

Abstract
Objective:
To evaluate (1) the frequency of aquaporin-4 antibody (AQP4-ab)-seropositive cases among patients treated with natalizumab (NAT) and previously diagnosed with MS (MSNAT) in a nationwide cohort, (2) the clinical course of NAT-treated AQP4-ab–seropositive neuromyelitis optica spectrum disorder (NMOSD) patients (NMONAT), (3) AQP4-ab titers in NMONAT and AQP4-ab–seropositive NMOSD treated with other immunotherapies (NMOIT), and (4) immune mechanisms influencing disease activity in NMONAT.
Methods:
MSNAT serum samples were retrospectively screened with a cell-based assay for AQP4-IgG and titers determined by ELISA. The annualized relapse rate (ARR) and disability progression were assessed. Serum levels of proinflammatory cytokines (interleukin [IL]-1β, IL-4, IL-6, IL-8, IL-10, IL-17, IL-21, and interferon [IFN]-γ) and the chemokine CXCL-10 of NMONAT patients identified in this (n = 4) and a previous study (n = 5) were measured by cytometric bead array and ELISA.
Results:
Of the 1,183 MSNAT patients (851 female, median 9 NAT infusions), only 4 (0.33%; 3 female, 1 male) had AQP4-IgG. Of these, 2 fulfilled the 2006 NMO criteria and all met the 2015 NMOSD criteria. The ARR was higher in NMONAT vs MSNAT (p = 0.0182). All 4 NMONAT patients had relapses and 2 had an increase of disability. AQP4-ab titers were higher in NMONAT (n = 9) vs NMOIT (n = 13; p = 0.0059). IL-8, IL-1β, and IFN-γ serum levels were significantly higher, and CXCL-10 was significantly lower in NMONAT vs NMOIT.
Conclusions:
Misdiagnosis of NMOSD with MS is rare. NAT was not able to control disease activity in NMONAT patients, who had higher serum levels of AQP4-IgG and proinflammatory cytokines than patients with NMOSD treated with other immunotherapies.
Neuromyelitis optica (NMO) is a relapsing autoimmune CNS disease which mainly affects the optic nerves and spinal cord and often leads to severe disability.1 The detection of a serum antibody targeting the astrocytic water channel aquaporin-4 antibody (AQP4-ab)2 led to the definition of revised diagnostic criteria3 and to renaming of the entity as NMO spectrum disorder (NMOSD).4 Although distinction between MS and NMOSD was facilitated by AQP4-ab testing, there is a substantial overlap between clinical phenotypes of MS and NMOSD, which caused diagnostic uncertainty or misdiagnosis, in particular prior to the availability of AQP4-ab testing. Moreover, current AQP4-ab assays differ significantly with regard to sensitivity.1 Distinguishing MS and NMOSD is of high clinical relevance, since optimum treatments for the 2 diseases differ. Most treatments found to be beneficial in MS have been suggested to be ineffective or even to cause disease exacerbation in NMOSD.5
Previously, we have shown that natalizumab (NAT), a very effective therapy for relapsing MS, had detrimental effects in 5 patients who had been misdiagnosed with MS and were treated with NAT prior to establishment of the correct diagnosis of AQP4-ab–seropositive NMOSD.6 Although this study suggested treatment failure, it could not rule out that other patients with known or undetected NMOSD might benefit from NAT.7
In the current study, we investigated a large set of serum samples from NAT-treated patients with the diagnosis of MS (MSNAT) for the presence of AQP4-IgG to identify AQP4-ab–seropositive patients with NMOSD (NMONAT). We hypothesized that some AQP4-ab–seropositive NMONAT patients might have been misdiagnosed with MS and that NAT might turn out to be unable to control disease activity in those patients. In addition, we investigated immune mechanisms possibly underlying disease activity in NMONAT patients and compared serologic markers with control NMOSD patients not treated with NAT.
METHODS
Standard protocol approvals, registrations, and patient consents.
Ethical approval was obtained from the institutional ethics review boards of the Universities of Bochum (no. 4390-12) and Düsseldorf (nos. 3419 and 3738). Prospectively evaluated patients provided written informed consent. A waiver for retrospective analysis of serum samples and unblinding of NMONAT patients to get further clinical information from treating doctors was issued by the institutional ethics review board, since previous data suggested deleterious effects of NAT treatment in AQP4-ab–seropositive NMO patients.6,8
Patients.
We retrospectively evaluated blinded frozen serum samples from MSNAT patients stored in a serum depository at the St. Josef Hospital Bochum.9 The sera were initially sent for analysis of anti-NAT-abs with neutralizing activity. The study population consisted of a nationwide cohort of patients with relapsing-remitting MS treated at all levels of care. We screened all samples obtained between February 2007 and August 2009. In case of several samples per patient, only the first eligible sample was considered. Inclusion criteria were ≥6 infusions of NAT within at least 6 months, absence of anti-NAT-abs, availability of clinical data for calculation of the annualized relapse rate (ARR), and availability of sufficient sample volume. Samples seropositive for AQP4-IgG were unblinded, and new serum samples were requested from the treating physician.
The following clinical data were assessed: the ARR during NAT therapy (total number of attacks divided through the total NAT treatment time in years) in MSNAT and NMONAT patients; the ARR prior NAT, calculated as the number of attacks within the last 12 months before start of NAT; and the expanded disability status scale (EDSS) score in NMONAT patients during and after NAT treatment. The clinical course of NMONAT patients was further evaluated by a standardized questionnaire and telephone interview of treating neurologists. The latter included detailed data about immunotherapies, MRI presentation, relapses, and disability progression before, during, and after NAT treatment.
As control group for studies of AQP4-ab titers, a prospective cohort of AQP4-ab–seropositive NMOSD patients not treated with NAT was tested. That cohort was matched for age, sex, disease duration, and disease activity (acute disease vs remission) against the identified cohort of NMONAT patients. Patients with an apheresis therapy in the last 60 days were excluded. Active disease was defined as relapse ±60 days from the day of serum sampling. To rule out the influence of immunotherapies when testing antibodies and cytokines, we divided the control group into NMOSD without immunotherapy (NMOno IT) and NMOSD with immunotherapy (NMOIT).
Serological studies.
The presence of serum AQP4-IgG was evaluated by a standardized immunohistochemical cell-based assay (CBA) according to the manufacturer's protocol (AQP4-CBA; Euroimmun, Lübeck, Germany).10 Staining of each sample was evaluated with a fluorescence microscope (EuroStar; Euroimmun) at ×200 magnification and 1,000 ms exposure time in comparison with appropriate negative and positive controls. Positive and ambiguous samples were sent in a masked fashion for confirmation to 2 independent reference laboratories (Euroimmun; Molecular Neuroimmunology Group, University Hospital Heidelberg, Germany).
Serum AQP4-ab concentrations were evaluated by a human M23-AQP4–based ELISA according to the manufacturer's protocol (anti-AQP4-Ab ELISA version 2; RSR, Cardiff, UK). The cut-off for positive values was 3.0 U/mL. To evaluate the presence of AQP4-IgG reactive only with the M1 isoform of human AQP4, additionally, a second, M1-AQP4–based ELISA was used (anti-AQP4-ELISA version 1; RSR; cut-off 3.0 U/mL).11 Absorbance was read at 450 and 405 nm by an ELISA plate reader (Emax; Molecular Devices, Sunnyvale, CA).
Cytokines (interleukin [IL]-1β, IL-4, IL-6, IL-8, IL-10, IL-17, IL-21, and interferon [IFN]-γ) and the chemokine CXCL-10 were evaluated by a multiplex bead-based immunoassay (cytometric bead array) according to the manufacturer's protocol (BD Biosciences, Heidelberg, Germany). Samples were acquired on a flow cytometer, and data were analyzed using FCAP Array software v3 (both BD Biosciences, Franklin Lakes, NJ). IL-8 and CXCL-10 additionally were evaluated by ELISA according to the manufacturer's protocol (IL-8 Quantikine ELISA, CXCL-10 Quantikine ELISA; both R&D, Minneapolis, MN). For IL-8, serum was diluted 1:10, and for CXCL-10, serum was diluted 1:2. Absorbance was read at 650 and 450 nm. All samples were stored at −80°C under standard conditions and investigated simultaneously in the respective assay.
Statistical analysis.
ARR between groups was compared by the 2-sided Mann-Whitney U test and ARR prior/during/after NAT and all serologic tests by the Kruskal-Wallis test with Dunn correction for multiple comparison using Prism v6 (Graph Pad, San Diego, CA).
RESULTS
Description of study population.
We screened a serum depository, which contained 2,787 serum samples from 2,245 individual patients treated with NAT for suspected relapsing-remitting MS. A total of 1,183 samples from 1,183 patients fulfilled the inclusion criteria (figure 1). The clinical characteristics of included patients were median age 40 years, 71.9% female, median disease duration 8 years, median NAT infusions 9, median ARR prior to NAT 2, and median EDSS at the time of serum sampling 3.5. Prior to NAT, 143 patients (12.1%) had received an immunosuppressive therapy at least once and 920 patients (77.8%) an immunomodulatory therapy.
Figure 1. Flowchart of study population and assays.
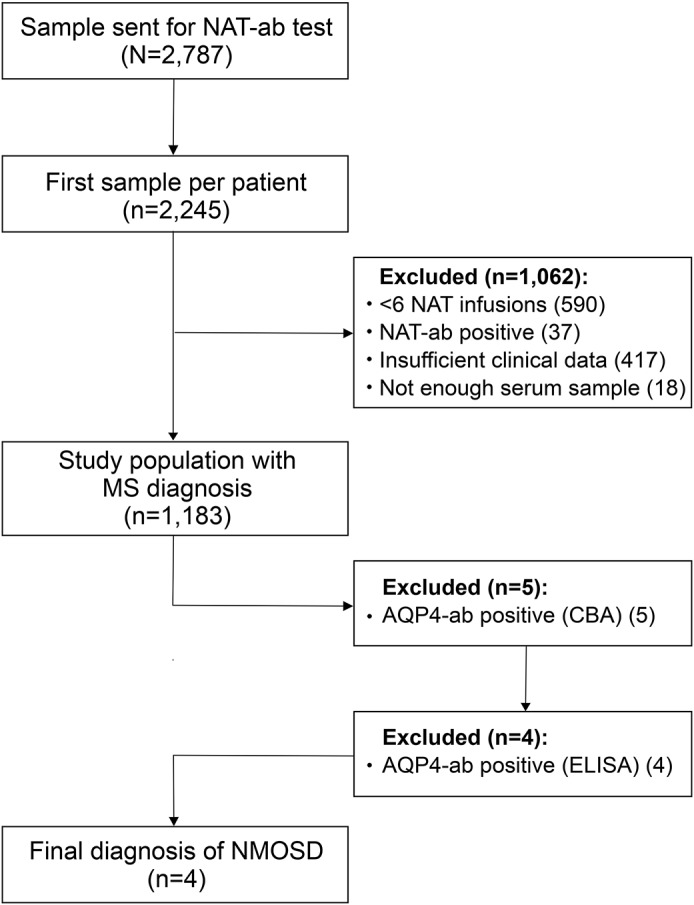
A total of 2,787 samples sent for analysis of antinatalizumab antibodies (NAT-ab) were screened. Samples from 1,183 patients with a diagnosis of MS were evaluated for AQP4-IgG by a cell-based assay (CBA). Positive samples were retested with further CBAs and ELISAs. NMOSD was diagnosed according to the 2015 criteria. AQP4-ab = aquaporin-4 antibody; NMOSD = neuromyelitis optica spectrum disorder.
Frequency of AQP4-abs in NAT-treated patients with suspected MS.
Of 1,183 patients, 5 (0.4%; 4 female) were positive for AQP4-IgG by a CBA (table 1). The results were confirmed in 2 independent laboratories, which use the same AQP4-transfected human embryonic kidney (HEK) 293 cells as test substrate. Signal strength at standard 1:10 starting dilution was intense in 3 and weak in 2 patients. The median AQP4-ab titer was 1:320 (range 1:10–1:1,000). Four of the samples identified by CBA were positive for AQP4-IgG by ELISA as well. Patient 1 with weak staining by CBA (titer 1:32) was negative for AQP4-IgG of the M23 isoform by ELISA. To rule out that the negative ELISA result was caused by the use of the M23-AQP4 isoform in the AQP4-ELISA, we tested that sample in addition in a second ELISA which uses human M1-AQP4 as antigenic substrate; however, the sample was negative as well. A follow-up sample from this patient taken 4.7 years after the first sample was again weakly positive in the CBA (titer 1:10) but negative in the 2 ELISAs. Given the repeatedly negative M1- and M23-ELISA results and the fact that the patient had clinical symptoms compatible with secondary-progressive MS rather than NMOSD (see below), patient 1 was finally classified as AQP4-IgG–seronegative and excluded from further analysis. Follow-up samples were also available from patient 4. This patient was positive by CBA and ELISA both in a sample taken 10 months after the start of NAT and in a sample obtained 4.9 years later during fingolimod therapy. For patient 2, an earlier sample taken after 5 infusions of NAT existed in the serum depository. Of interest, it was negative by CBA, but the sample initially used for the study taken 7 months later after 14 infusions of NAT was strongly positive by both CBA and ELISA.
Table 1.
Results of AQP4-ab testing

Disease course and diagnosis of AQP4-ab–seropositive patients treated with NAT.
Epidemiological and clinical features of the 5 AQP4-ab–seropositive patients are summarized in table 2. Median age was 39.7 years and median disease duration was 8.1 years. Four patients had previously received immunomodulatory therapies, 2 additionally immunosuppressants. Four patients were seropositive for JC virus (JCV)-abs. At the time of serum sampling, the median number of NAT infusions was 12. In total, patients were treated with NAT for median 2.8 years (range 1.0–7.3 years). Reasons for NAT withdrawal were diagnosis of NMOSD in 2 patients (after 1.0 and 2.2 years, respectively), seropositivity for JCV-abs in 2 patients (after 2.8 and 4.2 years, respectively), and clinical deterioration in 1 patient (after 7.3 years).
Table 2.
Characteristics of aquaporin-4 antibody–seropositive NAT-treated patients

The diagnosis of NMOSD had to be rejected on clinical grounds in 1 female patient (1; AQP4-IgG positive in CBA and negative in ELISA). She had a history of severe and incompletely remitting attacks with double vision, sensory defects, and paresis of the left leg, but no episodes of optic neuritis. Brain MRI was always typical for MS with supratentorial and infratentorial lesions. Unfortunately, no spinal MRI was performed throughout the clinical course. During therapy with IFN-beta-1b and glatiramer acetate 2–3 relapses per year occurred with relapse-associated disability up to EDSS 7.5. After 2 infusions of mitoxantrone, she was switched to NAT, which was given for a total of 7.3 years. During this time, 1 relapse with monoparesis of the left arm occurred. After some years of NAT treatment, walking gradually deteriorated, she had to use a walker, and later became wheelchair-bound, which is why NAT was suspended. Subsequent therapies with dimethyl fumarate and rituximab failed to control progression. The treating neurologist was convinced that this patient suffers from secondary-progressive MS.
Two female AQP4-ab–seropositive patients (3 and 5) had typical NMO according to the 2006 Wingerchuk criteria;3 both patients also met the 2015 Wingerchuk criteria.4 Clinicoradiological features included recurring episodes of myelitis and optic neuritis as well as longitudinally extensive lesions on spinal cord MRI. Both patients had ongoing disease activity (ARR 1.0 and 2.27, respectively) and a worsening of the EDSS (0.5 and 1.5, respectively) during NAT treatment. After the failure of NAT, both patients were switched to recommended NMOSD therapies (mitoxantrone and rituximab, respectively). In both patients, the ARR decreased and the EDSS improved by 1.0 until the end of follow-up.
Two further AQP4-ab–seropositive patients did not meet the 2006 criteria for NMO, but the revised 2015 criteria for NMOSD when treated with NAT. Patient 4 (male) had an initial episode of optic neuritis and 3 attacks of myelitis. The initial MRI showed several short spinal cord lesions and, in addition, 2 supratentorial brain lesions adjacent to the lateral ventricle, all compatible with the diagnosis of MS (figure e-1 at Neurology.org/nn). He had 2 relapses during 4.2 years of NAT therapy and an increase of short spinal lesions. After switching to fingolimod, he had 3 further relapses (1 optic neuritis and 2 episodes of myelitis) and subsequently, still under the assumption of MS, received alemtuzumab. Four and 5 months later, 2 severe relapses with longitudinally extensive transverse myelitis occurred, which left the patient wheelchair-bound and finally established the diagnosis of NMOSD.
Patient 2 (female) had been initially diagnosed with relapsing-remitting MS because of recurring episodes of sensory disturbances and double vision. IFN-beta-1b was ineffective. She later had 2 episodes of myelitis, but no optic neuritis. The only available MRI showed short spinal cord lesions and multiple supratentorial brain lesions. She had 1 relapse while on NAT therapy with a stable EDSS. NAT was discontinued due to JCV-ab seropositivity after 2.8 years. Subsequently, she was treated with glatiramer acetate and had 2 mild relapses with stable EDSS and 2 new small spinal lesions at the last MRI follow-up. After switching to dimethyl fumarate, she was relapse and progression free for the remaining follow-up of 2.8 years.
ARR in NAT-treated patients with NMOSD.
To evaluate the clinical efficacy of NAT in NMOSD, we compared the ARR before, during, and after NAT therapy. The ARR of the 4 identified AQP4-ab–seropositive NMOSD patients decreased from median 2.0 (range 2.0–3.0) before, to 0.735 (0.36–2.27) during, and 0.55 (0.30–1.0) after NAT therapy (the latter including time without immunotherapy and on immunotherapies recommended for NMOSD or not). The ARR clearly decreased in 2 patients with an early diagnosis of NMOSD and change from NAT therapy to mitoxantrone and rituximab, respectively (patients 3 and 5), whereas it did not change in the 2 patients switched to immunomodulatory therapies used for MS (patients 2 and 4) (table 2).
Next, we examined all patients from our NAT-treated cohort stratified for the AQP4-ab status. We found a significant difference between AQP4-ab–seropositive NMOSD (median ARR 0.7, range 0.2–2.2) and AQP4-IgG–seronegative MS (median ARR 0, range 0–6.6; p = 0.0182) (figure 2). Adding the 5 NAT-treated AQP4-ab–seropositive NMO patients previously identified6 to the analysis, the ARR in NMOSD still was significantly worse (AQP4-ab seropositive median ARR 2.4, range 0.2–6.0; p < 0.0001 vs AQP4-ab negative).
Figure 2. Annualized relapse rate during natalizumab therapy.
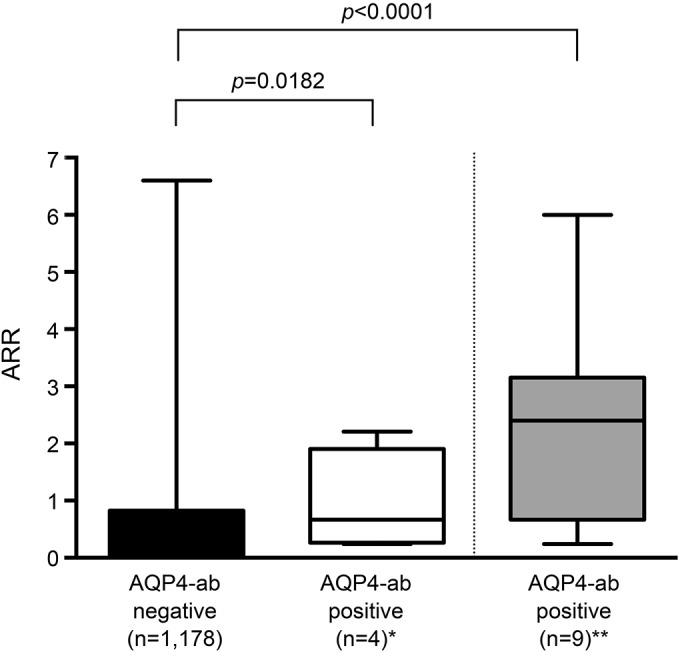
AQP4-ab–seronegative patients with relapsing-remitting MS were compared with AQP4-ab–seropositive patients with confirmed NMOSD* and additionally 5 previously identified** NAT-treated patients with NMO.6 Whisker box plots show minimum, Q1, median, Q3, and maximum values. Mann-Whitney U test. AQP4-ab = aquaporin-4 antibody; ARR = annualized relapse rate; NAT = natalizumab; NMOSD = neuromyelitis optica spectrum disorder.
Serological findings in AQP4-ab–seropositive NMOSD patients treated with NAT.
Since AQP4-IgG are pathogenic, we investigated the AQP4-ab titer in the 4 NMONAT patients identified in the current study and 5 previously identified NMONAT patients.6 As a comparator, we collected serum samples from patients with NMOSD who were not treated with NAT and divided this group into patients with immunotherapy (NMOIT, n = 13) and untreated patients (NMOno IT, n = 9) (table e-1). We found significantly higher AQP4-ab titers in NMONAT (median 479.1 U/mL, range 10.5–799.2 U/mL) vs NMOIT (median 30.7 U/mL, range 0–510.1 U/mL; p = 0.0058), but no difference to NMOno IT (median 124.71 U/mL, range 0–799.2 U/mL) (figure 3A).
Figure 3. Titers of aquaporin-4 IgG and of proinflammatory cytokines in aquaporin-4 antibody–seropositive neuromyelitis optica spectrum disorder patients.

Serological studies in NAT-treated patients with NMOSD (NMONAT, n = 9) and control NMOSD patients treated with immunotherapies (NMOIT, n = 13) or not (NMOno IT, n = 9). (A) Titer of AQP4-IgG measured by ELISA, serum was diluted 1:10. Some samples were saturated at the upper detection limit of 800 U/mL. Shown are individual patients and the median of each group. (B) Titer of IL-8 measured by ELISA, serum was diluted 1:10. (C–H) Other cytokines measured by cytometric bead array, serum was undiluted. Shown are individual patients and median. The Kruskal-Wallis test was used for statistical analysis. *Patients treated with tocilizumab were excluded from IL-6 analysis. AQP4 = aquaporin-4; IFN = interferon; IL = interleukin; NAT = natalizumab; NMOSD = neuromyelitis optica spectrum disorder.
Using the same experimental groups, we assessed whether alterations in proinflammatory cytokines and the chemokine CXCL-10 might contribute to increased disease activity in NMONAT patients (figure 3, B–H). We found significantly higher levels of serum IL-8 (median 2.8 vs 0 ng/mL, p = 0.0001), IFN-γ (median 5.9 vs 1.7 pg/mL, p = 0.0019), and IL-1β (median 7.5 vs 2.8 pg/mL, p = 0.0002), and significantly lower levels of CXCL-10 (median 32.8 vs 160.6 pg/mL, p = 0.0091) in NMONAT vs NMOIT patients, whereas no differences were found between NMONAT and NMOno IT patients. Levels of IL-4, IL-6, IL-10, IL-17, and IL-21 were generally very low and did not differ significantly (figure 3 and not shown).
DISCUSSION
We examined AQP4-ab in patients treated with NAT for suspected MS and determined their frequency and relevance for the clinical course. We found 4 of 1,183 patients (0.33%) to be unequivocally AQP4-ab seropositive. Before being identified by our study, 2 of the 4 AQP4-ab–seropositive patients already had been diagnosed with NMOSD and fulfilled classic diagnostic criteria,3 leaving 2 unrecognized AQP4-ab–seropositive patients. Both had the final diagnosis of NMOSD according to the 2015 criteria,4 which illustrates that NMOSD is only rarely misdiagnosed as MS nowadays when the clinical course is followed over several years.
The AQP4-ab status of the 5 patients who were seropositive in the screening CBA was analyzed in several additional, methodologically independent assays to reduce the risk of center or assay bias.11,12 Only 1 patient was classified as probably false positive for AQP4-IgG (0.08%). Previous reports suggested that NAT-containing serum might cause a false-positive signal in the AQP4-ab CBA, owing to cross reactivity of NAT with AQP4-expressing HEK cells used in this assay.13,14 Our results suggest that this might occur—if at all—only very rarely. However, since AQP4-abs are mainly IgG1 and NAT is an IgG4, it was proposed to use anti-human-IgG1 as a secondary antibody in the CBA.13 Another solution in NAT-treated patients would be to add an independent confirmation method, e.g., an AQP4-ab ELISA.
It is unknown whether all 1,178 AQP4-ab–seronegative patients were correctly diagnosed with MS prior to NAT treatment. Our study revealed a wrong diagnosis in at least 4 patients who were AQP4-ab seropositive. Given that the sensitivity of all immunoassays is limited for reasons inherent to the methods used, that AQP4-ab serum levels vary substantially over time,15 and that 10%–20% of patients with NMO never develop AQP4-IgG, we cannot rule out that more patients were falsely diagnosed with MS. Therefore, it is formally not possible to calculate assay specificity. However, given the low prevalence of NMOSD in Europe,16 it is highly likely that the majority of patients included in the MSNAT cohort had indeed MS. This would correspond to a very high specificity (1 false-positive result among >1,000 samples tested, or ≥99.9%) of the CBA used here. Our results are in line with a previous report, showing a false-positive rate of a fixed transfected CBA of 0.1% in a Northern Californian MS population.17 The high specificity of AQP4-abs for NMOSD was also confirmed in a recent meta-analysis.18
The clinical response to NAT therapy was dependent on the AQP4-ab status. As expected, the majority of patients with MS had no relapses, whereas none of the NMONAT patients was relapse free. Taking also the previous literature into account, it is apparent that NAT is not beneficial for most patients with NMOSD.6–8,19–24 A review of 19 patients published so far revealed that the majority of patients with NMOSD treated with NAT for at least 3 months deteriorated (table e-2). Moreover, early appearance of severe attacks with atypical cerebral manifestation was noted in a subset of NMONAT patients.6,21,22,24
As already suggested,6 we now could verify that AQP4-ab titers are indeed higher in NMONAT patients as compared to NMOIT patients. We also found an increase of the AQP4-ab titer in 1 patient during NAT therapy. Although it might be hypothesized that this elevated AQP4-ab serum level contributes to relapses in NMONAT patients,15 the underlying mechanisms remain speculative. NAT results in a rise in proinflammatory cytokines and distinct T- and B-cell subsets in the periphery.25–27 These changes of the peripheral immune milieu might promote high AQP4-ab titers and subsequent disease activity. Indeed, we found a higher level of proinflammatory cytokines, particularly IL-8, IFN-γ, and IL-1β, in the sera of NMONAT patients in comparison with NMOIT. An increase of T-helper (Th)17-, Th1- and Th2-related cytokines and chemokines in the serum and particularly the CNS of patients with NMOSD has been reported.28 Particularly, IL-8 and IL-1β were shown to be increased in the CSF of patients with NMOSD.29,30 CSF IL-8 correlates with disease activity, EDSS, and length of spinal cord lesions in NMOSD.28,31
The AQP4-ab and cytokine levels in NMONAT patients did not differ significantly in comparison with untreated patients with NMOSD. This indicates that NAT, unlike immunotherapies recommended for NMOSD, is not able to reduce proinflammatory cytokines which might be relevant for the control of disease activity.
The downregulation of serum CXCL-10 in NMONAT patients seems counterintuitive, since CXCL-10 might contribute to the recruitment of CXCR3+ inflammatory cells to the CNS in NMOSD.32,33 CXCL-10 is elevated in the CSF but not serum of patients with NMO, MS, and other neuroinflammatory diseases.31,34,35 It was shown that CXCL-10 levels in the CSF (and to a lesser degree in the serum) were reduced after 1 year of NAT treatment for MS.36
Although NAT reduces VLA-4 expression on T-cells, B-cells, and monocytes/macrophages and thereby prevents migration of these cells to the CNS,37 not all lymphocyte subsets are equally affected. Studies in the animal model experimental autoimmune encephalomyelitis (EAE) revealed that Th17 but not Th1 cells can invade the CNS in the absence of VLA-4 expression and cause primarily supraspinal infiltrates.38 It was proposed that differential integrin expression promotes the targeting of different Th-cell subsets to distinct niches of the CNS.38 In EAE, Th17 cells migrate to the CNS via the choroid plexus through specific interaction of their chemokine receptor CCR6 with CCL20.39 It is unknown whether human Th17 cells use the same route and mode of action for migration to the CNS, but of interest, NAT treatment increases peripheral Th17 responses27,40 and CCR6 expression of CD4+ T-cells,26,40 which potentially could worsen Th17-mediated diseases such as NMOSD.
Limitations of our study include the retrospective evaluation of the clinical course and the low amount of serum available for serologic studies, preventing analysis of further inflammatory markers.
We identified only few cases of AQP4-ab–seropositive NMOSD among patients with suspected MS, suggesting that the diagnostic criteria in daily practice are sufficient to discriminate between MS and NMOSD. Our data do not support routine assessment of AQP4-abs in typical patients with MS. However, in cases with recurrent optic neuritis or myelitis, opticospinal lesions on MRI, and particularly when persistent disease activity occurs despite optimal MS therapy, AQP4-ab testing should be performed. As shown in previous studies, NAT had limited efficacy in NMOSD, probably due to peripheral inflammatory mechanisms induced by NAT treatment.
ACKNOWLEDGMENT
The authors thank all patients and the treating neurologists, particularly Alexander Sieke, for their contributions and Christa Kraushaar-Sczesni, Marion Frickmann, Britta Haas, Anna Eschlbeck, and the Nikon Imaging Center at the University of Heidelberg for expert technical assistance.
GLOSSARY
- AQP4-ab
aquaporin-4 antibody
- ARR
annualized relapse rate
- CBA
cell-based assay
- EAE
experimental autoimmune encephalomyelitis
- EDSS
expanded disability status scale
- HEK
human embryonic kidney
- JCV
JC virus
- IL
interleukin
- IFN
interferon
- NAT
natalizumab
- NMOSD
neuromyelitis optica spectrum disorder
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
A.G. analyzed and interpreted data and drafted the manuscript. A.-K.T., S.H., and S.J. analyzed and interpreted data and revised the manuscript. M.R., O.A., A.B., and B.W. contributed patients and data and revised the manuscript. R.G. supervised the study and revised the manuscript. I.K. designed the study, obtained funding, analyzed and interpreted data, did statistical analysis, and drafted the manuscript. All authors were involved in revising the manuscript for intellectual content.
STUDY FUNDING
Part of the study was funded by an unrestricted research grant of Biogen Germany to I.K. The work of S.J. was supported by research grants from Merck Serono and from the Dietmar-Hopp-Stiftung to the Department of Neurology, University of Heidelberg (Prof. Brigitte Wildemann).
DISCLOSURE
A. Gahlen received travel funding from Sanofi Genzyme. A.-K. Trampe and S. Haupeltshofer report no disclosures. M. Ringelstein received travel funding from Novartis, Bayer Healthcare, Biogen Idec, Genzyme, Teva, and Merz. O. Aktas received travel funding and/or speaker honoraria from Bayer, Novartis, Biogen, Merck Serono, Teva, Genzyme, Roche, and Almirall; is an academic editor for PLoS ONE; consulted for Novartis; and received research support from Novartis, Bayer, Biogen, German Ministry of Science, German Research Foundation, Heinrich and Erna Schaufler-Foundation, and Walter and Ilse Rouse-Foundation. A. Berthele received travel funding and/or speaker honoraria from Bayer Healthcare, Biogen, Merck Serono, Mylan, Novartis, Roche, and Teva and received research support from Bayer Healthcare. B. Wildemann served on the scientific advisory board for Novartis, Merck Serono, and Sanofi Genzyme; received travel funding and/or speaker honoraria from Biogen, Merck Serono, Novartis, Teva, and Sanofi Genzyme; and received research support from Bundesministerium für Forschung und Technologie, Dietmar Hopp Stiftung, Klaus Tschira Stiftung, Biogen, Merck Serono, Novartis, Teva, and Sanofi Genzyme. R. Gold served on the scientific advisory board for Teva Laquinimod DSMB; received speaker honoraria from Biogen, Genzyme, Teva, Merck Serono, Bayer Schering, Ozgene, and Novartis; is on the editorial board for Therapeutic Advances in Neurologicals Disorders, Aktuelle Neurologie, and Experimental Neurology. S. Jarius reports no disclosures. I. Kleiter received travel funding and/or speaker honoraria from Bayer Healthcare, Roche, and Merck; consulted for Bayer Healthcare, Chugai, Roche, and Shire; and received research support from Biogen, Chugai, Diamed, and Affectis. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Jarius S, Wildemann B, Paul F. Neuromyelitis optica: clinical features, immunopathogenesis and treatment. Clin Exp Immunol 2014;176:149–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zekeridou A, Lennon VA. Aquaporin-4 autoimmunity. Neurol Neuroimmunol Neuroinflamm 2015;2:e110 doi: 10.1212/NXI.0000000000000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485–1489. [DOI] [PubMed] [Google Scholar]
- 4.Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015;85:177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kleiter I, Gold R. Present and future therapies in neuromyelitis optica spectrum disorders. Neurotherapeutics 2016;13:70–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kleiter I, Hellwig K, Berthele A, et al. Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol 2012;69:239–245. [DOI] [PubMed] [Google Scholar]
- 7.Govindarajan R, Salgado E. Is it too early to predict the failure of natalizumab in NMO? Arch Neurol 2012;69:1085; author reply 1086. [DOI] [PubMed] [Google Scholar]
- 8.Barnett MH, Prineas JW, Buckland ME, Parratt JD, Pollard JD. Massive astrocyte destruction in neuromyelitis optica despite natalizumab therapy. Mult Scler 2012;18:108–112. [DOI] [PubMed] [Google Scholar]
- 9.Trampe AK, Hemmelmann C, Stroet A, et al. Anti-JC virus antibodies in a large German natalizumab-treated multiple sclerosis cohort. Neurology 2012;78:1736–1742. [DOI] [PubMed] [Google Scholar]
- 10.Jarius S, Probst C, Borowski K, et al. Standardized method for the detection of antibodies to aquaporin-4 based on a highly sensitive immunofluorescence assay employing recombinant target antigen. J Neurol Sci 2010;291:52–56. [DOI] [PubMed] [Google Scholar]
- 11.Jarius S, Franciotta D, Paul F, et al. Testing for antibodies to human aquaporin-4 by ELISA: sensitivity, specificity, and direct comparison with immunohistochemistry. J Neurol Sci 2012;320:32–37. [DOI] [PubMed] [Google Scholar]
- 12.Granieri L, Marnetto F, Valentino P, et al. Evaluation of a multiparametric immunofluorescence assay for standardization of neuromyelitis optica serology. PLoS One 2012;7:e38896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen M, De Seze J, Marignier R, Lebrun C. False positivity of anti aquaporin-4 antibodies in natalizumab-treated patients. Mult Scler 2016;22:1231–1234. [DOI] [PubMed] [Google Scholar]
- 14.Sanchez Gomar I, Diaz Sanchez M, Ucles Sanchez AJ, et al. An immunoassay that distinguishes real neuromyelitis optica signals from a labeling detected in patients receiving natalizumab. BMC Neurol 2014;14:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jarius S, Aboul-Enein F, Waters P, et al. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain 2008;131:3072–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pandit L, Asgari N, Apiwattanakul M, et al. Demographic and clinical features of neuromyelitis optica: a review. Mult Scler 2015;21:845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pittock SJ, Lennon VA, Bakshi N, et al. Seroprevalence of aquaporin-4-IgG in a northern California population representative cohort of multiple sclerosis. JAMA Neurol 2014;71:1433–1436. [DOI] [PubMed] [Google Scholar]
- 18.Jarius S, Wildemann B. Aquaporin-4 antibodies (NMO-IgG) as a serological marker of neuromyelitis optica: a critical review of the literature. Brain Pathol 2013;23:661–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kleiter I, Hellwig K, Berthele A, et al. Is it too early to predict the failure of natalizumab in NMO?-reply. Arch Neurol 2012;69:1085–1086. [DOI] [PubMed] [Google Scholar]
- 20.Jacob A, Hutchinson M, Elsone L, et al. Does natalizumab therapy worsen neuromyelitis optica? Neurology 2012;79:1065–1066. [DOI] [PubMed] [Google Scholar]
- 21.Jurynczyk M, Zaleski K, Selmaj K. Natalizumab and the development of extensive brain lesions in neuromyelitis optica. J Neurol 2013;260:1919–1921. [DOI] [PubMed] [Google Scholar]
- 22.Kitley J, Evangelou N, Kuker W, Jacob A, Leite MI, Palace J. Catastrophic brain relapse in seronegative NMO after a single dose of natalizumab. J Neurol Sci 2014;339:223–225. [DOI] [PubMed] [Google Scholar]
- 23.Lee DH, Laemmer AB, Waschbisch A, et al. Neuromyelitis optica presenting with relapses under treatment with natalizumab: a case report. J Med Case Rep 2014;8:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kornberg MD, Newsome SD. Unmasking and provoking severe disease activity in a patient with NMO spectrum disorder. Neurol Neuroimmunol Neuroinflamm 2015;2:e66 doi: 10.1212/NXI.0000000000000066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krumbholz M, Meinl I, Kumpfel T, Hohlfeld R, Meinl E. Natalizumab disproportionately increases circulating pre-B and B cells in multiple sclerosis. Neurology 2008;71:1350–1354. [DOI] [PubMed] [Google Scholar]
- 26.Kivisakk P, Healy BC, Viglietta V, et al. Natalizumab treatment is associated with peripheral sequestration of proinflammatory T cells. Neurology 2009;72:1922–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bornsen L, Christensen JR, Ratzer R, et al. Effect of natalizumab on circulating CD4+ T-cells in multiple sclerosis. PLoS One 2012;7:e47578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uzawa A, Mori M, Kuwabara S. Cytokines and chemokines in neuromyelitis optica: pathogenetic and therapeutic implications. Brain Pathol 2014;24:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yanagawa K, Kawachi I, Toyoshima Y, et al. Pathologic and immunologic profiles of a limited form of neuromyelitis optica with myelitis. Neurology 2009;73:1628–1637. [DOI] [PubMed] [Google Scholar]
- 30.Uzawa A, Mori M, Arai K, et al. Cytokine and chemokine profiles in neuromyelitis optica: significance of interleukin-6. Mult Scler 2010;16:1443–1452. [DOI] [PubMed] [Google Scholar]
- 31.Matsushita T, Tateishi T, Isobe N, et al. Characteristic cerebrospinal fluid cytokine/chemokine profiles in neuromyelitis optica, relapsing remitting or primary progressive multiple sclerosis. PLoS One 2013;8:e61835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chihara N, Aranami T, Oki S, et al. Plasmablasts as migratory IgG-producing cells in the pathogenesis of neuromyelitis optica. PLoS One 2013;8:e83036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shimizu F, Nishihara H, Sano Y, et al. Markedly increased IP-10 production by blood-brain barrier in neuromyelitis optica. PLoS One 2015;10:e0122000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Narikawa K, Misu T, Fujihara K, Nakashima I, Sato S, Itoyama Y. CSF chemokine levels in relapsing neuromyelitis optica and multiple sclerosis. J Neuroimmunol 2004;149:182–186. [DOI] [PubMed] [Google Scholar]
- 35.Ando H, Sato T, Tomaru U, et al. Positive feedback loop via astrocytes causes chronic inflammation in virus-associated myelopathy. Brain 2013;136:2876–2887. [DOI] [PubMed] [Google Scholar]
- 36.Mellergard J, Edstrom M, Vrethem M, Ernerudh J, Dahle C. Natalizumab treatment in multiple sclerosis: marked decline of chemokines and cytokines in cerebrospinal fluid. Mult Scler 2010;16:208–217. [DOI] [PubMed] [Google Scholar]
- 37.Wipfler P, Oppermann K, Pilz G, et al. Adhesion molecules are promising candidates to establish surrogate markers for natalizumab treatment. Mult Scler 2011;17:16–23. [DOI] [PubMed] [Google Scholar]
- 38.Rothhammer V, Heink S, Petermann F, et al. Th17 lymphocytes traffic to the central nervous system independently of alpha4 integrin expression during EAE. J Exp Med 2011;208:2465–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reboldi A, Coisne C, Baumjohann D, et al. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol 2009;10:514–523. [DOI] [PubMed] [Google Scholar]
- 40.Benkert TF, Dietz L, Hartmann EM, et al. Natalizumab exerts direct signaling capacity and supports a pro-inflammatory phenotype in some patients with multiple sclerosis. PLoS One 2012;7:e52208. [DOI] [PMC free article] [PubMed] [Google Scholar]