment for five days post-injury (DPI) on multiple measures of hindlimb functional recovery following SCI. A. Serial BMS testing before injury through 28 days DPI showed that both vehicle-and Pio-treated animals displayed severe hindlimb paralysis following injury; however, at all time-points post-injury, Pio-treated mice scored higher than vehicle-treated animals. Pio-treated mice had greater recovery of fine locomotor skills as assessed by terminal BMS subscores (B) and the gridwalk test (C). Bars and symbols represent means ± SEM, n = 10–12 per group, *p<0.05 compared to vehicle; **p<0.01 compared to vehicle; #p<0.05 main effect of treatment.
Effects of pioglitazone on tissue sparing
Histological assessment of spinal cord sections revealed marked tissue sparing following pioglitazone treatment compared to vehicle, as shown in representative photomicrographs (Figure 3A). Analysis of cross-sectional areas revealed increased white matter sparing in pioglitazone-treated group compared to vehicle, particularly in the sections rostral and penumbral to the epicenter (Figure 3B). A two-way ANOVA revealed a main effect of treatment (F(1,340)=16.86, p<0.0001) and spinal level (F(16,340)=47.32, p<0.0001). Similarly, quantification of gray matter area showed increased spared gray matter sparing with pioglitazone treatment, and sparing was most evident in the cross-sections rostral to the epicenter were grey matter was effectively abolished (Figure 3C). A two-way ANOVA revealed main effects of treatment (F(1,340)=5.607, p<0.05) and spinal level (F(16,340)=10.58, p<0.0001). Pioglitazone treatment also resulted in reduced lesioned tissue compared to vehicle-treated animals across nearly all of the cord (Figure 3D). Accordingly, a two-way ANOVA revealed a main effect of treatment (F(1,340)=11.76, p<0.001) and spinal level (F(16,340)=76.77, p<0.0001). Analyses across the entire rostral-caudal extent of injury (3.2 mm of spinal cord) showed that the pioglitazone-treated group had significantly (p<0.05) less lesion volume, which corresponded to significantly (p<0.05) greater white and grey matter volumes (Figure 3E). Correlational analyses indicate that these anatomical measures significantly correlated with functional outcome measures (Figure 4). Collectively, these results show that pioglitazone treatment significantly reduces secondary injury evidenced by increased spared tissue and decreased lesion volume associated with improved functional recovery, specifically hindlimb-forelimb coordination.
Figure 3.
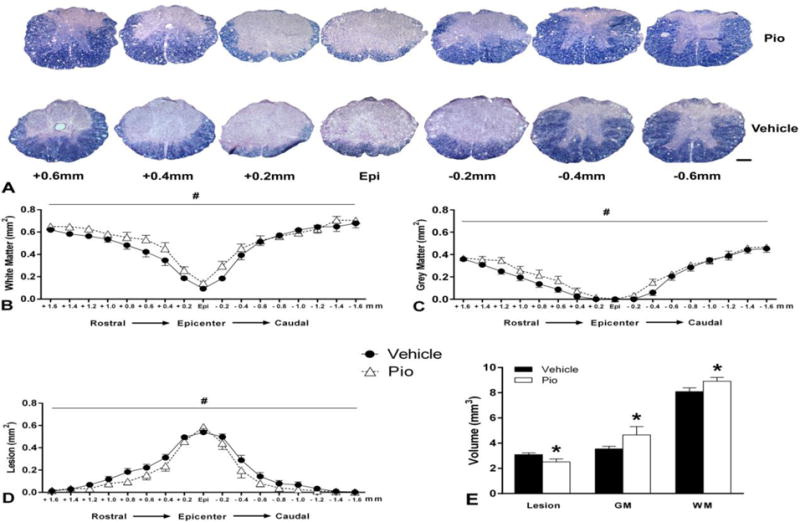
Effect of daily pioglitazone (Pio) treatment for five days post-injury (DPI) on histopathological outcome measures following SCI. A. Representative photomicrographs illustrate differences in tissue morphology between pioglitazone and vehicle-treated animals at 28 days after injury. The Pio-treated group demonstrated increased tissue sparing compared to vehicle treatment, particularly in the sections penumbral and/or rostral to the lesion epicenter. Analysis of evenly spaced tissue sections showed that Pio-treated mice displayed significant sparing of white (B) and grey (C) matter, and less injured tissue (D). E. Quantification of tissue volumes across 3.2mm of injured spinal cord showed that Pio treatment significantly decreased lesion volume while increasing spared white and grey matter volumes. Bars and symbols represent means ± SEM, *p<0.05 compared to vehicle; #p<0.05 main effect of treatment, n = 10–12 per group. Scale Bar = 200μm.
Figure 4.
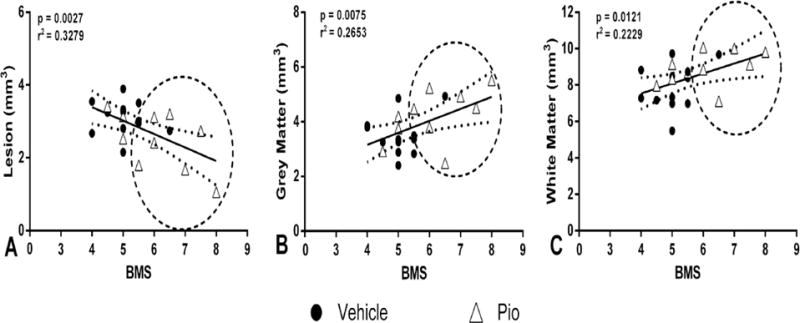
Correlation analyses: lesion volume and spared tissues versus terminal BMS scores. A. There was a significant negative correlation between terminal BMS scores and lesion volume, which corresponded to significant positive correlations between terminal BMS scores and spared grey (B) and white (C) matter. There was also a notable group separation, represented by encircled portions, indicating that pioglitazone (Pio) treatment showed significant neuroprotection associated with higher BMS scores, as compared to vehicle treatment. n = 10–12 per group.
Effects of pioglitazone on microglia/macrophage responses
Because pioglitazone may block PPAR on microglia/macrophages, we also examined the effects of pioglitazone treatment on the inflammatory response to SCI. Despite differences in spared tissues, demarcated by GFAP+ astroglial scars, IBA-1+ microglia/macrophage responses to SCI appeared the same in vehicle- vs. pioglitazone-treated mice at 28 DPI (Figure 5A). Such responses were concentrated at the injury epicenter and tapered off at distal regions, and there was a trend for reduced IBA-1+ densities after pioglitazone treatment. However, while a two-way ANOVA revealed a significant main effect of spinal level (F(4,100)=46.45, p<0.001), there was no treatment effect (F(1,100)=2.321, p>0.05) or treatment × spinal level interaction (F(4,100)=0.2667, p>0.05) (Figure 5B).
Figure 5.
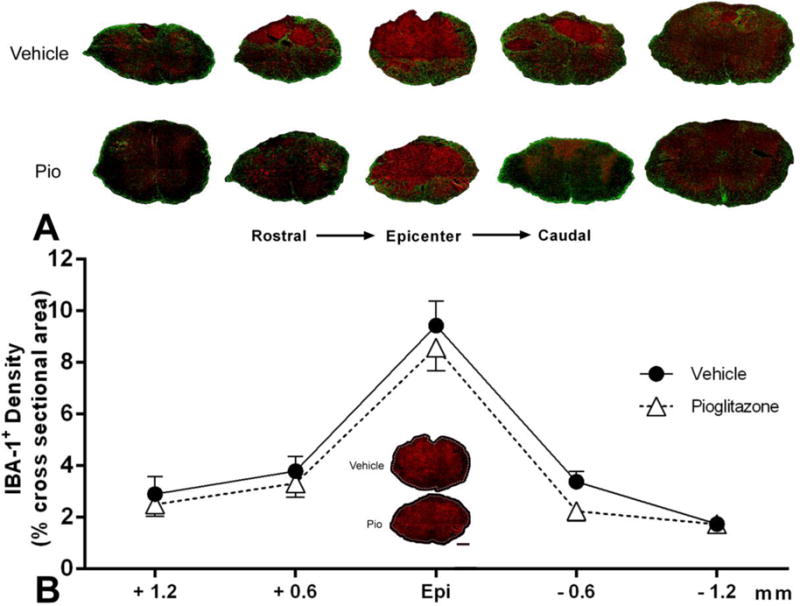
Effect of pioglitazone (Pio) treatment on microglia/macrophage activation 4 weeks after contusion SCI. A. Representative photomicrographs across injured spinal cord levels illustrate similarities in activated IBA-1+ microglia/macrophage responses (red) in pioglitazone-and vehicle-treated spinal cords, despite differences in spared tissue identifiable by the GFAP+ glial scar (green). B. Photomicrographs of IBA-1+ immunoreactivity at the injury epicenter are shown in the graph inset; the entire region of interest is outlined. Image analysis throughout the cross-sectional area of tissue sections at the injury epicenter (Epi), as well as 0.6 mm and 1.2 mm rostral and caudal, showed no significant differences (p>0.05) across spinal levels. Symbols represent means ± SEM, n = 10–12 per group. Scale Bar = 200μm.
DISCUSSION
Our results demonstrate for the first time that pioglitazone modulates mitochondrial function after SCI and this is associated with improved recovery and tissue sparing. Dosage as well as timings and duration of pioglitazone administration were based on our previous reports in a TBI model (Sauerbeck et al., 2011). As we hypothesized, by maintaining mitochondrial function acutely after SCI, daily treatment with pioglitazone resulted in increased tissue sparing and functional recovery at later stages. Furthermore, pioglitazone-treated animals displayed significantly improved hindlimb motor recovery throughout the post-injury period and at terminal assessments, which correlated with greater tissue sparing. These behavioral and morphological data are consistent with previous investigations of pioglitazone treatment for SCI in rats (McTigue et al., 2007; Park et al., 2007), and here we report for the first time that pioglitazone treatment improves gridwalk performance in injured mice. This is an important and refined behavioral consideration, as the gridwalk task discriminates between animals which display the same stepping frequency during open-field assessment (Cummings et al., 2007). Our seminal finding, however, is that similar treatment in SCI mice significantly protected acute mitochondrial function, both after immediate and 3 hr delayed administrations. Moreover, the tissue sparing afforded by pioglitazone was not associated with decreased levels of microgliosis, again in accordance with findings in a rat SCI model (McTigue et al., 2007).
Rescuing mitochondrial bioenergetics is linked to improved functional neuroprotection
A growing body of data indicates that preserving mitochondrial bioenergetics following CNS injury is a powerful therapeutic strategy for reducing tissue damage and attenuating locomotor deficits. Multiple interventions which target the mitochondria have shown promise, including administration of mitochondrial uncouplers (Patel et al., 2009), alternative biofuels (Patel et al., 2012; Patel et al., 2010), and glutathione precursors (Pandya et al., 2014; Patel et al., 2014). We have previously demonstrated that pioglitazone maintains mitochondrial integrity following TBI (Sauerbeck et al., 2011) and the current study extends these findings, demonstrating that pioglitazone preserves mitochondrial function after SCI as well. Likewise, improving mitochondrial function was associated with increased tissue sparing and improved behavioral performance.
While a number of SCI therapeutics have directly targeted downstream pathophysiological cascades such as oxidative damage, the present study and our previous data suggest that a more promising approach for clinical translation may be targeting upstream processes, specifically mitochondrial respiration. CNS trauma causes an influx of intracellular Ca2+, which increases Ca2+ absorption by mitochondria (Sullivan et al., 2005) and this Ca2+ sequestering increases production of reactive oxygen species (ROS) (Dugan et al., 1995). Free radical damage via lipid peroxidation is known to contribute significantly to the propagation of tissue damage after injury (Anderson and Hall, 1993; Hall, 1991; Hall and Braughler, 1993; Springer et al., 1997). These effects are compounded since oxidative stress instigates pro-inflammatory responses (Christman et al., 2000). Critically, we have previously demonstrated that maintaining mitochondrial bioenergetics reduces oxidative stress following contusion SCI (Patel et al., 2009).
We have reported that significant mitochondrial dysfunction occurs as early as 6 hr post-SCI in rats (Sullivan et al., 2007), indicating a clinically relevant window of opportunity to rescue mitochondrial dysfunction after injury. This may help to explain, in part, why there were no changes in OCR values after 15 min vs 3 hr post-injury administration of pioglitazone. Irrespective of targeting mitochondrial bioenergetics, antagonism of PPAR-γ by itself can reduce pro-inflammatory activities (Drew et al., 2006). Accordingly, low-dose pioglitazone treatment after SCI in rats (1.5 mg/kg i.p.; four doses at 5 min and 12, 24, and 48 h) significantly reduced the number of activated microglia at 7 days following injury (Park et al., 2007). In contrast, dosing throughout the first week after SCI (10 mg/kg i.p. at 15 min post-SCI, then every 12 hr for 7 days) did not reduce macrophage accumulation in the lesion at 7 DPI (McTigue et al., 2007). Consistent with the latter results, we detected no significant long-term changes in microglia/macrophage accumulation after pioglitazone treatment (10 mg/kg i.p. at 15 min post-SCI, then daily for 5 days) (Figure 5). Notably, the qualitative trends for reduced microglia/macrophage densities following pioglitazone treatment likely reflects increased tissue sparing compared to vehicle-treated groups (Figure 3).
We have also shown, however, that in the context of TBI pioglitazone treatment reduces microglial activation via PPAR-γ independent mechanisms. More specifically, delivery of a PPAR-γ antagonist (T0070907) in conjunction with pioglitazone elicited similar reduction in activated microglia as pioglitazone treatment alone (Sauerbeck et al., 2011). This could be due, in part, to reduced mitochondrial dysfunction and consequent tissue damage as a result of pioglitazone treatment leading to a reduction in downstream inflammatory cascades. Likewise, emerging evidence suggests that protective effects of pioglitazone following experimental TBI in mice are not solely related to PPAR-γ-dependent mechanisms (Thal et al., 2011). Although it has been established in TBI, it will be important to determine if the results of the current study are, in fact, PPAR-γ independent.
Interactions between pioglitazone and mitoNEET
Over a decade ago, pioglitazone was found to bind to a novel outer mitochondrial membrane protein which was then termed “mitoNEET” (Colca et al., 2004). Evidence now suggests that mitoNEET plays a critical role in energy homeostasis through regulating β-oxidation rates by controlling iron transport into the inter-membrane matrix (Kusminski et al., 2012). There are important differences between the effects of binding mitoNEET by pioglitazone, compared with other PPAR-γ agonists. For instance, compared with pioglitazone, rosiglitazone has a stronger binding affinity for PPAR-γ and a weaker affinity for mitoNEET, yet has demonstrated less robust neuroprotection following CNS trauma (Berger and Moller, 2002). Furthermore, data from our TBI experiments suggest that pioglitazone’s ability to improve mitochondrial function may be due, in part, to its acute effects on mitochondrial respiration (Sauerbeck et al., 2011). More specifically, we found that a single injection of pioglitazone 15 min after TBI results in a non-significant trend towards improved mitochondrial function when respiration was measured the next day. Conversely, when an additional injection was given 1 hour prior to analyzing the injured tissue, significant improvement in respiration was observed. Collectively, these data point towards our working hypothesis that neuroprotective effects of pioglitazone are at least partially independent of PPAR-γ activation and rely, instead, on direct modification of mitochondrial integrity.
Overall, while this study represents an independent replication of reports that pioglitazone treatment following contusion SCI in rats improves long-term neuroprotection (McTigue et al., 2007; Park et al., 2007), we further identified that pioglitazone treatment after contusion SCI in mice significantly attenuates acute mitochondrial dysfunction. We also found comparable maintenance of mitochondrial bioenergetics following both early and delayed treatment, setting the stage for testing more delayed administration. Accordingly, ongoing studies are assessing different dosages and clinically relevant therapeutic windows of administration (3–6 hr post-injury) to potentially repurpose pioglitazone for neurotrauma and/or develop novel therapeutics that target mitochondrial dysfunction. In addition to planned stereological studies to evaluate neuroprotection, comprehensive assessments of neuroinflammatory markers and microglia/macrophage phenotypes will also help to elucidate the mechanisms by which pioglitazone confers both bioenergetics homeostasis and tissue sparing.
Highlights.
Pioglitazone (Pio) maintains acute mitochondrial bioenergetics after spinal cord injury.
Prolonged Pio treatment improves functional recovery following spinal cord injury.
Improved functional recovery is correlated with increased tissue sparing.
Functional neuroprotection is not associated with reduced inflammatory microgliosis.
Acknowledgments
The authors extend special thanks to Dr. Bei Zhang and Mr. Michael Orr for helping with gridwalk, and Mr. Taylor Smith for helping with BMS assessments. This study was supported by NIH/NINDS R01 NS069633 (AGR & PGS); KSCHIRT 15-14A (PGS); SCoBIRC Chair Endowment (AGR); NIH/NINDS 2P30NS051220
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson DK, Hall ED. Pathophysiology of spinal cord trauma. Ann Emerg Med. 1993;22:987–992. doi: 10.1016/s0196-0644(05)82739-8. [DOI] [PubMed] [Google Scholar]
- Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma. 2006;23:635–659. doi: 10.1089/neu.2006.23.635. [DOI] [PubMed] [Google Scholar]
- Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- Bracken MB. Methylprednisolone in the management of acute spinal cord injuries. The Medical journal of Australia. 1990;153:368. [PubMed] [Google Scholar]
- Bracken MB. Methylprednisolone and acute spinal cord injury: an update of the randomized evidence. Spine. 2001;26:S47–54. doi: 10.1097/00007632-200112151-00010. [DOI] [PubMed] [Google Scholar]
- Bracken MB, Collins WF, Freeman DF, Shepard MJ, Wagner FW, Silten RM, Hellenbrand KG, Ransohoff J, Hunt WE, Perot PL, Jr, et al. Efficacy of methylprednisolone in acute spinal cord injury. Jama. 1984;251:45–52. [PubMed] [Google Scholar]
- Bracken MB, Shepard MJ, Collins WF, Holford TR, Young W, Baskin DS, Eisenberg HM, Flamm E, Leo-Summers L, Maroon J, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. Results of the Second National Acute Spinal Cord Injury Study. The New England journal of medicine. 1990;322:1405–1411. doi: 10.1056/NEJM199005173222001. [DOI] [PubMed] [Google Scholar]
- Bracken MB, Shepard MJ, Collins WF, Jr, Holford TR, Baskin DS, Eisenberg HM, Flamm E, Leo-Summers L, Maroon JC, Marshall LF, et al. Methylprednisolone or naloxone treatment after acute spinal cord injury: 1-year follow-up data. Results of the second National Acute Spinal Cord Injury Study. Journal of neurosurgery. 1992;76:23–31. doi: 10.3171/jns.1992.76.1.0023. [DOI] [PubMed] [Google Scholar]
- Bracken MB, Shepard MJ, Hellenbrand KG, Collins WF, Leo LS, Freeman DF, Wagner FC, Flamm ES, Eisenberg HM, Goodman JH, et al. Methylprednisolone and neurological function 1 year after spinal cord injury. Results of the National Acute Spinal Cord Injury Study. Journal of neurosurgery. 1985;63:704–713. doi: 10.3171/jns.1985.63.5.0704. [DOI] [PubMed] [Google Scholar]
- Bracken MB, Shepard MJ, Holford TR, Leo-Summers L, Aldrich EF, Fazl M, Fehlings M, Herr DL, Hitchon PW, Marshall LF, Nockels RP, Pascale V, Perot PL, Jr, Piepmeier J, Sonntag VK, Wagner F, Wilberger JE, Winn HR, Young W. Administration of methylprednisolone for 24 or 48 hours or tirilazad mesylate for 48 hours in the treatment of acute spinal cord injury. Results of the Third National Acute Spinal Cord Injury Randomized Controlled Trial. National Acute Spinal Cord Injury Study. Jama. 1997;277:1597–1604. [PubMed] [Google Scholar]
- Center, N.S.C.I.S. Spinal Cord Injury (SCI) Facts and Figures at a Glance. 2016 doi: 10.1080/10790268.2016.1160676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XR, Besson VC, Palmier B, Garcia Y, Plotkine M, Marchand-Leroux C. Neurological recovery-promoting, anti-inflammatory, and anti-oxidative effects afforded by fenofibrate, a PPAR alpha agonist, in traumatic brain injury. J Neurotrauma. 2007;24:1119–1131. doi: 10.1089/neu.2006.0216. [DOI] [PubMed] [Google Scholar]
- Christman JW, Blackwell TS, Juurlink BH. Redox regulation of nuclear factor kappa B: therapeutic potential for attenuating inflammatory responses. Brain Pathol. 2000;10:153–162. doi: 10.1111/j.1750-3639.2000.tb00252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colca JR, McDonald WG, Waldon DJ, Leone JW, Lull JM, Bannow CA, Lund ET, Mathews WR. Identification of a novel mitochondrial protein (“mitoNEET”) cross-linked specifically by a thiazolidinedione photoprobe. Am J Physiol Endocrinol Metab. 2004;286:E252–260. doi: 10.1152/ajpendo.00424.2003. [DOI] [PubMed] [Google Scholar]
- Conlan AR, Axelrod HL, Cohen AE, Abresch EC, Zuris J, Yee D, Nechushtai R, Jennings PA, Paddock ML. Crystal structure of Miner1: The redox-active 2Fe-2S protein causative in Wolfram Syndrome 2. Journal of molecular biology. 2009;392:143–153. doi: 10.1016/j.jmb.2009.06.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings BJ, Engesser-Cesar C, Cadena G, Anderson AJ. Adaptation of a ladder beam walking task to assess locomotor recovery in mice following spinal cord injury. Behav Brain Res. 2007;177:232–241. doi: 10.1016/j.bbr.2006.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly DJ, Gensel JC, Ankeny DP, van Rooijen N, Popovich PG. An efficient and reproducible method for quantifying macrophages in different experimental models of central nervous system pathology. Journal of neuroscience methods. 2009;181:36–44. doi: 10.1016/j.jneumeth.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew PD, Xu J, Storer PD, Chavis JA, Racke MK. Peroxisome proliferator-activated receptor agonist regulation of glial activation: relevance to CNS inflammatory disorders. Neurochem Int. 2006;49:183–189. doi: 10.1016/j.neuint.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Dugan LL, Sensi SL, Canzoniero LM, Handran SD, Rothman SM, Lin TS, Goldberg MP, Choi DW. Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-D-aspartate. J Neurosci. 1995;15:6377–6388. doi: 10.1523/JNEUROSCI.15-10-06377.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel E. The mitochondrion: is it central to apoptosis? Science. 2001;292:624–626. doi: 10.1126/science.292.5517.624. [DOI] [PubMed] [Google Scholar]
- Frazier-Wood AC, Ordovas JM, Straka RJ, Hixson JE, Borecki IB, Tiwari HK, Arnett DK. The PPAR alpha gene is associated with triglyceride, low-density cholesterol and inflammation marker response to fenofibrate intervention: the GOLDN study. Pharmacogenomics J. 2013;13:312–317. doi: 10.1038/tpj.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geldenhuys WJ, Funk MO, Awale PS, Lin L, Carroll RT. A novel binding assay identifies high affinity ligands to the rosiglitazone binding site of mitoNEET. Bioorganic & medicinal chemistry letters. 2011;21:5498–5501. doi: 10.1016/j.bmcl.2011.06.111. [DOI] [PubMed] [Google Scholar]
- Hall ED. Inhibition of lipid peroxidation in CNS trauma. J Neurotrauma. 1991;8(Suppl 1):S31–40. discussion S41. [PubMed] [Google Scholar]
- Hall ED, Braughler JM. Free radicals in CNS injury. Res Publ Assoc Res Nerv Ment Dis. 1993;71:81–105. [PubMed] [Google Scholar]
- Hall ED, Springer JE. Neuroprotection and acute spinal cord injury: a reappraisal. NeuroRx. 2004;1:80–100. doi: 10.1602/neurorx.1.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X, Liu R, Ross S, Smart EJ, Zhu H, Gong W. Crystallographic studies of human MitoNEET. The Journal of biological chemistry. 2007;282:33242–33246. doi: 10.1074/jbc.C700172200. [DOI] [PubMed] [Google Scholar]
- Kapadia R, Yi JH, Vemuganti R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front Biosci. 2008;13:1813–1826. doi: 10.2741/2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusminski CM, Holland WL, Sun K, Park J, Spurgin SB, Lin Y, Askew GR, Simcox JA, McClain DA, Li C, Scherer PE. MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nature medicine. 2012;18:1539–1549. doi: 10.1038/nm.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry AP, Ding H. Redox control of human mitochondrial outer membrane protein MitoNEET [2Fe-2S] clusters by biological thiols and hydrogen peroxide. J Biol Chem. 2014;289:4307–4315. doi: 10.1074/jbc.M113.542050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Zhou T, Ye K, Wang J. Crystal structure of human mitoNEET reveals distinct groups of iron sulfur proteins. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:14640–14645. doi: 10.1073/pnas.0702426104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Bachstetter AD, Cass WA, Lifshitz J, Bing G. Pioglitazone Attenuates Neuroinflammation and Promotes Dopaminergic Neuronal Survival in the Nigrostriatal System of Rats after Diffuse Brain Injury. J Neurotrauma. 2016 doi: 10.1089/neu.2015.4361. [DOI] [PubMed] [Google Scholar]
- Martin HL, Mounsey RB, Mustafa S, Sathe K, Teismann P. Pharmacological manipulation of peroxisome proliferator-activated receptor gamma (PPARgamma) reveals a role for anti-oxidant protection in a model of Parkinson’s disease. Experimental neurology. 2012;235:528–538. doi: 10.1016/j.expneurol.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen ML, Sullivan PG, Rabchevsky AG, Springer JE. Targeting mitochondrial function for the treatment of acute spinal cord injury. Neurotherapeutics. 2011;8:168–179. doi: 10.1007/s13311-011-0031-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTigue DM, Tripathi R, Wei P, Lash AT. The PPAR gamma agonist Pioglitazone improves anatomical and locomotor recovery after rodent spinal cord injury. Experimental neurology. 2007;205:396–406. doi: 10.1016/j.expneurol.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalik L, Desvergne B, Wahli W. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nat Rev Cancer. 2004;4:61–70. doi: 10.1038/nrc1254. [DOI] [PubMed] [Google Scholar]
- Michalik L, Wahli W. Peroxisome proliferator-activated receptors: three isotypes for a multitude of functions. Curr Opin Biotechnol. 1999;10:564–570. doi: 10.1016/s0958-1669(99)00030-0. [DOI] [PubMed] [Google Scholar]
- Paddock ML, Wiley SE, Axelrod HL, Cohen AE, Roy M, Abresch EC, Capraro D, Murphy AN, Nechushtai R, Dixon JE, Jennings PA. MitoNEET is a uniquely folded 2Fe 2S outer mitochondrial membrane protein stabilized by pioglitazone. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:14342–14347. doi: 10.1073/pnas.0707189104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya JD, Readnower RD, Patel SP, Yonutas HM, Pauly JR, Goldstein GA, Rabchevsky AG, Sullivan PG. N-acetylcysteine amide confers neuroprotection, improves bioenergetics and behavioral outcome following TBI. Experimental neurology. 2014;257:106–113. doi: 10.1016/j.expneurol.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SW, Yi JH, Miranpuri G, Satriotomo I, Bowen K, Resnick DK, Vemuganti R. Thiazolidinedione class of peroxisome proliferator-activated receptor gamma agonists prevents neuronal damage, motor dysfunction, myelin loss, neuropathic pain, and inflammation after spinal cord injury in adult rats. J Pharmacol Exp Ther. 2007;320:1002–1012. doi: 10.1124/jpet.106.113472. [DOI] [PubMed] [Google Scholar]
- Patel SP, Sullivan PG, Lyttle TS, Magnuson DS, Rabchevsky AG. Acetyl-L-carnitine treatment following spinal cord injury improves mitochondrial function correlated with remarkable tissue sparing and functional recovery. Neuroscience. 2012;210:296–307. doi: 10.1016/j.neuroscience.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Sullivan PG, Lyttle TS, Rabchevsky AG. Acetyl-L-carnitine ameliorates mitochondrial dysfunction following contusion spinal cord injury. J Neurochem. 2010;114:291–301. doi: 10.1111/j.1471-4159.2010.06764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Sullivan PG, Pandya JD, Goldstein GA, VanRooyen JL, Yonutas HM, Eldahan KC, Morehouse J, Magnuson DS, Rabchevsky AG. N-acetylcysteine amide preserves mitochondrial bioenergetics and improves functional recovery following spinal trauma. Experimental neurology. 2014;257:95–105. doi: 10.1016/j.expneurol.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Sullivan PG, Pandya JD, Rabchevsky AG. Differential effects of the mitochondrial uncoupling agent, 2,4-dinitrophenol, or the nitroxide antioxidant, Tempol, on synaptic or nonsynaptic mitochondria after spinal cord injury. J Neurosci Res. 2009;87:130–140. doi: 10.1002/jnr.21814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedada KK, Zhou X, Jogiraju H, Carroll RT, Geldenhuys WJ, Lin L, Anderson DJ. A quantitative LC-MS/MS method for determination of thiazolidinedione mitoNEET ligand L-1 in mouse serum suitable for pharmacokinetic studies. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2014;945–946:141–146. doi: 10.1016/j.jchromb.2013.11.048. [DOI] [PubMed] [Google Scholar]
- Rabchevsky AG, Fugaccia I, Sullivan PG, Blades DA, Scheff SW. Efficacy of methylprednisolone therapy for the injured rat spinal cord. J Neurosci Res. 2002;68:7–18. doi: 10.1002/jnr.10187. [DOI] [PubMed] [Google Scholar]
- Rabchevsky AG, Patel SP, Springer JE. Pharmacological interventions for spinal cord injury: where do we stand? How might we step forward? Pharmacol Ther. 2011;132:15–29. doi: 10.1016/j.pharmthera.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Sauerbeck A, Gao J, Readnower R, Liu M, Pauly JR, Bing G, Sullivan PG. Pioglitazone attenuates mitochondrial dysfunction, cognitive impairment, cortical tissue loss, and inflammation following traumatic brain injury. Experimental neurology. 2011;227:128–135. doi: 10.1016/j.expneurol.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood HS, Hunt MJ, Tyagi SC. Peroxisome proliferator ameliorates endothelial dysfunction in a murine model of hyperhomocysteinemia. Am J Physiol Lung Cell Mol Physiol. 2003;284:L333–341. doi: 10.1152/ajplung.00183.2002. [DOI] [PubMed] [Google Scholar]
- Sood V, Colleran K, Burge MR. Thiazolidinediones: a comparative review of approved uses. Diabetes Technol Ther. 2000;2:429–440. doi: 10.1089/15209150050194297. [DOI] [PubMed] [Google Scholar]
- Springer JE, Azbill RD, Mark RJ, Begley JG, Waeg G, Mattson MP. 4-hydroxynonenal, a lipid peroxidation product, rapidly accumulates following traumatic spinal cord injury and inhibits glutamate uptake. J Neurochem. 1997;68:2469–2476. doi: 10.1046/j.1471-4159.1997.68062469.x. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Krishnamurthy S, Patel SP, Pandya JD, Rabchevsky AG. Temporal characterization of mitochondrial bioenergetics after spinal cord injury. J Neurotrauma. 2007;24:991–999. doi: 10.1089/neu.2006.0242. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Rabchevsky AG, Waldmeier PC, Springer JE. Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? J Neurosci Res. 2005;79:231–239. doi: 10.1002/jnr.20292. [DOI] [PubMed] [Google Scholar]
- Thal SC, Heinemann M, Luh C, Pieter D, Werner C, Engelhard K. Pioglitazone reduces secondary brain damage after experimental brain trauma by PPAR-gamma-independent mechanisms. J Neurotrauma. 2011;28:983–993. doi: 10.1089/neu.2010.1685. [DOI] [PubMed] [Google Scholar]
- Wiley SE, Murphy AN, Ross SA, van der Geer P, Dixon JE. MitoNEET is an iron-containing outer mitochondrial membrane protein that regulates oxidative capacity. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:5318–5323. doi: 10.1073/pnas.0701078104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JH, Park SW, Brooks N, Lang BT, Vemuganti R. PPARgamma agonist rosiglitazone is neuroprotective after traumatic brain injury via anti-inflammatory and anti-oxidative mechanisms. Brain Res. 2008;1244:164–172. doi: 10.1016/j.brainres.2008.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonutas HM, Pandya JD, Sullivan PG. Changes in mitochondrial bioenergetics in the brain versus spinal cord become more apparent with age. J Bioenerg Biomembr. 2015;47:149–154. doi: 10.1007/s10863-014-9593-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonutas HM, Sullivan PG. Targeting PPAR isoforms following CNS injury. Curr Drug Targets. 2013;14:733–742. doi: 10.2174/1389450111314070003. [DOI] [PubMed] [Google Scholar]
- Zhang B, Bailey WM, Braun KJ, Gensel JC. Age decreases macrophage IL-10 expression: Implications for functional recovery and tissue repair in spinal cord injury. Experimental neurology. 2015a;273:83–91. doi: 10.1016/j.expneurol.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Bailey WM, Kopper TJ, Orr MB, Feola DJ, Gensel JC. Azithromycin drives alternative macrophage activation and improves recovery and tissue sparing in contusion spinal cord injury. J Neuroinflammation. 2015b;12:218. doi: 10.1186/s12974-015-0440-3. [DOI] [PMC free article] [PubMed] [Google Scholar]