

Abstract
The SLC25 family member SLC25A38 (Hem25 in yeast) was recently identified as a mitochondrial glycine transporter that provides substrate to initiate heme/hemoglobin synthesis. Mutations in the human SLC25A38 gene cause congenital sideroblastic anemia. The full extent to which SLC25 family members coregulate heme synthesis with other mitochondrial functions is not clear. In this study, we surveyed 29 nonessential SLC25 family members in Saccharomyces cerevisiae for their ability to support growth in the presence and absence of HEM25. Six SLC25 family members were identified that were required for growth or for heme synthesis in cells lacking Hem25 function. Importantly, we determined that loss of function of the SLC25 family member Flx1, which imports FAD into mitochondria, together with loss of function of Hem25, resulted in inability to grow on media that required yeast cells to supply energy using mitochondrial respiration. We report that specific components of complexes of the electron transport chain are decreased in the absence of Flx1 and Hem25 function. In addition, we show that mitochondria from flx1Δ hem25Δ cells contain uncharacterized Cox2-containing high molecular weight aggregates. The functions of Flx1 and Hem25 provide a facile explanation for the decrease in heme level, and in specific electron transport chain complex components.
Keywords: heme, mitochondria, electron transport chain, SLC25 protein family, glycine import
Heme is a component of many cellular constituents including respiratory cytochromes, P450 cytochromes, catalase, peroxidase, myoglobin, and hemoglobin (Ajioka et al. 2006; Chiabrando et al. 2014; Yuan et al. 2013; Kardon et al. 2015). Heme biosynthesis is catalyzed by eight enzymes located in the cytoplasm and mitochondria (Figure 1A). The first enzymatic reaction in heme synthesis takes place in mitochondria through the condensation of glycine with succinyl-CoA to form 5-aminolevulinic acid (5-Ala) (Aivado et al. 2006; Bishop et al. 2012). In the yeast Saccharomyces cerevisiae this reaction is catalyzed by Hem1, while in humans it is catalyzed by two different aminolevulinic acid synthases (ALAS), one expressed ubiquitously (ALAS1) and the other expressed in erythroid precursor cells (ALAS2) (Ajioka et al. 2006; Chiabrando et al. 2014; Yuan et al. 2013; Fernández-Murray et al. 2016). ALAS requires the cofactor pyridoxal 5-phosphate (PLP) to catalyze its reaction (Astner et al. 2005). 5-Ala is exported to the cytoplasm, where, through a sequence of enzymatic reactions (Figure 1A), coproporphyrinogen III is synthesized. Coproporphyrinogen III is imported from the cytoplasm into mitochondria, where it is converted through a sequence of enzymatic reactions into heme (Figure 1A) (Chiabrando et al. 2014). Heme is subsequently incorporated into mitochondrial proteins, including those within respiratory cytochromes of the electron transport chain (ETC) and mitochondrial P450 enzymes. Heme is also transported across the mitochondrial membrane into the cytoplasm, where heme chaperons bind and transport heme for incorporation into various hemoproteins, including microsomal P450 enzymes and other detoxifying enzymes, and in erythroid cells with globin chains to form hemoglobin (Weatherall 2013).
Figure 1.

Mitochondrial glycine is used to synthesize heme and produce one-carbon units. (A) Heme biosynthesis pathway. (B) Mitochondrial glycine can be used to synthesize heme or catabolized by the GCV into nitrogen and one-carbon units. Hem25, the yeast homolog of human SLC25A38, is a mitochondrial glycine importer, and Lpd1 is a subunit of the yeast GCV. ALAS, aminolevulinic acid synthase; ALAD, aminolevulinic acid dehydratase; PBGD, porphobilinogen deaminase; UROS, uropoporphyrinogen III synthase; UROD, uroporphyrinogen III decarboxylase; CPOX, coproporphyrinogen oxidase; PPO, protoporphyrinogen oxidase; FECH, ferrochelatase; GCV, glycine cleavage complex.
Mitochondria possess both outer and inner membranes with differential permeabilities. The outer mitochondrial membrane is permeable to solutes up to ∼5 kDa, while the inner membrane is comparatively impermeable to enable efficient oxidative phosphorylation. Various transporters reside within the inner mitochondrial membrane to overcome its permeability barrier, including transporters required to facilitate the synthesis of heme. The identities of the transporters required for heme biosynthesis are still being unraveled. The exporter of 5-Ala out of the mitochondria has been hypothesized to be ABCB10 (Chen et al. 2009, 2010; Shintre et al. 2013; Tang et al. 2012; Yamamoto et al. 2014)—a member of the ABC transporter family that has an important role during erythroid differentiation, and whose over expression promotes hemoglobin synthesis. The mitochondrial importer of coproporphyrinogen III has been proposed to be ABCB6, which binds both porphyrins and heme; moreover, ABCB6 expression is positively regulated by the stimulation of erythroid differentiation and by heme levels (Paterson et al. 2007; Zhang et al. 2013; Zhao et al. 2013; Krishnamurthy et al. 2006; Schultz et al. 2010). Several lines of evidence point to the mitochondrial heme exporter being FLVCR1b (Chiabrando et al. 2012; Fleming and Hamza 2012). FLVCR1b is essential for erythroid differentiation, and overexpression of FLVCR1b promotes heme biosynthesis, whereas its silencing results in mitochondrial heme accumulation. In addition, FLVCR1a is a heme exporter that resides in the plasma membrane (Chiabrando et al. 2012). Other mitochondrial membrane transporters have recently been implicated in the synthesis of heme, in particular, members of the SLC25 protein family (Gutierrez-Aguilar and Baines 2013).
The SLC25 transporter family member SLC25A38 was recently identified as a glycine transporter that provides substrate for ALAS2 to initiate heme/hemoglobin synthesis in the mitochondria of erythropoietic cells (Fernández-Murray et al. 2016; Lunetti et al. 2016). Another SLC25 family member, SLC25A32, imports folate into mitochondria (Urano et al. 2014; Lawrence et al. 2011). SLC25A32 appears to be essential for glycine synthesis inside mitochondria as folate is required for the conversion of serine to glycine by mitochondrial serine hydroxymethyltransferase (Locasale 2013; Giardina et al. 2015; Saint-Marc et al. 2015). Thus, both the import of glycine into the mitochondria, and glycine synthesis by mitochondria, require SLC25 family members. Glycine is required for the first step in heme synthesis, as well as other mitochondrial processes, including its use as a substrate for the glycine cleavage complex (GCV) (Tibbetts and Appling 2010; Wang et al. 2013). Mitochondrial folate is also required for the GCV, which produces one-carbon units required for the synthesis of several macromolecules, including formylmethionine for mitochondrial translation initiation (Tibbetts and Appling 2010; Wang et al. 2013). It has also been suggested that SLC25A32 could transport FAD across the inner mitochondrial membrane (Spaan et al. 2005; Gutierrez-Aguilar and Baines 2013). Other SLC25 transporters, including SLC25A28 and SLC25A37 (also referred to as mitoferrin-2 and mitoferrin-1, respectively), contribute to heme metabolism while also contributing to other mitochondrial functions (Chen et al. 2009; Urano et al. 2014). SLC25A28 and SLC25A37 both transport iron into mitochondria. Iron is required for the last step of heme biosynthesis, and for the formation of Fe–S clusters. Fe–S clusters are incorporated into many mitochondrial enzymes, including several within the ETC (Ajioka et al. 2006; Gomez et al. 2014; Wingert et al. 2005). The SLC25 family members SLC25A4, SLC25A5, SLC25A6, and SLC25A31 are adenine nucleotide translocases that are the main transporters of ADP into the mitochondria and ATP to the cytoplasm; however, they have also been proposed to transport heme or heme precursors across mitochondrial membranes (Azuma et al. 2008; Fleming and Hamza 2012; Yuan et al. 2013). It is clear that SLC25 family members are important for the synthesis of heme while simultaneously regulating other mitochondrial processes.
The extent to which the mitochondrial SLC25 family members regulate heme synthesis is not known. In this study, new SLC25 family members whose function is required to maintain normal heme level were identified and characterized. In addition, we describe a previously unknown SLC25 family mediated relationship that simultaneously regulates heme synthesis and ETC function.
Materials and Methods
Yeast strains
Yeast strains used are listed in Supplemental Material, Table S1 in File S1. In some instances, the KanMX4 gene, which had been used to inactivate yeast genes obtained from the yeast deletion collection, was replaced with the nourseothricin acetyltransferase (NatMX4) gene. This was done by transformation of the corresponding strain with a linearized pAG25 plasmid as described (Voth et al. 2003). Briefly, the linearized plasmid was transformed into yeast cells, cells were plated and grown in yeast peptone dextrose (YPD) medium for 1 d, and were replica plated onto YPD medium containing nourseothricin. Antibiotic resistant colonies were restreaked to isolate single-cell-derived colonies. Proper antibiotic resistance gene replacement was assessed by the consequent loss of G-418 resistance character. BY4741 and BY4742 strains carry a Ty1 element inserted in the 3′ region of the HAP1 open reading frame (Buschlen et al. 2003; Gaisne et al. 1999). The HAP1 gene encodes a transcriptional regulator involved in regulation of gene expression in response to heme and oxygen levels, thus strains from the BY4741 and BY4742 backgrounds used in this study were transformed with a low-copy plasmid carrying a wild-type allele of HAP1 and a selectable URA3 marker.
To study the genetic interactions of 29 nonessential yeast SLC25 family members with the SLC25 family member HEM25, double gene deletion strains were constructed by standard genetic crosses of hem25Δ haploid cells with each SLC25 family member single gene deletion strain. Following diploid cell section, sporulation, and haploid cell selection, single colonies were isolated that contained a deletion of the HEM25 gene with each yeast SLC25 family member. During the process of construction of the double mutant strains, the media used were supplemented with 5 mM glycine and 0.38 mM 5-Ala.
Growth assays
Cells were grown overnight in liquid synthetic dextrose (SD) medium without uracil (SD–URA) containing 1 g/liter of ammonium sulfate. Cells were washed twice, and resuspended in liquid SD–URA medium containing 30 g/liter glycine as the sole nitrogen source. An OD600 nm 0.1 was standardized for every culture at time 0, cells were grown at 30°, and OD600 nm was monitored to determine growth rate.
The growth of yeast strains on plates was estimated using a serial dilution assay. Cells were grown to late log phase (0.9–1 OD600 nm) at 30° in SD–URA, the cell density of the culture was adjusted to OD600 0.4, serially diluted 1:10 four times, spotted onto appropriate solid medium using a replica pinner, and incubated at 30°. Plates were imaged using a Bio-Rad VersaDoc.
Isolation of mitochondria
Mitochondrial fractions were prepared by differential centrifugation as previously described (Diekert et al. 2001; Gaspard and McMaster 2015). Briefly, yeast strains were grown at 30° until they reached midlog phase. Cells were washed and inoculated in 200 ml of synthetic medium without uracil with raffinose as the carbon source at OD600 nm 0.2, and grown until OD600 nm 1–1.5. Cells were washed and transferred to synthetic medium without uracil with lactate as the carbon source for 5 hr. For mitochondrial samples to be analyzed by SDS-PAGE, cells were collected by centrifugation at 2500 × g for 5 min at 4°, and the cell pellet was resuspended in ice-cold 0.6 M sorbitol, 20 mM HEPES-KOH, 1 mM phenylmethanesulfonylfluoride at a density of 0.5 g cells/ml. Cells were lysed by vortexing twice for 15 sec at maximum speed in the presence of glass beads at 4°. Unbroken cells, cell debris and nuclei were spun down at 600 × g for 5 min at 4°. The supernatant was carefully removed from the pellet and centrifuged for 10 min at 10,000 × g at 4°. The resulting crude mitochondrial pellet was resuspended in 20 μl of 50 mM Tris-HCl, pH 7.0, 1% sodium dodecyl sulfate (SDS). Then, 10 μl of 3% SDS, 132 mM Na2CO3, 4% β-mercaptoethanol was added to the mitochondrial suspension followed by urea to a 6 M final concentration. This crude mitochondrial preparation, subjected to SDS-PAGE and western blot analysis, was used to estimate the relative abundance of several mitochondrial proteins.
To assess the integrity of mitochondrial respiratory supercomplexes by Blue Native (BN)-PAGE, mitochondria were isolated by differential centrifugation (Diekert et al. 2001; Gaspard and McMaster 2015) Briefly, harvested cells were washed once in distilled water, resuspended in 30 ml of freshly prepared TD buffer (100 mM Tris-sulfate, pH 9.4, 10 mM DTT) and incubated for 5 min at 30° with gentle shaking. These cells were then collected by centrifugation at 2500 × g for 5 min and resuspended in SP buffer (1.2 M sorbitol, 20 mM potassium phosphate, pH 7.4). Spheroplasts were generated by treatment with zymolase 100T (1.5 mg/g cells) for 30–60 min at 30° with gentle shaking. Spheroplasts were harvested by centrifugation at 2500 × g for 5 min at 4°, and then washed twice with 40 ml of ice-cold SP buffer. After the washes, spheroplasts were resuspended in 60 ml of ice-cold SH buffer (0.6 M sorbitol, 20 mM HEPES-KOH, pH 7.4) with 1 mM phenylmethanesulfonylfluoride and complete protease inhibitors (Roche). The suspension was transferred to a large piston glass homogenizer (Potter-Elvehjem-Type Tissue Grinders), and homogenized with 25 strokes; the homogenized mixture was then subjected to centrifugation twice at 2500 × g for 5 min at 4°. The supernatants were pooled together in a fresh 50 ml glass tube, and pelleted by centrifugation at 12,000 × g for 10 min at 4°. Using a small-scale glass homogenizer, the pellet was first carefully resuspended in 1 ml of SH buffer, and then resuspended in 25 ml of ice cold SH buffer. This suspension was further centrifuged at 2500 × g for 5 min at 4°, the supernatant was saved and then pelleted at 12,000 × g for 10 min at 4°. The resulting pellet (mitochondria) was resuspended in 0.5 ml of SH buffer and divided into aliquots containing 0.1–0.3 mg protein (estimated by a modified Lowry method; Markwell et al. 1978). The aliquots were flash-frozen in liquid nitrogen and stored at −70° until further analysis.
BN-PAGE
The status of yeast respiratory chain supercomplexes was assessed by BN-PAGE as described previously with minor modifications (Wittig et al. 2006). Frozen mitochondria (150 μg of protein) were thawed, collected by centrifugation at 12,000 × g for 10 min, and suspended in 15 μl of 50 mM NaC1, 2 mM 6-aminohexanoic acid, 50 mM imidazole/HC1, pH 7.0. To solubilize membranes, 6 μl of 5% digitonin (detergent/protein ratio of 2 g/g; Invitrogen) was added to this mixture, incubated for 10 min on ice, and centrifuged at 20,000 × g for 20 min at 4°. Alternatively, mitochondrial proteins were solubilized with dodecylmaltoside (detergent/protein ratio of 1 g/g). The supernatant containing the solubilized mitochondrial proteins was transferred to a new tube, and 2 μl of 50% glycerol and 0.8 μl of 5% Coomassie blue G-250 (Pierce) were added. Solubilized mitochondrial proteins were separated on a 3–12% precast Bis-Tris Native PAGE (Invitrogen) using a XCell sure lock Mini-Cell gel running tank (Invitrogen) by the application of constant current at 150 V for 3 hr in a cold room. To improve detection of faint protein bands and to maximize protein transfer to PVDF membranes, the dark cathode buffer with 0.02% Coomassie blue G-250 was replaced with light cathode buffer (0.002% Coomassie blue G-250) once the proteins reached one-third of the running distance in BN-PAGE. A diluted aliquot of ferritin, which runs as two visible (brown) bands with molecular masses of 440 and 880 kDa was separated along with the samples by BN-PAGE to serve as a molecular marker. For western blotting, the BN gel was soaked for 10 min in electrode buffer (50 mM tricine, 7.5 mM imidazole, pH 7.0) containing 1% SDS and the proteins were then transferred to PVDF membranes using a semidry electroblotting apparatus in the presence of electrode buffer at 4° for 3 hr under limiting current (0.5 mA/cm2 of gel area) and voltage set at 20 V. Supercomplexes containing complex IV were probed with a monoclonal anti-Cox2 antibody (1:1000; from MitoSciences) in the presence of 0.02% SDS. Complex V, detected with a polyclonal anti-F1α antibody (1:1000; Dr. C. Koehler), was used as loading control. Primary antibodies were detected with HRP-conjugated secondary antibodies (from Cell Signaling), followed by chemoluminescense.
Western blot analysis
Protein concentration was determined by a modified Lowry method (Markwell et al. 1978). This modified method utilizes the detergent deoxycholate to solubilize hydrophobic proteins present in membranes. The mitochondrial fractions were added to loading buffer (0.05% bromophenol blue, 25% glycerol, 6% SDS, 6 mM EDTA, and 150 mM Tris-HCl pH 8.8 and 0.5% β-mercaptoethanol), and incubated for 1 hr at 37°. Samples were resolved by 12% SDS-PAGE and the separated proteins were transferred to a nitrocellulose membrane. The membranes were incubated for 1 hr under constant shaking in Odyssey blocking buffer (LI-COR). The blots were then incubated with an appropriate dilution of primary antibody (Table S2 in File S1) in blocking buffer under constant shaking overnight at 4°. The following day, the membranes were washed three times for 10 min each with 0.1% Tween-20 in phosphate-buffered saline (PBS), and were then incubated with an appropriate dilution of secondary antibody in LI-COR blocking buffer for 1 hr. Excess antibody was washed off with 0.1% Tween-20 in PBS and proteins were detected and imaged by using a LI-COR Odyssey Infrared Imaging System. The membranes were stripped with New Blot Nitro Stripping Buffer (LI-COR) for 10 min under constant shaking at room temperature, and reprobed with anti-porin antisera (Por1) as a loading control.
Heme determination
Logarithmically growing cells in SD-Ura media were harvested, washed in ice-cold water, and resuspended in 10 mM Tris-HCl (pH 8.0), 150 mM NaCl. Cells were lysed by vortexing with glass beads for two periods of 1 min intercalated with 1 min on ice. Cell debris was removed by centrifugation at 500 × g for 3 min, and heme and protein content of the supernatant were assayed. Heme determination was carried out using the Hemin Assay Kit from BioVision (Atamna et al. 2015).
Data availability
The authors state that all data necessary for confirming the conclusions presented in the article are represented fully within the article.
Results
Growth impairment by simultaneous deletion of HEM25 and members of the SLC25 family
The ability of a yeast cell to utilize glycine as a nitrogen source depends on the transport of glycine inside the mitochondria, which is mediated mostly by Hem25 followed by the catabolism of glycine into ammonia through the GCV (Figure 1B). We generated 29 double deletion yeast strains, whereby the HEM25 gene was inactivated in cells lacking each nonessential SLC25 family member. We hypothesized that inactivation of these SLC25 family members could affect the utilization of glycine as a nitrogen source. We anticipated that the absence of any other member of the SLC25 family that contributed to glycine import, or toward the functionality of the GCV, would exacerbate the growth defect of a hem25Δ strain. For comparison, an lpd1Δ strain, defective in lipoamide dehydrogenase that renders the GCV inactive, and a wild-type strain, were included.
FLX1, MTM1, ORT1, SFC1, PET8, and AAC3 genes were selected based on the growth phenotype on glycine as a nitrogen source of the null mutants alone or in combination with inactivated HEM25 (Figure S1 in File S1 and Table 1). To better assess the possible synthetic interaction between these genes and HEM25, we determined their growth rates on glycine as nitrogen source (Figure 2). The ort1Δ hem25Δ showed a synthetic growth defect. Double mutant strain flx1Δ hem25Δ grew poorly compared to wild-type cells and to the single gene knockout strains hem25Δ and flx1Δ, with the growth impairment of the double mutant reflecting the cumulative effect of each single mutant. Single mutant strains mtm1Δ and pet8Δ showed a severe growth phenotype that was not exacerbated when HEM25 was inactivated. Interestingly, sfc1Δ and aac3Δ cells grew faster than wild type when glycine was the nitrogen source; however, this growth improvement was reduced proportionately by the loss of HEM25.
Table 1. Identified and known functions of SLC25 family members that genetically interact with HEM25.
| Standard Name | Systematic Name | Known Function(s) |
|---|---|---|
| FLX1 | YIL134w | Mitochondrial flavin adenine dinucleotide transporter; FAD is a synthesis product of riboflavin; human homolog SLC25A32 is implicated in multiple acyl-CoA dehydrogenase deficiency (MADD) or glutaric aciduria type II (GAII), and can complement yeast null mutant |
| MTM1 | YGR257c | Mitochondrial protein of the mitochondrial carrier family; high affinity pyridoxal 5′-phosphate (PLP) transporter, important for delivery of PLP cofactor to mitochondrial enzymes; involved in mitochondrial iron homeostasis |
| ORT1 | YOR130c | Ornithine transporter of the mitochondrial inner membrane; exports ornithine from mitochondria as part of arginine biosynthesis; functionally complemented by human ortholog, SLC25A15, which is associated with hyperammonaemia-hyperornithinaemia-homocitrullinuria (HHH) syndrome, but HHH-associated variants fail to complement |
| SFC1 | YJR095w | Mitochondrial succinate-fumarate transporter; transports succinate into and fumarate out of mitochondria; required for ethanol and acetate utilization |
| PET8 | YNL003c | S-adenosylmethionine transporter of the mitochondrial inner membrane; member of the mitochondrial carrier family; required for biotin biosynthesis and respiratory growth |
| AAC3 | YBR085w | Mitochondrial inner membrane ADP/ATP translocator; exchanges cytosolic ADP for mitochondrially synthesized ATP; expressed under anaerobic conditions; has roles in maintenance of viability and in respiration |
Figure 2.

Growth rates on glycine as sole nitrogen source for selected members of the SLC25 family in combination with HEM25 deficiency. Cells of the indicated genotypes were grown overnight on SD-Ura medium containing 1 g/liter of ammonium sulfate at 30°, washed twice and inoculated at OD600 nm 0.1 in SD-Ura medium containing 30 g/liter of glycine. Cells were cultivated at 30°, and growth was monitored spectrophotometrically. Differences between single deletion strains and double deletion strains were determined using ANOVA test with randomized factors. At least three independent experiments were done to calculate the p values, mean, and SEM. The numbers represented in the graph are growth rates calculated over a period of 4 d.
Heme content was decreased in cells lacking SLC25 family members
In mitochondria, glycine is a substrate for the synthesis of several molecules, including heme (Fernández-Murray et al. 2016; Schonauer et al. 2009; Tibbetts and Appling 2010; Wang et al. 2013). We determined if inactivation of FLX1, MTM1, ORT1, SFC1, PET8, and AAC3 genes, per se or in combination with ablation of HEM25 gene, altered heme level. Heme levels were compared to the heme level found in wild type, hem25Δ, and lpd1Δ cells. Lpd1 is a subunit of both the GCV and the KGD complex. The KGD complex catalyzes the synthesis of succinyl-CoA, which is used along with glycine to synthesize 5-Ala to initiate heme synthesis (Heublein et al. 2014; Schonauer et al. 2009; Ajioka et al. 2006; Yuan et al. 2013). The hem25Δ cells had a heme level 35% that of wild type, while lpd1Δ cells had a heme level 12% that of wild type (Figure 3). Similar to hem25Δ cells, there was a decrease in heme content in cells lacking FLX1, ORT1, SFC1, or PET8, to 38–49% of wild type, while cells lacking MTM1 had heme levels that were 22% wild type (Figure 3). Interestingly, cells lacking AAC3 had a heme content that was 150% that of wild type cells. Inactivation of the HEM25 gene in each of these single mutants to create double mutant cells all resulted in a further decrease in heme content compared to the single mutant strains. The double ort1Δ hem25Δ exhibited a synthetic phenotype having its heme levels reduced beyond the cumulative effect of each single mutation (Figure 3), consistent with the observed growth defect for this double mutant when using glycine as nitrogen source. In the double mutant strains lacking HEM25 and either FLX1, SFC1, or PET8, heme content was 12–17% that of wild type. The double mutant aac3Δ hem25Δ had a reduction in heme content to 57% that observed in wild type cells, which was significantly less than that observed in aac3Δ cells but was higher than the level observed in hem25Δ cells. When MTM1 was deleted in hem25Δ cells, the heme content did not significantly decrease beyond the low heme level already present upon inactivation of just the MTM1 gene.
Figure 3.

Heme content for selected members of the SLC25 family in combination with HEM25 deficiency. Cells of the indicated genotypes were grown at OD600 nm 0.6–1 in SD-Ura. Cells were harvested and processed for heme determination. Differences between single deletion strains and double deletion strains were determined using ANOVA test with randomized factors. At least three independent experiments were done to calculate the p values, mean, and SEM. Values are normalized to wild type (WT). WT heme level was 28.9 ± 4.2 fmol/μg of protein.
Glycine and 5-Ala restore growth to strains lacking selective SLC25 family members
We sought to determine if the genes whose inactivation, per se or in combination with inactivated HEM25 cells that resulted in impaired growth on glycine as nitrogen source and reduced heme content, had a role specific to heme synthesis, or if they participated with glycine in a mitochondrial function beyond heme synthesis. We assessed if growth was restored by the addition of excess glycine (5 mM), or by the heme-specific biosynthetic metabolite 5-Ala (0.38 mM) (Figure 4) (Fernández-Murray et al. 2016). To enable determination of the effect of glycine and 5-Ala, these experiments were carried out on yeast minimal medium containing dextrose as the carbon source and ammonium sulfate as the nitrogen source. Under normal growth conditions, loss of Hem25 function did not affect growth, compared to the 25% decrease in growth rate when glycine was used as the sole nitrogen source. In addition, in hem25Δ cells, inactivation of AAC3, MTM1, and PET8 all slightly decreased growth beyond that of each single mutant, inactivation of ORT1 almost completely abrogated growth, while inactivation of FLX1 and SFC1 did not substantially alter growth. Thus, unlike when glycine was used as the sole nitrogen source, a decrease in growth was observed when ammonium sulfate was used as the sole nitrogen source only for hem25Δ cells lacking AAC3, MTM1, PET8, and ORT1, but not for FLX1 and SFC1. Interestingly, only the hem25Δ mtm1Δ strain growth defect was alleviated by the addition of 5-Ala, but not by glycine. The hem25Δ pet8Δ strain was the only one whose (slight) growth impairment was relieved by the addition of either 5-Ala or glycine. Glycine and 5-Ala supplementation did not affect the growth of any of the other strains.
Figure 4.

Glycine and 5-Ala alleviate the growth defect of a subset of SLC25 family members upon inactivation of the mitochondrial glycine importer HEM25. Yeast strains of the indicated genotypes were grown to early stationary phase in SC-Ura medium supplemented with 5 mM glycine and 0.38 mM 5-Ala to keep the double mutant cells growing without impediment. Cells were washed and resuspended in sterilized water to OD600 nm 0.4, then serially diluted (1:10) and spotted on SD-Ura solid media containing dextrose as carbon source. Plates were imaged after 3 d incubation at 30°.
Four out of the six genes whose inactivation decreased cell growth and heme level in concert with loss of Hem25 function are required for growth on nonfermentable carbon sources. These are SFC1, ORT1, MTM1, and PET8 (Figure 5). An inability to grow on nonfermentable carbon sources generally delineates a poorly functioning ETC. Heme is a component of several components of the ETC (Ajioka et al. 2006; Yuan et al. 2013).
Figure 5.

Growth rescue by glycine and 5-Ala on nonfermentable medium for cells lacking specific SLC25 family members in concert with HEM25. Yeast strains of the indicated genotypes were grown to early stationary phase in SC-Ura medium supplemented with 5 mM glycine and 0.38 mM 5-Ala to keep the double mutant cells growing without impediment. Cells were washed and resuspended in sterilized water to OD600 nm 0.4, then serially diluted (1:10) and spotted on SD-Ura solid media containing lactate as carbon source. Plates were imaged after 7 d incubation at 30°.
We determined if the addition of glycine or 5-Ala could enable growth on nonfermentable medium for each of the single mutants identified in our screen, as well as each of these mutants in concert with loss of function of Hem25. Both glycine and 5-Ala enabled growth of sfc1Δ strain, while only glycine enabled growth of ort1Δ cells. Glycine, and, to a smaller extent, 5-Ala also enabled growth of sfc1Δ hem25Δ cells. The ability of glycine and 5-Ala to restore growth to sfc1Δ cells implies that their inability to grow on a nonfermentable carbon source could be due to their decreased capacity to synthesize heme.
Single mutants flx1Δ and aac3Δ grew on lactate. Remarkably, the simultaneous loss of Hem25 function completely prevented growth on this nonfermentable carbon source, and it could not be restored by supplementation with glycine or 5-Ala.
Ablation of HEM25 and FLX1 affects electron transport chain subunit stability
The synthetic growth defect of the flx1Δ hem25Δ double mutant on a nonfermentable carbon source, but not on a fermentable carbon source, implied the ETC could be compromised. Hem25 imports glycine into the mitochondria, which is required for heme synthesis (Fernández-Murray et al. 2016; Lunetti et al. 2016). Heme molecules are required to form cytochromes for electron transfer by the ETC (Chiabrando et al. 2014; Yuan et al. 2013; DiMauro and Schon 2003). Flx1 transports FAD, which is a prosthetic group for flavoproteins, the majority of which are found in the mitochondria where they participate in redox processes of the ETC (Bafunno et al. 2004; Giancaspero et al. 2014; Spaan et al. 2005; Tzagoloff et al. 1996; Gudipati et al. 2014; Iwata et al. 2012; Kim et al. 2012; Koch et al. 2004). We hypothesized that there would be a decreased stability of FAD and heme containing components of the ETC based on the presence or absence of Flx1 and Hem25. To test this hypothesis, we determined the level of nine different ETC proteins within ETC complexes in wild type, hem25Δ, flx1Δ, and flx1Δ hem25Δ strains. The strains were grown in defined media with raffinose as carbon source, and then transferred to lactate medium for 5 hr prior to mitochondria isolation and western blotting to determine the level of each ETC protein examined. Porin (Por1) was used as a load control.
NADH-ubiquinone oxidoreductase, Ndi1, is a component of ETC complex I (Iwata et al. 2012). Its level was affected in cells that contain the flx1Δ hem25Δ double deletion. A significant reduction of Ndi1 levels was observed in cells with the double deletion flx1Δ hem25Δ compared with the levels observed in the mitochondrial fractions of the single mutant cells (Figure 6A).
Figure 6.

Specific components of the electron transport chain are significantly decreased in hem25Δ cells lacking the FAD importer FLX1. Cells of the indicated genotypes were grown to an OD of 1.0 in defined medium with raffinose. Cells were then transferred and grown in lactate for 5 hr. Cells were harvested and mitochondrial fractions were prepared and analyzed by western blotting using antibodies specific for (A) Ndi1, (B) Sdh1 and Sdh2, (C) Cor2 and Cyt1, (D) Cox2 and Cox4, and (E) F1α and F1β. Three independent segregant strains of flx1Δ and flx1Δ hem25Δ were used. The figure shown is representative of three independent analyses. Pixel intensity was measured and calculated by using Odyssey Software. Numbers under the lanes represent the mean of protein abundance normalized by the loading control Por1 and then to WT. The mean and the SEM were calculated from three independent western blot analyses. The significance of the differences on protein levels observed between the strains was calculated using ANOVA test with randomized factors (Table S3 in File S1). The deduced protein molecular weight (MW) of the bands revealed by western blotting was consistent with their MW: Ndi1 (57 kDa); Sdh1 (69 kDa) and Sdh2 (30 kDa); Cor2 (40 kDa) and Cyt1 (34 kDa); Cox2 (28 kDa) and Cox4 (17 kDa); F1α (58 kDa) and F1β (55 kDa).
Sdh2 and Sdh1 are subunits of succinate dehydrogenase, and form part of complex II of the ETC (Goffrini et al. 2009; Kim et al. 2012). Cells with the double deletion flx1Δ hem25Δ showed a decrease in Sdh1 levels compared with the single mutant cells (Figure 6B). A significant decrease in Sdh2 level was also observed in the double mutant in comparison with the single mutants. Sdh1 and Sdh2 levels were also reduced in hem25Δ cells (Figure 6B) but not to the extent of that observed in the double mutant cells.
We determined the abundance of subunit 2, Cor2, and cytochrome c1, Cyt1, for complex III of the ETC (di Rago et al. 1997; Hunte et al. 2000; Schneider and Guarente 1991). Cor2 and Cyt1 levels were significantly decreased in the double mutant flx1Δ hem25Δ cells (Figure 6C). Both Cor2 and Cyt1 levels were also reduced in hem25Δ cells compared with wild-type cells, but not to the extent observed in the double mutants. We also determined the level of the complex IV cytochrome oxidase subunits Cox2 and Cox4 (Ostrander et al. 2001; Bottinger et al. 2013; Cui et al. 2014; Stiburek et al. 2012; Su et al. 2014). Cox2 and Cox4 were significantly reduced in the hem25Δ cells compared with wild-type cells, but there was no reduction in protein levels in the double mutant flx1Δ hem25Δ compared with the hem25Δ cells (Figure 6D). We observed no decrease in F1α and F1β (Su et al. 2014) levels for complex V in the double mutant cells in comparison with flx1Δ, hem25Δ, or wild-type cells (Figure 6E).
There are clear differences in the levels of specific proteins within the ETC upon inactivation of the HEM25 and FLX1 genes (Table 2). The complex V subunits F1α and F1β were not affected by loss of function of Hem25, Flx1, or both, while complex IV subunits Cox2 and Cox4 were reduced in cells lacking Hem25 function, but this was not exacerbated by loss of Flx1 function. Interestingly, loss of function of both genes significantly impacted the level of the complex I subunit Ndi1, the complex II subunits Sdh1 and Sdh2, and the complex III subunits Cyt1 and Cor2, compared to loss of either HEM25 or FLX1 alone.
Table 2. Summary of phenotypes for HEM25 SLC25 family genetics interactors.
| Growth | Heme Level (% WT) | ETC Complex Stability | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Strain | Glycine as N Source | hem 25Δ | Dextrose as C Source | hem 25Δ | Lactate as C Source | hem25Δ | Heme Content | hem25Δ | I | hem25Δ | II | hem25Δ | III | hem25Δ | IV | hem25Δ | V | hem25Δ |
| Wild type | ++++a | ++++ | +++ | 100 | +++ | +++ | +++ | +++ | +++ | |||||||||
| hem25Δ | +++ | ++++ | +++ | 35 | +++ | ++ | ++ | ++ | +++ | |||||||||
| flx1Δ | +++ | ++ | ++++ | ++++ | —b | — | 49 | 10 | ++++ | + | +++ | + | ++++ | + | ++++ | ++ | +++ | +++ |
| mtm1Δ | +++ | ++ | ++ | +c | — | — | 22 | 19 | ||||||||||
| ort1Δ | +++ | + | ++++ | — | —d | — | 42 | 5 | ||||||||||
| sfc1Δ | ++++ | +++ | ++++ | ++++ | —b | —b | 40 | 8 | ||||||||||
| pet8Δ | ++ | + | ++++ | ++b | — | — | 38 | 8 | ||||||||||
| aac3Δ | +++++ | +++ | +++++ | ++++ | ++++ | — | 150 | 57 | ||||||||||
Relative level.
Alleviated by glycine and 5-Ala.
Alleviated by 5-Ala.
Alleviated by glycine.
To gain a deeper understanding about the inability of double mutant flx1Δ hem25Δ cells to grow on nonfermentable carbon source, the integrity of the respiratory supercomplexes from digitonin-solubilized mitochondria was assessed by BN-PAGE and western blotting. Consistent with the levels of Cox2 estimated by SDS-PAGE followed by western blotting (see Figure 6D) the status of supercomplexes III2IV2 and III2IV for flx1Δ did not differ appreciably compared to wild type cells (Figure 7), whereas, for both hem25Δ and flx1Δ hem25Δ cells a strong reduction of their levels was evident, with supercomplex III2IV2 barely detectable. Remarkably, very slow mobility forms were observed only in flx1Δ hem25Δ mitochondrial extracts. The levels of complex V, detected as dimeric and monomeric forms, were not affected by the ablation of HEM25, FLX1, or both genes, in agreement with data presented in Figure 6E. When mitochondria were solubilized with dodecylmaltoside (Figure S2 in File S1) instead of digitonin, complexes IV and V collapsed into their monomeric forms; however, the Cox2-containing low-mobility forms were still specifically detected in flx1Δ hem25Δ mitochondrial extracts.
Figure 7.
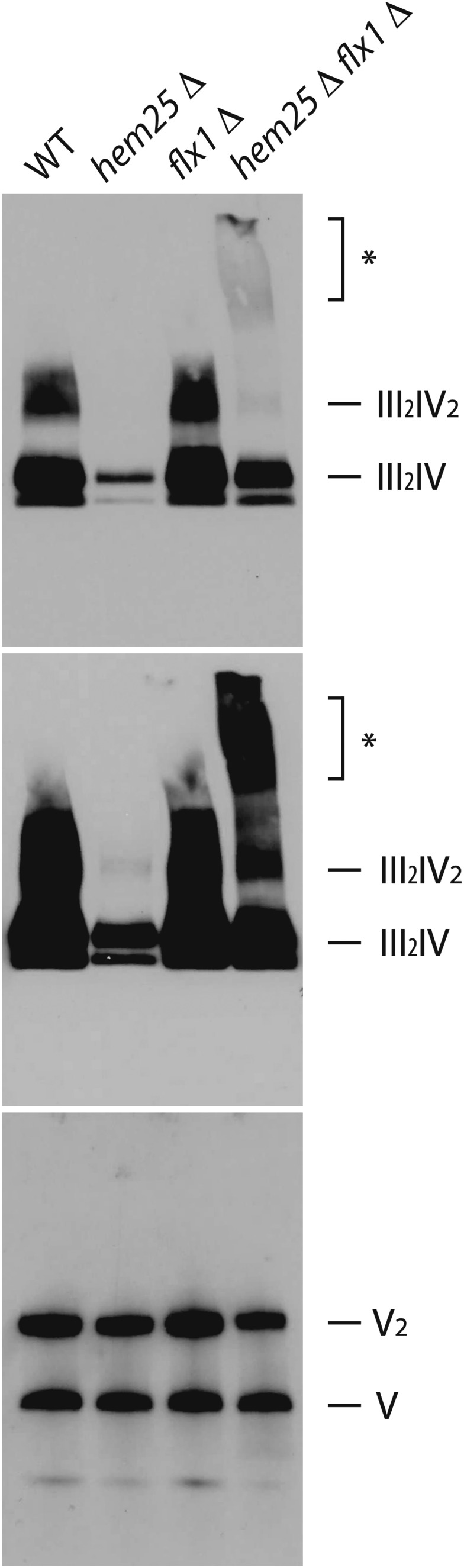
High molecular weight aggregates are present in mitochondria from flx1Δ hem25Δ cells. Cells of the indicated genotypes were grown to an OD of 1.0 in defined medium with raffinose. Cells were then transferred and grown in lactate for 5 hr. Cells were harvested, mitochondria were isolated and mitochondrial protein complexes were solubilized with digitonin. Protein complexes were resolved by BN-PAGE, transferred into PVDF and analyzed by western blotting using antibodies specific for Cox2 (upper and middle panels; they are identical, middle panel was a longer exposure) or F1α (lower panel). Asterisk denotes the position of high molecular weight aggregates.
Discussion
We identified new SLC25 family member genetic interactors with HEM25—a mitochondrial SLC25 family member that imports glycine into mitochondria to provide substrate for heme synthesis and the GCV. The genetic interactions between HEM25 and FLX1, ORT1, MTM1, and SFC1 or AAC3 have not been reported, while the genetic interaction of HEM25 with PET8 had been reported in the Saccharomyces Genome Database (SGD). Of the HEM25 genetic interactions with SLC25 family members we identified, we studied the genetic interaction between HEM25 and FLX1 in depth.
FLX1 encodes a mitochondrial FAD transporter; the absence of Flx1 affects the import of FAD needed for functional flavoproteins (Bafunno et al. 2004; Giancaspero et al. 2014; Spaan et al. 2005; Tzagoloff et al. 1996; Gudipati et al. 2014). We found that inactivation of FLX1 exacerbated the growth defect of hem25Δ cells when they were grown using glycine as a sole nitrogen source. Lpd1, which is one of the subunits of the GCV, is a flavoenzyme that uses FAD as a cofactor (Gudipati et al. 2014). The reduction of growth on glycine when Flx1 and Hem25 are absent could be due to reduced availability of FAD in the mitochondria, which is required for GCV, and a decrease of glycine inside the mitochondria, which in turn decreases the substrate glycine used by the GCV. Furthermore, when heme content was measured in the single and double mutants, flx1Δ hem25Δ showed a decrease in heme content compared to either single mutant alone. Protoporphyrinogen oxidase, Hem14, is a mitochondrial flavoprotein (Koch et al. 2004) that catalyzes the seventh reaction in the heme synthesis pathway. Thus, the combined defect of Hem25 and Flx1 together could decrease the supply of the initial substrate for heme synthesis, glycine, and also decrease the efficiency of the FAD requiring enzyme Hem14, resulting in a decrease in heme level upon loss of function of both Flx1 and Hem25.
Both heme and FAD are prosthetic groups required to form ETC complexes (Sazanov 2015; Chaban et al. 2014). The single mutants flx1Δ and hem25Δ can proliferate on nonfermentable carbon source, whereas the double mutant could not. Importantly, the decreased ability to incorporate FAD into components of the ETC, together with the strong reduction in heme level observed for flx1Δ hem25Δ cells, appears to severely compromise the function of the ETC.
Like mammalian cells, the ETC complexes of yeast ETC assemble to form supercomplexes (Cui et al. 2014; Pfeiffer et al. 2003; Gaspard and McMaster 2015). Our data suggest that Flx1 and Hem25 together are required for stability of specific ETC subunits. The abundance of Ndi1 subunit of complex I decreased in the flx1Δ hem25Δ cells compared with the levels in the single mutant cells. Complex I is the NADH-ubiquinone oxidoreductase, which has a FAD prosthetic group (Feng et al. 2012). The succinate-ubiquinone oxidoreductase is complex II, formed by four subunits, one of which is the flavoprotein Sdh1, which contains FAD bound covalently (Maklashina et al. 2016; Kim et al. 2012). Sdh2 contains a Fe-S group (Chaban et al. 2014; Gudipati et al. 2014; Sazanov 2015). The four complex II subunits bind cytochrome b, which contains a heme group (Chaban et al. 2014; Sazanov 2015; Chiabrando et al. 2014). In the western blot analyses, Sdh1 and Sdh2 subunits were significantly decreased in the double mutant cells compared to single mutant cells. The flx1Δ hem25Δ cells analyzed here also showed a decrease in the abundance of subunit 2 of ubiquinol cytochrome-c reductase (Cor2) and cytochrome c1 (Cyt1) proteins. Cor2 and Cyt1 form part of complex III, and both subunits contain heme molecules (Chaban et al. 2014).
Interestingly, we determined that the level of Cox2 and Cox4 proteins from complex IV was decreased in hem25Δ cells, but was not further reduced in flx1Δ hem25Δ cells. This result showed that the absence of Hem25 per se affects the stability of Cox4 and Cox2. Cox4 is essential for the assembly and function of the cytochrome c oxidase complex (Bottinger et al. 2013; Chaban et al. 2014). The decrease in heme levels observed in hem25Δ cells could affect the expression of Cox4, as its expression is regulated by heme level. If Cox4 is decreased, the Cox2 subunit cannot assemble and is degraded (Nakai et al. 1994). A similar situation was observed in yeast cells with a mutation in Cox4, in the absence of Cox4 subunits, Cox2 and Cox3 were not assembled into the higher order complex, and were degraded.
The subunits of the ATP synthase, F1α and F1β, were not diminished in abundance in flx1Δ hem25Δ cells compared to wild-type cells. The ATP synthase does not contain a heme or FAD molecule in the complex, which might explain why the abundance of these two proteins was not affected. The combined inability to synthesize heme at a wild-type rate and import FAD into the mitochondria results in a decreased ability to assemble ETC complexes that require these cofactors, but does not affect the assembly of ETC complexes where these cofactors are absent. The decreased levels of the ETC proteins studied is likely due to a decrease in the capacity of ETC supercomplexes to properly assemble, due to irregularities in the proper proportions of the proteins within these supercomplexes.
We analyzed the status of Cox2-containing supercomplexes by BN-PAGE. Consistent with the reduced steady state level of Cox2 observed for hem25Δ and flx1Δ hem25Δ cells, these cells showed a strong reduction in supercomplexes formation. Whereas this reduction did not compromise the ability of hem25Δ cells to proliferate on nonfermentable sources, flx1Δ hem25Δ cells did not grow on lactate. We detected, in mitochondria isolated from flx1Δ hem25Δ cells, the presence of uncharacterized Cox2-containing high molecular weight species. We propose that the simultaneous deficiency of FAD and heme could prompt the accumulation of precursors of the ETC in their apo-forms, leading to the aggregation of improper folded molecules, and saturation of the proteolytic capacity of the mitochondria. These combined effects would prevent the normal assemble and function of the ETC, thus impairing cell respiration.
Beyond FLX1, several other novel genetic interactions between HEM25 and other SLC25 family members were observed. MTM1 encodes a putative mitochondrial high affinity PLP transporter (Whittaker et al. 2015). PLP is a cofactor required by Lpd1, which is one of the subunits of GCV. The growth defect of mtm1Δ hem25Δ cells using glycine as the sole nitrogen source was not significantly different from mtm1Δ cells, and it could be due to reduction of PLP levels in the mitochondria. This reduction would affect GCV function. A reduction of mitochondrial glycine levels upon HEM25 inactivation would barely impact growth, as Hem25 lies upstream of GCV. When heme was measured, a decrease in heme content was observed in mtm1Δ cells, but the double mutant hem25Δ mtm1Δ cells had equal heme levels to the mtm1Δ cells. PLP is an essential cofactor of Hem1 (Astner et al. 2005), which is the first enzyme in the heme synthesis pathway (Ajioka et al. 2006; Chiabrando et al. 2014). The absence of Mtm1 would be expected to impact the function of Hem1 due to the decreased availability of the cofactor PLP. The double mutant cells probably did not have a heme level significantly lower than mtm1Δ cells, because, under the situation where Hem1 function is impaired by decreased PLP levels, the availability of glycine would not determine the rate of heme synthesis, as Hem25 lies upstream of Hem1 in the heme biosynthetic pathway. In contrast, mtm1Δ hem25Δ cells also showed a growth defect relative to single mutants when cells grew in dextrose medium without supplementation. The growth phenotype of the double mutant cells was improved by the addition of 5-Ala. 5-Ala is a downstream metabolite of Hem1, the first enzymatic reaction for heme synthesis, and it would bypass the deficiency of PLP and glycine. A decreased ability to import PLP clearly affects growth of cells when mitochondrial glycine import is also decreased. This appears to be due to the fact that glycine is a substrate for the GCV and for heme synthesis, and both pathways contain PLP-dependent enzymes. Simultaneous restriction of both GCV and heme synthesis is a likely explanation for the decreased growth of cells lacking Hem25 and Mtm1 function.
SFC1 encodes a mitochondrial succinate-fumarate transporter, which transports succinate into, and fumarate out of, mitochondria (Lin et al. 2011). When heme content was measured in cells lacking Sfc1 and Hem25, a severe decrease in heme content was observed compared to sfc1Δ or hem25Δ cells. The import of succinate and export of fumarate would increase the availability of succinate in the mitochondria for synthesis of succinyl-CoA by the tricarboxylic acid (TCA) cycle. Succinyl-CoA and glycine are the substrates for the first reaction of heme synthesis (Ajioka et al. 2006; Chiabrando et al. 2014). A deletion of SFC1 and HEM25 would deprive the mitochondria of the two substrates required for heme synthesis. Our phenotypic analysis of growth in lactate showed that the decrease in growth observed in sfc1Δ hem25Δ cells was partially restored by the addition of 5-Ala or glycine. This result could indicate that decreased succinyl-CoA and glycine limit heme synthesis, and possibly other mitochondrial processes in addition to the synthesis of heme.
ORT1 encodes an ornithine transporter of the mitochondrial inner membrane, which exports ornithine from mitochondria in exchange for protons (Marobbio et al. 2015). ORT1 had a strong genetic interaction with HEM25. When ort1Δ hem25Δ cells were grown in glycine as sole nitrogen source, there was a synthetic growth impairment compared to the respective single mutants. In growth on dextrose, the double mutant cells also showed a strong growth impairment. When heme content was measured, the double mutant ort1Δ hem25Δ cells had a severe decrease in heme levels that exceeded the mere cumulative effect of both single mutations. The ORT1 gene has been reported to genetically interact with PET8 (Szappanos et al. 2011). PET8 encodes a mitochondrial S-adenosylmethionine (SAM) transporter (Marobbio et al. 2003), and pet8Δ cells had their heme levels reduced by 50%. The pet8Δ hem25Δ cells showed a further decrease in heme content compared with their respective single mutants. There is a clear genetic link between ORT1, PET8, and HEM25 that revolved around the role of heme in mitochondrial function; however, more work will be required to ascertain the biochemical underpinnings of these genetic interactions.
Our phenotypic analysis also showed that in nonfermentable media, aac3Δ hem25Δ cells had a severe growth defect. AAC3 encodes a mitochondrial ADP/ATP translocator, which exchanges cytosolic ADP for mitochondrially synthesized ATP (Kolarov et al. 1990; Smith and Thorsness 2008). It has a role in maintenance of viability in respiratory conditions. However, during exponential growth on dextrose under aerobic conditions, it acts in the opposite direction, importing ATP into mitochondria (Azuma et al. 2008). Disruption of the AAC genes in yeast had previously been determined to result in a reduction of heme biosynthesis by blocking the translocation of heme precursors into the matrix. Although the reason why a reduction in growth is observed when Hem25 and Aac3 are absent in the cells is not known, a possible explanation of for the growth defect is that heme synthesis could be impaired (Azuma et al. 2008). The function of AAC3 has to be further studied to understand why it has a genetic interaction with HEM25.
Defects in heme synthesis can lead to various disease states in humans. Congenital sideroblastic anemias are caused by defects in the early steps in heme synthesis, including those encoding the human homologs of yeast HEM25 (SLC25A38 in humans) and HEM1 (ALAS2 in humans) (Aivado et al. 2006; Boycott et al. 2013, 2014; Guernsey et al. 2009; Aoki et al. 1973; Cotter et al. 1992). It could be interesting to determine if defects (polymorphisms or mutations) in any of the genes identified here that affect the growth, level of heme, and ETC stability in yeast cells lacking Hem25 function, modify the phenotype of congenital sideroblastic anemia patients.
Supplementary Material
Supplemental material is available online at http://www.g3journal.org/lookup/suppl/doi:10.1534/g3.117.041194/-/DC1.
Acknowledgments
We thank Carla Koehler, Dennis Winge, and Takao Yagi for providing antibodies used in this study, and Li Zhang for the plasmid carrying the HAP1 gene. This work was supported in part by Genome Canada and Genome Atlantic.
Footnotes
Communicating editor: B. J. Andrews
Literature Cited
- Aivado M., Gattermann N., Rong A., Giagounidis A. A., Prall W. C., et al. , 2006. X-linked sideroblastic anemia associated with a novel ALAS2 mutation and unfortunate skewed X-chromosome inactivation patterns. Blood Cells Mol. Dis. 37(1): 40–45. [DOI] [PubMed] [Google Scholar]
- Ajioka R. S., Phillips J. D., Kushner J. P., 2006. Biosynthesis of heme in mammals. Biochim. Biophys. Acta 1763(7): 723–736. [DOI] [PubMed] [Google Scholar]
- Aoki Y., Urata G., Takaku F., 1973. Aminolevulinic acid synthetase activity in erythroblasts of patients with primary sideroblastic anemia. Nippon Ketsueki Gakkai Zasshi 36(1): 74–77. [PubMed] [Google Scholar]
- Astner I., Schulze J. O., van den Heuvel J., Jahn D., Schubert W. D., et al. , 2005. Crystal structure of 5-aminolevulinate synthase, the first enzyme of heme biosynthesis, and its link to XLSA in humans. EMBO J. 24(18): 3166–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atamna H., Brahmbhatt M., Atamna W., Shanower G. A., Dhahbi J. M., 2015. ApoHRP-based assay to measure intracellular regulatory heme. Metallomics 7(2): 309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma M., Kabe Y., Kuramori C., Kondo M., Yamaguchi Y., et al. , 2008. Adenine nucleotide translocator transports haem precursors into mitochondria. PLoS One 3(8): e3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bafunno V., Giancaspero T. A., Brizio C., Bufano D., Passarella S., et al. , 2004. Riboflavin uptake and FAD synthesis in Saccharomyces cerevisiae mitochondria: involvement of the Flx1p carrier in FAD export. J. Biol. Chem. 279(1): 95–102. [DOI] [PubMed] [Google Scholar]
- Bishop D. F., Tchaikovskii V., Hoffbrand A. V., Fraser M. E., Margolis S., 2012. X-linked sideroblastic anemia due to carboxyl-terminal ALAS2 mutations that cause loss of binding to the beta-subunit of succinyl-CoA synthetase (SUCLA2). J. Biol. Chem. 287(34): 28943–28955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottinger L., Guiard B., Oeljeklaus S., Kulawiak B., Zufall N., et al. , 2013. A complex of Cox4 and mitochondrial Hsp70 plays an important role in the assembly of the cytochrome c oxidase. Mol. Biol. Cell 24(17): 2609–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott K. M., Vanstone M. R., Bulman D. E., MacKenzie A. E., 2013. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat. Rev. Genet. 14(10): 681–691. [DOI] [PubMed] [Google Scholar]
- Boycott K. M., Dyment D. A., Sawyer S. L., Vanstone M. R., Beaulieu C. L., 2014. Identification of genes for childhood heritable diseases. Annu. Rev. Med. 65: 19–31. [DOI] [PubMed] [Google Scholar]
- Buschlen S., Amillet J. M., Guiard B., Fournier A., Marcireau C., et al. , 2003. The S. cerevisiae HAP complex, a key regulator of mitochondrial function, coordinates nuclear and mitochondrial gene expression. Comp. Funct. Genomics 4(1): 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaban Y., Boekema E. J., Dudkina N. V., 2014. Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim. Biophys. Acta 1837(4): 418–426. [DOI] [PubMed] [Google Scholar]
- Chen W., Paradkar P. N., Li L., Pierce E. L., Langer N. B., et al. , 2009. Abcb10 physically interacts with mitoferrin-1 (Slc25a37) to enhance its stability and function in the erythroid mitochondria. Proc. Natl. Acad. Sci. USA 106(38): 16263–16268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W., Dailey H. A., Paw B. H., 2010. Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis. Blood 116(4): 628–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiabrando D., Marro S., Mercurio S., Giorgi C., Petrillo S., et al. , 2012. The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J. Clin. Invest. 122(12): 4569–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiabrando D., Mercurio S., Tolosano E., 2014. Heme and erythropoieis: more than a structural role. Haematologica 99(6): 973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter P. D., Baumann M., Bishop D. F., 1992. Enzymatic defect in “X-linked” sideroblastic anemia: molecular evidence for erythroid delta-aminolevulinate synthase deficiency. Proc. Natl. Acad. Sci. USA 89(9): 4028–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui T. Z., Conte A., Fox J. L., Zara V., Winge D. R., 2014. Modulation of the respiratory supercomplexes in yeast: enhanced formation of cytochrome oxidase increases the stability and abundance of respiratory supercomplexes. J. Biol. Chem. 289(9): 6133–6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diekert K., de Kroon A. I., Kispal G., Lill R., 2001. Isolation and subfractionation of mitochondria from the yeast Saccharomyces cerevisiae. Methods Cell Biol. 65: 37–51. [DOI] [PubMed] [Google Scholar]
- DiMauro S., Schon E. A., 2003. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 348(26): 2656–2668. [DOI] [PubMed] [Google Scholar]
- di Rago J. P., Sohm F., Boccia C., Dujardin G., Trumpower B. L., et al. , 1997. A point mutation in the mitochondrial cytochrome b gene obviates the requirement for the nuclear encoded core protein 2 subunit in the cytochrome bc1 complex in Saccharomyces cerevisiae. J. Biol. Chem. 272(8): 4699–4704. [DOI] [PubMed] [Google Scholar]
- Feng Y., Li W., Li J., Wang J., Ge J., et al. , 2012. Structural insight into the type-II mitochondrial NADH dehydrogenases. Nature 491(7424): 478–482. [DOI] [PubMed] [Google Scholar]
- Fernández-Murray J. P., Prykhozhij S. V., Dufay J. N., Steele S. L., Gaston D., et al. , 2016. Glycine and folate ameliorate models of congenital sideroblastic anemia. PLoS Genet. 12(1): e1005783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming M. D., Hamza I., 2012. Mitochondrial heme: an exit strategy at last. J. Clin. Invest. 122(12): 4328–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaisne M., Becam A. M., Verdiere J., Herbert C. J., 1999. A ‘natural’ mutation in Saccharomyces cerevisiae strains derived from S288c affects the complex regulatory gene HAP1 (CYP1). Curr. Genet. 36(4): 195–200. [DOI] [PubMed] [Google Scholar]
- Gaspard G. J., McMaster C. R., 2015. The mitochondrial quality control protein Yme1 is necessary to prevent defective mitophagy in a yeast model of Barth syndrome. J. Biol. Chem. 290(14): 9284–9298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancaspero T. A., Dipalo E., Miccolis A., Boles E., Caselle M., et al. , 2014. Alteration of ROS homeostasis and decreased lifespan in S. cerevisiae elicited by deletion of the mitochondrial translocator FLX1. BioMed Res. Int. 2014: 101286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardina G., Brunotti P., Fiascarelli A., Cicalini A., Costa M. G., et al. , 2015. How pyridoxal 5′-phosphate differentially regulates human cytosolic and mitochondrial serine hydroxymethyltransferase oligomeric state. FEBS J. 282(7): 1225–1241. [DOI] [PubMed] [Google Scholar]
- Goffrini P., Ercolino T., Panizza E., Giache V., Cavone L., et al. , 2009. Functional study in a yeast model of a novel succinate dehydrogenase subunit B gene germline missense mutation (C191Y) diagnosed in a patient affected by a glomus tumor. Hum. Mol. Genet. 18(10): 1860–1868. [DOI] [PubMed] [Google Scholar]
- Gomez M., Perez-Gallardo R. V., Sanchez L. A., Diaz-Perez A. L., Cortes-Rojo C., et al. , 2014. Malfunctioning of the iron-sulfur cluster assembly machinery in Saccharomyces cerevisiae produces oxidative stress via an iron-dependent mechanism, causing dysfunction in respiratory complexes. PLoS One 9(10): e111585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudipati V., Koch K., Lienhart W. D., Macheroux P., 2014. The flavoproteome of the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 1844(3): 535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guernsey D. L., Jiang H., Campagna D. R., Evans S. C., Ferguson M., et al. , 2009. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat. Genet. 41(6): 651–653. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Aguilar M., Baines C. P., 2013. Physiological and pathological roles of mitochondrial SLC25 carriers. Biochem. J. 454(3): 371–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heublein M., Burguillos M. A., Vogtle F. N., Teixeira P. F., Imhof A., et al. , 2014. The novel component Kgd4 recruits the E3 subunit to the mitochondrial alpha-ketoglutarate dehydrogenase. Mol. Biol. Cell 25(21): 3342–3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunte C., Koepke J., Lange C., Rossmanith T., Michel H., 2000. Structure at 2.3 A resolution of the cytochrome bc(1) complex from the yeast Saccharomyces cerevisiae co-crystallized with an antibody Fv fragment. Structure 8(6): 669–684. [DOI] [PubMed] [Google Scholar]
- Iwata M., Lee Y., Yamashita T., Yagi T., Iwata S., et al. , 2012. The structure of the yeast NADH dehydrogenase (Ndi1) reveals overlapping binding sites for water- and lipid-soluble substrates. Proc. Natl. Acad. Sci. USA 109(38): 15247–15252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardon J. R., Yien Y. Y., Huston N. C., Branco D. S., Hildick-Smith G. J., et al. , 2015. Mitochondrial ClpX activates a key enzyme for heme biosynthesis and erythropoiesis. Cell 161(4): 858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. J., Jeong M. Y., Na U., Winge D. R., 2012. Flavinylation and assembly of succinate dehydrogenase are dependent on the C-terminal tail of the flavoprotein subunit. J. Biol. Chem. 287(48): 40670–40679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch M., Breithaupt C., Kiefersauer R., Freigang J., Huber R., et al. , 2004. Crystal structure of protoporphyrinogen IX oxidase: a key enzyme in haem and chlorophyll biosynthesis. EMBO J. 23(8): 1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolarov J., Kolarova N., Nelson N., 1990. A third ADP/ATP translocator gene in yeast. J. Biol. Chem. 265(21): 12711–12716. [PubMed] [Google Scholar]
- Krishnamurthy P. C., Du G., Fukuda Y., Sun D., Sampath J., et al. , 2006. Identification of a mammalian mitochondrial porphyrin transporter. Nature 443(7111): 586–589. [DOI] [PubMed] [Google Scholar]
- Lawrence S. A., Hackett J. C., Moran R. G., 2011. Tetrahydrofolate recognition by the mitochondrial folate transporter. J. Biol. Chem. 286(36): 31480–31489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A. P., Anderson S. L., Minard K. I., McAlister-Henn L., 2011. Effects of excess succinate and retrograde control of metabolite accumulation in yeast tricarboxylic cycle mutants. J. Biol. Chem. 286(39): 33737–33746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale J. W., 2013. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat. Rev. Cancer 13(8): 572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunetti P., Damiano F., De Benedetto G., Siculella L., Pennetta A., et al. , 2016. Characterization of human and yeast mitochondrial glycine carriers with implications for heme biosynthesis and anemia. J. Biol. Chem. 291(38): 19746–19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maklashina E., Rajagukguk S., Starbird C. A., McDonald W. H., Koganitsky A., et al. , 2016. Binding of the covalent flavin assembly factor to the flavoprotein subunit of complex II. J. Biol. Chem. 291(6): 2904–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markwell M. A., Haas S. M., Bieber L. L., Tolbert N. E., 1978. A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Anal. Biochem. 87(1): 206–210. [DOI] [PubMed] [Google Scholar]
- Marobbio C. M., Agrimi G., Lasorsa F. M., Palmieri F., 2003. Identification and functional reconstitution of yeast mitochondrial carrier for S-adenosylmethionine. EMBO J. 22(22): 5975–5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marobbio C. M., Punzi G., Pierri C. L., Palmieri L., Calvello R., et al. , 2015. Pathogenic potential of SLC25A15 mutations assessed by transport assays and complementation of Saccharomyces cerevisiae ORT1 null mutant. Mol. Genet. Metab. 115(1): 27–32. [DOI] [PubMed] [Google Scholar]
- Nakai T., Mera Y., Yasuhara T., Ohashi A., 1994. Divalent metal ion-dependent mitochondrial degradation of unassembled subunits 2 and 3 of cytochrome c oxidase. J. Biochem. 116(4): 752–758. [DOI] [PubMed] [Google Scholar]
- Ostrander D. B., Zhang M., Mileykovskaya E., Rho M., Dowhan W., 2001. Lack of mitochondrial anionic phospholipids causes an inhibition of translation of protein components of the electron transport chain. A yeast genetic model system for the study of anionic phospholipid function in mitochondria. J. Biol. Chem. 276(27): 25262–25272. [DOI] [PubMed] [Google Scholar]
- Paterson J. K., Shukla S., Black C. M., Tachiwada T., Garfield S., et al. , 2007. Human ABCB6 localizes to both the outer mitochondrial membrane and the plasma membrane. Biochemistry 46(33): 9443–9452. [DOI] [PubMed] [Google Scholar]
- Pfeiffer K., Gohil V., Stuart R. A., Hunte C., Brandt U., et al. , 2003. Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 278(52): 52873–52880. [DOI] [PubMed] [Google Scholar]
- Saint-Marc C., Hurlimann H. C., Daignan-Fornier B., Pinson B., 2015. Serine hydroxymethyltransferase: a key player connecting purine, folate and methionine metabolism in Saccharomyces cerevisiae. Curr. Genet. 61(4): 633–640. [DOI] [PubMed] [Google Scholar]
- Sazanov L. A., 2015. A giant molecular proton pump: structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 16(6): 375–388. [DOI] [PubMed] [Google Scholar]
- Schneider J. C., Guarente L., 1991. Regulation of the yeast CYT1 gene encoding cytochrome c1 by HAP1 and HAP2/3/4. Mol. Cell. Biol. 11(10): 4934–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonauer M. S., Kastaniotis A. J., Kursu V. A., Hiltunen J. K., Dieckmann C. L., 2009. Lipoic acid synthesis and attachment in yeast mitochondria. J. Biol. Chem. 284(35): 23234–23242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz I. J., Chen C., Paw B. H., Hamza I., 2010. Iron and porphyrin trafficking in heme biogenesis. J. Biol. Chem. 285(35): 26753–26759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintre C. A., Pike A. C., Li Q., Kim J. I., Barr A. J., et al. , 2013. Structures of ABCB10, a human ATP-binding cassette transporter in apo- and nucleotide-bound states. Proc. Natl. Acad. Sci. USA 110(24): 9710–9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. P., Thorsness P. E., 2008. The molecular basis for relative physiological functionality of the ADP/ATP carrier isoforms in Saccharomyces cerevisiae. Genetics 179(3): 1285–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaan A. N., Ijlst L., van Roermund C. W., Wijburg F. A., Wanders R. J., et al. , 2005. Identification of the human mitochondrial FAD transporter and its potential role in multiple acyl-CoA dehydrogenase deficiency. Mol. Genet. Metab. 86(4): 441–447. [DOI] [PubMed] [Google Scholar]
- Stiburek L., Cesnekova J., Kostkova O., Fornuskova D., Vinsova K., et al. , 2012. YME1L controls the accumulation of respiratory chain subunits and is required for apoptotic resistance, cristae morphogenesis, and cell proliferation. Mol. Biol. Cell 23(6): 1010–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C. H., McStay G. P., Tzagoloff A., 2014. Assembly of the rotor component of yeast mitochondrial ATP synthase is enhanced when Atp9p is supplied by Atp9p-Cox6p complexes. J. Biol. Chem. 289(45): 31605–31616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szappanos B., Kovacs K., Szamecz B., Honti F., Costanzo M., et al. , 2011. An integrated approach to characterize genetic interaction networks in yeast metabolism. Nat. Genet. 43(7): 656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L., Bergevoet S. M., Bakker-Verweij G., Harteveld C. L., Giordano P. C., et al. , 2012. Human mitochondrial ATP-binding cassette transporter ABCB10 is required for efficient red blood cell development. Br. J. Haematol. 157(1): 151–154. [DOI] [PubMed] [Google Scholar]
- Tibbetts A. S., Appling D. R., 2010. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 30: 57–81. [DOI] [PubMed] [Google Scholar]
- Tzagoloff A., Jang J., Glerum D. M., Wu M., 1996. FLX1 codes for a carrier protein involved in maintaining a proper balance of flavin nucleotides in yeast mitochondria. J. Biol. Chem. 271(13): 7392–7397. [DOI] [PubMed] [Google Scholar]
- Urano T., Shiraki M., Saito M., Sasaki N., Ouchi Y., et al. , 2014. Polymorphism of SLC25A32, the folate transporter gene, is associated with plasma folate levels and bone fractures in Japanese postmenopausal women. Geriatr. Gerontol. Int. 14(4): 942–946. [DOI] [PubMed] [Google Scholar]
- Voth W. P., Jiang Y. W., Stillman D. J., 2003. New ‘marker swap’ plasmids for converting selectable markers on budding yeast gene disruptions and plasmids. Yeast 20(11): 985–993. [DOI] [PubMed] [Google Scholar]
- Wang W., Wu Z., Dai Z., Yang Y., Wang J., et al. , 2013. Glycine metabolism in animals and humans: implications for nutrition and health. Amino Acids 45(3): 463–477. [DOI] [PubMed] [Google Scholar]
- Weatherall D. J., 2013. The role of the inherited disorders of hemoglobin, the first “molecular diseases,” in the future of human genetics. Annu. Rev. Genomics Hum. Genet. 14: 1–24. [DOI] [PubMed] [Google Scholar]
- Whittaker M. M., Penmatsa A., Whittaker J. W., 2015. The Mtm1p carrier and pyridoxal 5′-phosphate cofactor trafficking in yeast mitochondria. Arch. Biochem. Biophys. 568: 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingert R. A., Galloway J. L., Barut B., Foott H., Fraenkel P., et al. , 2005. Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. Nature 436(7053): 1035–1039. [DOI] [PubMed] [Google Scholar]
- Wittig I., Braun H. P., Schagger H., 2006. Blue native PAGE. Nat. Protoc. 1(1): 418–428. [DOI] [PubMed] [Google Scholar]
- Yamamoto M., Arimura H., Fukushige T., Minami K., Nishizawa Y., et al. , 2014. Abcb10 role in heme biosynthesis in vivo: Abcb10 knockout in mice causes anemia with protoporphyrin IX and iron accumulation. Mol. Cell. Biol. 34(6): 1077–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X., Fleming M. D., Hamza I., 2013. Heme transport and erythropoiesis. Curr. Opin. Chem. Biol. 17(2): 204–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Li D., Zhang J., Chen X., Huang M., et al. , 2013. Mutations in ABCB6 cause dyschromatosis universalis hereditaria. J. Invest. Dermatol. 133(9): 2221–2228. [DOI] [PubMed] [Google Scholar]
- Zhao S. G., Chen X. F., Wang L. G., Yang G., Han D. Y., et al. , 2013. Increased expression of ABCB6 enhances protoporphyrin IX accumulation and photodynamic effect in human glioma. Ann. Surg. Oncol. 20(13): 4379–4388. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors state that all data necessary for confirming the conclusions presented in the article are represented fully within the article.