

Abstract
Familial exudative vitreoretinopathy (FEVR) is an inherited blinding disorder characterized by the abnormal development of the retinal vasculature. The majority of mutations identified in FEVR are found within four genes that encode the receptor complex (FZD4, LRP5, and TSPAN12) and ligand (NDP) of a molecular pathway that controls angiogenesis, the Norrin-β-catenin signaling pathway. However, half of all FEVR-affected case subjects do not harbor mutations in these genes, indicating that further mutated genes remain to be identified. Here we report the identification of mutations in CTNNB1, the gene encoding β-catenin, as a cause of FEVR. We describe heterozygous mutations (c.2142_2157dup [p.His720∗] and c.2128C>T [p.Arg710Cys]) in two dominant FEVR-affected families and a de novo mutation (c.1434_1435insC [p.Glu479Argfs∗18]) in a simplex case subject. Previous studies have reported heterozygous de novo CTNNB1 mutations as a cause of syndromic intellectual disability (ID) and autism spectrum disorder, and somatic mutations are linked to many cancers. However, in this study we show that Mendelian inherited CTNNB1 mutations can cause non-syndromic FEVR and that FEVR can be a part of the syndromic ID phenotype, further establishing the role that β-catenin signaling plays in the development of the retinal vasculature.
Keywords: familial exudative vitreoretinopathy, CTNNB1
Main Text
Beta-catenin is an essential protein that orchestrates many key processes during human development and throughout adulthood. It fulfills these functions through its roles as both a cell adhesion molecule and a mediator of cell signaling.1 It is encoded by CTNNB1 (MIM: 116806) and somatic mutations in this gene, which predominantly result in the inhibition of the degradation of β-catenin, are well characterized in many different cancers (MIM: 114500, 114550, 155255, 167000, 132600).2 More recently, large-scale sequencing studies in individuals with syndromic intellectual disability (ID [MIM: 615075]) or autism spectrum disorder (ASD [MIM: 209850]) have identified heterozygous de novo mutations in this gene as the cause of these overlapping disorders.3, 4 In this report, we describe inherited Mendelian mutations in CTNNB1 in the blinding disorder familial exudative vitreoretinopathy (FEVR).
FEVR is an inherited developmental disorder caused by incomplete retinal angiogenesis (MIM: 133780).5 The primary defect, and the hallmark phenotype of this disease, is the absence of vasculature in the peripheral retina but this feature alone often causes no symptoms. In a subset of case subjects, however, retinal ischemia ensues, resulting in a variety of sight-threatening secondary complications including neovascularization, vitreoretinal traction, exudation, retinal folds, and retinal detachment.6 The expressivity of FEVR is remarkably variable, even within families, ranging from asymptomatic individuals who are unaware that they have the condition to severely affected individuals who lose their vision in infancy (see GeneReviews in Web Resources).
In addition to its phenotypic variability, FEVR is also genetically heterogeneous showing dominant, recessive, and X-linked modes of inheritance. Mutations in eight genes have so far been found to cause FEVR and a ninth has been mapped but remains to be identified: NDP (MIM: 300658), FZD4 (MIM: 604579), LRP5 (MIM: 603506), TSPAN12 (MIM: 613138), ATOH7 (MIM: 609875), ZNF408 (MIM: 616454), KIF11 (MIM: 148760), RCBTB1 (MIM: 607867), and EVR3 on chromosome 11p12-13 (MIM: 605750).7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18 Mutation screening has shown that only ∼50% of FEVR-affected families have mutations in these genes.19 Of these, the vast majority (∼90% of case subjects with a molecular diagnosis) harbor mutations in four genes—NDP, FZD4, LRP5, and TSPAN12—all of which encode components of the same molecular pathway, the Norrin-β-catenin signaling pathway (see GeneReviews in Web Resources).19
The Norrin-β-catenin pathway (also referred to as the Norrin/Fz4 pathway) is a derivative of the Wnt-β-catenin pathway but it is activated by a non-Wnt ligand and utilizes a specific set of receptors (Figure 1).20 Norrin (encoded by NDP) is the extracellular ligand that binds to a receptor complex composed of Frizzled-4 (FZD4), Low-density lipoprotein receptor-related protein-5 (LRP5), and Tetraspanin-12 (TSPAN12).21 The ligand-receptor signaling complexes are believed to cluster through the multimerization of TSPAN12, and possibly Norrin, to form specialized membrane microdomains. In the absence of Norrin binding, cytoplasmic β-catenin is targeted for ubiquitination and proteasomal degradation through the action of the “destruction complex.” This complex is composed of the scaffolding proteins Axin and Adenomatous polyposis coli (APC) and the Ser/Thr kinases Glycogen synthase kinase 3 (GSK3) and Casein kinase 1 (CK1). CK1 and GSK3 sequentially phosphorylate β-catenin, resulting in β-catenin recognition and ubiquitination by β-transducin repeat containing protein (β-Trcp) and subsequent degradation.22 However, upon Norrin binding, the resulting signal directs the destruction complex to relocate to the receptors at the cell membrane through the action of Dishevelled (DSH). This inhibits the degradation of β-catenin, allowing its cytoplasmic levels to increase. Subsequently, β-catenin translocates into the nucleus and interacts with the T cell factor (TCF)/Lymphoid enhancing factor (LEF) family of transcription factors in order to control the expression of target genes.21
Figure 1.

Schematic Overview of the Norrin-β-Catenin Pathway
Mutations in the genes encoding the ligand Norrin, the receptors FZD4, LRP5, and TSPAN12, and now the transcriptional activator β-catenin all cause FEVR. For pathway description see main text.
The genetic investigation of families with FEVR-related retinopathies has been pivotal in helping to define the components of this unique angiogenesis pathway and it is hoped that the remaining unidentified mutated genes will provide further insight. With this in mind, the aim of this study was to identify new genes mutated in FEVR.
Our experiment focused on an international multi-ethnic cohort of 36 families, with at least one member diagnosed with FEVR, in whom we had previously excluded the presence of mutations in the genes known to be mutated in FEVR using Sanger sequencing and/or targeted next generation sequencing. The study was approved by the Leeds East (Project number 03/362) Research Ethics Committee and adhered to the tenets of the Declaration of Helsinki. Individuals participated in the study after giving their informed consent. Blood samples were obtained from both affected and unaffected family members and genomic DNA was extracted via standard protocols.
Whole-exome sequencing (WES) was performed in 1 affected individual from each of 12 families from the cohort. The SureSelect All Exon V5 Capture Reagent (Agilent Technologies) was used to enrich exonic regions from genomic DNA and processed according to Agilent’s SureSelectQXT Library Prep protocol. The processed libraries were subsequently pooled and subjected to 150-bp paired-end sequencing on a HiSeq3000 sequencer (Illumina). The resulting reads were aligned to the human reference genome (GRCh37) using Novoalign short-read alignment software (Novocraft Technologies) and processed in the SAM/BAM format using Picard and the Genome Analysis Toolkit (GATK).23, 24, 25 SNPs and indels were called in the VCF format using the HaplotypeCaller function of GATK, with variants being filtered on the basis of mapping quality, strand bias, and genotype quality. After further filtering to exclude polymorphisms with a minor allele frequency > 1% in dbSNP142, ExAC, or EVS (see Web Resources), the remaining variants were annotated and their functional consequences determined with ANNOVAR software (version July 2014).26 For the full WES dataset, the average depth of coverage was 60×, with an average 96% of targeted bases covered at a depth of 5× and 90% at 10×. These data confirmed that no pathogenic nucleotide variants or copy number variations (screened for via ExomeDepth software27) were present in the genes known to cause FEVR when mutated. The remaining lists of variants were subsequently compared to determine whether any mutations were present within the same gene in two or more families. This analysis led to the discovery of different heterozygous CTNNB1 mutations. We therefore designed primers (primer pairs are shown in Table S1) and used PCR and Sanger sequencing to confirm and segregate mutations and to screen all 14 coding exons and flanking intronic sequences in 1 affected member from the 24 remaining families in the cohort. Three CTNNB1 mutations were identified that were not present in the dbSNP, EVS, or ExAC databases. These mutations accounted for 8% (3/36) of FEVR-affected case subjects in this series, although it should be noted that the known mutated genes causing FEVR had previously been excluded in the majority of this cohort.
A heterozygous 16-bp duplication in exon 15 was found in a Hawaiian FEVR-affected family (family F258) of Japanese origin, c.2142_2157dupTAGCTATCGTTCTTTT (p.His720∗) (Figure 2). Three family members were affected by FEVR and the mutation segregated perfectly with the disease phenotype in the pedigree. The proband (II:2) was assessed in her late teens. Her left eye had a retinal detachment with proliferative vitreoretinopathy and temporal traction resulting in a severe retinal fold through the macula. She had a vitrectomy with silicone oil but her vision remained poor (limited to Hand Movements). Her right eye had temporal macular dragging but this was stabilized by peripheral laser treatment and she maintained good vision in this eye (20/40). Her father (I:1) had nystagmus and severe retinal scarring and was registered blind. When last assessed, in his fifth decade, his vision was limited to Hand Movements in one eye and No Light Perception in the other. The proband’s affected younger brother (II:3) was diagnosed at age 10 when he started to have active retinal disease in both eyes. This was stabilized with bilateral laser treatment allowing him to maintain good vision. The mother and two unaffected siblings had normal eyes.
Figure 2.

Mutations in CTNNB1 Cause FEVR
(A) The pedigrees of the families and CTNNB1 mutation segregation data. The genotypes for all tested family members are shown below each individual, with M representing the mutant allele and + representing the wild-type allele. Affected individuals are shaded black. The grandmother (I:2) in family F410 is shaded gray because fluorescein angiography examination showed no evidence of peripheral retina avascularity (the hallmark feature of FEVR), although she did show focal retinal degeneration in one eye. The symbol with a dot represents an asymptomatic individual.
(B) Schematic representation of CTNNB1 (GenBank: NM_001904.3) showing the location and sequence traces of the three disease-causing variants identified in this study.
(C) Schematic representation of the β-catenin protein domains showing the variants identified in this study. The amino-terminal domain (NTD) spans amino acids 1–137, 12 armadillo repeats (ARM) span amino acids 138–664, the carboxy-terminal domain (CTD) spans 665–781, and the Helix-C domain spans amino acids 667–683.
In a Japanese family segregating the FEVR phenotype (family F410), a heterozygous missense change in exon 14 was identified, c.2128C>T (p.Arg710Cys) (Figure 2). The mutation was present in all affected family members. The proband (III:2) presented at 11 years of age with very low vision. Fundus examination showed retinal avascularity, exudation, retinal holes, and bilateral retinal detachment, for which he underwent buckle surgery in both eyes (Figure 3). His affected brother (III:1) had a similar phenotype with retinal avascularity, exudation, and bilateral tractional folds, while their father (II:1) showed abnormal retinal vessels and retinal avascularity. The paternal grandmother (I:2) showed no evidence of retinal avascularity upon fluorescein angiography examination but she did have unilateral focal retinal degeneration. As this phenotype was not typical for an FEVR diagnosis but was clearly abnormal, she was given a diagnosis of “query FEVR” but does carry the mutation. The proband’s asymptomatic brother (III:3) was also found to carry the mutation but fundus examination at age 9 revealed no signs of disease. Analysis of Arg710 among homologs of CTNNB1 revealed this residue to be conserved down to Danio rerio and Xenopus (Figure S2). Furthermore, this mutation was predicted to be disease causing on various bioinformatic pathogenicity prediction tools (Table S2).
Figure 3.

Phenotype of FEVR-Affected Case Subjects with Heterozygous CTNNB1 Mutations
(A and B) Color fundus photograph of the peripheral retina of (A) the right eye and (B) the left eye of the proband (III:2) from family F410 at age 11 years showing retinal holes and tractional retinal detachment.
(C and D) Anterior segment photograph of (C) the right eye and (D) the left eye of the proband from family F1321 aged 1 month showing the absence of retinal view or red reflex due to dense vitreous hemorrhage in the right eye and neovascularization of the iris and a retrolenticular fibrovascular plaque obscuring the view of the retina in the left eye.
A 1-bp insertion in exon 9 was identified in a simplex Chinese case subject, a child with congenital FEVR (family F1321), c.1434_1435insC (p.Glu479Argfs∗18) (Figure 2). He presented at 1 month of age and assessment under general anesthesia showed that his left eye had a total retinal detachment with anterior fibrovascular proliferation, vitreous hemorrhage, and 360° rubeosis iridis. His right eye showed shallow retinal detachment with fibrovascular traction at the optic disc and vitreous hemorrhage (Figures 3 and S1). Neuroimaging and skeletal survey were unremarkable and no facial dysmorphology was noted. Both parents had normal eyes. Segregation analysis revealed that this mutation was de novo, as neither of the unaffected parents carried the mutation but the inheritance of variants in unrelated genes was consistent with the child being related to both parents.
Human β-catenin contains 781 amino acids and is subdivided into three domains: an amino-terminal domain (NTD), a central region containing 12 armadillo (ARM) repeats (residues 138–664), and a carboxy-terminal domain (CTD).28 The NTD contains the binding site for α-catenin and the phosphorylation sites for GSK3 and CK1 and is the location of the mutations responsible for the oncogenic form of β-catenin.2 The ARM repeats form the binding sites for the majority of the interacting partners of β-catenin and are crucial for its roles in cell adhesion and signaling.1 At the start of the CTD is an α-helix motif (residues 667–683), termed Helix-C, that facilitates the binding of proteins to the ARM repeats and is essential for β-catenin signaling.28 The role of the remaining CTD is less well characterized as many of the experiments on this section of the protein also included the last ARM repeat and Helix-C.29 However, it has been suggested that this domain influences signaling by preventing the self-aggregation of the ARM repeat domain and increasing the binding specificity of ARM repeat interactions.28
The effects of the mutations identified in this study are predicted to produce two different outcomes. The exon 9 frameshift mutation identified in family F1321 is expected to lead to nonsense-mediated mRNA decay (NMD) of the mutated transcript and hence to haploinsufficiency of CTNNB1. However, the truncating mutation identified in family F258 is in the last coding exon of CTNNB1, which would indicate that it would not be targeted for NMD and would create a truncated 719 amino acid polypeptide.30 This truncated form of β-catenin is predicted to be missing the very end of the protein beyond Helix-C. Similarly, the missense mutation identified in family F410 (p.Arg710Cys) alters an amino acid residue in this uncharacterized terminal region of the CTD (Figure 2). These findings indicate that FEVR is part of the phenotypic spectrum caused by CTNNB1 haploinsufficiency and that non-syndromic FEVR, which is at the milder end of this spectrum, is caused by mutations altering the uncharacterized CTD of β-catenin.
De Ligt et al.3 first reported de novo CTNNB1 mutations in three subjects with ID, microcephaly, and spasticity. Subsequent reports followed showing that de novo CTNNB1 mutations lead to a recognizable clinical entity that comprises ID with speech impairment, abnormal muscle tone, ASD, microcephaly, distinctive facial features, and brain abnormalities such as corpus callosum hypoplasia.31, 32, 33, 34 This form of syndromic ID appears to be the result of haploinsufficiency of CTNNB1. While full gene deletions have been reported in two individuals,32, 33 the majority of intragenic mutations (Figure S3) are predicted to result in a truncated transcript that will undergo NMD and this mechanism has been confirmed experimentally in two subjects.31 In a parallel study, two de novo CTNNB1 mutations were found in individuals with ASD.4 At the moment it is not clear whether the ASD-affected case subjects have additional features consistent with them having a diagnosis of syndromic ID, because only limited clinical information was provided. However, these individuals did have low non-verbal IQ scores, indicating that they also had ID.4 Furthermore, the reported CTNNB1 mutations in ASD result in a nonsense change (p.Trp504∗) and a missense variant in the crucial ARM repeat domain (p.Thr551Met),4 suggesting that these represent null CTNNB1 alleles similar to mutations that cause syndromic ID.
All the affected individuals in the current study had a diagnosis of non-syndromic FEVR, clearly linking mutations in CTNNB1 with this phenotype. However, given that the affected child from family F1321 is predicted to have haploinsufficiency of CTNNB1 and was only 4 weeks old when originally examined, the family was re-contacted after this molecular result. The child is now 3 years old and displays many clinical features associated with syndromic ID: global developmental delay, motor delay (he can stand briefly with support), significant speech delay (he only uses 1–2 single words and most of his communication is at a non-verbal level), and dysmorphic facial features (squint, long face, and prominent nasal tip). Interestingly, a recent report has described a 22-month-old boy with a history of lipomyelomeningocele, failure to thrive, short stature, developmental delay, and ASD along with an FEVR ocular phenotype. This child was reported to have a de novo heterozygous mutation in CTNNB1 (c.2112_2116dupAGAAC [p.Pro706Glnfs∗31]) but no other genetic information was provided. It is therefore unclear whether all the child’s clinical features are attributable to the β-catenin defect but it appears to be another report of FEVR in an infant with a predicted null allele in CTNNB1.35 The presence of FEVR in these two case subjects supports the hypothesis that FEVR is part of the CTNNB1 haploinsufficiency phenotype.
Although mild visual defects have been reported in 19/30 of the CTNNB1-related syndromic ID-affected case subjects reviewed to date,31, 32, 33, 34 these were not part of the typical FEVR phenotypic spectrum and frequently included either strabismus, hyperopia, myopia, or poor eye contact. In addition, a single case of photophobia has been reported and another subject had slight optic atrophy in one eye and moderate optic papilla hypoplasia in the other.31, 32, 33 Given our findings, we suspect that many of these case subjects may also have subtle retinal avascularity. In all dominant forms of FEVR, the severity of the disease is highly variable and asymptomatic individuals carrying mutations are frequently reported (see GeneReviews in Web Resources).9, 11, 36 The asymptomatic case present in family F410 in this study indicates that CTNNB1-related FEVR follows the same pattern of variable expression so it would be interesting to perform fluorescein angiography examinations on a cohort of CTNNB1-related syndromic ID- and ASD-affected individuals to look for mild FEVR.
The familial mutations identified in this study caused only an FEVR phenotype. It is unlikely that additional defects will have been missed in these individuals given their age but they clearly display the full spectrum of FEVR severity. One could speculate that the mutations are only altering a tissue-restricted isoform of CTNNB1 but there is no evidence to support the existence of such a transcript. There are currently 15 isoforms of CTNNB1 on Ensembl (accessed 03/17) and not all of these contain exons 14 and 15 or encode proteins. However, all four of the annotated RefSeq coding transcripts encode proteins with identical CTDs and are predicted to be altered by the mutations (Genome Browser accessed 03/07). The importance of this part of the transcript is reflected by the lack of variants in the exome databases in this part of the gene. ExAC shows only two loss-of-function alleles in CTNNB1 and one of these is of low quality while the other is a splice defect that is predicted to alter the exons encoding the uncharacterized CTD. Similarly, there are only 28 variants predicted to cause missense substitutions in the CTD beyond Helix-C and these are all rare, the most common one having an allele frequency of 0.0001.
Undoubtedly, the fact that both of the familial mutations are predicted to produce β-catenin with a defective CTD is the likely explanation why these mutations are sufficiently mild to be passed from one generation to the next and why they cause a restricted ocular phenotype. We did attempt to identify additional familial mutations in this region of the gene by sequencing exons 14 and 15 of CTNNB1 (which encode the CTD beyond Helix-C) in an additional cohort of 36, partially pre-screened, predominantly Northern European FEVR-affected case subjects. However, no more variants were identified, indicating that these mutations are rare.
Previously, Cox et al.37 examined the effects of truncating the Drosophila β-catenin homolog, armadillo, in an in vivo model. One of the mutants investigated was almost identical to the predicted truncated protein present in family F258. Their results showed that the truncation had no effect on cell adhesion but caused a reduction in armadillo (β-catenin) signaling.37 To investigate whether the familial mutations identified in this study caused a similar reduction in signaling, the mutations were modeled using the TOPflash β-catenin transcriptional reporter assay. Untagged expression constructs for wild-type (WT) or mutant β-catenin (p.Arg710Cys or p.His720∗) in pDEST40 (Life Technologies) were created using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies) and the expression capability of the constructs was verified by western blot (Figure S4). The assays were performed in STF cells, which are HEK293 cells stably transfected with the TOPflash firefly luciferase construct (a kind gift from Jeremy Nathans).38 Experiments were performed in triplicate in a 24-well plate and repeated in three independent experiments. 90,000 cells/well were transfected with 399 ng of construct DNA plus 1 ng of Renilla luciferase control plasmid (pRL-TK from Promega) using 1.5 μL of FuGENE 6 transfection reagent (Promega). Luciferase activity was measured after 48 hr using the Dual-Luciferase Reporter assay system (Promega) on a Mithras LB 940 plate reader (Berthold Technologies). The data were analyzed using GraphPad PRISM 7.0 software (one-way ANOVA and Dunnett’s test). The results showed that both of the variant CTNNB1 constructs produced significantly different TOPflash activity levels when compared to the WT CTNNB1 construct but that they had opposite effects (Figure 4). The p.His720∗ variant produced significantly reduced levels of TOPflash activation, almost at null allele levels, whereas the p.Arg710Cys variant produced significantly higher levels of TOPflash activation.
Figure 4.
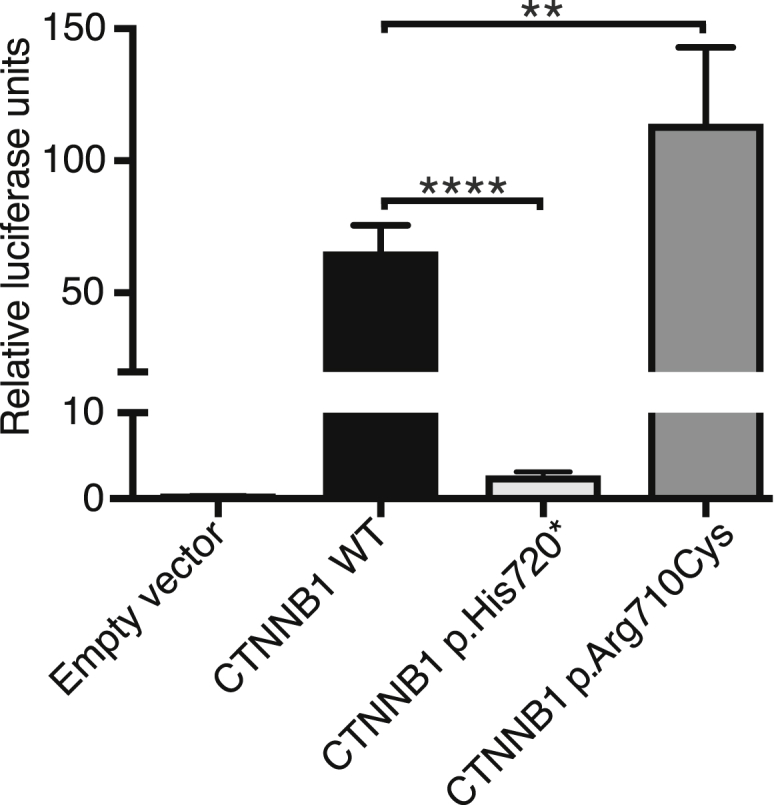
Functional Assessment of Mutant β-Catenin on Transcription using the TOPflash Luciferase Activity Assay
STF cells were transiently transfected with expression constructs for wild-type (WT) or mutant β-catenin (p.Arg710Cys or p.His720∗) or empty vector (pDEST40) alongside a Renilla luciferase plasmid and luciferase activity was measured 48 hr later. The bars represent the firefly/Renilla luciferase ratios for the different constructs. The results are from three independent experiments performed in triplicate. Error bars depict SEM; ∗∗p < 0.01, ∗∗∗p < 0.0001.
It is difficult to envisage a mechanism explaining these contradictory findings. However, contrasting TOPflash results like these have been previously reported for NDP mutations.38, 39 Similarly, although a reduction in Norrin-β-catenin signaling is usually associated with the angiogenesis defects associated with FEVR, ectopic over-activation of Norrin signaling has also been shown to disrupt embryonic angiogenesis.40 Nevertheless, this experiment is unlikely to be an accurate biological replicate of the in vivo consequences of these mutations. For example, if the p.His720∗ variant reduced β-catenin signaling levels to that same extent in family F258, they would be expected to have additional clinical features associated with CTNNB1-related ID syndrome. The molecular mechanisms that control β-catenin signaling are extremely complex and involve a growing number of tissue-specific co-factors, inhibitors, and activators that vary in a time- and context-dependent manner and are unquestionably modulated by feedback loops and other signaling pathways. Although a great deal of research into the function of β-catenin has been performed in animal and cell-based models,1, 41 it is clear that they do not always accurately model the function of β-catenin in humans. A clear example of this is the heterozygous β-catenin knockout mice. These should be a model for CTNNB1-related syndromic ID but are reported to have a wild-type phenotype.42
The fact that the FEVR phenotype is also present in individuals who are haploinsufficient for CTNNB1 suggests that these mutations may cause a partial loss of function of β-catenin and therefore act in a hypomorphic fashion. However, it’s unclear whether the defects alter a retina- or Norrin-β-catenin-specific function or whether they reduce a ubiquitous fundamental function of β-catenin and the retinal vasculature is the tissue most susceptible to this reduction. The location of the mutations and the abnormal TOPflash assay results point to the mutations altering the transcriptional activity of β-catenin. However, in addition to its well-established roles in cell adhesion and transcription, β-catenin has also been reported to play an uncharacterized role at the centrosome and in mitotic spindle formation.43 The links between asymmetric cell division and angiogenesis,44 and the fact that the gene encoding the mitotic protein KIF11 is mutated in FEVR,16 suggest that this potential role warrants further investigation.
Conditional knockout studies in mice have shown that β-catenin depletion in endothelial cells leads to angiogenesis defects throughout the central nervous system (CNS) including the retina.45, 46, 47 Although FEVR is characterized as a disorder that affects only the retinal vasculature, the FEVR knockout mouse models suggests that there may be mild vascular defects in other parts of the CNS. For example, the Ndp−/− and Fzd4−/− mice have capillary defects in the ear (stria vascularis) and additional vasculature defects have been identified in the cerebellum of the Fzd4−/− mouse.38, 48, 49 The retina is the most accessible part of the CNS and this allows detailed examination of its vasculature. It is this accessibility that allows us to diagnose subtle vascular defects in asymptomatic individuals harboring FEVR mutations, but defects in the wider CNS could easily be missed. If present in humans, these could potentially contribute to some of the neurological features associated with syndromic ID and ASD. Consistent with this suggestion, defects in angiogenesis have been reported in ASD, and there are many reports linking vascular defects with brain development, ID, and dementia.50, 51, 52, 53
In summary, we have shown that heterozygous mutations in CTNNB1 can cause non-syndromic FEVR and that FEVR is part of the CTNNB1 haploinsufficiency phenotype. These findings confirm the importance of β-catenin signaling in the pathogenesis of FEVR but also raise questions as to how mutations in such a ubiquitously essential protein can lead to such a restricted phenotype.
Acknowledgments
We thank the FEVR-affected families for their participation in this study and Professor Jeremy Nathans (John Hopkins University School of Medicine) for kindly providing the STF cells. This project has received funding from the European Union’s Seventh Framework Programme for research, technological development, and demonstration under grant agreement no. 317472 (EyeTN), Fight For Sight (grants 1493/1494 and GR586), and RP Fighting Blindness (grant GR586). We acknowledge the support of the UK Inherited Retinal Disease Consortium.
Published: June 1, 2017
Footnotes
Supplemental Data include four figures and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.ajhg.2017.05.001.
Accession Numbers
The variants reported in this paper have been deposited into the ClinVar database at the National Centre for Biotechnology Information under accession numbers ClinVar: SCV000266847–SCV000266849.
Web Resources
Ensembl, http://grch37.ensembl.org
ExAC Browser, http://exac.broadinstitute.org/
GenBank, http://www.ncbi.nlm.nih.gov/genbank/
GeneReviews, Toomes, C., and Downey, L. (2011). Familial exudative vitreoretinopathy, autosomal dominant. https://www.ncbi.nlm.nih.gov/books/NBK1147/
MutationTaster, http://www.mutationtaster.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
OMIM, http://www.omim.org/
VCF hacks, https://github.com/gantzgraf/vcfhacks
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
Supplemental Data
References
- 1.Valenta T., Hausmann G., Basler K. The many faces and functions of β-catenin. EMBO J. 2012;31:2714–2736. doi: 10.1038/emboj.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anastas J.N., Moon R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer. 2013;13:11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- 3.de Ligt J., Willemsen M.H., van Bon B.W., Kleefstra T., Yntema H.G., Kroes T., Vulto-van Silfhout A.T., Koolen D.A., de Vries P., Gilissen C. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 4.O’Roak B.J., Vives L., Fu W., Egertson J.D., Stanaway I.B., Phelps I.G., Carvill G., Kumar A., Lee C., Ankenman K. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Criswick V.G., Schepens C.L. Familial exudative vitreoretinopathy. Am. J. Ophthalmol. 1969;68:578–594. doi: 10.1016/0002-9394(69)91237-9. [DOI] [PubMed] [Google Scholar]
- 6.Benson W.E. Familial exudative vitreoretinopathy. Trans. Am. Ophthalmol. Soc. 1995;93:473–521. [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Z.Y., Battinelli E.M., Fielder A., Bundey S., Sims K., Breakefield X.O., Craig I.W. A mutation in the Norrie disease gene (NDP) associated with X-linked familial exudative vitreoretinopathy. Nat. Genet. 1993;5:180–183. doi: 10.1038/ng1093-180. [DOI] [PubMed] [Google Scholar]
- 8.Robitaille J., MacDonald M.L.E., Kaykas A., Sheldahl L.C., Zeisler J., Dubé M.P., Zhang L.H., Singaraja R.R., Guernsey D.L., Zheng B. Mutant frizzled-4 disrupts retinal angiogenesis in familial exudative vitreoretinopathy. Nat. Genet. 2002;32:326–330. doi: 10.1038/ng957. [DOI] [PubMed] [Google Scholar]
- 9.Toomes C., Bottomley H.M., Jackson R.M., Towns K.V., Scott S., Mackey D.A., Craig J.E., Jiang L., Yang Z., Trembath R. Mutations in LRP5 or FZD4 underlie the common familial exudative vitreoretinopathy locus on chromosome 11q. Am. J. Hum. Genet. 2004;74:721–730. doi: 10.1086/383202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiao X., Ventruto V., Trese M.T., Shastry B.S., Hejtmancik J.F. Autosomal recessive familial exudative vitreoretinopathy is associated with mutations in LRP5. Am. J. Hum. Genet. 2004;75:878–884. doi: 10.1086/425080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poulter J.A., Ali M., Gilmour D.F., Rice A., Kondo H., Hayashi K., Mackey D.A., Kearns L.S., Ruddle J.B., Craig J.E. Mutations in TSPAN12 cause autosomal-dominant familial exudative vitreoretinopathy. Am. J. Hum. Genet. 2010;86:248–253. doi: 10.1016/j.ajhg.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nikopoulos K., Gilissen C., Hoischen A., van Nouhuys C.E., Boonstra F.N., Blokland E.A., Arts P., Wieskamp N., Strom T.M., Ayuso C. Next-generation sequencing of a 40 Mb linkage interval reveals TSPAN12 mutations in patients with familial exudative vitreoretinopathy. Am. J. Hum. Genet. 2010;86:240–247. doi: 10.1016/j.ajhg.2009.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poulter J.A., Davidson A.E., Ali M., Gilmour D.F., Parry D.A., Mintz-Hittner H.A., Carr I.M., Bottomley H.M., Long V.W., Downey L.M. Recessive mutations in TSPAN12 cause retinal dysplasia and severe familial exudative vitreoretinopathy (FEVR) Invest. Ophthalmol. Vis. Sci. 2012;53:2873–2879. doi: 10.1167/iovs.11-8629. [DOI] [PubMed] [Google Scholar]
- 14.Khan K., Logan C.V., McKibbin M., Sheridan E., Elçioglu N.H., Yenice O., Parry D.A., Fernandez-Fuentes N., Abdelhamed Z.I., Al-Maskari A. Next generation sequencing identifies mutations in Atonal homolog 7 (ATOH7) in families with global eye developmental defects. Hum. Mol. Genet. 2012;21:776–783. doi: 10.1093/hmg/ddr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collin R.W., Nikopoulos K., Dona M., Gilissen C., Hoischen A., Boonstra F.N., Poulter J.A., Kondo H., Berger W., Toomes C. ZNF408 is mutated in familial exudative vitreoretinopathy and is crucial for the development of zebrafish retinal vasculature. Proc. Natl. Acad. Sci. USA. 2013;110:9856–9861. doi: 10.1073/pnas.1220864110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robitaille J.M., Gillett R.M., LeBlanc M.A., Gaston D., Nightingale M., Mackley M.P., Parkash S., Hathaway J., Thomas A., Ells A. Phenotypic overlap between familial exudative vitreoretinopathy and microcephaly, lymphedema, and chorioretinal dysplasia caused by KIF11 mutations. JAMA Ophthalmol. 2014;132:1393–1399. doi: 10.1001/jamaophthalmol.2014.2814. [DOI] [PubMed] [Google Scholar]
- 17.Wu J.H., Liu J.H., Ko Y.C., Wang C.T., Chung Y.C., Chu K.C., Liu T.T., Chao H.M., Jiang Y.J., Chen S.J., Chung M.Y. Haploinsufficiency of RCBTB1 is associated with Coats disease and familial exudative vitreoretinopathy. Hum. Mol. Genet. 2016;25:1637–1647. doi: 10.1093/hmg/ddw041. [DOI] [PubMed] [Google Scholar]
- 18.Downey L.M., Keen T.J., Roberts E., Mansfield D.C., Bamashmus M., Inglehearn C.F. A new locus for autosomal dominant familial exudative vitreoretinopathy maps to chromosome 11p12-13. Am. J. Hum. Genet. 2001;68:778–781. doi: 10.1086/318790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salvo J., Lyubasyuk V., Xu M., Wang H., Wang F., Nguyen D., Wang K., Luo H., Wen C., Shi C. Next-generation sequencing and novel variant determination in a cohort of 92 familial exudative vitreoretinopathy patients. Invest. Ophthalmol. Vis. Sci. 2015;56:1937–1946. doi: 10.1167/iovs.14-16065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clevers H. Eyeing up new Wnt pathway players. Cell. 2009;139:227–229. doi: 10.1016/j.cell.2009.09.027. [DOI] [PubMed] [Google Scholar]
- 21.Ye X., Wang Y., Nathans J. The Norrin/Frizzled4 signaling pathway in retinal vascular development and disease. Trends Mol. Med. 2010;16:417–425. doi: 10.1016/j.molmed.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stamos J.L., Weis W.I. The β-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 2013;5:a007898. doi: 10.1101/cshperspect.a007898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and samtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Plagnol V., Curtis J., Epstein M., Mok K.Y., Stebbings E., Grigoriadou S., Wood N.W., Hambleton S., Burns S.O., Thrasher A.J. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28:2747–2754. doi: 10.1093/bioinformatics/bts526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xing Y., Takemaru K., Liu J., Berndt J.D., Zheng J.J., Moon R.T., Xu W. Crystal structure of a full-length beta-catenin. Structure. 2008;16:478–487. doi: 10.1016/j.str.2007.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mosimann C., Hausmann G., Basler K. Beta-catenin hits chromatin: regulation of Wnt target gene activation. Nat. Rev. Mol. Cell Biol. 2009;10:276–286. doi: 10.1038/nrm2654. [DOI] [PubMed] [Google Scholar]
- 30.Kervestin S., Jacobson A. NMD: a multifaceted response to premature translational termination. Nat. Rev. Mol. Cell Biol. 2012;13:700–712. doi: 10.1038/nrm3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tucci V., Kleefstra T., Hardy A., Heise I., Maggi S., Willemsen M.H., Hilton H., Esapa C., Simon M., Buenavista M.T. Dominant β-catenin mutations cause intellectual disability with recognizable syndromic features. J. Clin. Invest. 2014;124:1468–1482. doi: 10.1172/JCI70372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dubruc E., Putoux A., Labalme A., Rougeot C., Sanlaville D., Edery P. A new intellectual disability syndrome caused by CTNNB1 haploinsufficiency. Am. J. Med. Genet. A. 2014;164A:1571–1575. doi: 10.1002/ajmg.a.36484. [DOI] [PubMed] [Google Scholar]
- 33.Kuechler A., Willemsen M.H., Albrecht B., Bacino C.A., Bartholomew D.W., van Bokhoven H., van den Boogaard M.J., Bramswig N., Büttner C., Cremer K. De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: expanding the mutational and clinical spectrum. Hum. Genet. 2015;134:97–109. doi: 10.1007/s00439-014-1498-1. [DOI] [PubMed] [Google Scholar]
- 34.Kharbanda M., Pilz D.T., Tomkins S., Chandler K., Saggar A., Fryer A., McKay V., Louro P., Smith J.C., Burn J., DDD Study Clinical features associated with CTNNB1 de novo loss of function mutations in ten individuals. Eur. J. Med. Genet. 2017;60:130–135. doi: 10.1016/j.ejmg.2016.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dixon M.W., Stem M.S., Schuette J.L., Keegan C.E., Besirli C.G. CTNNB1 mutation associated with familial exudative vitreoretinopathy (FEVR) phenotype. Ophthalmic Genet. 2016;37:468–470. doi: 10.3109/13816810.2015.1120318. [DOI] [PubMed] [Google Scholar]
- 36.Toomes C., Bottomley H.M., Scott S., Mackey D.A., Craig J.E., Appukuttan B., Stout J.T., Flaxel C.J., Zhang K., Black G.C. Spectrum and frequency of FZD4 mutations in familial exudative vitreoretinopathy. Invest. Ophthalmol. Vis. Sci. 2004;45:2083–2090. doi: 10.1167/iovs.03-1044. [DOI] [PubMed] [Google Scholar]
- 37.Cox R.T., Pai L.M., Kirkpatrick C., Stein J., Peifer M. Roles of the C terminus of Armadillo in Wingless signaling in Drosophila. Genetics. 1999;153:319–332. doi: 10.1093/genetics/153.1.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu Q., Wang Y., Dabdoub A., Smallwood P.M., Williams J., Woods C., Kelley M.W., Jiang L., Tasman W., Zhang K., Nathans J. Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004;116:883–895. doi: 10.1016/s0092-8674(04)00216-8. [DOI] [PubMed] [Google Scholar]
- 39.Qin M., Kondo H., Tahira T., Hayashi K. Moderate reduction of Norrin signaling activity associated with the causative missense mutations identified in patients with familial exudative vitreoretinopathy. Hum. Genet. 2008;122:615–623. doi: 10.1007/s00439-007-0438-8. [DOI] [PubMed] [Google Scholar]
- 40.Ye X., Wang Y., Cahill H., Yu M., Badea T.C., Smallwood P.M., Peachey N.S., Nathans J. Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell. 2009;139:285–298. doi: 10.1016/j.cell.2009.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grigoryan T., Wend P., Klaus A., Birchmeier W. Deciphering the function of canonical Wnt signals in development and disease: conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes Dev. 2008;22:2308–2341. doi: 10.1101/gad.1686208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haegel H., Larue L., Ohsugi M., Fedorov L., Herrenknecht K., Kemler R. Lack of beta-catenin affects mouse development at gastrulation. Development. 1995;121:3529–3537. doi: 10.1242/dev.121.11.3529. [DOI] [PubMed] [Google Scholar]
- 43.Mbom B.C., Nelson W.J., Barth A. β-catenin at the centrosome: discrete pools of β-catenin communicate during mitosis and may co-ordinate centrosome functions and cell cycle progression. BioEssays. 2013;35:804–809. doi: 10.1002/bies.201300045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Costa G., Harrington K.I., Lovegrove H.E., Page D.J., Chakravartula S., Bentley K., Herbert S.P. Asymmetric division coordinates collective cell migration in angiogenesis. Nat. Cell Biol. 2016;18:1292–1301. doi: 10.1038/ncb3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stenman J.M., Rajagopal J., Carroll T.J., Ishibashi M., McMahon J., McMahon A.P. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science. 2008;322:1247–1250. doi: 10.1126/science.1164594. [DOI] [PubMed] [Google Scholar]
- 46.Daneman R., Agalliu D., Zhou L., Kuhnert F., Kuo C.J., Barres B.A. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc. Natl. Acad. Sci. USA. 2009;106:641–646. doi: 10.1073/pnas.0805165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou Y., Wang Y., Tischfield M., Williams J., Smallwood P.M., Rattner A., Taketo M.M., Nathans J. Canonical WNT signaling components in vascular development and barrier formation. J. Clin. Invest. 2014;124:3825–3846. doi: 10.1172/JCI76431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Y., Huso D., Cahill H., Ryugo D., Nathans J. Progressive cerebellar, auditory, and esophageal dysfunction caused by targeted disruption of the frizzled-4 gene. J. Neurosci. 2001;21:4761–4771. doi: 10.1523/JNEUROSCI.21-13-04761.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rehm H.L., Zhang D.S., Brown M.C., Burgess B., Halpin C., Berger W., Morton C.C., Corey D.P., Chen Z.Y. Vascular defects and sensorineural deafness in a mouse model of Norrie disease. J. Neurosci. 2002;22:4286–4292. doi: 10.1523/JNEUROSCI.22-11-04286.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Azmitia E.C., Saccomano Z.T., Alzoobaee M.F., Boldrini M., Whitaker-Azmitia P.M. Persistent angiogenesis in the autism brain: an immunocytochemical study of postmortem cortex, brainstem and cerebellum. J. Autism Dev. Disord. 2016;46:1307–1318. doi: 10.1007/s10803-015-2672-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pimentel-Coelho P.M., Rivest S. The early contribution of cerebrovascular factors to the pathogenesis of Alzheimer’s disease. Eur. J. Neurosci. 2012;35:1917–1937. doi: 10.1111/j.1460-9568.2012.08126.x. [DOI] [PubMed] [Google Scholar]
- 52.Newton S.S., Fournier N.M., Duman R.S. Vascular growth factors in neuropsychiatry. Cell. Mol. Life Sci. 2013;70:1739–1752. doi: 10.1007/s00018-013-1281-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Silva T.M., Faraci F.M. Microvascular dysfunction and cognitive impairment. Cell. Mol. Neurobiol. 2016;36:241–258. doi: 10.1007/s10571-015-0308-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.